原子吸收光谱法的定量分析方法和测定条件的选择
原子吸收光谱分析-下

体元素不同可能带来影响。
(2)标准溶液浓度应使 A ~ C 在直线的范围内, C
不能太大,一般控制A在0.2~0.8之间。
(3) 测定过程中应保持测定条件不变。 • 标准曲线法简便、快速,适用于组分比较简单的样 品,适用于大批量的样品分析。但样品的情况不清 或很复杂时分析误差较大,可用其他方法定量。
检测限 (Detection limit, DL)
• 检出限不仅与灵敏度有关,而且还考虑 到仪器噪声!因而检测限比灵敏度具有更 明确的意义,更能反映仪器的性能。只有 同时具有高灵敏度和高稳定性时,才有低 的检出限。
——测定条件的选择 • 分析方法的精密度和准确度除了与仪器的性能有 关外,还与测定条件有关,注意选择: 1、试样取量及处理
用有机溶剂
(二)化学干扰及其抑制
指待测元素与其它组分之间的化学作用所引起的干扰效应 ,主要 影响到待测元素的原子化效率,是选择性干扰,为主要干扰源
1. 化学干扰的类型
(1)待测元素与其共存物质作用生成难挥发的化合物,致使参 与吸收的基态原子减少。 a、铝、硅、硼、钛、铍在火焰中易生成难熔化合物 b、硫酸盐、磷酸盐与钙生成难挥发物。 (2)待测原子发生电离反应,生成离子,不产生共振吸收,总 吸收强度减弱,电离电位≤6eV的元素易发生电离,火焰温度越高 ,干扰越严重,(如碱及碱土元素)。
• 氘灯是连续光谱( 190-360nm ),它和空心阴极灯的锐线
光源通过切光器交替照射在原子化器上。 氘灯的能量被背景和被测元素吸收,但被测元素是线吸收,
它占整个连续光谱的吸收信号很小,可以忽略。因此可以
认为,氘灯测得的就是背景吸光度。 A氘=A背 • 空心阴极灯测得的是被测元素吸光度和背景吸光度,
例如:钙电离,在溶液中加入大量易电离的 钾或铯,有大量电子存在,抑制钙的电离,提高 测定灵敏度。 K ---- K+ + e
原子吸收光谱的定量依据

原子吸收光谱的定量依据
原子吸收光谱是一种分析化学方法,它基于原子对特定波长的光的吸收来定量分析样品中的元素。
该方法的基本原理是,当原子处于基态时,它们可以通过吸收电磁辐射而被激发到激发态,这种激发态是非常短暂的,常常只有几微秒的寿命。
在激发态中,原子会吸收特定波长的光,并且其吸收量与元素浓度成正比。
因此,通过测量所吸收光的强度,可以计算出样品中的元素浓度。
原子吸收光谱定量分析的精度和准确性取决于很多因素,其中最重要的是所选用的谱线。
对于每个元素,都有一些谱线可以用来进行定量分析。
这些谱线的选择应该基于它们的灵敏度、准确性、稳定性和实验条件等因素。
一般来说,具有较高灵敏度和稳定性的谱线应该被优先选择。
除了谱线的选择外,还有一些其他因素也会影响到原子吸收光谱的精度和准确性。
这些因素包括样品制备、仪器校准和环境条件等。
为了获得准确的分析结果,必须严格控制这些因素。
总之,原子吸收光谱是一种非常有用的分析化学方法,它可以用于定量分析广泛的元素。
对于这种方法的应用,我们需要选择适当的谱线,并严格控制所有影响分析结果的因素。
- 1 -。
第四章原子吸收光谱分析3
景吸收的总吸光度AT,在分析线邻近选一条非共振线, 非共振线不会产生共振吸收,此时测出的吸收为背景吸 收AB 。两次测量吸光度相减,所得吸光度值即为扣除背 景后的原子吸收吸光度值A。
AT = A + AB
A = AT - AB = k c
08:39:38
4.4.1 光谱干扰
光散射 ➢ 光散射是待测元素的吸收度增大,光散射出现在各个方
向,效果是入射光减弱,似乎被组分吸收了,因而使T 减小、A增大。 ➢ 消除:一般可用“调零”的方法消除。
08:39:38
背景干扰校正方法
背景干扰可采用仪器校正背景方法,有邻近非共振线、 连续光源、Zeeman效应等校正方法。
➢ 对于曲线斜率太小的测定,易引进较大的误差。
08:39:38
定量分析方法
(2)标准加入法
标准加入法有时只用单标准加入,即取两分相同量的被测
试液,其中一分加入一定量的标准溶液,稀释到相同体积后
测定吸光度。根据吸收定律,可得:
Ax = k Cx As = k(Cx + Cs )
Cx
Ax A0 Ax
本法适用于分析线附近背景吸收变化不大的情况, 否则准确度较差。
08:39:38
背景干扰校正方法
① 邻近非共振线校正法 参比谱线选择: ➢ 参比线与测量线很近(保证二者经过的背景一致) ➢ 待测物基态原子不吸收参比线。 ➢ 参比线是测量原子的非共振线或元素灯内惰性气体元素
的谱线。
08:39:38
1
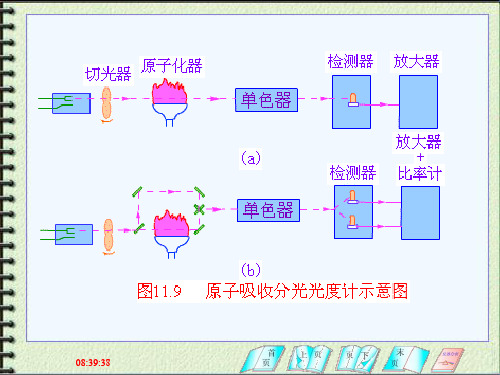
原子
光电池 光电倍增管 光敏晶体管 (2)放大器:将光电倍增管输出的较弱信号,经 电子线路进一步放大。 (3)对数变换器:光强度与吸光度之间的转换。 (4)显示、记录:新仪器配置:原子吸收计算机 工作站
原子吸收光谱定量分析方法

原子吸收定量分析方法一、定量分析方法(P145)⑴标准曲线法:配制一系列浓度不同的标准溶液,在相同测定条件下,测定标准系列溶液和待测试样溶液的吸光度,绘制A-c标准曲线,由待测溶液的吸光度值在标准曲线上得到其含量。
(2)标准加入法当试样组成复杂,待测元素含量很低时,应采用标准加入法进行定量分析。
取若干份体积相同的试液(cX),依次按比例加入不同量的待测物的标准溶液(cO):浓度依次为:cX,cX+cO,cX+2cO,cX+3cO,cX+4cO …分别测得吸光度为:AX ,A1 ,A2 ,A3 ,A4 …直线外推法:以对浓度做图得一直线,图中cX点即待测溶液浓度。
(3)稀释法:⑷内标法:在标准试样和被测试样中,分别加入内标元素,测定分析线和内标线的吸光度比,并以吸光度比与被测元素含量或浓度绘制工作曲线。
内标元素的选择:内标元素与被测元素在试样基体内及在原子化过程中具有相似的物理化学性质,样品中不存在,用色谱纯或者已知含量二、灵敏度和检出限(1)灵敏度1、定义:在一定浓度时,测定值(吸光度)的增量(△ A)与相应的待测元素浓度(或质量)的增量(△ c或A m)的比值(即分析校正曲线的斜率)PS:习惯上用特征浓度和特征质量表征灵敏度2、特征浓度定义:能产生1%吸收或产生0.0044吸光度时所对应的被测元素的质量浓度定义为元素的特征浓度3、特征质量定义:能产生1%吸收或产生0.0044吸光度时所对应的被测元素的质量定义为元素的特征质量。
(2)检出限定义:适当置信度下,能检测出的待测元素的最低浓度或最低质量。
用接近于空白的溶液,经若干次重复测定所得吸光度的标准偏差的3倍求得。
(3)测定条件的选择1.分析线的选择每种元素都有几条可供选择使用的吸收线。
一般选待测元素的共振线作为分析线,可以得到最好的灵敏度。
在测量高含量元素时,也可选次灵敏线。
2.单色器光谱通带的选择(调节狭缝宽度)光谱通带的选择以排除光谱干扰和具有一定透光强度为原则。
仪器分析 复习 重修 自学 预习5 原子吸收光谱分析法

原子吸收光谱分析法
原子吸收基本原理
第一节
一、共振线 二、基态原子数与原子化温度 三、定量基础
历史
原子吸收光谱法是一种基于待测基态原子对特征谱线的 吸收而建立的一种分析方法。这一方法的发展经历了3个发 展阶段:
原子吸收现象的发现
1802年Wollaston发现太阳光谱的暗线; 1859年Kirchhoff和 Bunson解释了暗线产生的原因;
试样雾滴在火焰中,经蒸发,干燥,离解(还原)等过 程产生大量基态原子。火焰原子化的方法就是使试样变成 原子蒸汽。 火焰温度的选择: (a)保证待测元素充分离解为基态原子的前提下,尽量 采用低温火焰;因为火焰温度越高,产生的热激发态原子 越多,则基态原子数量减少;但太低温就会使盐类无法解
离,降低灵敏度。
I
Ve
I 0V e KV L dv;当发射线宽《吸收线宽时,可以认为
0 Ve
KV 是常数,相当峰值吸收系数K 0:I e K 0 L 于是A lg 1 e
K0L
I
0
0V
dv
0.4343 K 0 L
K0=?
吸收线轮廓仅取决于多普勒变宽时 1 KV dv 2 ln 2 K 0v,结合积分吸收式 KV dv的值 2 ln 2 e 2 解得:K 0 fN 0 v mc
太阳光
暗 线
第一激发态
E
热能
基态
E = h = h
C
发现钠蒸汽发出的光线通过温度比较低的钠蒸汽,会引起 钠光的吸收,并且钠发射线和暗线在光谱中位置相同,由此 判断太阳连续光谱中的暗线是太阳外层中的钠原子对太阳光 谱中钠辐射吸收的结果
原子吸收光谱基本原理:
35 原子吸收光谱分析的测量条件和测量灵敏度

2、灵敏度和检出限(四) 、灵敏度和检出限(
光谱分析中,往往也指明方法的测定限 指明方法的测定限, 光谱分析中 , 往往也 指明方法的测定限, 又称测定下限, 又称测定下限,它是指定量分析方法能够测定 到的最低量或最低浓度。 到的最低量或最低浓度。它与检出限有不同的 定义。一般认为,检出限是指定性检测 指定性检测, 定义。一般认为, 检出限是 指定性检测,即断 定样品中确实存在着高于空白值的待测物质。 定样品中确实存在着高于空白值的待测物质。 高于空白值的待测物质 而测定限实际上是可以测定的极限。 测定限实际上是可以测定的极限。
2、灵敏度和检出限 、
灵敏度。1975年 a) 灵敏度。1975年,国际纯粹和应用化学联合会 校正曲线的斜率S IUPAC)建议把校正曲线的斜率 称为灵敏度, (IUPAC)建议把校正曲线的斜率S称为灵敏度, S=dA/dc。 即 S=dA/dc 。 它表示当被测元素浓度或含量改 变一个单位时,吸光度的变化量。 越大, 变一个单位时,吸光度的变化量。S 越大,灵 敏度越高, 敏度越高,必须注意的是不同浓度区域校正曲 线有不同的斜率,因此灵敏度必须指明浓度或 线有不同的斜率,因此灵敏度必须指明浓度或 含量范围。 含量范围。
3.6 原子吸收光谱分析的 特点和应用
原子吸收光谱分析的特点和应用
原子吸收分析法与其他一些仪器分析法( 原子吸收分析法与其他一些仪器分析法 ( 如 经典发射光谱、 极谱、 分光光度法) 比较, 经典发射光谱 、 极谱 、 分光光度法 ) 比较 , 有一 些突出的特点: 些突出的特点: a)选择性高。大多数情况下共存元素对原子吸 a)选择性高。 选择性高 收分析不产生干扰, 收分析不产生干扰 , 所以一般不需要分离共 存元素。 存元素。 b)灵敏度高 一般说, 灵敏度高。 b)灵敏度高 。一般说 ,原子吸收的灵敏度比较 高 。 火焰原子化法灵敏度在mg/L级以上,少 火焰原子化法灵敏度在mg/L级以上, mg/L 级以上 数达到ug/L ug/L级 数达到 ug/L 级 。 无火焰原子化有更高的灵敏 度。
第四章 原子吸收光谱法测定条件的选择

第四章原子吸收光谱法测定条件的选择1.空心阴极灯测量条件的选择1.1 吸收线选择为获得较高的灵敏度、稳定性、宽的线性范围和无干扰测定 , 须选择合适的吸收线。
选择谱线的一般原则:a)灵敏度一般选择最灵敏的共振吸收线, 测定高含量元素时 , 可选用次灵敏线。
例如在测定高浓度钠时,不选择最灵敏线(589.0nm),而选择次灵敏线(330.2 nm)。
具体可参考Z-5000分析软件中提供各元素的谱线信息。
b)干扰谱线干扰当分析线附近有其他非吸收线存在时 , 将使灵敏度降低和工作曲线弯曲 , 应当尽量避免干扰。
例如 ,Ni230.Om 附近有 Ni231.98nm 、 Ni232.14 nm 、 Ni231.6nm 非吸收线干扰,因此,可选择灵敏度稍低的吸收线(341.48 nm)作为分析线。
而测定铷时,为了消除钾、钠的电离干扰,可用798.4nm代替780.0nm。
c)仪器条件大多数原子吸收分光光度计的波长范围是190 900 nm,并且一般采用光电倍增管作为检测器,它在紫外区和可见区具有较高的灵敏度.因此,对于那些共振线在这些区域附近或以外的元素,常选用次灵敏线作为分析波长。
例如测定铅时,为了克服短波区域的背景吸收和吸收和噪声,一般不使用217.0nm灵敏线而用283.3nm谱线。
1.2 电流的选择选择合适的空心阴极灯灯电流 , 可得到较高的灵敏度与稳定性,图4-1为Cd 灵敏对水灯电流变化的曲线。
从灵敏度考虑 , 灯电流宜用小 , 因为谱线变宽及自吸效应小 , 发射线窄 , 灵敏度增高。
但灯电流太小 , 灯放电不稳定,光输出稳定性差,为保证必要的信号输出,势必增加狭缝宽度或提高检测器的负高压,这样就会引起噪声增加,使谱线的信噪比降低,导致精密度降低。
从稳定性考虑 , 灯电流要大 , 谱线强度高 , 负高压低 , 读数稳定 , 特别对于常量与高含量元素分析 ,灯电流宜大些。
灯电流的选择原则是:保证稳定放电和合适的光强输出的前提下,尽可能选用较低的工作电流。
第03章 原子吸收光谱分析

7
• 各种元素的基态至第一激发态跃迁最易发生,吸收最强,最灵 敏线——主共振吸收线。 • 各种元素的原子结构和外层电子排布不同,由基态至第一激发 态跃迁吸收能量不同,共振线不同——具有特征性。
• 利用基态的原子蒸气对光源辐射的特征谱线(共振线)的吸收
可以进行定量分析。 • 光谱位于光谱的紫外区和可见区。
• 准确度高,分析速度快;
• 应用广泛。 • 局限:不能对多元素同时测定(需更换光源)、对难 熔元素测定灵敏度和精密度较低、对于成分复杂样品 干扰较严重、对多数非金属元素不能直接测定。
5
元素周期表中可用原子吸收光谱法分析的元素
6
3.2 原子吸收光谱法的基本原理
3.2.1 原子吸收光谱的产生
• 基态原子吸收其共振辐射,外层电子由基态跃迁至激发态 而产生原子吸收光谱。
收定律,有:
I I 0e
Kvl
• 或
I0 A lg 0.434 K v l I
21
• 采用锐线光源进行测量,则Δv发< < Δv
吸
,在辐射线宽度范围内,Kν可近似
发射线
认为不变,并近似等于峰值时的吸收 系数K0,则:
I0 A lg 0.434 K 0l I
22
• 峰值吸收系数K0与谱线的宽度有关,在通常原子吸收测定条
• 由于原子在空间作无规则热运动所导致的,故也称为热变宽。
2v0 vD c
2(ln 2) RT T 7 7.1610 v0 Ar Ar
• Doppler 变宽随温度升高、谱线频率升高和相对原子质量减小而 变宽。
11
3.压力变宽( 10-3nm)
• 当原子吸收区气体压力变大时,相互碰撞引起的变宽是 不可忽略的。原子之间的相互碰撞导致能级变化,激发 态原子平均寿命缩短,引起谱线变宽。 • 劳伦兹(Lorentz)变宽:待测元素原子和其他粒子碰撞。
火焰原子吸收光谱法测定饮用水中的钙.

火焰原子吸收光谱法测定饮用水中的钙一、实验目的1、了解原子吸收光谱仪的结构及其操作。
2、掌握以原子吸收光谱法进行定量测定的方法。
3、学会优选测定条件方法。
二、实验要求1、要求同学利用所学原子吸收光谱知识,设计出用火焰原子化法对钙元素的测定,选择出最佳测试条件。
2、设计出合理的实验方法(两种)测定出饮用水中的钙。
三、实验条件1、仪器:日立180-80型原子吸收光谱仪;电子天平(0.0001g);空心阴极灯(钙);空气压缩机;容量瓶;移液管;烧杯。
2、试剂:盐酸(优级纯)溶液;HCl(1+2)。
钙标准溶液的配制:Ca=1000μg/mL准确称取2.5000g(优级纯)CaCO3(在120℃,烘2小时),加去离子水50mL,滴加HCl溶液(1+2)至CaCO3完全溶解,移入1000 mL容量瓶中,用去离子水稀释至刻度,摇匀。
工作液的配制:Ca=100μg/mL取10.0 mL钙的标准溶液于100 mL容量瓶中,用去离子水稀释至刻度,摇匀。
四、实验方案1、原理:根据原子吸收定量的原理 A=KLC2、定量的方法:标准曲线法采用标准曲线法定量饮用水中微量钙,以钙标准系列溶液浓度为横坐标,以对应的吸光度为纵坐标绘制一通过原点的直线,在相同的条件下测得样品溶液的吸光度值,进而计算出样品中钙的含量。
标准加入法以钙的标准加入法工作溶液测得吸光度,绘制工作曲线,将其外推,求得饮用水中钙的含量。
3、实验方法3.1系列标准溶液的配制取5个100 mL容量瓶,依次加入0.00, 1.00, 3.00,5.00 ,7.00mL100μg/mL钙的工作标准溶液,用去离子水稀释至刻度,摇匀。
此标准系列钙的浓度为0.00, 1.00, 3.00,5.00 ,7.00 μg/mL。
3. 2 未知样溶液的配制取20mL饮用水于100mL容量瓶中,用去离子水稀释至刻度,摇匀。
3.3标准加入法工作液的配制取4个100 mL容量瓶,各加入10 mL 未知试样溶液,然后依次加入0.0, 1.0, 3.0, 5.0 mL100μg/mL的钙工作标准溶液,用去离子水稀释至刻度,摇匀。
原子吸收光谱法习题解答

缺点:共存化合物的干扰大,由于取样量少,所以进样量及注入管内位置的变 动会引起误差,因而重现性较差.
ห้องสมุดไป่ตู้.说明在原子吸收分析中产生背景吸收的原因及影响,如 何避免这一类影响?
解:背景吸收是由于原子化器中的气态分子对光的吸收或高浓 度盐的固体微粒对光的散射而引起的,它们属于一种宽频带吸 收.而且这种影响一般随着波长的减短而增大,同时随着基体元 素浓度的增加而增大,并与火焰条件有关.可以针对不同情况采 取不同的措施,例如火焰成分中OH,CH,CO等对光的吸收主要影 响信号的稳定性,可以通过零点调节来消除,由于这种吸收随 波长的减小而增加,所以当测定吸收波长位于远紫外区的元素 时,可以选用空气-H2,Ar-H2火焰.对于火焰中金属盐或氧 化物、氢氧化物引起的吸收通常利用高温火焰就可消除。
6.石墨炉原子化法的工作原理是什么?与火焰原子化法相比较,有什么 优缺点?为什么?
解:石墨炉原子化器是将一个石墨管固定在两个电极之间而制成的,在惰性 气体保护下以大电流通过石墨管,将石墨管加热至高温而使样品原子化.
与火焰原子化相比,在石墨炉原子化器中,试样几乎可以全部原子化,因而测 定灵敏度高.对于易形成难熔氧化物的元素,以及试样含量很低或试样量很 少时非常适用.
化学与化学工程学院分析化学精品课程组制
6-原子吸收光谱

二、原子化器:
作用:
原子化器的功能是提供能量,使试样干燥、蒸发并原 子化,产生原子蒸气。 要求: ●原子化效率要高。 ●稳定性要好。雾化后的液滴要均匀、粒细; ●低的干扰水平。背景小,噪声低; ●安全、耐用,操作方便。
火焰原子化系统 原子化系统类型 非火焰原子化系统
1、火焰原子化系统:
火焰原子化系统是由化学火焰热能提供能量。
★ 分析速度快,仪器比较简单,操作方便,应用比较广。
缺点:
1. ★除了一些现成、先进的仪器可以进行多元素的测定外,
目前大多数仪器都不能同时进行多元素的测定; 2. ★由于原子化温度比较低,对于一些易形成稳定化合物的 元素,如W、Ni、Ta等稀土等以及非金属元素,原子化效 率低,检出能力差; 3. ★非火焰的石墨炉原子化器虽然原子化效率高,检测限低 ,但是重现性和准确性较差; 4. 对复杂样品分析干扰也较严重。
宽度(mm)。
四、检测系统
定量分析方法
1.标准曲线法
配制一系列不同浓度的标准试样,由低到高依次分析, 将获得的吸光度A数据对应于浓度作标准曲线,在相同条件下 测定试样的吸光度A数据,在标准曲线上查出对应的浓度值;
应注意的问题:
1. 所配置的标准溶液的浓度,应在吸光度和浓度呈直 线关系的范围内。 2. 由于雾化效率和火焰状态经常变动,标准曲线的斜 率也随之变动,每次测定前应用标准溶液对吸光度 进行检测。 3. 在整个分析过程中操作条件应保持不变。 4. 标准样品与待测试样的组成应保持一致。
(3)压力变宽(劳伦兹变宽,赫鲁兹马克变宽)ΔVL
由于原子相互碰撞使能量发生稍微变化。 劳伦兹(Lorentz)变宽: 待测原子和其他原子碰撞。 赫鲁兹马克(Holtsmark)变宽(共振变宽): 同种原子碰撞。浓度高时起作用,在原子吸收中可忽略 (4)场致变宽 外界电场、带电粒子、离子形成的电场及磁场的作用使 谱线变宽的现象;影响较小; 在一般分析条件下温度变宽和劳伦兹为主
原子吸收光谱法测量条件的选择

原子吸收光谱法测量条件的选择
原子吸收光谱法测量条件的选择主要考虑以下几个方面:
1. 波长:选择与元素对应谱线的波长,使吸收峰的信号强度达到最大,同时避开可能受到干扰的谱线。
2. 稀释度:稀释度应适中,既要保证峰高足以显示,又要避免过浓引起信号饱和。
3. 标准品:选择与待测物质相同的标准品进行测量,即应选择含有相同量的待测元素的标准品。
4. 干扰元素的消除:如有干扰元素,则在选择波长时需要避开其谱线,或加入掩蔽剂或使用背景校正技术等方法进行干扰消除。
5. 试剂或样品预处理:某些元素需要特殊的试剂处理或样品预处理才能进行测量,如钠可通过加入碳酸钠等试剂后预处理,然后进行测量。
总之,选择合适的测量条件是保证原子吸收光谱法测量精度和准确度的关键。
原子吸收光谱分析法

对于物理干扰,最好的消除方法 就是配制与试样溶液组成相似的 标准溶液。也可用标准参加法来 进行测定。
三,测定条件的选择: 1.分析线的选择:一般选用共
振线作分析线。 2.灯电流:保正稳定和适当光
强度输出的条件下,尽量选 用较低的工作电流。
5.狭缝宽度:由于原子吸收光谱法谱 线的重叠较少,一般可用较宽的狭 缝,以增强光的强度。但当存在谱 线干扰和背景吸收较大时,那么宜 选用较小的狭缝宽度。
SCV0.0044(g/1% 吸 收 ) A
式中:S为绝对灵敏度;C为试液 中 待 测 元 素 的 浓 度 〔g能检 出的元素的最低浓度或最小质 量。
定义为:能给出信号强度 等于3倍噪声信号强度标准偏差 时所对应的元素浓度或质量。
当在正负电极上施加适当电 压〔一般为200~500伏〕时,在 正负电极之间便开始放电,这时, 电子从阴极内壁射出,经电场加 速后向阳极运动。
电子在由阴极射向阳极的过程中, 与载气〔惰性气体〕原子碰撞使其 电离成为阳离子。带正电荷的惰性 气体离子在电场加速下,以很快的 速度轰击阴极外表,使阴极内壁的 待测元素的原子溅射出来,在阴极 腔内形成待测元素的原子蒸气云。
三.光学系统: 分光系统一般用光栅来进行分光。
光谱通带: W=D×S×10-3
其中:W为光谱通带〔单位nm〕;D为 光 栅 的 倒 线 色 散 率 〔 单 位 nm/mm-1〕 ; S为狭缝宽度〔单位μm〕。
四.检测系统: 检测系统包括检测器、放大器、
对数转换器、显示器几局部。
原子吸收光谱法的分析过程:
计算式为:D c 3 ( g / m L )
A
或 D g 3 ( g )
A
式 中 D 为 检 出 极 限 〔μg/mL 或 g〕 ; σ 为 对 空 白 溶 液 进 行 不 少 于 10 次 测 量时的标准偏差;A为吸光度;g为 质量〔g〕。
大连理工分析化学课件-第7章 原子吸收光谱法

火焰原子化法:
3
cDL A
石墨炉原子化法:
V 3
mDL
A
第六节
原子吸收光谱法的应用
元素的原子吸收法测定
碱金属
碱土金属
有色金属
贵金属
如:人发中钴的测定。浓硝酸消解,萃取分离。 灯电流为6 mA,光谱通带为0.2 nm。测定Co 240.7 nm的吸光度。检出限为0.02 mg/L, RSD<6%,回收率 94.0% ~ 102.0%。
化学干扰的抑制(了解)
通过在标准溶液和试液中加入某种光谱化学缓 冲剂来抑制或减少化学干扰:
释放剂:与干扰元素生成更稳定化合物使待测 元素释放出来。
例:锶和镧可有效消除磷酸根对钙的干扰,因 为锶和镧与磷酸根形成更稳定的化合物。
保护剂:与待测元素形成稳定的络合物,防止 干扰物质与其作用。
例:测定钙时,加入EDTA生成EDTA-Ca2+,避 免磷酸根与钙作用。
洛伦兹变宽(碰撞变宽):由于原子相互 碰撞使能量发生稍微变化。
定量基础
钨丝灯光源和氘灯,经分光后,光谱通带0.2 nm 。而原子吸收线的半宽度:10-3 nm。
若用一般光源照射时,吸收光的强度变化仅为 0.5%,灵敏度极差。
若将原子蒸气吸收的全部
能量,即谱线下所围面积
测量出(积分吸收),则
是一种绝对测量方法,现
在的分光装置无法实现。
定量基础(续)
峰值吸收:峰值吸收系数K0与火焰中待测元素的 基态原子数N0成正比。
前提是:采用温度不太高的稳定的火焰原子化器 和使用锐线光源辐射。
(1)光源的发射线与吸收线的v0一致。 (2)发射线的Δv<吸收线的 Δv。 空心阴极灯可发射锐线光源。
第八章原子吸收光谱

换用纯度较高的单元素灯减小干扰。
(3)灯的辐射中有连续背景辐射, 用较小通带或更换灯。
二 物理干扰(基体效应) 指试液与标准溶液物理性质(如粘度、
表面张力、雾化气体的压力等)有差别 而产生的干扰。是非选择性干扰。
消除方法:
1. 采用与被测试样组成相似的标准样 品制作工作曲线。 2. 标准加入法。
基态第一激发态,吸收一定频率的辐射能量。
产生共振吸收线(简称共振线)
激发态基态
吸收光谱
发射出一定频率的辐射。
产生共振吸收线(也简称共振线) 发射光谱
特征吸收 基态元素M E 特征辐射 激发态M*
1.原子吸收与原子发射法的对比
(1)吸收辐射后基态的原子数减少,辐射吸收值与基
态原子的数量有关,也即由吸收前后辐射光强度的变化
I
e
0
I d
将 It=I0e-KvL 代入上式: I e I e-K L d 0
则: A lg
e
e
0
0
I 0 d
0
I 0 e -K L d
用锐线光源测量,则Δve< Δva ,
由图可见,在辐射线宽度范围内,Kν
可近似认为不变,近似等于峰值时的 吸收系数K0。
气体测量管中进行吸光度测量。 特点:常温测量; 灵敏度、准确度较高(可达10-8 g汞)。
五、单色器
1.作用 2.组件 将待测元素的共振线与邻近线分开。 色散元件(棱镜、光栅)、凹凸镜、狭缝等。 两条谱线间的距离与波长差的比
3.单色器性能参数 (1)线色散率(D)
值ΔX/∆λ 。实际工作中常用其倒数 ∆λ/ ∆X 。
将待测试样在专门的氢化物生成器中产生氢化物,送
(完整版)原子吸收光谱的定量分析

(完整版)原子吸收光谱的定量分析
介绍
原子吸收光谱是一种常用的定量分析方法,用于测量样品中特定元素的浓度。
本文档旨在介绍原子吸收光谱的基本原理和定量分析的步骤。
原理
原子吸收光谱利用原子吸收特定波长的光来测量样品中特定元素的浓度。
当光通过样品中的原子气体时,原子会吸收与其原子结构相关的特定波长的光线。
通过测量吸收光的强度,可以确定样品中特定元素的浓度。
步骤
以下是进行原子吸收光谱定量分析的基本步骤:
1. 样品制备:将待分析的样品转化为原子气态。
常用的方法包括火焰法、电感耦合等离子体法等。
2. 选择波长:根据待分析元素的吸收峰进行波长选择。
可以通过参考相关文献或经验来确定。
3. 校准曲线:准备一系列浓度已知的标准溶液,测量它们的吸光度,并绘制校准曲线。
4. 测量样品:将样品引入原子吸收光谱仪器,测量其吸光度。
5. 数据分析:利用校准曲线,计算出样品中特定元素的浓度。
6. 重复测量:进行重复测量,确保结果的准确性和可靠性。
7. 结果报告:将测得的浓度结果整理并报告。
结论
原子吸收光谱是一种可靠的定量分析方法,能够有效测量样品中特定元素的浓度。
正确的样品制备、波长选择和数据分析步骤对于获得准确结果至关重要。
通过遵循上述步骤,可以进行原子吸收光谱的定量分析。
*注意:本文档仅为介绍原子吸收光谱的基本原理和步骤,具体实验细节和参数设置需要根据实际情况进行调整。
*。
【材料研究与测试方法】原子吸收光谱法-2

(3)选择性比较好,谱线较简单,谱线数目比AES法少得多,谱 线干扰少,大多数情况下共存元素对被测定元素不产生干扰。
(4)分析速度快,能够测定的元素多达70多种,不仅可以测定 金属元素,也可以用间接法测定某些非金属元素和有机化合物。
11
3、碰撞变宽(压力变宽)
是由于微粒间相互碰撞的结果。
被测原子与蒸汽中的原子或其它粒子相互碰撞引
起激发态原子的平均寿命发生变化,导致吸收线
变宽,这种变宽与吸收区气体的压力有关,因此
也称压力变宽。根据与其碰撞粒子的不同,又分
为劳伦兹(Lorents)变宽和赫鲁兹马克(Holtsmark)
变宽两种
使被测组分变成变成气态基态原子。 • (3)用被测元素的同种原子发射的特征辐射照射原子
化器中的原子蒸汽,则该辐射部分被吸收,强度减小。 • (4)分光。把待测元素的共振线与其他谱线分离,只
让待测元素的共振线通过。 • (5)检测特征辐射被吸收的情况。
6
原子吸收光谱法的特点:
(1)灵敏度高、检测限低,火焰原子化法的检测限可达ng/cm3 级,石墨炉原子化法更低,可达10-10~10-14g;
AAS与AES之比较:
相似之处——产生光谱的对象都是原子; 不同之处——AAS:选择性吸收光辐射能(h)-共振吸收线;
AES:原子从基态跃迁至激发态,返回到基态时所产生的光 谱(共振发射线和非共振发射线)。
AAS 实现多元素的同时测定较困难!
5
AAS的分析过程
• (1)把分析试样经适当的化学处理后变为试液。 • (2)把试液引入原子化器中进行蒸发离解及原子化,
7-4原子吸收光谱法的几种常用的定量方法

n
浙
D = 3σ C
σ=
∑ ( A − Ai )2 1
江
A
n −1
师
范 大
C—标准溶液溶液浓度
学 仪
A—浓度为C的标准溶液的吸光度值
器
A —为浓度为C的标准液的吸光度的平均值
分 析
S—空白溶液的吸光度值的标准偏差。
¾ 通常以ppm,ppb表示检测极限,检测极限越
小,表示可测定的浓度越低,灵敏度越高。
a.干扰谱线的影响
¾ 由于分析线周围的干扰线也进入了检测器,
结果使测得的吸光度降低,且浓度越大,干扰
谱线的影响越严重,曲线向浓度轴弯曲。
b.电离效应
¾ 某些电离电位较低的元素,如:Li、K、Na、
浙 江
Cs等在火焰中易电离,且低浓度时的电离度比
师
高浓度时大,故出现校正曲线向上弯曲。
范 大
c.碰撞变宽的影响
¾ 检测极限不仅表示各元素的测定特性,也表
示仪器噪音的大小,所以,对同一元素的不同
仪器上测定检测极限不同,而且各种书上的报
浙
道也不尽相同。
江 师
¾ 检测极限不能直接用来表示原了吸收光谱法
范
所能测定的下限,测定下限指在满足分析误差
大 学
要求情况下方法可测的最低浓度。通常,测定
仪
下限约为检测极限的5-10倍。
第四节 原子吸收光谱法的 几种常用的定量方法
一、灵敏度的表示方法
¾ 在原子吸收分析中,据1975年IUPAC规定,灵
敏度定义为:校正曲线的斜率,即被测元素的单
位浓度或质量改变时引起的吸光度的变化。
浙 江 师 范
S = dA dC
or S = dA dm
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Ax c 当A=0时, k
cx
A kc Ax
A—c曲线
方法
特点
适用范围
注意事项
横 向 比 较
标准 曲线 法
简便、快 速、可扣 除空白值
1.所用标准溶液系列浓度应在 A-C曲线的线性范围内 2.标准溶液与试样溶液要用相 组成简单、 同的试剂处理。 大量试样 3.扣除空白值。 的快速分 4.测定过程中,操作条件不变。 析 5.标准试样的组成应尽量与待 测溶液相同。
火焰的氧化性随火焰高度 的变化而变化
Mg Ag
Cr
原则:使测量光束从自由 原子浓度最大的火焰区通 过,保证最大的吸收灵敏 度。
相对吸收值 自由原子在火焰中的分布
5.狭缝宽度的选择
单色器分辨能力大,或光源辐射弱或共振线吸收 小,应选择较宽的狭缝宽度。 单色器分辨能力小,火焰的背景发射强,或吸收 线附近有干扰时,应选择较窄的狭缝宽度。 合适的狭缝宽度应通过实验确定 原则:能将吸收线与邻近的干扰线分开
一、AAS的定量分析方法
定量依据 标准曲线法
标准加入法
定量依据
强度为 I0 的某一波长的辐射通过均匀的原 子蒸气时,根据吸收定律,有 I I 0 exp( K 0l )
I0 和I分别为入射光和透射光的强度,K0为峰值吸收系数, l为原子蒸气层厚度
当在原子吸收线中心频率附近一定频率范围 Δv测量,则 v I 0 Ivdv
E K S lg ai
二、测定条件的选择
分析线 的选择 放大倍 数的选 择
狭缝宽 度
火焰原 子化法 仪器工 作条件
燃烧器 高度
空心阴 极灯电 流
火焰
1.分析线的选择
(1)一般选择最灵敏线(主共振线) (2)最灵敏线受干扰较大或测定高含量元素时,选 择次灵敏线或其它谱线 最适宜的分析线应视具体情况通过实验决定,其 原则是选用干扰小的谱线作为分析线。
670.78,323.26
335.96,328.17 285.21,279.55
Sn
Sr Ta
224.61,520.69
460.73,407.77 271.47,277.59
Cd
Ce
228.80,326.11
520.00,369.70
Mn
Mo
279.48,403.68
313.26,317.04
Tb
Te
432.65,431.89
214.28,225.90
2.灯电流的选择
灯电流过大:发射谱线变宽,自吸收增大,灵敏 度下降,灯寿命缩短。 灯电流过小:辐射锐线光谱窄,灵敏度高,但光 强小,信噪比降低。
通常选择最大灯电流的1/2~2/3为工作电流,实际 实验中,最合适的工作电流需通过试验确定。
原则:在保证稳定和合适光强输出的情况下,尽 量选用最小的灯电流。
3.火焰的选择
分析线200nm以下采用氢气—空气火焰 易离解的物质用低温火焰 易生成难离解化合物的物质用高温火焰,如乙 炔—空气火焰,乙炔—氧化亚氮火焰。
选定火焰类型后,应通过实验确定燃助比,才可 得到所需特点的火焰。
4.燃烧器高度的选择
火 焰 高 度
各元素在火焰 中都有合适的 测量位置
Thank you!
标准 加入 法
1.校正曲线是一条不过原点的 消除基体 曲线 效应,化 2.最少应采用4个点来做曲线 学干扰和 组成复杂, 3.第一份加入的标准溶液浓度 电离干扰,且不完全 与试样浓度应尽量接近 不能直接 确定组分 4.斜率太小的曲线易引起较大 消除背景 的试样 误差 干扰 5.应该扣除试剂的空白值。 (必须是标准加入法的空白值)
由峰值吸收中
基态原 子数
2 K0 vD
In2 e N0 f mc
2
N0 c
A=k·c
标准曲线法
配制一系列已知浓度的标准溶液(可等差可不 等差) 在选定条件下,用空白溶液调吸光度为零。 (扣除空白值) 依次测吸光度A1、A2……An(由低到高) 绘制A-C曲线 测出待测液吸光度A',从曲线上找出对应浓度
标准加入法 直接计算法
取相同体积的试样溶液两份,分别移入容 量瓶A、B中 另取一定量的标准溶液加入到B中,然后将 两份溶液稀释至刻度 测出A、B的吸光度 Ax Ax=k∙Cx cx c0 A0 Ax A0=k(C0+Cx)
外推法
取若干份体积相同的样品溶液 从第二份开始加入不同量的待测元素的标液,稀 释至一定体积,各溶液的浓度分别cx+c1,cx+c2…… 依次测吸光度 做出A-c曲线
6.放大倍数的选择
原则:在保证最佳灵敏度的前提下,尽量 选择最大的放大倍数
石墨炉原子化条件的选择
合理的选择干燥、灰化、原子化及去残等阶段的 温度和时间
干燥
灰化
原子化
• 水溶液一般在105~120℃ 条件下,有机溶剂稍高 于其沸点即可 • 原则:既能消除试样中的 有机物或基体,又不会造 成被测元素的损失 • 原则:选择可达到待测元 素最大吸光度的最低温度
成员:杨 靖 邢忠银 班级:应化0902
原子吸收光谱法是基于被测元素蒸气对其原子的共 振辐射吸收,而进行定量分析的方法。
• 大量基体存在,盐蒸发与离解消耗 物理 大量热 干扰 • 待测元素与共存物生成难挥发化合 化学 物
干扰 干扰
干 扰
• 与光源有关的、光谱线重叠干扰、 光谱 与原子化器有关的
在实际工作中,这些干扰问题不可忽视, 某些情况下,干扰甚至是很严重的。
元素
Ru
Sb Sc Se Si Sm
λ/nm
349.89,372.80
217.58,206.83 391.18,402.04 196.09,703.99 251.61,250.69 429.67,520.06
Be
Bi Ca
234.86
223.06,222.83 422.67,239.86
Li
Lu Mg
d VsCshx 波高h成正比 cx (Vx Vs ) H Vxhx
•极限浓度i 与
纵向比较
lg I lg a b lg c
•准确,只适用 于纯物质中低 含量成分分析 •背景干扰很严 重 仅用一种标准 溶液,简单快 速 •有大量络合剂 存在时,测得 是离子总浓度
消除基 体影响, 一定程 度上消 除其它 干扰
标准曲线法
特点 AAS •曲线理论上过 原点,实际上 极谱 •适用 存在干扰 于组 法 成简 单, 大批 AES 量试 样的 测定 •标准曲线容易 •简便、 电位 快速 发生移动(K值 易受T、搅拌速 分析 度、电极表面 法 性质影响)可 代两点标准确 定曲线的位置 定量依据
标准加入法
特点
A kc h kc x
原子吸收光谱法中常用的分析线
元素
Ag
Al As Au B Ba
λ/nm
328.07,338.29
309.27,308.22 193.64,197.20 242.80,267.60 249.68,249.77 253.55,455.40
元素
Hg
Ho In Ir K La
λ/nm
253.65
410.38,405.39 303.94,325.61 209.26,208.88 766.49,769.90 move1().13,418. 73
三、思考题
问题1: 标准加入法为什么能消除基体效应?
答:基体效应:试样的物理变化(试液粘度;试样喷 入火焰速率;表面张力:雾滴大小及分布;雾化气体 压力:喷入量;基体元素:原子化效率)。 它们的改变一般可以用一个系数校正。
Ax cx c0 A0 Ax
因此,对浓度测定不会有什么影响。
问题2: 现拟用原子吸收法测定碳灰中微量Si,为了选 择适宜的分析条件,进行了初步试验,当Si浓 度为5.0ug/mL时测得Si251.61nm、251.43nm和 251.92nm的吸光度分别为0.44、0.044和0.022。 试计算各谱线灵敏度,选择最适宜测量的谱 线。
解:对于Si251.61nm:C特=5×0.00434/0.44=0.049(ug/mL/1%) 对于Si251.43nm:C特=5×0.00434/0.044=0.49(ug/mL/1%) 对于Si251.43nm:C特=5×0.00434/0.022=0.99(ug/mL/1%) 由于Si251.61nm的灵敏度最高,因此选用 该谱线
I Iv exp(kvl )dv
0
v
0
使用锐线光源, Δv很小,用中心频率 处K0表示原子对辐射的吸收。
I0 A lg lg I
v
v
0
Ivdv
0
Iv exp( K 0l )dv
lgv 0
exp( K 0l ) Ivdv
0.43K 0l