高分子化学-3(聚合速率2)
高分子化学第三章

(二)阴离子聚合
在链式聚合反应中,活性中心为阴离子的聚 合反应。常用的引发剂有碱金属、丁基锂等亲核 试剂。
阴离子聚合反应的通式可表示如下:
A B M BM A M M n
其中B-为阴离子活性中心,A+为反离子,一般 为金属离子。与阳离子聚合不同,阴离子聚合中 ,活性中心可以是自由离子、离子对,以及处于 缔合状态的阴离子。
酸根的亲核性不能太强,否则会与活性中心结合成 共价键而终止,如HCl
CH3 CH A X
A CH3 CH
X
不同质子酸的酸根的亲核性不同
氢卤酸的X-亲核性太强,不能作为阳离子聚合引发剂, 如HCl引发异丁烯
(CH3)3C Cl
(CH3)3C Cl
HSO4- H2PO4-的亲核性稍差,可得到低聚体。 HClO4,CF3COOH,CCl3COOH的酸根较弱,可生成高聚 物。
Lewis酸引 发
傅-克(俗称Friedel-Grafts催化剂)反应中的各种
金属卤化物,都是电子的接受体,称为Lewis酸。
从工业角度看,是阳离子聚合最重要的引发剂。
Lewis酸包括: 金属卤化物:
BF3 , AlCl3, SnCl4 , TiCl4, SbCl5, PCl5, ZnCl2 金属卤氧化物:
离子聚合:活性中心是离子的聚合。
根据中心离子电荷性质的不同 阳离子聚合 阴离子聚合
离子聚合的理论研究开始于五十年代:
1953年,Ziegler在常温低压下制得PE 1956年,Szwarc发现了“活性聚合物”
多数烯烃单体都能进行自由基聚合,但是 离子聚合却有极高的选择性。 原因: 离子聚合对阳离子和阴离子的稳定性要求 比较严格。
《高分子化学》习题答案

《高分子化学》习题答案(王槐三第2版)第1章1、解释下列概念(1) 高分子化合物:由众多原子或原子团主要以共价键结合而成的相对分子质量在1万以上的化合物。
(2) 重复结构单元:将大分子链上化学组成和结构可重复的最小单位称为重复结构单元(在高分子物理里也称为链节)。
(3) 结构单元:由1个单体分子通过聚合反应而进入聚合物重复单元的那一部分叫结构单元。
(4) 平均相对分子质量:高分子化合物中同系物相对分子质量的统计平均值。
(5) 平均聚合度:所有大分子链上所含重复结构单元数量的统计平均值。
(6) 多分散性和分散指数:多分散性是指聚合物材料中所含大分子同系物的相对分子质量不相等的这一特性。
分散指数是指重均相对分子质量与数均相对分子质量的比值。
2、写出合成下列聚合物的聚合反应方程式并标出结构单元 (1) 涤纶n HOOCCOOH n HO(CH 2)2OH (2n -1)H 2OHO[OCCOO(CH 2)2O]n H +=+结构 结构单元单元(2) 尼龙-610n HOOC COOH n H 2N(CH 2)6NH 2(2n -1)H 2OHO [ OC(CH 2)8COHN(CH 2)6NH ]+=+(CH 2)8n H 结构单元结构单元(3) 有机玻璃n CH 2CH 3COOCH 3CCH 2CH 3C COOCH 3=[]n CH 2CH 3C COOCH 3结构单元:(4) 聚乙烯醇n CH 2 = CHOCOCH 3CH 2CH []OCOCH 3n水解聚合[]CH 2CH OHn(5) 环氧树脂 (见P8) (6) 聚碳酸酯HOCH 3CH 3C Cl C O ClH OC CH 3CH 3OCCl + (2n - 1)HCl=+n n []OH n O(7) 聚己二氨基甲酸丁二酯n OCN(CH 2)6NCO + n HO(CH 2)2OH = []OCNH(CH 2)6NHCOO(CH 2)4O n(8) 维尼纶[]CH 2CH OHn + CH 2O CH 2CHCH 2CH CH 2CH OCH 2OOH(9) 丁腈橡胶nCH 2CHCN CH 2CHCH CH CH 2CHCH 2CHCHCH 2CNnn +[](10) ABS 树脂nCH 2CHCN CH 2CHCH CH 2n ++ nCH 2CH+ BPOCH 2CHCH 2CHCHCH 2CH 2CH CNn[]3、写出合成下列聚合物的聚合反应方程式并命名聚合物 (1) HO OC(CH 2)8CONH(CH 2)6NH n H []HO OC(CH 2)8CONH(CH 2)6NH n H[]n HOOC(CH 2)8COOH + n H 2N(CH 2)6NH 2 =尼龙-610 (2)[]OCNH(CH 2)6NHCOO(CH 2)4O n见第2题(7)小题 聚己二氨基甲酸丁二酯(3) H O(CH 2)5CO OH[]nn HOOC(CH 2)5OH = HO []OC(CH 2)5O H + (n-1)H 2On聚6-羟基己酸酯 4、参见教材p3315、分别写出单独或与别的单体进行聚合的反应方程式并命名聚合物。
高分子化学-第3章 自由基聚合
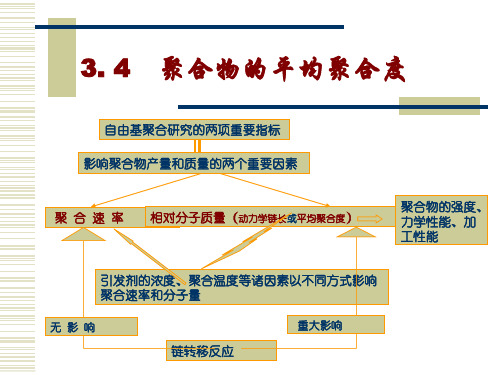
3. 4
聚合物的平均聚合度
1、动力学链长和聚合度
(1)动力学链长υ (kinetic chain length)的定义
每个活性种从引发阶段到终止阶段所消耗单体分子数。无 链转移时,动力学链长为增长速率和引发速率的比。 依据稳态时引发速率等于终止速率,则动力学链长可表 示为增长速率与终止速率的比: 即为单体消耗速率与
自由基产生(或消失) 速率之比
3. 4
聚合物的平均聚合度
如将稳态时的自由基浓度 入上式,可得下式:
,代
3. 4
聚合物的平均聚合度
若自由基聚合反应由引发剂引发时,
引发速率Ri = 2 f kd[I],则:
3. 4
聚合物的平均聚合度
可知动力学链长与引发速率存在以下关系:
1) 动力学链长与单体浓度的一次方成正比,与 引发剂浓度平方根成反比。 2) 说明了在自由基聚合体系中,增加引发剂用 量虽然可以提高聚合速率,但又使聚合物相对分子 质量降低。由此说明引发剂在自由基聚合中的重要
(1)温度对聚合速率的影响
总聚合速率常数k与温度T(K)遵循Arrhenius经验公式: 由前面推导可知: k=Ae-E/RT
k=kp(kd/kt)1/2
因此:
3.5 影响自由基聚合反应的因素
从而可知,总活化能E=(Ep-Et/2)+Ed/2
由Ep、 Et和Ed的大小可以得到总活化能E约为83 kJ/mol,为正值,表明温度升高,速率常数增大k增大。
3.5 影响自由基聚合反应的因素
1. 链自由基的双基终止过程的三步曲:
1) 链自由基的平移;
2) 链段重排,使活性中心靠近;
3) 双基相互反应而使链终止。
第二步(链段重排)是 控制步骤,受体系粘度 影响显著。
高分子化学复习简答题(五)--聚合方法(精)

高分子化学复习简答题(五)---聚合方法学校名称:江阴职业技术学院院系名称:化学纺织工程系时间:2017年3月10日1、比较自由基聚合的四种聚合方法。
实施方法本体聚合溶液聚合悬浮聚合乳液聚合配方主要成分单体、引发剂单体引发剂、溶剂单体、引发剂、分散剂、水单体、引发剂、乳化剂、水聚合场所单体内溶剂内液滴(单体)内胶束内聚合机理自由基聚合一般机理,聚合速度上升聚合度下降容易向溶剂转移,聚合速率和聚合度都较低类似本体聚合能同时提高聚合速率和聚合度生产特征设备简单,易制备板材和型材,一般间歇法生产,热不容易导出传热容易,可连续生产。
产物为溶液状。
传热容易。
间歇法生产,后续工艺复杂传热容易。
可连续生产。
产物为乳液状,制备成固体后续工艺复杂产物特性聚合物纯净。
分子量分布较宽。
分子量较小,分布较宽。
聚合物溶液可直接使用较纯净,留有少量分散剂留有乳化剂和其他助剂,纯净度较差2、悬浮聚合的配方至少有哪几个组分?单靠搅拌能不能得到聚合物颗粒?加入悬浮稳定剂的目的和作用是什么?常用的悬浮稳定剂有哪几种?影响聚合产物粒径大小因素有哪些?悬浮聚合的主要缺点是什么?答:①悬浮聚合的配方一般至少有四个组分,即单体,引发剂,水和悬浮稳定剂。
②搅拌的剪切力可使油状单体在水中分散成小液滴。
当液滴分散到一定程度后,剧烈搅拌反而有利于细小液滴的并和(成大液滴),特别是当聚合反应发生后,由于液滴中含有一定量的聚合物,此时搅拌增大了这些液滴的碰撞粘结概率,最后导致聚合物结块,所以单靠搅拌不能得到稳定的悬浮体系,因而体系中必须③加入悬浮剂,以降低表面张力,使分散的小液滴表面形成一层保护膜,防止彼此并和和相互粘结,从而使聚合在稳定的悬浮体系中的液滴中进行。
如果只加悬浮剂,而不进行搅拌,则单体就不会自动分散成小液滴;同样不能形成稳定的悬浮体系。
④可作悬浮剂的物质有:水溶性聚合物如聚乙烯醇,明胶和苯乙烯-马来酸酐共聚物等;水不溶性无机物如磷酸钙,碳酸镁,碳酸钡和硫酸钡等。
高分子化学名词解释

1、高分子(聚合物):合成高分子多半是许多结构单元重复连接而成的聚合物。
2、碳链聚合物:大分子主链完全由碳原子组成,绝大部分烯类和二烯类的加成聚合物。
3、杂链聚合物:大分子主链中除了碳原子外,还有氧、氮、硫等杂原子。
如-O-,-OCO-,-NHCO-4、结构单元:单体通过聚合反应转变成大分子的一部分结构。
5、反应程度P:参与反应的基团数(N0-N)占起始基团数N的分数。
6、体型缩聚:指某-2官能度与另一官能度大于2的单体先进行酯化而后形成交联结构的缩聚过程。
7、界面缩聚:两种单体分别溶于水和有机溶剂中,在界面处进行聚合,具有明显的表面反应的特征,而且不必严格等基团数之比。
8、官能度:一分子中能参与反应的官能团数。
9、偶合终止:两自由基的独电子相互结合成共价键的终止方式,偶合终止的结果是大分子的聚合度为链自由基重复单元数的两倍。
10、歧化终止:某自由基夺取另一自由基的氢原子或其他原子而终止的方式,歧化终止的结果是聚合度与链自由基的单元数相同。
11、动力学链长v:一个活性种从引发开始到链终止所消耗的单体分子数。
12、链转移常数:是链转移速率常数与增长速率常数之比,代表这两反应的竞争能力。
13、竞聚率r:均聚和共聚链增长速率常数之比、自增长速率常数与交叉增长速率常数的比值。
14、无规共聚物:两结构单元M1,M2按概率无规排布。
-CO-15、交替共聚物:共聚物中M1,M2两单元严格交替相间。
-alt-16、嵌段共聚物:由较长的M1链段和另一较长的M2链段构成的大分子。
-b-17、接枝共聚物:主链由M1单元组成,支链则由另一种M2单元组成。
-g-18、悬浮聚合:单体以小液滴状悬浮在水中的聚合方法(单体、水、油溶性引发剂、分散剂)场所:液滴内19、乳液聚合:单体在水中分散成乳液状态的聚合(单体、水、水溶性引发剂、水溶性乳化剂)场所:胶束或胶粒20、摩尔系数:令Na、Nb分别为官能团a、b的起始数,则两种单体的官能团之比r=Na/Nb<1称为摩尔系数。
高分子化学导论第3章_自由基聚合机理及分子量链转移
链转移与链终止反应
链转移 自由基与其他非自由基分子的反应
链终止 自由基与自由基的反应
引发 增长
E (kJ/mol)
k
特点
Ed:105~150 Ei: 21~34
Ep=20~34
kd: 10- 4~10- 6s-1 慢引发 kp=102~104l/mol·s 快增长
终止 Et=8~21
kt=106~108l/mol·s 速终止
如:过氧化乙酰环己烷磺酰(ACSP)
2) 无机过氧化物——过硫酸盐 过硫酸钾,过硫酸铵
O
O
KO S O O S OK
O
O
O 2 KO S O
O
K2S2O8
2KSO4
水溶性引发剂
可单独使用,还可与适当的还原剂构成氧化 还原体系,在室温或更低温度下引发聚合
3. 氧化-还原体系引发剂
由氧化剂与还原剂组合在一起,通过电子转移 反应(氧化-还原反应),产生自由基而引发单 体进行聚合 特点: 活化能低,可在室温或更低温度下引发聚合 引发速率快,即活性大 种类多
歧化终止的结果:
Xn与链自由基中的单体单元数相同。
每个大分子只有一端为引发剂残基,
另一端为饱和或不饱和(两者各半)。
终止方式与单体种类、聚合条件有关 St:偶合终止为主 MMA:>60℃歧化终止为主
< 60℃两种终止方式均有
链终止的特点: Et(终止活化能)很低,8-21KJ/mol Rt(终止速率)极高 双基终止受扩散控制
均裂(homolysis) 共价键上一对电子分属两个基团,带独 电子的基团呈中性,称为自由基
RR
2R
异裂(heterolysis) 共价键上一对电子全部归属于某一基团, 形成阴离子,另一缺电子的基团,称做阳 离子
高分子化学 自由基聚合讲解

高分子化学
3.1 连锁聚合反应
3.1.2 连锁聚合反应分类
活性中心(reactive center)
可以是自由基、阳离子和阴离子,它进攻单体的双键,使单 体的π键打开,与之加成,形成单体活性种,而后进一步与单体 加成,促使链增长。
自由基聚合
连锁聚合反应
依据活性种的不同
阳离子聚合
阴离子聚合
11
高分子化学
30
高分子化学
3.2
自由基聚合的基元反应
自由基聚合机理
链引发 链增长 链终止 链转移反应
自由基聚合属于一
种链锁聚合反应,符 合一般连锁反应特征
3.2.1 链引发反应 initiation reaction
实现自由基聚合反应的首要条件是在聚合体系中产生自由 基。链引发反应是形成单体自由基活性种的反应。
2.转移反应 3.偶合反应 4.歧化反应
R·+ R'H R· + R'·
R-H + R'· R-R'
5.氧化反应
HO· + Fe2+
HO―+ Fe3+
27
高分子化学
3.2
自由基聚合机理
三.自由基的稳定性
说明:①共轭效应使自由基稳定;和空间效应。
②斥电子诱导效应使自由基稳定性降低; ③共轭效应和诱导效应矛盾时,共轭效应为主; ④空间位阻大,自由基稳定性高。 ⑤电性效应和空间效应矛盾时,空间效应为主。
3.1 连锁聚合反应
三种自由基聚合示例
R
+CH2 CH
X
R
CH2
CH X
n CH
2CHຫໍສະໝຸດ XRCH2
高分子化学第三章 自由基聚合

• 链转移反应前后,自由基的数目未变。
35
1. 向单体转移
· ~~CH2-CH + CH2=CH Cl Cl
· ~~CH=CH + CH3-CH Cl Cl
• 注意CH2=CHCl单体
36
2. 向溶剂或链转移剂转移
X ~~CH2CH · + YS X ~~CH2CHY + S ·
• 溶剂:
• 链转移剂:有较强的链转移能力的化合
1 2
[I ]
1
2
[M ] (3—35式)
注意本方程的适用范围
73
二、温度对聚合速率的影响
• 阿累尼乌斯公式:K=Ae–Ea/RT
其中:K=kp(kd/kt)½ 则:Ea=Ep+Ed/2–Et/2
74
一般情况下: Ep≈29kJ•mol–1, Ed≈126kJ•mol–1 Et≈17kJ•mol–1
10
一、 聚合的可能性
• 主要取决于双键上取代基的空间 效应
11
1.烯类单体: CXY=CMN
(1)一取代( CH2=CHX)
可均聚合
12
(2)二取代
(CH2=CXY、CHX=CHY) (a)1,1——二取代:一般不考虑空 间位阻效应,可均聚合。
注意:CH2=C(Ar)2只能形成二聚体
13
(b)1,2——二取代
54
2.半衰期
[I] ln = Kd t [I0]
• 60℃
ln2 t½ = K d
(3—17)
t½ >6h,低活性引发剂 1h< t½ <6h,中活性引发剂 t½ <1h,高活性引发剂
55
3. 引发效率
高分子化学复习简答题(三)---自由基聚合(精)

精品好资料——————学习推荐高分子化学复习简答题(三)---自由基聚合学校名称:江阴职业技术学院院系名称:化学纺织工程系时间:2017年3月10日1、自由基聚合反应转化率-时间曲线特征。
答:诱导期:初级自由基为阻聚杂质所终止,无聚合物形成,聚合速率零。
若严格取除杂质,可消除诱导期。
初期:单体开始正常聚合,转化率在5%~10%以下(研究聚合时)或10%~20%(工业上)以下阶段称初期;此时转化率与时间近似呈线性关系,聚合恒速进行。
中期:转化率达10%~20%以后,聚合速率逐渐增加,出现自动加速现象,直至转化率达50%~70%,聚合速率才逐渐减慢。
后期: 自动加速现象出现后聚合速率逐渐减慢,直至结束,转化率可达90%~100%。
2、自由基聚合与缩聚反应的特征比较。
答:自由基聚合:(1)由基元反应组成,各步反应的活化能不同。
引发最慢。
(2)存在活性种。
聚合在单体和活性种之间进行。
(3)转化率随时间增长,分子量与时间无关。
(4)少量阻聚剂可使聚合终止。
线形缩聚:(1)聚合发生在官能团之间,无基元反应,各步反应活化能相同。
(2)单体及任何聚体间均可反应,无活性种。
(3)聚合初期转化率即达很高,官能团反应程度和分子量随时间逐步增大。
(4)反应过程存在平衡。
无阻聚反应。
3、为什么自由基聚合时聚合物的相对分子质量与反应时间基本无关,缩聚反应中聚合物的相对分子质量随时间的延长而增大?答:自由基聚合遵循连锁聚合机理:链增加反应的活化能很低,Ep=20~34KJ/mol,聚合反应一旦开始,在很短的时间内(0.01s~几秒)就有成千上万的单体参加了聚合反应,也就是生成一个相对分子质量几万~几十万的大分子只需要0.01s~几秒的时间(瞬间可以完成),体系中不是聚合物就是单体,不会停留在中间聚合度阶段,所以聚合物的相对分子质量与反应时间基本无关。
缩聚反应遵循逐步聚合机理:单体先聚合成低聚体,低聚体再聚合成高聚物。
链增加反应的活化较高,Ep=60KJ/mol生成一个大分子的时间很长,几乎是整个聚合反应所需的时间,缩聚物的相对分子质量随聚合时间的延长而增大。
高分子化学期末常考简答题

高分子化学期末常考简答题1.为什么在缩聚反应中不用转化率雨用反应程度来描述反应过程?因缩聚反应本质是官能团之间的反应,只有官能团之间充分反应才能生成大分子,故应用反应程度描述其反应过程。
而转化率一开始就很高。
2.欲使逐步聚合成功,必须考虑哪些原则和措施?有哪些实施方法?原料尽可能纯净;按化学计量或等摩尔比投料:尽量提高反应程度;采用减压或其他手段打破化学平衡,使反应向聚合物方向移动。
实施方法有:熔融聚合:溶液聚合和界面缩聚。
3.传统自由基聚合时,单体转化率和聚合度随时间的变化有何特征?画出简图自由基聚合时,转化率随时间延长而逐渐增加,而对产生的一个活性中心来说,它与单体间反应的活化能很低,k值很大,因此瞬间内就可生成高聚物。
因此,从反应一开始有自由基生成时,聚合物分子量就很大,反应过程中任一时刻生成的聚合物分子量相差不大。
4.竟聚率的定义?说明其物理意义?理想恒比共聚和交替共聚的竞聚率分别是么?竟聚率系单体均聚链增长速率常数和共聚链增长速率常数之比。
即它表征两单体的相对活性,r1=2=1,则两单体为理想恒比共聚体系。
r1=r2=0时,则两单体为交替共聚体系。
5.解释笼蔽效应和诱导分解,它们对引发效率有什么影响?笼蔽效应:引发剂分解成游离基后,处在溶剂的笼子中,来不及引发单体的聚合,就发生了副反应,形成了稳定的分子,消耗了引发剂。
诱导分解:诱导分解是指链自由基向引发剂的转移反应,原来的来的链自由基终止,只生成一个新的自由基,无偿消耗一个引发剂分子。
二者都使引发效率降低。
6.何为自动加速现象?它主要是由什么引起的?某些聚合反应到达一定程度后,聚合速度自动增加,直到后期,聚合速度又减慢。
它主要是由体系粘度增加所引起的,称为自动加速现象。
7.为什么乳液聚合可以同时获得高的聚合速度和聚合度?聚合速率与聚合度均与乳胶粒数成正比。
因此增加乳胶粒数可以同时增加聚合速率与聚合度。
8.用 Ziegler- Natta催化剂催化丙烯和乙烯聚合,分别用什么催化剂组分,为什么这样选择?催化丙烯用:TiCl3(α,Y,δ)- AletAI,综合考虑立构规整度,聚合速率和分子量。
3.3 聚合速率

阻聚剂
(1)阻聚剂和阻聚反应
阻聚剂的种类很多 分子型的阻聚剂
苯醌、硝基化合物、芳胺、酚类、 含硫化合物、氧等
稳定自由基型
1,1-二苯基-2-三硝基苯肼(DPPH)等
象氯化铁、氯化铜等
电荷转移型阻聚剂
苯醌是最重要的阻聚剂
芳族硝基化合物
氧有显著的阻聚作用
自由基型高效阻聚剂
DPPH,1,1-二苯基-2-三硝基苯肼 在 10-4mol/L 的低浓度下,就足以完全阻聚。一个 DPPH 能化学 计量地消灭一个自由基,是理想的自由基捕捉剂,还可用来测 定引发速率。DPPH自由基呈深紫色,反应后变成无色
总活化能为正值,表明随温度升高,速 率常数增大,总的聚合速率也提高 E值愈大,温度对聚合速率影响愈显著
聚合反应活化能 [E = EP - 1/2Et + 1/2Ed]
活化能中,引发剂分解活化能 Ed 占主要地位 选择 Ed 较低的引发剂,则可显著加速聚合 引发剂种类和用量是控制聚合速率的主要手段 热引发聚合活化能约为 80~96 KJ/mol, 温度对聚合速率的影响很大 光和辐射引发体系的活化能很低, 约 20 kJ/mol 温度对聚合速率的影响较小,在较低的温度 下(0℃)也能聚合
单体浓度随反应时间的减少
实际测定的是转化率 随时间的变化
3.3.1 聚合速率及其测定方法
聚合速率也可用 单位时间内的转化率变化表示:
或
C%
t
图3-1 转化率~聚合时间关系
转化率随聚合时间变化的测定
分为直接法和间接法两类 常用的直接法为沉淀法 一定温度下聚合,定时取样,求得不同t时的 聚合物量 也可通过分析单体的浓度而求得某时刻的转化 率 间接方法,即测定聚合体系的比容、粘度、折光 率、吸收光谱等物理化学性质的变化,推算出反 应体系中单体浓度的减少,或聚合物量的增加
高分子化学重点课后习题解答
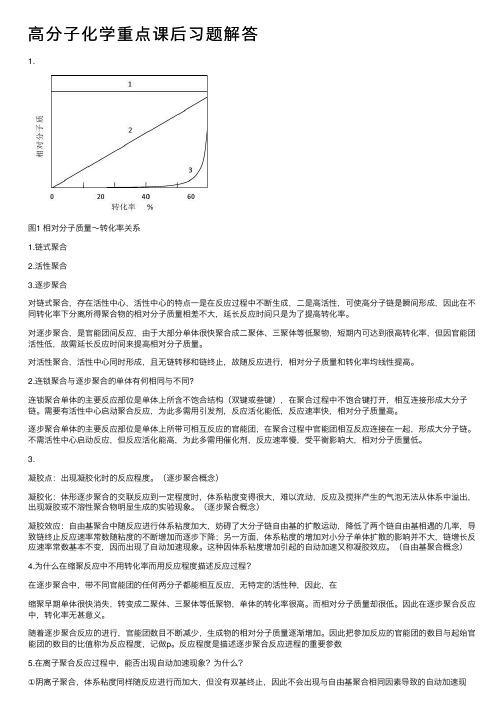
⾼分⼦化学重点课后习题解答1.图1 相对分⼦质量~转化率关系1.链式聚合2.活性聚合3.逐步聚合对链式聚合,存在活性中⼼,活性中⼼的特点⼀是在反应过程中不断⽣成,⼆是⾼活性,可使⾼分⼦链是瞬间形成,因此在不同转化率下分离所得聚合物的相对分⼦质量相差不⼤,延长反应时间只是为了提⾼转化率。
对逐步聚合,是官能团间反应,由于⼤部分单体很快聚合成⼆聚体、三聚体等低聚物,短期内可达到很⾼转化率,但因官能团活性低,故需延长反应时间来提⾼相对分⼦质量。
对活性聚合,活性中⼼同时形成,且⽆链转移和链终⽌,故随反应进⾏,相对分⼦质量和转化率均线性提⾼。
2.连锁聚合与逐步聚合的单体有何相同与不同?连锁聚合单体的主要反应部位是单体上所含不饱合结构(双键或叁键),在聚合过程中不饱合键打开,相互连接形成⼤分⼦链。
需要有活性中⼼启动聚合反应,为此多需⽤引发剂,反应活化能低,反应速率快,相对分⼦质量⾼。
逐步聚合单体的主要反应部位是单体上所带可相互反应的官能团,在聚合过程中官能团相互反应连接在⼀起,形成⼤分⼦链。
不需活性中⼼启动反应,但反应活化能⾼,为此多需⽤催化剂,反应速率慢,受平衡影响⼤,相对分⼦质量低。
3.凝胶点:出现凝胶化时的反应程度。
(逐步聚合概念)凝胶化:体形逐步聚合的交联反应到⼀定程度时,体系粘度变得很⼤,难以流动,反应及搅拌产⽣的⽓泡⽆法从体系中溢出,出现凝胶或不溶性聚合物明显⽣成的实验现象。
(逐步聚合概念)凝胶效应:⾃由基聚合中随反应进⾏体系粘度加⼤,妨碍了⼤分⼦链⾃由基的扩散运动,降低了两个链⾃由基相遇的⼏率,导致链终⽌反应速率常数随粘度的不断增加⽽逐步下降;另⼀⽅⾯,体系粘度的增加对⼩分⼦单体扩散的影响并不⼤,链增长反应速率常数基本不变,因⽽出现了⾃动加速现象。
这种因体系粘度增加引起的⾃动加速⼜称凝胶效应。
(⾃由基聚合概念)4.为什么在缩聚反应中不⽤转化率⽽⽤反应程度描述反应过程?在逐步聚合中,带不同官能团的任何两分⼦都能相互反应,⽆特定的活性种,因此,在缩聚早期单体很快消失,转变成⼆聚体、三聚体等低聚物,单体的转化率很⾼。
高分子化学第三章 自由基聚合Radical Polymerization

链终止反应:在一定条件下,增长链自由基失去活性形成 稳定聚合物分子的反应。可分为偶合终止和歧化终止。 ①偶合终止(Coupling termination):
两链自由基的独电子相互结合成共价键的终止反应。
CH2 CH + X CH X CH2 CH2 CH X CH X CH2
腈基对阴ቤተ መጻሕፍቲ ባይዱ子的稳定作用是使负电荷离域在碳-氮两原子上:
具有共轭体系的烯类单体 电子云流动性大,易诱导极化,可随进攻试剂性质的不同而取不 同的电子云流向,可进行多种机理的聚合反应。因此既可进行自 由基聚合,也可进行阴、阳离子聚合。如苯乙烯、 α-苯乙烯、丁 二烯、异戊二烯等。
R
+
H2C CH +
依据单烯CH2=CHX中取代基X电负性次序和聚合
倾向的关系排列如下:
阳离子聚合
取代基 X:
NO2
CN
COOCH3
阴离子聚合
CH
CH2
C6H5
CH3
OR
自由基聚合
表2-1 常见烯类单体的聚合类型
单体 中文名称 分子式 自由基 聚合类型 阴离子 阳离子 配位
乙烯
丙烯 正丁烯 异丁烯 丁二烯 异戊二烯 氯丁二烯 苯乙烯 α-苯乙烯 氯乙烯
CH3 C CH2
异丁烯
O C O CH3
甲基丙烯酸甲酯
1,2双取代的烯类化合物XCH=CHY,因结构对称,极化程度低, 位阻效应大,一般不能聚合或只能形成二聚体。但有时能与其 他单体共聚,如马来酸酐能与苯乙烯共聚。 三取代、四取代的烯类化合物一般不能聚合,但氟代乙烯例外。 例如:氟乙烯、1,1-二氟乙烯、1,2-二氟乙烯、三氟乙烯、四
续表
高分子化学公式

第一章绪论(Introduction)(1)分子量的计算公式:M0:重复单元数的分子量M1:结构单元数的分子量(2)数均分子量:N1,N2…N i分别是分子量为M1,M2…M i的聚合物分子的分子数。
x i表示相应的分子所占的数量分数。
(3)重均分子量:m1,m2…m i分别是分子量为M1,M2…M i的聚合物分子的重量W i表示相应的分子所占的重量分数(4)Z均分子量:(5)粘均分子量:α:高分子稀溶液特性粘度—分子量关系式中的指数,一般在 0.5~0.9之间(6)分布指数:分布指数第二章自由基聚合(Free-Radical Polymerization)(1)引发剂分解动力学:引发剂的分解速率:引发剂的浓度引发剂分解一般属于一级反应,因而分解速率为的一次方。
将上式积分得:进而得到半衰期(引发剂分解至起始浓度一半时所需的时间)对应半衰期时:,由前面的推导有:半衰期(2)自由基聚合微观动力学链引发速率:链增长速率:链终止速率:式中:kd、kp、kt分别为引发、增长及终止速率常数;[M]为体系中单体总浓度;为体系中活性种(自由基)的总浓度;f为引发剂效率。
推导如下:链引发反应由以下两个基元反应组成:式中:为初级自由基;为单体自由基。
若第二步的反应速率远大于第一步反应(一般均满足此假设),有:引入引发剂效率后,得引发速率的计算式如下:一般用单体的消失速率来表示链增长速率,即:链增长反应如下式:引入自由基聚合动力学中的第一个假定:等活性理论,即链自由基的活性与链长基本无关,即各步速率常数相等,kp1=kp2=kp3=…kp x=kp推得:自由基聚合一般以双基终止为主要的终止方式,在不考虑链转移反应的情况下,终止反应方程式如下:偶合终止:歧化终止:终止总速率:式中:Rtc为偶合终止速率;Rtd为歧化终止速率;Rt为总终止速率;ktc、ktd、kt为相应的速率常数。
在以上公式的基础上,引入处理自由基动力学的三个假设,得到以单体消耗速率表示的总聚合速率,其计算公式为:以及单体浓度随时间的变化关系为:若引发剂浓度可视为常数,则上式还原为:以上公式推导如下:自由基浓度较难测定,也很难定量化,因而无实用价值,引入处理自由基动力学的第二个假定——稳态假定,假定体系中自由基浓度在经过一段很短的时间后保持一个恒定值,或者说引发速率和终止速率相等,Ri=Rt即:解出:再引入处理自由基动力学的第三个假定:大分子的聚合度很大,用于引发的单体远少于增长消耗的单体,Ri <<Rp由此,用单体消失速率来表示的聚合总速率就等于链增长速率代入引发速率的表达式得:代入引发剂浓度随时间的变化关系得到:积分得:两边同时变号当引发剂的浓度可看作常数时即:即:此时:可略去高阶无穷小量得:(3)动力学链长及平均聚合度1)不考虑链转移反应自由基聚合过程中双基终止有两种方式,一种为双基偶合终止,另一种为双基歧化终止,二者所占的分率的不同将会引起平均聚合度的改变,但两种终止方式不会改变动力学链长的大小,二者的计算公式为:式中:Rtc为双基偶合终止的反应速率;Rtd为双基歧化终止的反应速率;Rp为链增长速率。
湖北大学高分子化学第三章 自由基聚合 总结

fkd kt
1/2 [I]1/2 [M]
[I] [M]
聚合反应速率
控制方向 RP RP
ERp = EP + Ed/2 - Et/2 >0
T反
RP
阻聚剂、缓聚剂
CZ 、[ Z ]
RP
引发剂
活性
RP
要求能从理论上进行解释和综合应用
总结
聚合反应速率
习题9
从理论上对引发剂引发自由基聚合的 聚合工艺及实验现象进行解释
小样2号比3号的相对分子质量稍低(?) 6、动力学分析表明:Rp = K [ M ]1.5[ I ]0.8(?) 7、如要进一步提高生产效率,可采取的有效措施(?)
聚合度 控制
总结
一、自由基寿命、动力学链长
聚合度控制
ν= Rp/Ri = Rp/Rt τ= [M·]/Rt
_
kP (2k t )1/2
聚合反应 速率控制
总结
基元反应
链引发:形成单体自由基活性种的反应
二步反应:
I
2R· (初级自由基)
R· + M → RM·(单体自由基)
链引发?
一 、
链增长:形成高分子链自由基的反应 n步反应: RM· + M → → → → ~~~~~~~~~~M·
基
决定大分子链的结构(序列、立构……)
自由基聚合 对结构的 控制能力
动力学方程:
1、三个假设
1/2
RP
k
P
fkd kt
[I]1/2 [M]
等活性、稳态、聚合度很大
2、与[I]成1/2次方, [M]成1次方的讨论(P78)
3、应用范围
高分子化学复习简答题(三)---自由基聚合(精)

高分子化学复习简答题(三)---自由基聚合学校名称:江阴职业技术学院院系名称:化学纺织工程系时间:2017年3月10日1、自由基聚合反应转化率-时间曲线特征。
答:诱导期:初级自由基为阻聚杂质所终止,无聚合物形成,聚合速率零。
若严格取除杂质,可消除诱导期。
初期:单体开始正常聚合,转化率在5%~10%以下(研究聚合时)或10%~20%(工业上)以下阶段称初期;此时转化率与时间近似呈线性关系,聚合恒速进行。
中期:转化率达10%~20%以后,聚合速率逐渐增加,出现自动加速现象,直至转化率达50%~70%,聚合速率才逐渐减慢。
后期: 自动加速现象出现后聚合速率逐渐减慢,直至结束,转化率可达90%~100%。
2、自由基聚合与缩聚反应的特征比较。
答:自由基聚合:(1)由基元反应组成,各步反应的活化能不同。
引发最慢。
(2)存在活性种。
聚合在单体和活性种之间进行。
(3)转化率随时间增长,分子量与时间无关。
(4)少量阻聚剂可使聚合终止。
线形缩聚:(1)聚合发生在官能团之间,无基元反应,各步反应活化能相同。
(2)单体及任何聚体间均可反应,无活性种。
(3)聚合初期转化率即达很高,官能团反应程度和分子量随时间逐步增大。
(4)反应过程存在平衡。
无阻聚反应。
3、为什么自由基聚合时聚合物的相对分子质量与反应时间基本无关,缩聚反应中聚合物的相对分子质量随时间的延长而增大?答:自由基聚合遵循连锁聚合机理:链增加反应的活化能很低,Ep=20~34KJ/mol,聚合反应一旦开始,在很短的时间内(0.01s~几秒)就有成千上万的单体参加了聚合反应,也就是生成一个相对分子质量几万~几十万的大分子只需要0.01s~几秒的时间(瞬间可以完成),体系中不是聚合物就是单体,不会停留在中间聚合度阶段,所以聚合物的相对分子质量与反应时间基本无关。
缩聚反应遵循逐步聚合机理:单体先聚合成低聚体,低聚体再聚合成高聚物。
链增加反应的活化较高,Ep=60KJ/mol生成一个大分子的时间很长,几乎是整个聚合反应所需的时间,缩聚物的相对分子质量随聚合时间的延长而增大。
高分子化学 第四章 自由基共聚合(2)-精选文档

1
三元(Tri-Component)共聚:
三种单体参加反应,共聚物由三个单体单元组成。
3种自由基;3个引发反应;9个增长反应;6个终止
反应;6个竞聚率
二元共聚: 2个引发反应;4个增长反应;3个终止反应;2个竞聚率
6个竞聚率:
M1-M2
r12 k 11 k 12
M2-M3
r 23 k 22 k 23
Valvassori-Sartori的稳态假定:
三元共聚物组成比为:
若三种单体的两两竞聚率已知,可估算其三元 共聚物组成。
4.6
一、竞聚率的测定 1、曲线拟合法
将多组组成不同的 单体配料(f1)进行共聚, 控制低转化率,共聚物分 离精制后,测定其组成F1, 作 F1 ~ f1 图,根据其图形 由试差法求得r1、r2。
13
4.7 单体和自由基的活性
回顾:
在均聚反应中,无法比较单体和自由 基的活性, 如
St St PS
k p 145
VAc VAc PVAc
单体活性 St>>VAc ????
k p 2300
原因:
1) 增长反应的kp的大小,不仅取决于M还 取决于M *; 2) 缺少比较的标准,参考体系不一致。
但这并不表示醋酸乙烯酯及其单体的活性 大于苯乙烯,因为均聚过程中,苯乙烯和醋酸 乙烯酯都只与自身的自由基进行共聚,因此相 互之间没有可比性。 事实上,苯乙烯的活性大于醋酸乙烯酯, 而它们的自由基的活性正好相反。 两种单体或两种自由基的活性只有与同种 自由基或单体反应才能比较。竟聚率可以用以 判别单体或自由基的相对活性。
d [ M ] [ M ] r [ M ] [ M ] 1 2 1 1 1 d [ M ] [ M ] r [ M ] [ M ] 2 2 2 2 1
高分子化学 科学出版社答案 (3)

第三章 自由基聚合1.以过氧化二苯甲酰作引发剂,在60℃进行苯乙烯聚合动力学研究,数据如下: (1)60℃苯乙烯的密度为0.887g/mL (2)引发剂用量为单体重的0.109% (3)R p =0.255×10-4 mol/ L·s (4)聚合度=2460 (5)f =0.80(6)自由基寿命τ=0.82s试求k d 、k p 、k t ,建立三个常数的数量级概念,比较[M]和[M·]的大小,比较R i 、R p 、R t 的大小。
全部为偶合终止,a =0.5解:设1L 苯乙烯中:苯乙烯单体的浓度[M]=0.887×103/104=8.53mol/L (104为苯乙烯分子量) 引发剂浓度[I]= 0.887×103×0.00109/242=4.0×10-3mol/L (242为BPO 分子量)代入数据 ⎪⎪⎩⎪⎪⎨⎧⨯=⨯⨯⨯=⨯⨯=⨯----)10255.0/53.8)(2/(0.8210255.0/53.8246053.8)104()/80.0(10255.044222/132/14t p t p t d p k k k k k k k解得:k d =3.23×10-6 s -1 10-4~10-6R p =k p (f k d k t)1/2[I]1/2[M]5.022][)(22=+==D Ca R aK M K pt p k n χpt p R M k k ][2⋅=τk p =1.76×102 L/mol·s 102~104 k t =3.59×107 L/mol·s 106~108[M·]=R p / k p [M]=0.255×10-4/(1.76×102×8.53)=1.70×10-8mol/L 而[M]=8.53mol/L 可见,[M]>>[M·]R i =2fk d [I]=2×0.80×3.23×10-6×4×10-3=2.07×10-8mol/L·s R t =2k t [M·]2=2×3.59×107×(1.70×10-8)2=2.07×10-8mol/L·s 而已知R p =2.55×10-5mol/L·s,可见R p >>R i =R t2.以过氧化二特丁基为引发剂,在60℃下研究苯乙烯聚合。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
聚合反应(%)
MMA聚合反应中,伴随着自加速作用,聚合物分子量增加的情况
链自由基的寿命随转化率增加而增加,即产物分子量随转 化率增加。
60
1
2
3
4
5
6
聚合速率(%)
40
7
8 9
10
20
0
400
800
1200
1600
2000
时间,min
溶剂对MMA聚合时自动加速效应的影响 沉淀剂: 1. 硬脂酸丁酯 2. 庚烷 3.环己烷 不良溶剂: 4、醋酸正戊酯 5、戊基氯 6、醋酸乙酯 7、四氯化碳 良溶剂: 8、苯 9、氯仿 10、二氯甲烷
稳定自由基类:自由基捕捉剂
. C6H5 . NO2 CH2-CH + N N C6H5 X NO2 C6H5 NO2
H NO2 CH=CH + N N C6H5 NO2 X
NO2
2,2-二苯基-2,4,6-三硝基苯肼(DPPH)
缓聚剂
活性小的阻聚剂,只能减缓聚合反应,不能停止 聚合反应。
对St:苯醌为阻聚剂, 硝基苯为缓聚剂。 阻聚或缓聚取决于单体或反应体系: 苯醌:St, 阻聚; MMA,缓聚。 硝基化合物:VAc, 阻聚;St, 缓聚。
链增长:
+ M + M + M ... . . . RM . RM2 RMx RM3
kp1 kp2 kp3
等活性理论(第一假设):链自由基的活性与链长无关
kp1= kp2 =... =kp
d[M] Rp=-( )p dt = kp[M] [RMi. ]
=kp[M] [M.] Rp=10-4-10-6 mol/L.S (2)
向单体的转移能力与单体的结构、温度等因素有关 。
向单体的链转移常数Cm×10-4
单体
30 50 0.15 0.27
温度(º C) 60 0.18 0.30 70 0.3 80 0.4
MMA AN
0.12 0.15
S
VAc VC
0.32
0.94 6.25
3、温度对聚合速率的影响 K=Ae-E/RT
1/2 1/2 fk d Rp=Kp( ) [I] [M] kt 1/2 K=Kp ( kd)? kt
?
E=(Ep-Et) + Ed 2 2
一般Ed=125 KJ/mol,Ep=29KJ/mol,Et=17KJ/mol 故,E=83KJ/mol,为正值。 温度升高,k增大。 E越大,温度对其影响较大。 引发剂的种类的选择和用量的确定是控制聚合速率的主要原因
时间 转化率
5.18
6.63
H
H
H
5.61 7.30 7.21 7.14
7.30
7.21
7 6 5 4 PPM 3 2 1 0
1.62 2.76 1.87 2.76 1.87 0.96 7.13 7.13 7.13
2.76
1.87 2.94 1.34 7.13 7.13
7.18 7.08
7.18 7.08
三、自动加速现象(autoacceleration)
80
40% 100% 80% 60% 20% 10%
聚合反应(%)
60 40
20
500
1000
1500 (min)
时间 在不同浓度的苯溶液中,MMA在50º C,BPO的聚合反应
当kdf及[I]、[M]等值不变或变化不大时,若有某种原因能使kp增大 或kt减小时,都能使Rp增大,而各方面的研究报道皆认为自动加速 作用是由于kt减少所致。
S-1 L/mol.S L/mol.S mol/L.S mol/L.S mol/L.S mol/l mol/l S mol/l -
10-4-10-6 102-104 106-108 10-8-10-10 10-4-10-6 10-8-10-10 10-7-10-9 10-0.1 0.1-10 10-2-10-4 10-4-10-6
电荷转 移型
芳香胺 阻碍酚
Mn.+ ArNAr H
Mn.+R'
MnH + ArNAr .
R OH R R' R O. R
CH3
MnH+
FeCl3 CuCl2
Mn.+ FeCl3
MnCl+FeCl2
O2:室温为阻聚剂,高温为引发剂 Mn. +O2 Mn-O-O. Mn-O-OR
高温
MnO. + RO.
RH +CH2=CH-CY H
.
CH2-CH=CHY
.
这类单体用自由基聚合得不到高分子量的聚合物.
Chapter 3 自由基聚合动力学
( Kinetics of Free Radical Polymerization )
一、聚合反应速率的表示与测定
1、 概述
转化率
1 2 3
4
时间
1、诱导期 2、初期 3、中期 4、后期
常用的阻聚剂
加 成 型
苯醌 硝基化合 物
对苯二醌 邻苯二醌 硝基苯 多硝基苯 2,4二硝 基氯苯 β苯基萘胺
C(CH3)3 OH C(CH3)3
Mn.+ O O
Cl NO2 R. NO2 Cl
MnO
O.
R. Mn O
OR
Mn.+
H Cl . H NO2 Mn NO2
RH + Mn
NO2
链 转 移 型
自动加速现象在良溶剂中较少出现
四、聚合过程中速率变化类型
3 2 1
转化率
时间
1、S型曲线 低活性引发剂
2、匀速反应 合适引发剂
3、前快后慢 高活性引发剂
五、聚合物的分子量与分子量分布
聚合物的分子量、分子量分布与聚合物的强度、力学性能、热稳定 性、加工性、溶液性质密切相关。
一、聚合物的分子量 平均聚合度是每生成一个聚合物分子所消耗的单体的分子的平均数 数均聚合度Xn= 单体分子转化数 聚合物分子生成数 单体转化速度 聚合物分子生成速度
1.179
1.062
20.6
14.5
0.906
44.4
C=k V C=1, 1 k= V V= V
简单膨胀计
V = Vm - Vp =Vm -Vm dm dp =Vm(1- dm ) dp C = Vm(1V dm ) dp
ΔV:反应时间t时体积收缩; ΔV∞:单体100%转化为聚合物时的体积收缩; Vm:纯单体起始体积; Vp:单体100%转化为聚合物时聚合物的体积; dp:聚合物的密度;dm:纯单体的密度。
7.18 7.08
7.18 7.08
7.18
7
6
5
4 PPM
3
2
1
0
膨胀计法测定聚合速率
原理: 聚合物的密度比单体大, 聚合后发生体积收缩。 测定聚合反应中随时间发生的体积变化。
单体和聚合物密度
单体 VC AN MMA St B 密度(g. cm-1, 25º c) 单体 0.919 0.800 0.940 0.905 0.6276 聚合物 1.406 1.170 聚合时体积收缩 (%) 34.4 31.0
1 2KtRp [S] [I] C + = + C + M Cs I xn Kp2 M2 [M] [M]
(双基歧化终止)
1
Kt Rp [S] [I] + + C = + C xn Kp2 M2 M I [M] Cs [M]
(双基偶合终止)
(1) 向单体的转移
1 2KtRp = + CM 2 2 xn Kp M
10-4 10-3
10-2
1/2 [M]o 1/2 fk d Ln kp( = ) [I] t [M] kt
(低转化率5-10%,低活性氧化剂)
10-1 [I] 1/2 ,mol/l
Mx10, mol/L MMA
讨论: (1) 单基终止: Rp=B[I] (2)单基和双基终止并存: Rp= A[I]1/2+ B [I]
Rp
不考虑向大分子转移 Rp Xn= Rt +ΣRtr
=
Rt + Rtr.M+ Rtr.I+ Rtr.S
双基终止部分暂作歧化终止考虑
2KtRp Ktr.M 1 K [S] tr.I[I] + Ktr.S + + = Kp2 M2 Kp xn Kp[M] Kp[M] Ktr = C Kp (链转移常数, 代表两反应的竞争能力)
3/2 1/2 初级自由基与单体引发反应较慢 Rp=Kp( fkd ) [I] [M] kt Rp= k[I]n[M]m (n=0.5-1.0, m=1-1.5) 综合情况:
( 3)
1/2
自由基聚合参数
参数 单位 数值范围
kd kp kt Ri Rp Rt [M.] [M] τ [I] Kp/kt
阻聚剂和缓聚剂
一、阻聚条件和阻聚剂
1 80 1:无阻聚剂 2 4 40 20 2000 4000 6000 3 2:0.1%苯醌 3:0.5%硝基苯
转化率 (%) 60
4:0.2%亚硝基苯
苯乙烯加热聚合反应 时间(min)
Mn.+ A
MnA.+ M
ktrA
MnA.
MnAM.
此步很易进行Ktr.A>>Kp 此步极难,Ka=0
1/2 Ri . ) [M ]=( 2kt
聚合度很大(假设三): 一般高分子聚合度很大, 用于引发的 单体远少于增长消耗的单体 Ri<<Rp R=Ri+Rp≈Rp