第一章卤化反应
《卤化反应 》课件

目 录
• 卤化反应概述 • 卤化反应机理 • 卤化反应的条件与影响因素 • 卤化反应的工业应用 • 卤化反应的安全与环保 • 卤化反应的前沿进展与展望
01
卤化反应概述
定义与分类
定义
卤化反应是指将其他元素或基团替换 为卤素(氟、氯、溴、碘)的反应。
分类
根据卤化反应中卤素的不同,可以分 为氟化、氯化、溴化和碘化等。
详细描述
亲核取代卤化反应中,亲核试剂(如醇、胺等)进攻卤代烃的碳原子,卤素原子被取代基取代。这个反应过程中 ,亲核试剂首先与卤代烃形成络合物,然后发生取代反应,生成新的碳-碳键和卤化物。
消除反应卤化
总结词
不饱和烃在加热条件下发生消除反应,同时生成碳-卤键。
详细描述
消除反应卤化中,不饱和烃在加热条件下发生消除反应,同 时生成碳-卤键。这个过程中,不饱和烃首先形成不稳定的消 除中间体,然后发生消除反应,生成新的碳-卤键和烯烃。
氟代烃的合成工艺难度较大, 且氟气具有剧毒和强腐蚀性, 因此研究和应用相对较少。
05
卤化反应的安全与环 保
卤化反应的危险性
卤化反应通常涉及高温、高压和有毒有害物质,操作不当可能导致火灾、爆炸等安 全事故。
卤化反应过程中产生的废气、废水和废渣等废弃物,如未经妥善处理,可能对环境 造成严重污染。
卤化反应过程中使用的原料和催化剂等物质,如对人体有害,可能对操作人员的健 康造成危害。
高选择性卤化反应的研究
研究高选择性卤化反应,以实现特定位置或特定结构的卤化,提高产物的纯度和 收率。
开发高选择性卤化反应的机理和动力学模型,为优化反应条件和提高产物选择性 提供理论支持。
卤化反应在绿色化学领域的应用
探索卤化反应在绿色合成中的实际应用,如药物合成、材料 制备和生物活性分子合成等。
《药物合成反应》第一章卤化反应课件

亲核卤化反应是一种常见的有机合成方法,具有操作简便、产物纯度高、产率 较高等优点。
常见的亲核卤化试剂
氯化氢(HCl)、溴 化氢(HBr)、碘化 氢(HI)等氢卤酸。
氯化亚砜(SOCl₂) 、溴化钠(NaBr) 、碘化钾(KI)等卤 化物。
氯气(Cl₂)、溴( Br₂)、碘(I₂)等卤 素单质。
03
亲电卤化反应
定义与特点
总结词
亲电卤化反应是指卤素与带有部分正电荷的碳原子相 连的反应,其特点是卤素取代碳上的氢原子或取代基 。
详细描述
亲电卤化反应是一种常见的有机合成反应,其特点是 卤素(如氯、溴、碘)与有机分子中的碳原子相连, 形成新的碳-卤键。这种反应通常发生在带有部分正电 荷的碳原子上,因此被称为亲电卤化反应。在反应过 程中,卤素原子取代了碳上的氢原子或取代基,生成 新的有机化合物。亲电卤化反应是一种重要的有机合 成手段,在药物合成和其他化学领域中广泛应用。
卤化反应在药物合成中的应用
1 2
引入卤素官能团
在药物合成中,卤化反应常用于引入卤素官能团 ,如氟代、氯代等,以改变药物的理化性质和药 效。
增加药物稳定性
卤化反应可以增加药物的稳定性,如将烯醇式结 构转化为卤代烃,提高药物的化学稳定性。
3
调节药物的代谢和排泄
通过卤化反应可以调节药物的代谢和排泄,如将 羟基或氨基等代谢敏感基团替换为卤素,降低药 物的代谢速度和排泄速度。
实例
以苯酚的溴化为例,苯酚与溴在光照条件下发生自由基溴化反应,生成2-溴苯酚。在这个反应中,溴原子取代了 苯酚中的羟基氢原子,形成了一个新的碳-溴键,同时生成了一个苯氧自由基。
05
卤化反应的选择性与控制
选择性卤化反应的条件与影响因素
卤化反应在有机合成中的应用

第一章卤化反应周子豪药学院08级 20083022900261.卤化反应在有机合成中的应用?为什么常用一些卤代物作为反映中间体?a)制备特定活性的化合物:药物、兽药、农药、杀虫剂除草剂等b)构成新的有机化合物:成链,成环,官能团转换等c)提高有机物的反应性能:取代、加成、消除、缩合、聚合等d)引入卤素原子作为保护基、阻断基等X原子的引入可以使有机分子的理化性质、提高反应性能,为重要的有机合成中间体, C-X很容易转化成其它官能团2.归纳常用的氯化剂、溴化剂有哪些?它们的主要理化性质及适用对象和范围?答:氯化剂主要是氯气Cl2,具有足够的电负性,它本身在反应中发生极化而参加反应;也可用HOCl,CH3CO2Cl,氯化硫S2Cl2、硫酰氯SO2Cl2和次氯酸叔丁酯t-BuOCl,均可以释放氯正离子作为亲电试剂。
用分子溴的取代反应,通常在醋酸中进行,若在反应中加入碘,可以提高反应速度;其他溴化剂包括NBS、HOBr、酰基次溴酸酐(AcOBr、CF3CO2Br等),尤其后者特别有活性。
3.比较X2,HX,HOX对双键离子型加成反应机理有什么异同,如何判断加成产物的立体构型?答:X2:卤素负离子进攻碳正离子;HX对双键加成;HOX中的卤素正离子对烯烃的双键做亲电攻击,形成桥卤三圆环过渡态,再由水分子或者OH对其亲核进攻。
立体构型一般取决于反应中间体的结构,若中间体为离子形式,则最终产物正反各一半,如果中间体形成溴鎓离子,则最后产物为反式。
区域选择性卤化反应In chemistry, regioselectivity is the preference of one direction of chemical bond making or breaking over all other possible directions. It can often apply to which of many possible positions a reagent will affect, such as which proton a strong base will abstract from an organic molecule, or where on a substituted benzene ring a further substituent will add.A specific example is a halohydrin formation reaction with 2-propenylbenzene:The reaction product is a mixture of two isomers and the regioselectivity is said to be poor.Regioselectivity in ring-closure reactions is subject to Baldwin's rules.参考文献Reactivity–selectivity principleIn chemistry the reactivity–selectivity principle or RSP states that a more reactive chemical compound or reactive intermediate is less selective in chemical reactions. In this context selectivity represents the ratio of reaction rates.This principle was generally accepted until the 1970s when too many exceptions started to appear. The principle is now considered obsolete .A classic example of perceived RSP found in older organic textbooks concerns the free radical halogenation of simple alkanes. Whereas the relatively unreactive bromine reacts with2-methylbutane predominantly to 2-bromo-2-methylbutane, the reaction with much more reactive chlorine results in a mixture of all four regioisomers.Another example of RSP can be found in the selectivity of the reaction of certain carbocations with azides and water. The very stable triphenylmethyl carbocation derived from solvolysis of the corresponding triphenylmethylchloride reacts a 100 times faster with the azide anion than with water. When the carbocation is the very reactive tertiary adamantane carbocation (as judged from diminished rate of solvolysis) this difference is only a factor of 10.Constant or inverse relationships are just as frequent. For example a group of 3- and 4-substituted pyridines in their reactivity quantified by their pKa show the same selectivity in their reactions with a group of alkylating reagents.The reason for the early success of RSP was that the experiments involved very reactive intermediates with reactivities close to kinetic diffusion control and as a result the more reactive intermediate appeared to react slower with the faster substrate.General relationships between reactivity and selectivity in chemical reactions can successfully explained by the Hammond postulate.When reactivity-selectivity relationships do exist they signify different reaction modes. In one study the reactivity of two different free radical species (A, sulfur, B carbon) towards addition to simple alkenes such as acrylonitrile, vinyl acetate and acrylamide was examined.The sulfur radical was found to be more reactive (6*108 vs. 1*107 mole-1.s-1) and less selective (selectivity ratio's 76 vs 1200) than the carbon radical. In this case the effect can be explained by extending the Bell–Evans–Polanyi principle with a factor accounting for transfer of charge from the reactants to the transition state of the reaction which can be calculated in silico:with the activation energy and the reaction enthalpy change. With the electrophilic sulfur radical the charge transfer is largest with electron-rich alkenes such as acrylonitrile but the resulting reduction in activation energy (βis negative) is offset by a reduced enthalpy. With the nucleophilic carbon radical on the other hand both enthalpy and polar effects have the same direction thus extending the activation energy range.[edit] External linksReactivity–selectivity principle Gold Book Link[edit] References1.^ Minireview The Reactivity-Selectivity Principle: An Imperishable Myth in OrganicChemistry Herbert Mayr, Armin R. Ofial Angewandte Chemie International Edition Volume 45, Issue 12 , Pages 1844 - 1854 Abstract2.^ Search for High Reactivity and Low Selectivity of Radicals toward Double Bonds: TheCase of a Tetrazole-Derived Thiyl Radical Jacques Lalevée, Xavier Allonas, and Jean Pierre Fouassier J. Org. Chem.; 2006; 71(26) pp 9723 - 9727; (Article) doi:10.1021/jo061793w3.^ Sulfur tetrazole radical derived from photolysis of disulfide and carbon radical derivedfrom photolysis of t-butylperoxide followed by proton abstraction from triethylamineElectrophilic halogenationIn organic chemistry, an electrophilic aromatic halogenation is a type of electrophilic aromatic substitution. This organic reaction is typical of aromatic compounds and a very useful method for adding substituents to an aromatic system.A few types of aromatic compounds, such as phenol, will react without a catalyst, but for typical benzene derivatives with less reactive substrates, a Lewis acid catalyst is required. Typical Lewis acid catalysts include AlCl3, FeCl3, FeBr3, and ZnCl2. These work by forming a highly electrophilic complex which attacks the benzene ring.[edit] Reaction mechanismThe reaction mechanism for chlorination of benzene is the same as bromination of benzene. Ferric bromide and ferric chloride become inactivated if they react with water, including moisture in the air. Therefore, they are generated in situ by adding iron fillings to bromine or chlorine.The mechanism for iodination is slightly different: iodine (I2) is treated with an oxidizing agent such as nitric acid to obtain the electrophilic iodine (2 I+). Unlike the other halogens, iodine does not serve as a base since it is positive. In one study the iodinization reagent is a mixture of iodine and iodic acid.[1]In another series of studies the powerful reagent obtained by using a mixture of iodine and potassium iodate dissolved in concentrated sulfuric acid was used. Here the iodinating agent is the tri-iodine cation I3+ and the base is HSO4-. In these studies both the kinetics of the reaction and the preparative conditions for the iodination of strongly deactivated compounds, such as benzoic acid and 3-nitrobenzotrifluoride, were investigated.[2][3]Halogenation of aromatic compounds differs from the halogenation of alkenes, which do not require a Lewis Acid catalyst. The formation of the arenium ion results in the temporary loss of aromaticity, which has a higher activation energy compared to carbocation formation in alkenes. In other words, alkenes are more reactive and do not need to have the Br-Br or Cl-Cl bond weakened.[edit] ScopeIf the ring contains a strongly activating substituent such as -OH, -OR or amines, a catalyst is not necessary, for example in the bromination of p-cresol:[4]However, if a catalyst is used with excess bromine, then a tribromide will be formed.Halogenation of phenols is faster in polar solvents due to the dissociation of phenol, with phenoxide ions being more susceptible to electrophilic attack as they are more electron-rich.Chlorination of toluene with chlorine without catalyst requires a polar solvent as well such as acetic acid. The ortho to para selectivity is low:[5]No reaction takes place when the solvent is replaced by tetrachloromethane. In contrast, when the reactant is 2-phenyl-ethylamine, it is possible to employ relatively apolar solvents with exclusive ortho- regioselectivity due to the intermediate formation of a chloramine making the subsequent reaction step intramolecular.The food dye erythrosine can be synthesized by iodination of another dye called fluorescein:This reaction is driven by sodium bicarbonate.[6][edit] References1.^ Regioselective iodination of hydroxylated aromatic ketones Bhagwan R. Patila, SudhakarR. Bhusarec, Rajendra P. Pawara, and Yeshwant B. Vibhute b Arkivoc 2006 (i) 104-108. Online Article2.^ The kinetics of aromatic iodination by means of the tri-iodine cation, J. Arotsky, A.C. Darby and J. B. A. Hamilton, J. Chem. Soc. B, 1968, 739 - 7423.^ Iodination and iodo-compounds Part IV, Judah Arotsky, A. Carl Darby and John B. A.Hamilton, J. Chem. Soc., Perkin Trans. 2, 1973, 595 - 5994.^ A. Sankaranarayanan and S. B. Chandalia (2006). "Process Development of the Synthesisof 3,4,5-Trimethoxytoluene". Org. Process Res. Dev. 10 (3): 487–492.doi:10.1021/op0502248.5.^ J. L. O'Connell, J. S. Simpson, P. G. Dumanski, G. W. Simpson and C. J. Easton (2006)."Aromatic chlorination of ω-phenylalkylamines and ω-phenylalkylamides in carbontetrachloride and α,α,α-trifluorotoluene". Organic & Biomolecular Chemistry 4 (14): 2716–2723. doi:10.1039/b605010g.6.^ Synthesis of Triarylmethane and Xanthene Dyes Using Electrophilic Aromatic SubstitutionReactions James V. McCullagh and Kelly A. Daggett J. Chem. Educ. 2007, 84, 1799. Abstract。
第一章 卤化反应

-
X R3 R1 C C R2 R4 X
R1 R2
X R3 C C R4 X
注:卤负离子究竟从三员环背面进攻哪一个碳原子, 取决于形成碳正离子的稳定性。 碳正离子的稳定性:叔>仲>伯 连有烷基、烷氧基、苯基等给电子基团的烯键 碳原子是卤负离子优先进攻的位置。
R1 R3 C C R2 R4 X (2)
δ X
-
R2 R1
X C C X
R3 R4
立体化学问题
H H CH3 + CH3 Br2 Br H H CH3
(a)
+ Br
CH3
(b)
(a)
(b)
H Br H
CH3 Br H
H
Br
CH3
CH3
CH3 Br
CH3 H Br H Br CH3
CH3 H Br Br H CH3
四种常用的N-卤代酰胺:
N-溴(氯)代乙酰胺
O H3C C NHBr (NBA)
O H3C C NHCl (NCA)
O
O N Cl O (NCS)
N-溴(氯)代丁二酰亚胺
N Br O (NBS)
•
定位:遵循马氏规则
H Ph C CH2 NBS / H2O H Br C CH2 Ph OH
R1 H
I O I2/KI/NaHCO3 O
CH2 COOH
H C C
H Ph Br2 第二步
Br Ph
O
C
O
OH
O
四、烯烃与次卤酸(酯)及N-卤代酰胺的加成
1. 次卤酸与烯烃加成,按照马氏规则,卤素加成在双键 的取代较少的一端,生成β -卤醇。
第一章 有机化学-卤化反应

X
H
CH3
Cl2 /Fe
例
+
OH H2O 3Br2 OH H2O 2Br2 Br
OH Br
OH Br 2Br2/Bu-NH2 °C -70 OH Br
1molBr2 /CS2 0°C
OH Br
Br OH
OH
Br
Br
OH Br2/CS2 CH3
OH
CH3 Br
OH Br2 CH3
OH Br
CH3
NH2 NBS/DMF
O BrH2C C CH2CH2CH3 (1.5%) +
α-羰基自由基取代
O
O R' C
H C R''' Br2 R''
O
+
Br2
+ 光
O
光
Br C R''' R''
R'
C
2Br OH O + HBr Br
选择性溴化剂
O Br2 Br Br O
副反应
O Br Br
+
O
O Br
+
OH Br Br
Br
O CH3 BuH2C C
Si CH 2
Bu
OSi C C CH3
+
H Br2
-78℃
O BuH2 C C
△
O BuHC C Br
CH2 Br +
CH 3
反应机理
X C C OSi X-X C X C O- Si X C O C + XSi
例
OHC
COOEt ClSi
1-卤化反应

Cl CM e3
桥式(环状)卤正离子
M e3C C H C Cl CM e2 Me H
反式(anti)加成
M e3C H H CH2 Me C Me
H
重排产物
M e3C
Cl C C
H CH2
Me C Me (40%*) H
过硫酸氢钾(oxone, 2KHSO5· KHSO4· K2SO4)与NaCl或NaBr 反应能迅速产生氯或溴,进而与不饱和键加成。 这种方法可避免直接使用氯或溴
在脂环烯的卤素加成反应中,由于存在邻位取代基等
空间位阻,卤素必然进攻在平面位阻较小的一方而形成桥卤正离 子,然后卤负离子也从环背面进攻有利于碳正离子的部位,生成 反式1,2-双直立键的二卤化物
C1 6H30
Me HO
C16H30
Br2/AcOH AcONa/Et 2O 20-25
o
Me HO Br
C16H30
苯环亲电取代反应的一般模式
+ E+
亲电试剂
E+
p -络合物
+
H E
E
+ H+
-络合物
-络合物的表达方式
H
E + +
H
E
+
H
E
H
+ E
共振式
离域式
反应机理
+ Cl-Cl 快 Cl
+
Cl Cl
- AlCl3
慢
Cl
+
_
H
ClAlCl3
快
+ AlCl3 + HCl
+
Br-Br
快
第一章卤化反应-第三节

2、与氯化亚砜的反应
ROH
SOCl2
RCl
HCl
SO2
优点: ①反应活性较高。 ②产物容易分离纯化,且异构化等副反应少,收 率较高。 ③选用不同溶剂,可得到指定构型的产物。 ④可以与其他试剂合用增强其选择性等。 缺点: ①反应中大量的HCl和SO2气体逸出会污染环境。 ②氯化亚砜易水解,需在无水条件下反应
碱催化与酸催化相反,卤原子是吸电子基, 它所连的α-碳上在碱的作用下更容易离去, 若在过量卤素存在下,所有的α-H原子都被 取代。
在a位具卤素等吸电子基时,卤代反应受到 阻滞,故在同一个α位碳原子上欲引入第二 个卤原子相对比较困难。若在α`位具活性 氢,则第二个卤素原子优先取代α`位氢原 子。如2-丁酮在和2mol溴素反应时,只得 到α,α'-二溴代丁酮。
卤化试剂(19)用于对α,β-不饱和酮的α′-卤取代反应中,能 够减少双键加成副反应。 卤化试剂(20)和(21)的特点是亲电活性大,不需要任何催 化剂,反应条件温和,只得到单溴代物,且在反应中不生成 卤素分子和卤化氢,特别适用于对酸、碱敏感的酮。 卤化试剂(22)可在温和条件下对羰基α位及苄位、烯丙位进 行氯代反应
PPh3
(PhO)3P
X2
RX
Ph3PX2
(PhO)2P
ROH
RX
R'OH
Ph3P O
R'X (PhO)2P O R
RX
该类卤化试剂具有活性大,反应条件温和、不易 发生由卤化氢引起的副反应。
Ph3P催化卤化机理
O Ph3P + X2 Ph3PX2 + ROH ROPPh3X + HX RX + PPh3
四溴环己二烯酮(不发生双键加成反应) 在少量HCl或HBr气体催化下,反应中以生成稳 定的三溴苯酚为动力,促使4位碳一溴键异裂, 生成的溴正离子向α,β-不饱和酮的α'位C-H作亲 电取代;同时,能有效地消除x-,于是可得到收 率良好的α'-溴代-α,β-不饱和酮。
第一章 卤化反应

2. 苄位、烯丙位的卤取代 苄位、
烯丙位、苄位氢原子较活泼,在较高温度及存在自由基引 发剂条件下,可用卤素、N-卤代酰胺、次卤酸酯等卤化剂于非 极性惰性溶剂中进行。 其中以N-卤代酰胺,尤其是NBS(N-溴代丁二酰亚胺)效果最 好,反应主要为三步: ①
X2 hν 或或或 引引卤 hν 或或或或 引引卤 X
R1 R2 X
R3 X R4 OH H2O R1 R2 OH R3 X R4 OH R1 R2
OH R3 X R4
4. 与卤化氢的加成
I2/KI/NaHCO3 H2O/r.t.4h
H2C H O C O I H H I H O O
88%
反应两步完成:① I2从位阻小的双键方向进攻,生成过渡态; ② 羧酸氧负离子于β方向进攻三元环发生亲核进攻生成酯。
H
H C C Ph CO2H
Br2/CHCl3 。 0 r.t.20min
Br
H
H
Cl C 2H5 Cl2/CH3CO2H C C CH3CH CH C 2H5 。 25 H H Cl
H3C
OCOCH3 Cl + CH3CH CH C2H5 + CH3CH CH C2H5 Cl OCOCH3
H
I2/AcOAg/Et2O
OAc I H
80%
2. 与N-卤代酰胺的加成 卤代酰胺的加成
Me C16H30 Br2/AcOH AcONa/Et2O HO
Me HO H Br
20-25°
Br
HO H Br M e C 16H30 Br
(84~85%)
当卤加成发生在亲核性溶剂(如H2O、ROH、RCO2H) 中时,因亲核试剂中的亲核基团也可进攻碳正离子过渡 态,故反应可得除1,2-二卤化和物外的其它产物。
1-卤化反应

HO(CH 2)6OH
I(CH2)6I
物质,除去生成的HI,促使反应顺利进行.
eg. 醋酸可的松等甾体抗炎激素的半合成,其C17
位的β-甲基酮一般在碱CaO或NaOH存在下,于有 机溶剂中加I2液,产物碘代酮不分离,接着与 I KOAc反应,结果在C12位引入乙酰氧基.
CaO + H2O NaOH + HI
Ca(OH)2 NaI + H2O CH3 C=O
RCH=CH + Br 2
.
RCHCH Br 2
.
.
加成取向:碳自由基稳定性(反马氏规则的应用)
HBr (g) CH2=CH-(CH2)8-COOEt (PhCOO)2, 0℃ Br-(CH2)10-COOEt (70%)
CH2=CHCH2Cl
HBr, NaBr (PhCOO)2, -5℃
BrCH2CH2CH2Cl
CH3COCH2CH3
BrCH2COCHBrCH3
(55~58%)
(4)溴化氢 反应中生成的HBr有两个作用: a,加快烯醇化速度 b,具有还原作用,消除α-溴酮中的溴原 子,使α-溴化效率降低. 为此常在反应中条加适量NaAc或吡啶,中 和HBr.
2,碱催化下的 卤代 ,碱催化下的α-卤代
a. 反应机理
eg:
(CH3)3CCOCH3 NaOH (CH3)3CCONa H2O H
+
(CH3)3CCOH
CO—CH3
NaOCl, Cl2
H2O, H +
COOH
(CH3)2=CHCOCH3
Cl2, NaOH, H2O H + 49~53%
第1章 卤化

注意事项: 1) 氢原子和卤原子的定位符合马尔科夫尼科夫(Mark ovnikov)规则,即氢原子加到含氢原子多的碳原子 上 2) 当烯烃上带有强的吸电性取代基,如-COOH、-CN, -CF3,-N(CH3)3时,使烯烃的电子云向取代基方向 转移,烯烃与卤化氢加成时活性降低,且定位方向 正与马尔科夫尼科夫规则相反 3) 卤化氢的活性次序为:HI > HBr > HCl
+ Ph2C=C-CH2Br Ph 23%
二、不饱和羧酸的卤内酯化反应
• 某些不饱和羧酸的双键上形成环状卤正离子时, 若立体条件许可,亲核性羧酸负离子向其进攻可 生成卤代五元或六元内酯称为卤内酯化反应 (halolactonization)。此反应与烯烃的对向卤加成 历程相似,在碱性条件下是高度立体选择性的。 • 在有机合成上,利用这一方法,可将不饱和羧酸 转化成用其他方法难以制得的内酯
R1 H
C
C
R2 H
NBS/DMSO
Br R1 R2 C C H H Me O S Me
Br R1 H
R2
C
C O SMe2
H
H2O
Br C H R1 C β OH H
R2
Dalton反应 β-溴醇
-消 除 (在 燥 MSO中 干 D )
R2 Br α-溴酮 C R1 C O α H
四、卤化氢对不饱和烃的加成反应
3)位阻的影响 无位阻,机会均等,形成外消旋混合物; 有位阻,在位阻小的一侧形成三元环 例3:
HO ACONa/Et2O Me Br HO Br Me Br2/AcOH HO + Br BrMe
对于刚性环烯,立体选择性主要取决于中间体卤鎓 离子的稳定性
4) 有重排产物生成,生成更稳定的C+离子
[医学]药物合成反应第一章卤化反应-0
![[医学]药物合成反应第一章卤化反应-0](https://img.taocdn.com/s3/m/7fc42647b84ae45c3b358cf9.png)
孙丽萍 张灿 孙宏斌 黄文龙 张惠斌
赖宜生
张大永 钱海 柳红
考试科目
药物化学- 有机合成综合
业务课复试参考书目(仅供参考、不作业务课命题依据)
参考书目咨询qq:2233244831
参考书目,编著者及出版者、版本
《药物合成反应》闻韧主编,化学工业出版社,2003年,第二版。 《新编有机合成化学》黄宪等主编,化学工业出版社,2003年,第一版。 《药物化学》(第二版)尤启冬主编,化学工业出版社,2008年。(此参考书目也适用于面试
8
药物合成反应
Organic Reactions for Drug Synthesis
课程教学目的: 本课程的教学目的是使学生在学习有关基础课后(如有机化
学等),能较系统地掌握常见的重要有机药物合成反应、反应 的影响因素、反应的选择性及其实际应用,培养学生在药物合 成中的实际工作能力,并具有发现问题、分析问题和解决问题 的初步能力,为学生学习“药物化学”和“制药工艺学”奠定 基础。
10
学习方法和要求
学习方法: 在学好有机化学的基础上,掌握重要药物合成反应,将官能团
反应性、试剂活性、反应条件之间的关系进行联系、比较,以达 到牢固掌握药物合成的方法及其重要反应。
学习安排: 课堂讲授为主、自学为辅。课堂上重点突出,讲解主要内容及
难点,因课时限制,部分内容要求同学自学,仍属大纲中要求掌 握的内容。本课程进行期终考试。
药物合成反应_第一 章___卤化反应-0
前途有木有?
2
3
4
中 国 药 科 大 学二○一二年硕士研究生招生简章与招生目录
院系所、专业、研究方向
导师
001药学院
100701药物化学
药物合成反应(闻韧_第三版)第一章课后答案Chapter_1_Halogenation_Reaction
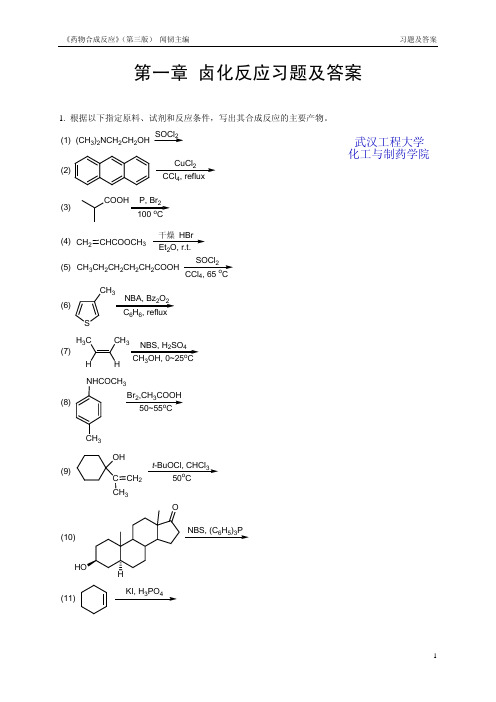
第一章 卤化反应习题及答案1. 根据以下指定原料、试剂和反应条件,写出其合成反应的主要产物。
(1)(CH 3)2NCH 2CH 2OHSOCl 2(2)CuCl 24(3)P, Br 2o(4)CH 2CHCOOCH 3干燥 HBr 2 (5)CH 3CH 2CH 2CH 2CH 2COOHSOCl 24o(6)S CH 32266(7)H 3C CH 3NBS, H 2SO 43o(8)NHCOCH 3CH 3Br 2,CH 3COOH o(9)OHC CH 3CH 2t -BuOCl, CHCl 3o(10)NBS, (C 6H 5)3P(11)KI, H 3PO 4武汉工程大学化工与制药学院(12)C 6H 5H 3Br 2,Cl 4o (13)CH 3CH CH CH 3232(14)(CH 3)3CCH 2OHHBr(15)OOP2(16)NBS, Et 3N ·3HF 22o(17)OHBr 24o(18)O23o2. 在下列指定原料和产物的反应式中分别填入必需的化学试剂(或反应物)和反应条件。
(1)CH 3CH 2CH 2CH 2CH CHCH 3CH 3CH 2CH 2CHCHCHCH 3Br(2)COOHBr(3)(4)OHBr(5)2CH 2BrBr(6)(7)(CH 3)3CCH 2OH(CH 3)3CCH 2Br(8)OOBocHNO OBocHNBr(9)OOBr OO BrBr2. 在下列指定原料和产物的反应式中分别填入必需的化学试剂(或反应物)和反应条件。
(参考答案)题号答案注释(1) NBS/(PhCO)2O, CCl4, △(2) Br2/HgO/tetrachloroethaneNaNO2, HCl, H2O; 2. HPF6; 3. △ (168℃)(3) 1.(4) Ph3P, Br2, CH3CN, △ (200-340℃)(5) NBS/(PhCO)2O, CCl4refluxing (10min. )acetone,(6) NaI,(7) Bu3P, Br2, DMF(8) NBS/hv, CCl4(9) NBS/(PhCO)2O, CCl4, reflux3. 阅读(翻译)以下有关反应操作的原文,请在理解基础上写出:(1)此反应的完整反应式(原料、试剂和主要反应条件);(2)此反应的反应机理(历程)。
《药物合成反应》第1章卤化反应

1. 醇和卤化氢或氢卤酸的反应 反应机理:
R1
R1
R2
H+ C OH
R2
C
+
OH2
R3
R3
R1
-H2O 慢
R2
C+
R3
R1 R2 C X
R3
(SN1机理)
X慢
R1
R1
XR2
C
+ -H2O
OH2 R3
XC R2
R3
(SN2机理)
主要影响因素: ①水
加入去水剂除去生成的水使反应向有机氯化物 方向移动 ②醇的结构
CH3 – CH = CH2 + Br2 CH2Br
Hλ 或(PhCOO)2
CH3 – CH2 –
例如,11-溴十一酸乙酯的制备及消炎镇痛药苄 达明合成原料的制备:
(过氧苯甲酸酐)
HBr,2 (,N Ph aC BrOO)
C2 = HC2 C Hl-- C 5 oH C
Br2 C C 2 C H H 2 C Hl (8% 5 )
σ键 :原子轨道重叠部分对键轴(两原子 的核间连线)具有圆柱形对称时所 形成的键;
π键 :原子轨道重叠部分对键轴所在的某 一平面具有反对称性时所形成的键。
p原子轨道的角度分布剖面图:
z(或y)
+
x
-
y(或z)
注:其中+、-号不表示正、负电荷,而是表示原子轨道角度分布图形 的对称关系
若两原子成键是由p轨道重叠形成的,且形成的是σ 键,则成键图形为:
③向反应介质中加入含卤素负离子的添加剂,可减
少溶剂引起的副反应。如:
④具有季碳取代基的烯烃加成反应中,还可能存在 重排反应。例如:
第一章卤化反应-第三节

Br O
Br (19)
O Br Br
Br Br O
OH
O CH3 Br
N
O CH3 BrBiblioteka NOH(20)
(21)
O CI
O
CI
N
N
O
N
O
CI
(22)
卤化试剂(19)用于对α,β-不饱和酮的α′-卤取代反应中,能 够减少双键加成副反应。
卤化试剂(20)和(21)的特点是亲电活性大,不需要任何催
化剂,反应条件温和,只得到单溴代物,且在反应中不生成
快
C H B:
C
B: 慢
C
OH
(-BH ) X
C OH X
X2
( -HX ) 快
CC XO
▪ 在酸或碱催化的α-卤取代反应中,羰基α 位取代基的影响是不同的。
▪ 对于酸催化的反应来说,若α位上具推电 子取代基,则有利于烯醇的稳定化,卤取 代反应比较容易,如环状酮的反应,均主 要得到在烷基较多的α位上卤取代的酮。
卤素分子和卤化氢,特别适用于对酸、碱敏感的酮。
卤化试剂(22)可在温和条件下对羰基α位及苄位、烯丙位进 行氯代反应
▪ 四溴环己二烯酮(不发生双键加成反应)
▪ 在少量HCl或HBr气体催化下,反应中以生成稳 定的三溴苯酚为动力,促使4位碳一溴键异裂, 生成的溴正离子向α,β-不饱和酮的α'位C-H作亲 电取代;同时,能有效地消除x-,于是可得到收 率良好的α'-溴代-α,β-不饱和酮。
CHO
O Br
O
Br O
O
OAc
C3H7CHCH
Br
OAc
H+ C3H7-C CHO Br
药物合成反应习题 第一章 卤化反应ppt课件

C r O /P y /C H C l 3 2 2
L i A l H / E t O 4 2
O H O H
C l C H C H C H ( C H ) C H O H 2 2 2 4 2 C l C H C H C H ( C H ) C O O C H 2 2 2 4 2 5
N a B H / E t O H 4
N R X N N+ N N X N NR N
环 六 亚 甲 基 四 胺 ( 乌 洛 托 品 )
四、药物合成反应课程的学习方法
3. 了解一些新试剂,新反应的特点、应用 范围,并与类似反应进行比较. 4.课后要做练习; 5.重视《药物合成实验》提高自己的动手 能力。
五.药物合成反应授课的要求和安排
H O 2
N H 2
B r
B r
(3)阻断基
• 阻断基的引入使反应物分子中某一活性部位被 封闭,让分子中其他活性低的部位发生反应并 顺利引入所需的基团,等目的达到后再除去阻 断基。
N H 2
( C H C O ) O 3 2
N H C O C H 3
H S O 9 8 % 2 4
N H C O C H 3
• 课堂讲授为主、自学为辅。课堂上的重 点突出,讲解主要内容及难点,因课时 有限,有部分内容要求同学自学。 • 课前要预习.
讨论与练习
1.学好本课程对从事药物及其中间体合 成工作有何意义? 2.药物合成反应有哪些特点?应如何学 习和掌握? 3.什么是导向基?具体包括哪些类型? 举例说明。 4.查阅报道药物合成领域的新技术及发 展动态资料?
药物合成反应习 题 第一章 卤化 反应
绪论
一、药物合成课程的目的 二、药物合成的发展趋势与新技术 三、药物合成反应课程教授内容 四、药物合成反应课程的学习方法 五、药物合成反应授课的要求和安排
药物合成反应复习题

第一章卤化反应1 卤化反应在有机合成中的应用?为什么常用一些卤代物作为反应中间体?2 归纳下常用的氯化剂、溴化剂都有哪些?它们的主要理化性质及应用范围?3 根据反应历程的不同,讨论一下卤化反应的类型、机理及对反应的影响。
(1)卤素对双键的离子型加成(2)芳香环上的取代(3)方向化合物侧链上的取代(4)卤化氢对醇羟基的置换(5)NBS 的取代反应4 比较X2、HX 、HOX 对双键的离子型加成反应的机理又何异同点。
怎样判断加成方向5 在-OH 得置换反应中各种卤化剂各有何特点?他们的应用范围如何?6 预测Br2/CCl4 于下列各种烯烃进行溴化反应的相对速度的次序。
CH2=CH2 (CH3)2C=CH2 HOOC-CH=CH-COOH (CH3)2C=C(CH3)2 CH3CH2=CH2 CH2=CH-CN7 对比下列反应的条件有何不同?结合反应机理加以说明:H3C CH2 CH 2Br(1)H3C CH CH 2H3C CHBr CH 3CH 3Cl CH 3(2)CH 2ClRH 2C CH CH 2(3) R H 2C CH CH 2BrRH 2C C CH 2OH8 下列反应选用何种氯化剂为好?说明原因。
(1) H3C C CH CH3 H3CC CH CH 2BrCH 3 CH 3(2)H3 C HC CH COOH H3C HC CH COCl(3) HO (CH 2 )6 OH IH2C(CH 2)4 CH 2 OH(4) H3CCO C H 2CH 2 COOH H3 CCO CH2CHCOBrBr(5) H3 CO CH2 OHH3CO CH 2 ClH2C C (CH 2 )6 COOHCl(6)H2 C HC (CH 2)6 COOHBr H2C (CH 2)6 COOHCOOHCOOHCOCl(7)Cl Cl HO OHNCl Cl NNCOClHO OHN9 完成下列反应,写出主要的试剂及反应条件:OHOH(1)COOH COClCH 3 CF3(2)(3) CH3COOHFXH2COOH(4) O COOH O I10 完成下列反应,写出其主要生成物(1) H3C C CHCH3Ca(OCl) 2/HOAc/H 2OH3C(2)HC CH2NBS/H 2O(3) CH3 C CH 2HBr/Bz2O2(4) OH 48%HBr(5) C H 2CH=CHCH2CH=CHBrCH 2=CH-COO-CH-CH 2 1molBr2/CCl4(CH 3)2C=CHCH 2CH=CH 2第二章烃化反应一烃化剂的种类有哪些?进行甲基化和乙基化反应时,应选用那些烃化剂?引入较大烃基时选用那些烃化剂为好?二用卤代烃对氨基和羟基的烃化反应各有何特点?烃化剂及被烃化物的结构对反应有何影响?三用于制备较纯的伯胺的方法有那几种?四举例说明“还原烃化”、“羟乙基化”的机理、特点及反应中的注意事项。
药物合成反应-第一章-卤化反应讲课讲稿

E tOO
t-B u O C l/R O H
C l R O H
C l
E tOO
O R
E tOO
与 次 卤 酸 酯 的 加 成
3
反应机理: N-卤代酰胺只提供卤正离子,环状卤桥正离子需要靠溶剂去进攻以完成加成。
应用特点
常见的N-卤代酰胺
O
O
O H3C C NHBr
(NBA)
O H3C C NHCl
卤 素
对向加成比例
对
碳正离子越稳定,环状卤桥正离子的比重
Br
烯
越低,对向加成相对越低;
烃
Br Br
的
溴的极化能力比氯强,更容易形成环状卤桥正离子,对向加成更多;
加
无位阻时,环状卤桥上下朝向概率相
Br Br
成
同,加成产生外消旋体混合产物。有 位阻时,依位阻来确定三元环朝向;
Br Br
1
次要反应机理:自由基加成
实际上是自身酸根负离子替代了
卤素负离子完成对环状卤桥正离
子的进攻,最终完成加成。
应用特点
O H X2/K I/N aH C O 3 O H 2O /r.t.
可制造五~六元环状内酯,进一步还可还原为半缩醛。
X O
O
X
O O
X 2 /K I/N a H C O 3
XD IB A H
X
H 2 O /r .t . C H 2 C O O H
OO
- 7 2 ℃H OO
不 饱 和 羧 酸 的 卤 内 酯 化
2
反应机理:与卤素加成类似
δ+δXOH
反应条件 次卤酸很不稳定,需现制现用。可用 氯气或溴与中性或含汞盐的碱性水溶 液反应而得到。
药物合成反应第一章卤化反应-PPT课件

反应机理
在亲电取代卤化反应中,卤 素离子首先与芳香环上的电 子云密度较高的区域结合, 形成正碳离子中间体。随后 ,正碳离子中间体发生重排 和消除质子,最终形成卤代 芳香烃。
影响因素
亲电取代卤化的反应速度和 选择性受多种因素的影响, 包括底物结构、反应条件( 如温度、催化剂、溶剂等) 、卤素原子的性质等。
药物合成反应第一章卤化反应ppt课件
目录
• 卤化反应简介 • 亲电取代卤化 • 亲核取代卤化 • 自由基卤化 • 其他卤化方式
01
卤化反应简介
卤化反应的定义
卤化反应
在有机化学中,卤化反应通常指 的是将氢原子替换为卤素(如氟 、氯、溴、碘)的反应。
卤化反应的分类
根据卤素和氢原子的取代位置, 卤化反应可以分为芳香族取代、 脂肪族取代和乙烯基取代等类型 。
非芳香族化合物的亲电取代卤化
01
非芳香族化合物的亲电取代卤化
对于非芳香族化合物,亲电取代卤化反应通常发生在具有电子富集基团
的碳-氢键上。这些基团可以是醇、醚、硫醇等。
02 03
反应机理
在非芳香族化合物的亲电取代卤化反应中,卤素离子首先与具有电子富 集基团的碳-氢键结合,形成正碳离子中间体。随后,正碳离子中间体 发生重排和消除质子,最终形成卤代烃。
HI>HBr>HCl。
溶剂和酸碱度
选择合适的溶剂和调整酸碱度 可以促进或抑制亲核取代卤化
的反应。
温度和压力
温度和压力也是影响亲核取代 卤化反应的重要因素。
04
自由基卤化
芳香族化合物的自由基卤化
总结词
芳香族化合物的自由基卤化是卤化反应的一种重要类型,主 要通过卤素与芳香族化合物发生自由基取代反应来实现。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
转1800C
R3 C X R4
• ③位阻的影响
• 无位阻,机会均等,形成外消旋混合物; • 有位阻:
Me C16H30 Br2/AcOH AcONa/Et2O HO
Me HO H Br
Me C16H30 Br HO H Br
(84~85%)
对于环烯、桥卤正离子在位阻小的一面形成:
2X .
CH2 CH CH2 + HX
CH2
CH
CH2
X2
CH2
CH
CH2 X
X
CH3 CH3
Br2 125
hν
oC,2h
CH2 Br CH2 Br
(48~53%)
(3)影响因素 ①a位亚甲基一般比a位甲基容易取代:
CH3(CH2)2 NBS (PhCO)2O2 CH2 CH CH CH3 CCl4 △ .2h Br CH3(CH2)2 CH CH CH CH3
• ②缺p芳杂环不利于卤代
• 4. 应用特点 • ①制备卤代芳烃
• • • • •
②氟代反应 反应剧烈,低温、稀释。 ③氯代反应 HOCl、CH3CO2Cl、Cl2O、S2Cl2、 O2Cl2、tBuOCl
• • • •
④溴代反应 加入I2可加速反应,因I2Br-比Br3-容易生成 其他溴化剂:NBS、HOBr、CF3CO2Br Lewis酸可加速反应
① 桥型卤正离子或离子对的过渡态形式
Q
Q R1 R3 C C R4 R2 X Q R1 R3 C C R4 R2 X Q R2 R3 C C R4 R1 X
(3)
R1 R2 R1 R2 R3 QX C C R4 R1
R3 C C R4 X
(1)
Q
(3)
R3 C C R4 R2 (2) X
(4)
Q=X、HO、RO、H、RCONH等
二、不饱和羧酸的卤内酯化反应
• (1)反应通式
(2)反应机理
I2/KI/NaHCO3 CH2CO2H O C H2C H O I H2O/r.t.4h I
H
H
*
*
H
O O
88%
H C C CO2H Br H
H Ph Br2/CHCl3 。 0 r.t.20min
H
Br CH CH Ph Br CO2H
R1 H C C R2 H Br R1 H R2 H NBS/DMSO
Br R1 R2 C C H H Me O S Me
C
C O SMe2
H2O
Br C H R1 C OH H R2
R2
b-消 除 (在 燥 MSO中 干 D )
Br C R1 C O H
OAc NBS/DMSO AcOCH2 O OgluAc4 OAc Br O AcOCH2 O OgluAc4 r.t
• 链增长
C C
Q-X
C X
· C
C X
· C
Q-X C X C Q
+X·
• 链终止
2
C X C X · C
· C
C X +X·
C C
C X
C X Q-X
C X
Q· X· +
• 2. 自由基取代
烯丙位卤代、羧酸脱羧卤置换、芳香重氮盐卤置换
• 链引发
• 链增长
• 链终止
第二节
不饱和烃的卤加成反应
0
CH3 CH CH2
CH2 CH CH3 OCOCH3
(80%)
Woodward、Prevost双羟化
• 2.卤素对炔烃的加成
• (1)反应通式
• (2)反应机理 • 亲电加成,反式产物
• (3)影响因素 • 同离子效应,可减少副反应
• (4)应用特点 • 应用烯烃直接制备二卤代烯,困难。炔烃 的卤加成,方便。
SN2
R-L + X X R
δ
L
δ
R-X
+ L
L=OH、OSO2R'、Cl、Br等
•新键的生成和旧键的断裂同时进行; •瓦尔登反转; •一个过渡态; •反应速度与反应物及试剂均有关;
二、自由基反应机理
• 1. 自由基加成
• 链引发
hv Q-X → Q· X· +
RO:OR → 2 RO·
RO· +Q-X → ROQ+X·
• 和卤素的加成反应 • 不饱和羧酸的卤内酯化反应 • 和次卤酸(酯)的加成反应
• 和N-卤代酰胺的加成反应 • 和卤化氢的加成反应
一
不饱和烃卤加成反应
1.卤素对烯烃的加成
(加成)
F2:加成反应激烈,副产物多,实用性小; I2:C-I键不稳定,易消除,不实用;
Cl2和Br2常用,重要,资源丰富,且活泼
同向(syn) 12% 37%
(MeO的供电子作用)
H C
CH3 O
C2H5
>
H C
C2H5
(稳定性)
• 当双键上有Ph基时,同向加成比例增加,(使 C 离子稳定) C -- C单键来得及旋转,按 三圆环过渡态进行的可能性减小。
• ②不同卤素的影响 • 溴加成,极化能力强,易形成鎓离子,以对 向加成为主(anti);氯加成,极化性小, 不易形成桥氯正离子,同向为主。
6-10h ,51-55%
CH3
CH Br
CH Br
(Br为吸电子基,使中间体不 稳定)
• (4)应用特点 • ①制备烯丙位或苄位卤化物
②重排反应
ph 3C CH2 CH CH2
ph ph ph Br2 CH CH CH2
NBS/CCl4 hν △ .4h
ph3C CH
CH CH2Br (94%)
• 3. 影响因素 • (1)芳环取代基电子效应的影响 • ①给电子有利-控制条件,卤化
• ②吸电子不利-提高卤化剂活性
NO2 Cl2O/(CF3SO2)2O 。 0 /POCl3
NO2
97%• 实际亲电Fra bibliotek剂为三氟甲磺酰次氯酸酐
Cl
• (2)芳杂环卤代 • ①多p芳杂环利于卤代 • Pyrole>furane>thiophene>benzene
第三节
烃类的卤代反应
一、脂肪烃的卤代反应 • 1.饱和烃-自由基历程 • 2.不饱和烃
R H
X2 高 / 温
h/过氧化物
h
R X
Cl2/CH2Cl2 Cl
3. 烯丙位和苄位碳原子上的卤代反应
CH2 CH CH3 X2 hν CH2 CH CH2 X
NBS
(2)反应机理
hν
X2
CH2 CH CH2 H + X
②三分子协同亲电加成
H C C HX C H C X X H + C X H C +X
•实际上,三分子碰撞几率小。一般认为, 先形成p络合物,然后再与另一分子反应
(2)亲电取代
亲电试剂取代其它官能团的化学反应,这种被取代的基 团通常是氢,但其他基团被取代的情形也是存在的
• ①芳烃的卤取代反应 • 芳环上的氢被亲电试剂取代的反应称为芳香亲电 取代反应 苯环亲电取代反应的一般模式
程度适中,反应相对易控制; Cl2来自于氯碱工业,Br2来自于海洋。
• (2)反应机理 • 以anti为主,但比例影响因素较多
• (3)影响因素 • ①烯烃结构影响
Br x CH HC CH3 Br2/CCl4 2-5 c
0
H C Br CH3 + X
Br CH3 C C H Br H
X
C H
对向(anti) X=H 88% X=OCH3 63%
• • • • •
机理: 亲电加成(不饱和烃和卤素加成) 亲电取代(芳烃和羰基a位卤代) 亲核取代(醇羟基、羧羟基等的卤置换) 自由基反应(苄位、烯丙位卤代,羧基、 重氮基卤置换)
第一节 卤化反应机理
• 一、电子反应机理 • 1.亲电反应 (1)亲电加成
通过化学键异裂产生的带正电的原子或基团进攻 不饱和键而引起的加成反应称为亲电加成反应。 亲电加成反应可以按照“环正离子中间体机 理”、“碳正离子中间体机理”、“离子对中间体 机理”和“三中心过渡态机理”四种途径进行。
ph3C CH CH CH2
(立体位阻大,不易 接近)
二
芳烃的卤代反应
δ δ X X
H X H X
X X 或 他 化 形 其 卤 剂 式 H X X
H
• • • • • •
X-L 常用卤化剂: X2、Cl2O、S2Cl2、SO2Cl2、tBuOCl NBS、HOBr、AcOBr、CF3COOBr 催化剂 AlCl3、SbCl5、FeCl3、SnCl4、TiCl4、 ZnCl2 • 溶剂 • 极性溶剂:HOAc、HCl、CHCl3
+ E+ E+ + H E
E
亲电试剂
p -络合物
-络合物
反应机理
+ Cl-Cl 快 Cl
+
Cl Cl
- AlCl3
慢
Cl
+
_
H
ClAlCl3
快
+ AlCl3 + HCl
+
Br-Br
快
+
Br Br
Br
- Br2
慢
Br
+
Br
-
Br 2
慢
_
+
H
Br
+ Br2
Br 快 + H+ + Br 3
Ph + O O
69%