Gromacs命令锦集
GROMACS教程

GROMACS程序DEMO例程####################### 概述 #######################----------------------------------------------------------------------------------------------------------------------------------该例程来自Gromacs程序/share/tutor/目录下。
整个例程大概只需要十分钟就可以完成,非常适合初学者学习。
该例程是一个完整的分子动力学模拟过程,涵盖了Gromacs程序基本的使用方法。
模拟内容是一个水环境下的小肽链。
模拟唯一要求是该小肽链的PDB文件。
文档翻译如果有错,请你给我发信:sen@。
相关内容请参阅Gromacs文档,或者给Gromacs开发组询问。
Gromacs官方网站:Email: gromacs@----------------------------------------------------------------------------------------------------------------------------------############################ 环境变量设置############################----------------------------------------------------------------------------------------------------------------------------------在以下的例程中,所有命令都直接运行,没有添加绝对路径。
所以,必须将Gromacs安装路径的bin文件夹加入到系统PATH变量中。
如果不加入PATH变量,那么运行时要加入命令的绝对路径。
gromacs结果 温度因子b-gactor

GROMACS(GNU Molecular Simulation)是一款强大的分子动力学模拟软件,广泛应用于生物分子的模拟和计算。
在GROMACS模拟中,温度因子(B-gactor)是一个重要的参数,它描述了分子系统的无序程度。
B因子的计算方法通常是通过RMSD(残基序列差异)来实现的。
具体来说,对于一个给定的分子系统,我们首先计算每个残基与其平均位置之间的RMSD,然后将所有残基的RMSD 平方相加,最后取平均值,得到B因子。
B因子的值通常在0到1之间,其中0表示完全有序,1表示完全无序。
在实际应用中,B 因子的值对于分子系统的分析和比较非常重要,例如在分子动力学模拟中,我们通常会通过比较不同系统的B因子来评估它们的无序程度和动力学稳定性。
如果你想在GROMACS中计算温度因子(B因子),可以使用以下命令:
md_0_10.xvg -rmsf -s md_0_10 -o bfactor.xvg -g bfactor.pdb -res bfactor.res
其中,md_0_10.xvg是模拟输出文件的名称,-rmsf选项表示计算残基序列差异的RMSF值,-s选项表示输入文件的名称,-o选项表示输出文件的名称,-g选项表示生成GROMACS格式的PDB文件,-res选项表示生成RES格式的结果文件,包括B因子。
请注意,具体的操作方法可能会因具体的系统和环境而有所不同,因此建议参考GROMACS 的官方文档或相关教程进行详细的学习和使用。
GROMACS使用教程要点
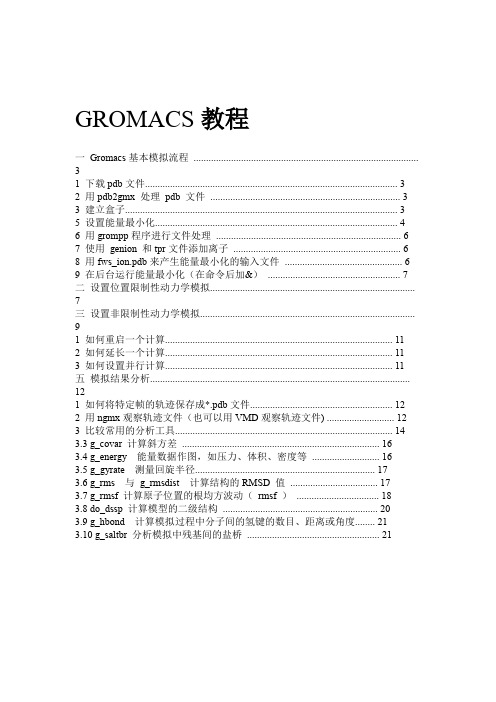
GROMACS教程一Gromacs基本模拟流程 (3)1 下载pdb文件 (3)2 用pdb2gmx 处理pdb 文件 (3)3 建立盒子 (3)5 设置能量最小化 (4)6 用grompp程序进行文件处理 (6)7 使用genion 和tpr文件添加离子 (6)8 用fws_ion.pdb来产生能量最小化的输入文件 (6)9 在后台运行能量最小化(在命令后加&) (7)二设置位置限制性动力学模拟 (7)三设置非限制性动力学模拟 (9)1 如何重启一个计算 (11)2 如何延长一个计算 (11)3 如何设置并行计算 (11)五模拟结果分析........................................................................................................121 如何将特定帧的轨迹保存成*.pdb文件 (12)2 用ngmx观察轨迹文件(也可以用VMD观察轨迹文件) (12)3 比较常用的分析工具 (14)3.3 g_covar 计算斜方差 (16)3.4 g_energy 能量数据作图,如压力、体积、密度等 (16)3.5 g_gyrate 测量回旋半径 (17)3.6 g_rms 与g_rmsdist 计算结构的RMSD 值 (17)3.7 g_rmsf 计算原子位置的根均方波动(rmsf ) (18)3.8 do_dssp 计算模型的二级结构 (20)3.9 g_hbond 计算模拟过程中分子间的氢键的数目、距离或角度 (21)3.10 g_saltbr 分析模拟中残基间的盐桥 (21)1GROMACS 是一个使用经典分子动力学理论研究蛋白质动力学的高端的高效的工具。
GROMACS是遵守GNU许可的免费软件,可以从以下站点下载:,并且可以在linux和Windows上使用。
在本教程中,将研究一个从漏斗形蜘蛛的毒液中分离的毒素。
Gromacs5.1安装教程 亲测无错全过程

1.下载安装 fftw
wget /fftw-3.3.4.tar.gz %%可自行下载
tar -xvf ~test/fftw-3.3.4.tar.gz
Qingdao , China
Key Lab of New Energy Physics & Materials Science in Universities, Shandong
cmake .. -DCMAKE_INSTALL_PREFIX=/opt/gromacs-5.1 -DGMX_FFT_
n
export PATH=/opt/cmake-2.8.12.2/bin/:$PATH
3. 下载安装 Gromacs (官网 / )
tar xvf gromacs-5.1.tar.gz
cd gromacs-5.1
mkdir build
cd build
China University of Petroleum
0
8
make install
3
9
安装完成后设置环境变量(/etc/profile) 可直接改.bashrc
@
s
export PATH=/opt/gromacs-5.1/bin:$PATH
.
u
export LD_LIBRARY_PATH=/opt/gromacs-5.1/lib64:$LD_LIBRARY_PATH
LIBRARY=fftw3 -DFFTWF_LIBRARY="/opt/fftw-3.3.4/lib/libfftw3f.so"
(完整版)Gromacs命令锦集

目录1、MAKE_NDX (2)2、G_TRAJ (3)3、G_ENERGY另外一种用法 (3)4、PDB2GMX (3)5、GENION (5)6、EDITCONF (6)7、G_RAMA (6)8、G_CLUSTER (7)9、GENBOX (7)1、make_ndxGromacs的索引文件,即index文件,由make_ndx程序生成,文件后缀为.ndx。
索引文件是gromacs最重要的概念文件,使用它可以在模拟过程中为所欲为。
举一个简单的例子,比如想详细了解HIV整合酶切割DNA的反应机理,使用量子力学模拟反应位点的反应过程,而分子其他部位使用一般分子动力学模拟。
于是我们就面临一个对模拟系统进行分割定义的问题,在gromacs中,就要用到索引文件。
基本的思路是这样的,在索引文件中,定义一个独立的组,这个组包括反应位点处所有原子。
在模拟的.mdp文件中,对这个组定义量子力学模拟,事情就是这么简单。
对蛋白进行量子力学模拟时,一般使用洋葱模型。
所谓洋葱模型,就是对反应位点使用量子机制,在反应位点一定的半径内,使用半量子力学机制,然后分子部分使用分子机制。
那么索引文件就定义一个使用量子力学的组,把需要引进量子机制的原子都放到这个组中;再定义一个半量子机制的组,同时放进需要半量子力学机制模拟的原子,再在.mdp文件中独自定义即可。
再举一个例子,比如说在进行SMD(Steered MolecularDynamics,这个我一直没有想到或者找到切恰的中文翻译方法,或许可以叫做牵引分子动力学??别扭!!)中,要对蛋白莫一个原子或者残基作用力,那么可以建立一个索引文件,在该文件中定义一个组,把要施力的残基或者原子放到该组中。
然后在.ppa文件中使用该组就行了。
如果我还没有说明白,那么看看gromacs的参考文件吧。
如果还是不明白,可以来找我,我免费培训。
^_^索引文件使用make_ndx命令产生,"make_ndx -h"可以看到全部的参数。
Gromacs

Gromacs 4.5+ 续跑部分内容转⾃Sensenbobo⼤神的⽂章1.利⽤cpt⽂件mpirun -n 27 mdrun_mpi -s md.tpr -cpi md.cpt -append -o md.trr -x md.xtc -c md.gro -e md.edr -g md.log⼤致命令如上,即连着之前所输出的⽂件进⾏继续写⼊,如果有相同的帧会被覆盖掉。
⽐较特别的地⽅是需要输⼊-x md.xtc⽂件。
2. tpbconvtpbconv -s topol.tpr -f traj.trr -e ener.edr -o newtopol.tprmdrun -s newtopol.tpr -o newtopol.trr -c newtopol.gro -e newtopol.edr -g newtopol.logtpbconv除了断点续跑之外还可以进⾏延长,-extend。
tpbconv不能更改你原来tpr⽂件中并⾏计算的节点数,⽐如你原来的tpr⽂件是8个节点的,那么使⽤tpbconv得到的重启tpr⽂件也是8个节点的。
如果想更改使⽤节点数,那只能⽤grompp重新做⼀个了。
但是使⽤grompp做重启模拟⽂件时,就算你指定了原来的轨迹⽂件和能量⽂件,它还是会根据麦克斯韦分布重新给各个原⼦指定速度.3.使⽤grompp提取上⼀次模拟最后速度和能量使⽤grompp可以制作⼀个新的.tpr⽂件,从上⼀步模拟的轨迹⽂件中提取速度,并从上⼀步能量⽂件中提取能量,也可以⽆缝的链接重启模拟计算。
要做到从上⼀步的最后的⼀个系统状态开始新的模拟计算。
⾸先要在.mdp⽂件中把“ gen_vel”参数定义为" no ",这样做是为了告诉grompp不要重新为系统中的原⼦指定随机速度。
指定新模拟开始的时间,即修改" tinit "参数。
然后可以使⽤⼀下命令制作⼀个从上⼀步模拟⽂件中提取速度和能量的.tpr⽂件:grompp -f [.mdp⽂件] -c [上⼀步模拟最后的系统坐标⽂件] -p [拓扑⽂件]-t [上⼀步的trr轨迹⽂件] -e [上⼀步能量⽂件] -time [坐标⽂件对应的模拟时间] -o [输出tpr⽂件] -np [CPU数⽬]提取上⼀步模拟系统的速度时使⽤trr⽂件,是因为xtc为单精度,没有trr⽂件精确。
(完整版)gromacs命令

1 Generating topologies and coordinates 生成拓扑和坐标文件1-1 pdb2gmx PDB文件转换到拓扑文件(.top)和坐标文件(.gro)1-2 g_x2top 从坐标文件(.gro)生成一个原始拓扑文件(.top)1-3 editconf编辑盒子及写入子组(subgroups)1-4 genbox体系溶剂化1-5 genion加入抗衡离子1-6 genconf 增加一个随机方向的构象1-7 genrestr 生成索引组的位置限制或距离限制1-8 g_protonate 质子化结构2 Running a simulation 模拟运行2-1 grompp生成一个运行输入文件2-2 tpbconv 从一个停止的运行生成一个重新运行的输入文件2-3 mdrun 执行模拟、正态分析及能量最小化3 Viewing trajectories 轨迹查看3-1 ngmx显示一条轨迹3-2 g_highway X Window System小工具,用于highway模拟3-3 g_nmtraj 从一个本征矢量eigenvector生成一个虚拟轨迹4 Processing energies 能量处理4-1 g_energy将能量写入xvg文件并显示平均值4-2 g_enemat 从能量文件中提取能量矩阵4-3 mdrun -rerun(重新)计算轨迹帧的能量5 Converting files 文件转换5-1 editconf 转换和编辑结构文件5-2 trjconv 转换和编辑轨迹文件5-3 trjcat连接轨迹文件5-4 eneconv 转换能量文件5-5 xpm2ps5-6 g_sigeps6 Tools 工具6-1 make_ndx制作索引文件6-2 mk_angndx 生成索引文件,用于g_angle6-3 gmxcheck 检查并比较文件6-4 gmxdump 生成人可读的二进制文件6-5 g_traj 从轨迹文件中绘制选定的原子或组的x、v、f6-5 g_analyze 分析数据集6-6 trjorder 根据与一个组的距离定义分子序数6-7 g_filter 轨迹频率过滤,制平滑的动画6-8 g_lie 线性拟合自由能评估6-9 g_dyndom 内插和外推结构旋转6-10 g_morph线性内插构象6-11 g_wham伞形抽样后加权直方分析6-12 xpm2ps convert XPM (XPixelMap) file to postscript6-13 g_sham读/写xmgr和xvgr数据集6-14 g_spatial 计算空间分布函数6-15 g_select selects groups of atoms based on flexible textual selections 6-16 g_tune_pme time mdrun as a function of PME nodes to optimize settings7 Distances between structures 结构间的差距7-1 g_rms 计算与参考结构之间的均方根偏差及其矩阵7-2 g_confrms叠合两个结构,并计算其rmsd7-3 g_cluster 团簇结构7-4 g_rmsf计算原子波动值8 Distances in structures over time 随时间变化,结构间差距8-1 g_mindist 计算两组间的最小距离8-2 g_dist 计算两组之间的质量中心的距离8-3 g_bond 计算原子间的距离8-4 g_mdmat 计算残留联系地图contact maps8-5 g_polystat 计算聚合物的静态属性8-6 g_rmsdist calculates atom pair distances averaged with power -2, -3 or -69 Mass distribution properties over time 随时间变化,质量分布性质9-1 g_traj plots x, v, f, box, temperature and rotational energy9-2 g_gyrate 计算回转半径9-3 g_msd 计算均方位移9-4 g_polystat 计算聚合物的静态属性9-5 g_rotacf 计算分子转动的相关函数9-6 g_rdf 径向分布函数的计算9-7 g_rotmat 根据参考结构绘制旋转矩阵图9-8 g_vanhove 计算凡霍夫位移函数10 Analyzing bonded interactions 分析键相互作用10-1 g_bond 计算键长分布10-2 mk_angndx 生成索引文件g_angle10-3 g_angle 计算角度和二面角的分布及相关性10-4 g_dih 二面角转换分析11 Structural properties 结构特性11-1 g_hbond 计算和分析氢键11-2 g_saltbr 计算盐桥11-3 g_sas 计算溶剂可及表面面积11-4 g_order computes the order parameter per atom for carbon tails11-5 g_principal 计算一组原子惯性轴11-6 g_rdf 计算径向分布函数11-7 g_sgangle 计算两组间的角度和距离11-8 g_sorient 分析溶质周围溶剂取向11-9 g_spol 分析溶质周围溶剂偶极取向和极化11-10 g_bundle 分析捆轴,如螺旋11-11 g_disre 分析距离限制11-12 g_clustsize 计算原子团簇尺寸分布11-13 g_anadock cluster structures from Autodock runs12 Kinetic properties 动力学性质12-1 g_traj plots x, v, f, box, temperature and rotational energy12-2 g_velacc 计算速度自相关函数12-3 g_tcaf 计算液体粘度12-4 g_bar 通过Bennett’s acceptance ratio接受率计算估计自由能差12-5 g_current 计算当前系统的自相关函数12-6 g_vanhove 计算凡霍夫相关函数12-7 g_principal calculate principal axes of inertion for a group of atoms13 Electrostatic properties 静电性质13-1 genion 添加带电离子13-2 g_potential 计算盒子中的静电势13-3 g_dipoles 计算总偶极波动13-4 g_dielectric 根据介电常数计算频率13-5 g_current 根据电荷计算体系介电常数13-6 g_spol 分析溶质周围的偶极子14 Protein specific analysis 蛋白质特殊分析14-1 do_dssp 计算分配二级结构和溶剂可及表面面积14-2 g_chi 计算所有需要的chi和其他二面角14-3 g_helix 计算α螺旋结构的基本性质14-4 g_helixorient 计算本地pitch/弯曲/旋转/内螺旋方向14-5 g_rama computes Ramachandran plots14-6 g_xrama shows animated Ramachandran plots14-7 g_wheel plots helical wheels15 Interfaces 界面15-1 g_potential计算盒子静电势15-2 g_density 计算体系密度15-3 g_densmap计算二维平面或轴向-径向密度图15-4 g_order computes the order parameter per atom for carbon tails 15-5 g_h2order 计算水分子的方向15-6 g_bundle 分析捆轴,如跨膜螺旋15-7 g_membed 蛋白质嵌入脂双层16 Covariance analysis 协方差分析16-1 g_covar 计算和对角化协方差矩阵16-2 g_anaeig 分析特征向量16-3 make_edi 生成输入文件用于本质动力学抽样17 Normal modes 正态模式17-1 grompp 生成运行输入文件17-2 mdrun 发现一个潜在的能量最低17-3 mdrun 计算Hessian17-4 g_nmeig 对角化Hessian17-5 g_nmtraj 产生本征模的振荡轨迹17-6 g_anaeig 正态模式分析17-7 g_nmens 从正常模式生成的结构合奏分析命令1 Groups in Analysis1-1 make_ndx To generate an index file consisting of a series of atom numbers 1-2 mk_angndx To generate an index file with angles or dihedralsDefault GroupsSelections1-3 g_select2 Looking at trajectory2-1 ngmx3 General properties3-1 g_energy3-2 g_traj。
gromacs基础知识

(18)gromacs基础知识Gromacs的pdb2gmx命令使用gromacs做分子动力学模拟时,第一个要用到的命令一般都是pdb2gmx。
这个命令吧pdb分子文件转化成gromacs独特的gro分子结构文件类型,同时产生分子拓扑文件。
Gromacs是典型的GPL软件,每一个命令都有很多命令参数。
这对熟悉windows环境的人来说有一点烦,但是如果熟悉了Linux环境,也就慢慢喜欢啦。
(建议多使用命令,就像VMD,Pymol,rasmol和Chimera等等分子可视化软件,如果接合命令使用,功能都非常强大。
另外一个比较bt的软件叫做WHATIF的,完全建立在bt的命令菜单上,心理承受能力不强者多半吐血而终。
使用“pdb2gmx -h”可以得到pdb2gmx的所有参数及简单说明(gromacs的任何命令都可以使用-h参数得到类似帮助)。
pdb2gmx的参数很多,但是常用的只有以下几个:------------------------------------------------------------------------f 指定你的坐标文件,可以是pdb、gro、tpr等等包含有分子坐标的文件;-o 输出文件,也就是处理过的分子坐标文件,同样可以是pdb、gro、g96等文件类型; -p 输出拓扑文件。
pdb2gmx读入力场文件,根据坐标文件建立分子系统的拓扑;-water 指定使用的水模型,使用pdb2gmx的时候最好加这个参数,不然后面会吃苦头。
它会提前在拓扑文件中添加水分子模型文件;-ff 指定力场文件(下文讨论),也可以不用这个参数,再自行选择;-ignh 舍弃分子文件中的H原子,因为H原子命名规则多,有的力场不认;-his 独个指定HIS残基的质子化位置。
------------------------------------------------------------------------其他的参数还不少,可以好好看一个pdb2gmx的帮助文件,一般的pdb2gmx的执行格式如下(假设你的分子坐标文件为sen.pdb):pdb2gmx -f sen.pdb -o sen.gro -p sen.top -water tip4p -ignh -his该命令读入分子文件,使用tipp水模型,等等,然后pdb2gmx会让你选择力场文件。
中心Gromacs使用指南

中心Gromacs使用指南一、作业提交•1、在电脑上用Xshell客户端(或其他类似软件)登陆高性能账号,若不知道自己的高性能帐号在哪个分区,登陆帐号以后输入:pwd,可以看到如下图,home-后面的就是高性能帐号所在的分区(图中为YW分区):•2、如需直接提交作业,在高性能账号下创建放计算任务的文件夹test/gromacs/run-12(该名称可以根据自己习惯命名):•3、用FTP工具登录高性能帐号,将计算所需的算例文件通过XFTP(或类似工具)上传至账号目录下test/gromacs/run-12:•4、切换至Xshell界面,进入算例文件夹中:•5、创建或拷贝提交作业的脚本文件gromacs-515_YW.lsf (也可在Windows系统写好以后用FTP工具上传;现以YW分区直接从公共目录中拷贝为例),Gromacs软件脚本的路径在/home-yw/soft/jobscripts/中。
先查看脚本是否存在,图中gromacs-515_YW.lsf即为5.1.5版本所需脚本(如需2018.4,请拷贝对应的脚本):•6、采用您最熟悉的方式将上述脚本拷贝至需要提交任务的文件夹中(注意cp后、./前有空格):•7、修改模板脚本:•8、输入字母“i”,进入编辑模式。
脚本内容如下,部分内容需要根据情况进行修改:•9、按下键盘上的esc键后,输入:wq保存脚本文件,并退出。
•10、将脚本文件转换为UNIX格式(如从Windows系统上传的话必须要转换,不然提交作业时会报错;若直接拷贝公共目录中的脚本,并在Linux环境中进行修改,则可以省略步骤10、11。
若提交作业的脚本名称不为gromacs-515_YW.lsf,则需要修改为对应的脚本名称):•11、赋予脚本文件可执行权限:•12、用bsub命令提交作业脚本:hpc-user@login node:~/test/gromacs/run-12> bsub gromacs-515_YW.lsf•13、如果提交正确,则会出现如下内容(其中Job后面的数字为:JobID,每个任务的JobID不一样,可根据JobID查看该任务情况,出问题时,请及时告知JobID,保留计算的输出文件):Job <> is submitted to queue <intelY_mid>.•14、查看任务是否计算完成,可以使用bjobs命令(当出现:Done successfully. The CPU time used is xxxx seconds说明计算结束):•15、任务计算结束后,可以查看输出文件,检查任务是否计算成功(作业脚本中指定的输出文件):•16、如果计算成功,在输出文件MD.log的最后会出现如下部分:•17、计算完成后,继续进行后续操作或用FTP工具将文件下载后,进行分析。
gromacs示例

gromacs示例本示例来自gromacs网站上的示例:gromacs tutorial for drug-enzyme complex.有些步骤按其上面所述服务器无法运行,自己稍微做了修改。
1.从下载1az8蛋白的pdb文件。
2.用UltraEdit打开1az8.pdb文件,将HETATM一段(除去其中的水)拷入到The Dundee PRODRG2 Server,在“Chirality”,“Full charges”,“Energy Minimization”三个选项分别选为“Yes”,“Yes”,“No”,点击“Run PRODRG”生成抑制剂文件,解压,将DRGGMX.ITP文件重命名为drg.itp文件;3. 编辑蛋白质坐标文件trp_minv.pdb导入工作站;4. 在进行任何分子动力学模拟之前,必须建立分子的拓扑文件。
Gromacs的分子拓扑文件是用pdb2gmx命令生成,文件后缀名为 .top。
pdb2gmx的输入唯一文件是分子的PDB文件。
把pdb分子文件转化成gromacs独特的gro分子结构文件类型,同时产生分子拓扑文件,使用命令:pdb2gmx -ignh -ff gmx -f trp_minv.pdb -o trp2.pdb -p trp.top -water spce解释:-f :指定你的坐标文件,可以是pdb、gro、tpr等等包含有分子坐标的文件;-o :输出文件,也就是处理过的分子坐标文件,同样可以是pdb、gro、g96等文件类型;-p :输出拓扑文件。
pdb2gmx读入力场文件,根据坐标文件建立分子系统的拓扑;-water :指定使用的水模型,使用pdb2gmx的时候最好加这个参数,不然后面会吃苦头。
它会提前在拓扑文件中添加水分子模型文件;-ff :指定力场文件(下文讨论),也可以不用这个参数,再自行选择;-ignh : 舍弃分子文件中的H原子,因为H原子命名规则多,有的力场不认;5. 打开生成的trp2.pdb文件,将DRGPOH.PDB文件复制到trp2.pdb文件的结尾,将残基数列改为224,将原子序列也作相应调整,保存(原文说接在trp.gro文件的ASN223后面,但现在还没有生成这个文件,感觉应该是trp2.pdb);6. 编辑tro.top文件:在力场部分添加#include “drg.itp”,在分子部分添加 DRG 1;7. 真空条件下进行分子模拟输出结果误差较大,所以模拟之前,必须给分子添加水环境。
Gromacs介绍

GROMACS运算流程
Step Three: Defining the Unit Cell & Adding Solvent
1. Define the box dimensions using editconf. 2. Fill the box with water using genbox.
90年代初制定和完善的一套并行语法 支持Fortran, C, C++ 简单易学
几种常见的分子动力学软件
NAMD AMBER CHARMM TINKER LAMMPS DL-POLY GROMACS
NAMD
主要针对与生物和化学软材料体系 优点 程序设计水平高,计算效率高,号称可以有效并行到上千个处理 器 兼容多种输入和输出文件格式,有很好的分析辅助软件VMD 有很好的维护服务 不需安装 免费 缺点 万一需要自己安装的话比较麻烦
/tinker/
LAMMPS
一般性分子模拟软件 优点 兼容当前大多数的势能模型 编程水平高,计算效率高(比NAMD差,强于其他所有类似软件) 可以模拟软材料和固体物理系统 免费 缺点 维护差
/~sjplimp/lammps.html
大型(复杂)体系和并行算法
必要性 体系越来越大 模拟时间越来越长 解决办法 制造更快的处理器 并行计算机
例子:~50000原子的生物 体系,1ns模拟
单个处理器:~12天 16个并行处理器:~1天
或者
MPI
Message Passing Interface
GROMACS运算流程
Step Four: Adding Ions
(完整版)gromacs命令

(完整版)gromacs命令1 Generating topologies and coordinates 生成拓扑和坐标文件1-1 pdb2gmx PDB文件转换到拓扑文件(.top)和坐标文件(.gro) 1-2 g_x2top 从坐标文件(.gro)生成一个原始拓扑文件(.top)1-3 editconf编辑盒子及写入子组(subgroups)1-4 genbox体系溶剂化1-5 genion加入抗衡离子1-6 genconf 增加一个随机方向的构象1-7 genrestr 生成索引组的位置限制或距离限制1-8 g_protonate 质子化结构2 Running a simulation 模拟运行2-1 grompp生成一个运行输入文件2-2 tpbconv 从一个停止的运行生成一个重新运行的输入文件2-3 mdrun 执行模拟、正态分析及能量最小化3 Viewing trajectories 轨迹查看3-1 ngmx显示一条轨迹3-2 g_highway X Window System小工具,用于highway模拟3-3 g_nmtraj 从一个本征矢量eigenvector生成一个虚拟轨迹4 Processing energies 能量处理4-1 g_energy将能量写入xvg文件并显示平均值4-2 g_enemat 从能量文件中提取能量矩阵4-3 mdrun -rerun(重新)计算轨迹帧的能量5 Converting files 文件转换5-1 editconf 转换和编辑结构文件5-2 trjconv 转换和编辑轨迹文件5-3 trjcat连接轨迹文件5-4 eneconv 转换能量文件5-5 xpm2ps5-6 g_sigeps6 Tools 工具6-1 make_ndx制作索引文件6-2 mk_angndx 生成索引文件,用于g_angle6-3 gmxcheck 检查并比较文件6-4 gmxdump 生成人可读的二进制文件6-5 g_traj 从轨迹文件中绘制选定的原子或组的x、v、f6-5 g_analyze 分析数据集6-6 trjorder 根据与一个组的距离定义分子序数6-7 g_filter 轨迹频率过滤,制平滑的动画6-8 g_lie 线性拟合自由能评估6-9 g_dyndom 内插和外推结构旋转6-10 g_morph线性内插构象6-11 g_wham伞形抽样后加权直方分析6-12 xpm2ps convert XPM (XPixelMap) file to postscript6-13 g_sham读/写xmgr和xvgr数据集6-14 g_spatial 计算空间分布函数6-15 g_select selects groups of atoms based on flexible textual selections 6-16 g_tune_pme time mdrun as a function of PME nodes to optimize settings7 Distances between structures 结构间的差距7-1 g_rms 计算与参考结构之间的均方根偏差及其矩阵7-2 g_confrms叠合两个结构,并计算其rmsd7-3 g_cluster 团簇结构7-4 g_rmsf计算原子波动值8 Distances in structures over time 随时间变化,结构间差距8-1 g_mindist 计算两组间的最小距离8-2 g_dist 计算两组之间的质量中心的距离8-3 g_bond 计算原子间的距离8-4 g_mdmat 计算残留联系地图contact maps8-5 g_polystat 计算聚合物的静态属性8-6 g_rmsdist calculates atom pair distances averaged with power -2, -3 or -69 Mass distribution properties over time 随时间变化,质量分布性质9-1 g_traj plots x, v, f, box, temperature and rotational energy9-2 g_gyrate 计算回转半径9-3 g_msd 计算均方位移9-4 g_polystat 计算聚合物的静态属性9-5 g_rotacf 计算分子转动的相关函数9-6 g_rdf 径向分布函数的计算9-7 g_rotmat 根据参考结构绘制旋转矩阵图9-8 g_vanhove 计算凡霍夫位移函数10 Analyzing bonded interactions 分析键相互作用10-1 g_bond 计算键长分布10-2 mk_angndx 生成索引文件g_angle10-3 g_angle 计算角度和二面角的分布及相关性10-4 g_dih 二面角转换分析11 Structural properties 结构特性11-1 g_hbond 计算和分析氢键11-2 g_saltbr 计算盐桥11-3 g_sas 计算溶剂可及表面面积11-4 g_order computes the order parameter per atom for carbon tails11-5 g_principal 计算一组原子惯性轴11-6 g_rdf 计算径向分布函数11-7 g_sgangle 计算两组间的角度和距离11-8 g_sorient 分析溶质周围溶剂取向11-9 g_spol 分析溶质周围溶剂偶极取向和极化11-10 g_bundle 分析捆轴,如螺旋11-11 g_disre 分析距离限制11-12 g_clustsize 计算原子团簇尺寸分布11-13 g_anadock cluster structures from Autodock runs12 Kinetic properties 动力学性质12-1 g_traj plots x, v, f, box, temperature and rotational energy12-2 g_velacc 计算速度自相关函数12-3 g_tcaf 计算液体粘度12-4 g_bar 通过Bennett’s acceptance ratio接受率计算估计自由能差12-5 g_current 计算当前系统的自相关函数12-6 g_vanhove 计算凡霍夫相关函数12-7 g_principal calculate principal axes of inertion for a group of atoms13 Electrostatic properties 静电性质13-1 genion 添加带电离子13-2 g_potential 计算盒子中的静电势13-3 g_dipoles 计算总偶极波动13-4 g_dielectric 根据介电常数计算频率13-5 g_current 根据电荷计算体系介电常数13-6 g_spol 分析溶质周围的偶极子14 Protein specific analysis 蛋白质特殊分析14-1 do_dssp 计算分配二级结构和溶剂可及表面面积14-2 g_chi 计算所有需要的chi和其他二面角14-3 g_helix 计算α螺旋结构的基本性质14-4 g_helixorient 计算本地pitch/弯曲/旋转/内螺旋方向14-5 g_rama computes Ramachandran plots14-6 g_xrama shows animated Ramachandran plots14-7 g_wheel plots helical wheels15 Interfaces 界面15-1 g_potential计算盒子静电势15-2 g_density 计算体系密度15-3 g_densmap计算二维平面或轴向-径向密度图15-4 g_order computes the order parameter per atom for carbon tails 15-5 g_h2order 计算水分子的方向15-6 g_bundle 分析捆轴,如跨膜螺旋15-7 g_membed 蛋白质嵌入脂双层16 Covariance analysis 协方差分析16-1 g_covar 计算和对角化协方差矩阵16-2 g_anaeig 分析特征向量16-3 make_edi 生成输入文件用于本质动力学抽样17 Normal modes 正态模式17-1 grompp 生成运行输入文件17-2 mdrun 发现一个潜在的能量最低17-3 mdrun 计算Hessian17-4 g_nmeig 对角化Hessian17-5 g_nmtraj 产生本征模的振荡轨迹17-6 g_anaeig 正态模式分析17-7 g_nmens 从正常模式生成的结构合奏分析命令1 Groups in Analysis1-1 make_ndx To generate an index file consisting of a series of atom numbers 1-2 mk_angndx To generate an index file with angles or dihedralsDefault GroupsSelections1-3 g_select2 Looking at trajectory2-1 ngmx3 General properties3-1 g_energy3-2 g_traj。
(完整版)Gromacs命令锦集
目录1、MAKE_NDX (2)2、G_TRAJ (3)3、G_ENERGY另外一种用法 (3)4、PDB2GMX (3)5、GENION (5)6、EDITCONF (6)7、G_RAMA (6)8、G_CLUSTER (7)9、GENBOX (7)1、make_ndxGromacs的索引文件,即index文件,由make_ndx程序生成,文件后缀为.ndx。
索引文件是gromacs最重要的概念文件,使用它可以在模拟过程中为所欲为。
举一个简单的例子,比如想详细了解HIV整合酶切割DNA的反应机理,使用量子力学模拟反应位点的反应过程,而分子其他部位使用一般分子动力学模拟。
于是我们就面临一个对模拟系统进行分割定义的问题,在gromacs中,就要用到索引文件。
基本的思路是这样的,在索引文件中,定义一个独立的组,这个组包括反应位点处所有原子。
在模拟的.mdp文件中,对这个组定义量子力学模拟,事情就是这么简单。
对蛋白进行量子力学模拟时,一般使用洋葱模型。
所谓洋葱模型,就是对反应位点使用量子机制,在反应位点一定的半径内,使用半量子力学机制,然后分子部分使用分子机制。
那么索引文件就定义一个使用量子力学的组,把需要引进量子机制的原子都放到这个组中;再定义一个半量子机制的组,同时放进需要半量子力学机制模拟的原子,再在.mdp文件中独自定义即可。
再举一个例子,比如说在进行SMD(Steered MolecularDynamics,这个我一直没有想到或者找到切恰的中文翻译方法,或许可以叫做牵引分子动力学??别扭!!)中,要对蛋白莫一个原子或者残基作用力,那么可以建立一个索引文件,在该文件中定义一个组,把要施力的残基或者原子放到该组中。
然后在.ppa文件中使用该组就行了。
如果我还没有说明白,那么看看gromacs的参考文件吧。
如果还是不明白,可以来找我,我免费培训。
^_^索引文件使用make_ndx命令产生,"make_ndx -h"可以看到全部的参数。
gromacs一些分析命令

Molecular Dynamics Hands-on Session IITutorial on MD simulations using Gromacs 3.3.3Tsjerk A. WassenaarAnalysis of data from molecular dynamics simulationWhen the simulation has finished, it is time to continue with the analysis of the data. The analysis comprises of two stages. First, it is necessary to perform some standard checks to assess the quality of the simulations. If the results from these analyses show that the simulations are fine, then the analyses required to answer the research question asked can be performed.As a side note, many of the analysis tools produce graphs in the form of .xvg files. these files can be viewed with the program xmgrace, but they can also be viewed in a terminal using the python script xvg2ascii.py.Quality assuranceThe quality assurance (QA) involves tests for the convergence of thermodynamic parameters, such as the temperature, the pressure, the potential and the kinetic energy. Convergence is also checked in terms of the structure, through the root mean square deviation (RMSD) against the starting structure and the average structure. Next to that, it has to be checked that there have not been interactions between adjacent periodic images, as such interactions could lead to unphysical effects. A final QA test involves the calculation of the root mean square fluctuations of atoms, which can be compared to crystallographic b-factors.Convergence of energy termsWe start of with the extraction of some thermodynamic parameters from the energy file. The following properties will be investigated: temperature, pressure, potential energy, kinetic energy, unit cell volume, density, and the box dimensions. Energy analysis is performed with the tool g_energy. This program reads in the energy file, which is produced during the simulation (1LW9-MD.edr). g_energy will ask for a term to extract from the energy file and will produce a graph from that. Give the following command to run g_energy:g_energy -f 1LW9-MD.edr -o temperature.xvgThis will give a list of the energy and related terms which are stored in the .edr energy file. In our case, there are 68 terms in the energy file, each of which may be extracted and graphed. The first nine entries correspond to different energy terms in the force field. Also note the entries from 47 onward, which list the terms split over the groups Protein and Non-Protein, including the interactions between the two. Type "temperature" or "14" and press Return twice. Have a look at the graph with the program xmgrace, and see how the temperature converges to the value specified (300 K). Also note the fluctuations in the temperature:xmgrace temperature.xvgReferencing the energy terms by name facilitates automatic processing of the energy file. Using 'echo' and a pipe ("|") to redirect output from one program to the other, the query from g_energy can be answered automatically. Copy and paste or type the following lines to extract the other terms:echo pressure 0 | g_energy -f 1LW9-MD.edr -o pressure.xvgecho potential kinetic 0 | g_energy -f 1LW9-MD.edr -o energy.xvgecho volume 0 | g_energy -f 1LW9-MD.edr -o volume.xvgecho density 0 | g_energy -f 1LW9-MD.edr -o density.xvgecho box-x box-y box-z 0 | g_energy -f 1LW9-MD.edr -o box.xvgHave a look at each of these files and see how the values converge. If the values have not converged, this indicates that the simulation has not yet reached thermal equilibrium and that it should be extended before doing further analysis. Moreover, the period over which equilibration takes place should not be included in the analysis. Here, for the sake of simplicity, we will neglect these considerations and use the results from the simulation as is.Minimum distances between periodic imagesOne of the most important things to check in terms of quality assurance is that there have not been direct interactions between periodic images. Since periodic images are identical, such interactions are unphysical self-interactions. Imagine that a protein with a dipolewould have direct interactions. Then the attraction between the two ends of the same molecule over the periodic boundary would affect and invalidate the results relating to the native behaviour of the protein. To verify that such interactions have not taken place, we calculate the minimal distance between periodic images at each time. This is done using g_mindist.g_mindist -f 1LW9-MD.xtc -s 1LW9-MD.tpr -od minimal-periodic-distance.xvg -piRoot mean square fluctuationsNext to inspection of energies and such, convergence of the simulation towards equilibrium should also be inspected from the relaxation of the structure. Usually, such relaxation is only considered in terms of the Cartesian distance from the structure to a reference structure, e.g. the crystal structure. This distance is termed the root mean square deviation (RMSD). However, it is recommended also to investigate the relaxation towards the average structure, i.e. the RMSD with respect to the average. The reasons for this will be set out in the next paragraph. But to calculate the RMSD against the average structure, requires first to obtain the average. This structure can be obtained as a side product from calculation the root mean square fluctuations (RMSF). The RMSF captures, for each atom, the fluctuation about its average position. This gives insight into the flexibility of regions of the protein and corresponds to the crystallographic b-factors (temperature factors). Usually, one would expect similar profiles for the RMSF and the b-factors and this can be used to investigate whether the simulation results are in accordance with the crystal structure. The RMSF (and the average structure) are calculated with g_rmsf. The -oqoptions allows to calculate b-factors and add them to a reference structure:g_rmsf -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o rmsf-all-atom.xvg -ox average.pdb -oq bfactors.pdbHave a look at the graph of the RMSF with xmgrace and identify the flexible and rigid regions. Also view the two pdb files. Color the structure bfactors.pdb according to b-factors and inspect the flexible regions. The average structure is an unphysical structure. Have a look at some of the side chains, and notice the effect of averaging over conformations.Convergence of RMSDNow that we also have the average structure, we can calculate the RMSD. The RMSD is commonly used as an indicator of convergence of the structure towards an equilibrium state. As mentioned above, the RMSD is merely a distance measure. The RMSD is calculated using the program g_rms. First calculate the RMSD for all protein atoms, using the starting structure as a reference:g_rms -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o rmsd-all-atom-vs-start.xvgHave a look at the graph, and note at what time the RMSD levels off and at which value. Then calculate the RMSD again, but now only selecting the backbone atoms:g_rms -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o rmsd-backbone-vs-start.xvgThis time the RMSD settles at a lower value, which is the result of excluding the, often flexible, side chain atoms. In both cases the RMSD increases to a plateau value. This means that the structure of the protein reaches a certain distance from the reference structure and then keeps that distance more or less. However, with increasing distance, the amount of conformations available also increases. This means that, although the RMSD has reached a plateau value, the structure may still be progressing towards its equilibrium state. For this reason, it is advisable also to check the convergence towards the average structure:g_rms -f 1LW9-MD.xtc -s average.pdb -o rmsd-all-atom-vs-average.xvgg_rms -f 1LW9-MD.xtc -s average.pdb -o rmsd-backbone-vs-average.xvgCompare the resulting graphs to the previous ones. Note at which point the RMSD values level off.Convergence of radius of gyrationAs a final part of the QA, we calculate the radius of gyration. This measure gives an indication of the shape of the molecule at each time. The radius of gyration compares to the experimentally obtainable hyrdodynamic radius. It can be calculated using g_gyrate. This program will also give the individual components, which correspond to the eigenvalues of the matrix of inertia. This means that the first individual component corresponds to the longest axis of the molecule, while the last corresponds to the smallest. In effect, the three axes give a global indication of the shape of the molecule. Issue the commandg_gyrate -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o radius-of-gyration.xvgHave a look at the radius of gyration and the individual components and note how each of these progress to an equilibrium value.Structural analysis: properties derived from configurationsHaving assured that the simulation has converged to an equilibrium state, it is time to do some real analysis. Analysis of simulation data can be divided into several categories. The first category comprises of the interpretation of single configurations according to some metric to obtain a value, or a number of values, for each time point. The RMSD and the radius of gyration are examples. Such properties can be called configurational, dependent or instantaneous properties. Next to that, one can analyze processes in the time domain, e.g. through averaging, as (auto)correlations or fluctutations. In this section, a number of common analyses will be performed, each of which yields a time series of values directly derived from the trajectory (the coordinates over time).Solvent accessible surface areaOne property which can be of interest is the surface area of the protein which is accessible to solvent, commonly referred to as the solvent accessible surface (SAS) or the solvent accessible surface area (SASA). This can be further divided into a hydrophilic SAS and a hydrophobic SAS. In addition, the SAS can be used together with some empirical parameters to obtain an estimate for the free energy of solvation. All four of theseparameters are calculated by the program g_sas. This program also allows to calculate the average SAS over time per residue and/or per atom. Issue the following command, specifying Protein both for the group to calculate the SAS for and for the output group, and have a look at the output files.g_sas -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o solvent-accessible-surface.xvg -oa atomic-sas.xvg -or residue-sas.xvgHydrogen bondsAnother property which can be informative is the number of hydrogen bonds, either internally (Protein - Protein) or between the protein and the surrounding solvent. The presence or not of a hydrogen bond is inferred from the distance between a donor-H - acceptor pair and the donor - H - acceptor angle. To calculate the hydrogen bonds issue the following commands, and have a look at the output files using xmgrace:g_hbond -f 1LW9-MD.xtc -s 1LW9-MD.tpr -num hydrogen-bonds-intra-protein.xvgg_hbond -f 1LW9-MD.xtc -s 1LW9-MD.tpr -num hydrogen-bonds-protein-water.xvgSalt bridgesBesides hydrogen bonds, proteins also often form salt bridges between oppositely charged residues. These can have an important stabilizing effect on the structure of the protein, in particular when they are embedded in a hydrophobic environment, such as the core of the protein. The existence of salt bridges can be investigated with g_saltbr. If requested(through the flag -sep) This program will generate an output file for every pair of oppositely charged residues which at some point in the trajectory are within a certain cut off distance from each other (here 1.0 nm, specified through the option -t). Execute the following command and look at the output generated.g_saltbr -f 1LW9-MD.xtc -s 1LW9-MD.tpr -t 1.0 -sepSecondary structureAmong the most common parameters to judge protein structure is the assignment of secondary structure elements, such as α-helices and β-sheets. One such assignment is provided by dssp. This program, which is not part of the gromacs distribution, but can be obtained at the CMBI, Radboud University. Gromacs does provide an interface to dssp, to allow the calculation of secondary structure for each frame of a trajectory. To use this, first set an environment variable DSSP, which points to the location of the program. Then run do_dssp as follows:setenv DSSP /home/coursead/Wassenaar/MD/dsspdo_dssp -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o secondary-structure.xpm -sc secondary-structure.xvgThe file secondary-structure.xvg contains a time series, listing the numbers of residues associated with each type of secondary structure per frame. More detailed information is in the .xpm file, which gives a color coded assignment of the secondary structure per residueover time. The .xpm file can be viewed with e.g. the Gimp, but some useful metadata can be added with the gromacs tool xpm2ps and the result can be viewed with gview or a similar program:xpm2ps -f secondary-structure.xpm -o secondary-structure.epsRamachandran (phi/psi) plotsThe phi and psi torsion angles of the protein backbone are two parameters which are useful to gain insight into the structural properties of a protein. The plot of phi against psi is called a Ramachandran plot and certain regions of this plot are characteristic of secondary structure elements or of amino acids. Other regions are considered forbidden (inaccessible). Projection of the phi/psi angles over time may give insight in structural transitions. These angles can be calculated by the program g_rama, albeit in a bit of a crude way, namely put all together in a single file. To investigate single residues, the linux progam 'grep' can be used to select them out of the plot.g_rama -f 1LW9-MD.xtc -s 1LW9-MD.tpr -o ramachandran.xvgAnalysis of dynamics and time-averaged propertiesAnalysis of interatomic distances, NOE's from simulationsRelaxation and order parametersConfigurational principal component analysisThe use of principal component analysis in molecular dynamics focuses on revealing thestructure of atomic fluctuations.An often used, but often little understood, method of analysis is principal component analysis (PCA) of the trajectory. This method, which is sometimes also referred to as 'essential dynamics' (ED), aims to identify large scale collective motions of atoms and thus reveal the structures underlying the atomic fluctuations. The fluctuations of particles in a molecular dynamics simulation are by definition correlated due to interactions between the particles. The degree of correlation will vary and notably particles which are directly connected through bonds or lie in the vicinity of each other will move in a concerted manner. The correlations between the motions of the particles give rise to structure in the total fluctuations in the system and for a macromolecule this structure is often directly related to its function or (bio)physical properties. Thus, the study of the structure of the atomic fluctuations can give valuable insight in the behaviour of such macromolecules. However, it does require a certain level of understanding of linear algebra methods and multivariate statistics to interpret the results and identify the shortcomings of the method. In particular, the aim of principal component analysis is to describe the original data in terms of new variables which are linear combinations of the original ones. This is also the most important problem with PCA: it only offers an interpretation in terms of linear relationships between atomic motions.The first step in PCA is the construction of the covariance matrix, which captures the degree of collinearity of atomic motions for each pair of atoms. The covariance matrix is, by definition, a symmetric matrix. This matrix is subsequently diagonalized, yielding a matrix of eigenvectors and a diagonal matrix of eigenvalues. Each of the eigenvectorsdescribes a collective motion of particles, where the values of the vector indicate how much the corresponding atom participates in the motion. The associated eigenvalue gives equals the sum of the fluctuation described by the collective motion per atom, and thus is a measure for the total motility associated with an eigenvector. Usually most of the motion in the system (>90%) is described by less than 10 eigenvectors or principal components.The construction and diagonalization of the covariance matrix can be performed with the program g_covar. Issue the following command to perform the analysis:g_covar -s 1LW9-MD.tpr -f 1LW9-MD.xtc -o eigenvalues.xvg -v eigenvectors.trr -ascii covariances.datSelect the backbone when asked for a selection. Constructing and diagonalizing the covariance matrix requires some time. When the program has finished, have a look at the plot of the eigenvalues and calculate how much of the total motility is explained by the first ten eigenvectors.本文主要讨论隐式溶剂模型和GB模型(广义Born模型)的各个方面。
gromacs中计算氢键结合能

gromacs中计算氢键结合能在Gromacs中计算氢键结合能,可以使用了Gromacs中的分析工具g_hbond。
以下是基本步骤:1. 通过Gromacs执行模拟,生成.trr或者.xtc文件。
确保拓扑文件(.top)中包含氢键描述信息。
2. 运行以下命令,创建氢键指纹文件(.ndx):```gmx make_ndx -f <input_file> -o <output_file>.ndx```这里,`<input_file>`是模拟输出的.trr或者.xtc文件,`<output_file>`是输出氢键指纹文件的路径和文件名。
3. 运行以下命令,使用g_hbond工具计算氢键:```gmx hbond -f <input_file> -s <tpr_file> -n <ndx_file> -num<output_file>_hbnum.xvg -dist <output_file>_hbdist.xvg -ang <output_file>_hbangle.xvg```这里,`<input_file>`是模拟输出的.trr或者.xtc文件,`<tpr_file>`是模拟输入的.tpr文件,`<ndx_file>`是氢键指纹文件(.ndx)。
`<output_file>`是输出文件的路径和文件名。
通过以上命令,将会生成三个输出文件:- `<output_file>_hbnum.xvg`:氢键数量的时间演化曲线。
- `<output_file>_hbdist.xvg`: 氢键长度的时间演化曲线。
- `<output_file>_hbangle.xvg`: 氢键角度的时间演化曲线。
4. 进一步处理以上生成的曲线数据,可以从氢键长度和氢键角度中计算氢键结合能。
GROMACS教程
GROMACS程序DEMO例程####################### 概述 #######################----------------------------------------------------------------------------------------------------------------------------------该例程来自Gromacs程序/share/tutor/目录下。
整个例程大概只需要十分钟就可以完成,非常适合初学者学习。
该例程是一个完整的分子动力学模拟过程,涵盖了Gromacs程序基本的使用方法。
模拟内容是一个水环境下的小肽链。
模拟唯一要求是该小肽链的PDB文件。
文档翻译如果有错,请你给我发信:sen@。
相关内容请参阅Gromacs文档,或者给Gromacs开发组询问。
Gromacs官方网站:Email: gromacs@----------------------------------------------------------------------------------------------------------------------------------############################ 环境变量设置############################----------------------------------------------------------------------------------------------------------------------------------在以下的例程中,所有命令都直接运行,没有添加绝对路径。
所以,必须将Gromacs安装路径的bin文件夹加入到系统PATH变量中。
如果不加入PATH变量,那么运行时要加入命令的绝对路径。
GROMACS使用教程

GROMACS教程一Gromacs基本模拟流程 (3)1 下载pdb文件 (3)2 用pdb2gmx 处理pdb 文件 (3)3 建立盒子 (3)5 设置能量最小化 (4)6 用grompp程序进行文件处理 (6)7 使用genion 和tpr文件添加离子 (6)8 用fws_ion.pdb来产生能量最小化的输入文件 (6)9 在后台运行能量最小化(在命令后加&) (7)二设置位置限制性动力学模拟 (7)三设置非限制性动力学模拟 (9)1 如何重启一个计算 (11)2 如何延长一个计算 (11)3 如何设置并行计算 (11)五模拟结果分析 (12)1 如何将特定帧的轨迹保存成*.pdb文件 (12)2 用ngmx观察轨迹文件(也可以用VMD观察轨迹文件) (12)3 比较常用的分析工具 (14)3.3 g_covar 计算斜方差 (16)3.4 g_energy 能量数据作图,如压力、体积、密度等 (16)3.5 g_gyrate 测量回旋半径 (17)3.6 g_rms 与g_rmsdist 计算结构的RMSD 值 (17)3.7 g_rmsf 计算原子位置的根均方波动(rmsf ) (18)3.8 do_dssp 计算模型的二级结构 (20)3.9 g_hbond 计算模拟过程中分子间的氢键的数目、距离或角度 (21)3.10 g_saltbr 分析模拟中残基间的盐桥 (21)GROMACS 是一个使用经典分子动力学理论研究蛋白质动力学的高端的高效的工具。
GROMACS是遵守GNU许可的免费软件,可以从以下站点下载:,并且可以在linux和Windows上使用。
在本教程中,将研究一个从漏斗形蜘蛛的毒液中分离的毒素。
我们将使用显性溶剂动力学的方法来进行研究。
首先比较真空中和溶解的模型。
我们将把毒素肽溶在水盒子里,紧接着用牛顿运动定律加以平衡。
我们还将比较偿离子在显性溶剂动力学中的影响。
gromac轨迹采样指令

GROMACS是一款功能强大的分子动力学模拟软件包,用于模拟生物大分子如蛋白质和脂质的行为。
在分子动力学模拟中,轨迹文件包含了系统中所有粒子的位置和速度信息,这些信息随模拟时间的进展而更新。
轨迹采样通常指的是从这些轨迹文件中提取特定时间点或时间间隔的数据进行分析。
在GROMACS中,轨迹采样通常涉及到处理轨迹文件(如.xtc或.trr文件),这可以使用GROMACS自带的一系列工具来完成。
以下是一些常用的GROMACS命令,这些命令可以帮助用户对轨迹文件进行采样和分析:1. `trjconv`:用于处理轨迹文件,包括改变坐标、去除周期性边界条件(PBC)效应、提取特定时间范围的轨迹等。
例如,提取特定时间范围的轨迹:```shgmx trjconv -s topol.tpr -f traj.xtc -o sampled_traj.xtc -b starttime -e endtime```其中`-b`和`-e`分别设定起始时间和结束时间。
2. `gmx trjcat`:用于合并轨迹文件或者连接轨迹片段。
例如,合并两个轨迹文件:```shgmx trjcat -f traj1.xtc traj2.xtc -o combined_traj.xtc```3. `gmx cluster`:用于根据结构相似性对轨迹中的帧进行聚类分析。
4. `gmx trjtool`:这是一个更通用的轨迹处理工具,可以用于数据的提取、格式转换等。
要执行上述任何命令,你需要确保GROMACS正确安装在你的系统上,并且你的环境变量设置允许你直接调用`gmx`命令。
在使用这些工具时,可能需要根据具体需求调整命令行参数,并且可能还需要使用其它工具进行后续分析,如`gmx rms`进行均方根偏差分析,`gmx gyrate`计算回转半径等。
请注意,上述命令和参数可能会随着GROMACS版本的不同而有所变化,所以建议参考具体使用的GROMACS版本的官方文档。
gromacs文件介绍and一些杂知识

1 1gromacsGMX 各种文件格式详细可以查阅GROMACS 手册第5章第6小节以下为简要介绍。
CPT文件该文件为模拟断点文件check point.cpt。
该文件为模拟过程固定时间间隔产生保存模拟系统所有信息。
该文件一部分可以在能量文件.edr找到一部分可以在双精度轨迹文件.trr中找到。
如果模拟不幸因为外界条件中断如断电模拟人发脾气砸电脑等可以使用该文件重新在断点处开始模拟以节省模拟时间。
同时也可以依靠该断点文件开始并延长模拟计算见tpbconv。
EDR文件系统能量文件energy.edr。
该文件记录模拟输入文件中定义的能量组的各种相互作用能量等。
EPS 文件封装文件格式.eps并不是GROMACS自身文件格式可以当图片打开。
LINUX系统下一般已经有默认打开程序WINDOWS要安装其他打开程序可以GOOGLE以下。
GROMACS的DSSP和罗麽占陀罗图等通过xpm2ps处理后都是这个文件格式。
习惯就好。
G87文件分子坐标文件.g87。
该文件记录并只记录原子坐标和速度不含原子序号。
并只记录常压强模拟系统的盒子信息。
G96文件分子坐标文件.g96。
GROMOS96程序的分子坐标文件模拟程序以15.9的C语言格式写入精度较高但是会比较大。
包含有文件头时间步原子坐标原子速度以及盒子信息等。
GRO文件分子坐标文件.gro。
GROMACS的最主要分子坐标文件明白这个文件就基本明白使用GROMACS了。
该文件类型的各个文本列字数固定C语言的写入格式为quot5d5s5s5d8.3f8.3f8.3f8.4f8.4f8.4fquot。
具体固定文本列有2 残基序号5位数残基名称5字母原子名称5字母原子序号5为数原子坐标三列XYZ坐标各8位数含3个小数位速度同坐标速度单位为nm/pskm/s。
ITP文件分子拓扑文件.itp。
被主拓扑文件.top 包含的分拓扑文件一般包含某个特定分子的类型。
于主拓扑文件区别有它不引用其他力场文件同时包含systemmolecule等拓扑字节。
动力学模拟gromacs(绝对详细)

结构特征
• LOX-1蛋白是分子量为50kDa的Ⅱ型穿膜糖 蛋,由273个氨基酸组成,属于清道夫受体 家族 。LOX-l蛋白由包含34个残基的N末胞 质区域、单个穿膜区域、由82个残基组成 的颈部紧接着的由130个残基组成的C型凝 集素样配体合区域的C末端胞外部分组成。
二,利用GROMACS进行深入的结构优化和
结果分析: 对模拟得到的轨迹文件.trr 或者.xtc文件进行分析。
9 , 用g_rms,计算蛋白质Ca原子随时间的 RMSD结果,画图rmsd.jpg
g_rms -s 1YPU_grompp_hot_8md.tpr -f 1YPU_grompp_hot_8md.trr -o rmsd9.xvg
• 结果构象和目标构象的的偏差统计RMSD计算前首先要 把模拟结果构型进行平动和转动,使之与目标构型(一 般为初始结构)进行尽量的重合或部分重合,然后计算 每个原子与目标构型的坐标的差值!计算每个原子与目 标构型的坐标的差值(r_i,c -r_i,r),c,r代表存储构型和目 标构型,i代表构型上的某个原子。RMSD就是这些差值 的平方的平均,再开方!
• RMSD是计算在某一时刻的构象与目标构象所有原子偏 差的加和,对原子数的平均!每一帧有一个RMSD!因此 是衡量体系是否稳定的重要
图二,root-mean-square deviation,均方根偏差,
• 10,用g_rmsf,计算Ca原子的RMSF变化幅度并画图
-ff :指定力场文件,也可以不用这个参数,再自行选择; -ignh : 舍弃分子文件中的H原子,因为H原子命名规则多,
有的力场不认;
2,Editconf, 为蛋白质加上1.5nm厚度的水层
editconf -bt cubic -f 1YPU.gro -o 1YPU_editconf.gro -d 1.5
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
目录1、MAKE_NDX (2)2、G_TRAJ (3)3、G_ENERGY另外一种用法 (3)4、PDB2GMX (3)5、GENION (5)6、EDITCONF (6)7、G_RAMA (6)8、G_CLUSTER (7)9、GENBOX (7)1、make_ndxGromacs的索引文件,即index文件,由make_ndx程序生成,文件后缀为.ndx。
索引文件是gromacs最重要的概念文件,使用它可以在模拟过程中为所欲为。
举一个简单的例子,比如想详细了解HIV整合酶切割DNA的反应机理,使用量子力学模拟反应位点的反应过程,而分子其他部位使用一般分子动力学模拟。
于是我们就面临一个对模拟系统进行分割定义的问题,在gromacs中,就要用到索引文件。
基本的思路是这样的,在索引文件中,定义一个独立的组,这个组包括反应位点处所有原子。
在模拟的.mdp文件中,对这个组定义量子力学模拟,事情就是这么简单。
对蛋白进行量子力学模拟时,一般使用洋葱模型。
所谓洋葱模型,就是对反应位点使用量子机制,在反应位点一定的半径内,使用半量子力学机制,然后分子部分使用分子机制。
那么索引文件就定义一个使用量子力学的组,把需要引进量子机制的原子都放到这个组中;再定义一个半量子机制的组,同时放进需要半量子力学机制模拟的原子,再在.mdp文件中独自定义即可。
再举一个例子,比如说在进行SMD(Steered MolecularDynamics,这个我一直没有想到或者找到切恰的中文翻译方法,或许可以叫做牵引分子动力学??别扭!!)中,要对蛋白莫一个原子或者残基作用力,那么可以建立一个索引文件,在该文件中定义一个组,把要施力的残基或者原子放到该组中。
然后在.ppa文件中使用该组就行了。
如果我还没有说明白,那么看看gromacs的参考文件吧。
如果还是不明白,可以来找我,我免费培训。
^_^索引文件使用make_ndx命令产生,"make_ndx -h"可以看到全部的参数。
运行make_ndx后,可以使用" r "命令选择残基," a "命令选择原子," name 命令多组进行改名。
可以使用" | "表示或运算," & "表示与运算。
下面是几个简单的例子:-----------------------------1.选择56号残基r 562.选择3至45号残基r 3-453.选择3至15,23至67号残基r 3-15 | r 23-674.选择3至15号残基的主干链原子r 3-15 & 4 #在索引文件中,4号组为默认的主干链。
-----------------------------组合是灵活的,使用的时候好好发挥聪明的大脑啦。
个人觉得gromacs把索引文件概念做得非常好,并独立成一类文件是一个不小的创举啊。
在这个概念很值钱的年代,基本上使gromacs多一个大大的卖点。
2、g_traj几乎gromacs的所有分析数据都可以输出为xmgrace的数据文件,g_traj可以产生gromacs轨迹的各个组的坐标,速度,受力和边界等等。
使用” -com “参数可以求出轨迹中各个组的质心的坐标,速度,受力等;使用” -mol “则可以求取系统中各个分子的信息;” -ot “则可以求出系统中各个组的温度。
还有几个其他参数,比如” -cv “可以求平均速度,” -cf “可以求平均受力等。
其他参数同其他命令无异,参加说明文件。
3、g_energy另外一种用法Gromacs的各个工具都很有个性,如果互相结合,可以做很多事情。
g_energy求系统轨迹各个能量的,一般跑完MD之后,使用g_energy处理ener.edr只能得到系统的各个能量项。
但是如果想求系统中两个不同部分在模拟过程中的相互作用能量,那就要使用一些小窍门。
以下是实现的一个方法:第一,根据原来的tpr文件建立一个新的tpr,在这个新的tpr中,明确定义感兴趣的组。
这要用索引文件,见上文。
第二,用mdrun的" -rerun "参数指定原来的轨迹文件再跑一次模拟,这个过程很快。
如果还想更快,可以使用trjconv把水分子去掉。
这一个重复的模拟也产生轨迹文件,重要的是,还产生一个新的ener.edr文件,这个文件中包含了tpr文件中定义的各个组能量及相互作用能量(库伦相互作用能,范德华相互作用能等)。
第三,再使用g_energy把各个能量项提出来,想要什么提什么。
嗯,结果非常好。
不信你试试。
4、pdb2gmx使用gromacs做分子动力学模拟时,第一个要用到的命令一般都是pdb2gmx。
这个命令吧pdb分子文件转化成gromacs独特的gro分子结构文件类型,同时产生分子拓扑文件。
Gromacs是典型的GPL软件,每一个命令都有很多命令参数。
这对熟悉windows环境的人来说有一点烦,但是如果熟悉了Linux环境,也就慢慢喜欢啦。
(建议多使用命令,就像VMD, Pymol, rasmol和Chimera等等分子可视化软件,如果接合命令使用,功能都非常强大。
另外一个比较bt的软件叫做WHATIF 的,完全建立在bt的命令菜单上,心理承受能力不强者多半吐血而终。
使用“pdb2gmx -h”可以得到pdb2gmx的所有参数及简单说明(gromacs的任何命令都可以使用-h参数得到类似帮助)。
pdb2gmx的参数很多,但是常用的只有以下几个:------------------------------------------------------------------------f 指定你的坐标文件,可以是pdb、gro、tpr等等包含有分子坐标的文件;-o 输出文件,也就是处理过的分子坐标文件,同样可以是pdb、gro、g96等文件类型;-p 输出拓扑文件。
pdb2gmx读入力场文件,根据坐标文件建立分子系统的拓扑;-water 指定使用的水模型,使用pdb2gmx的时候最好加这个参数,不然后面会吃苦头。
它会提前在拓扑文件中添加水分子模型文件;-ff 指定力场文件(下文讨论),也可以不用这个参数,再自行选择;-ignh 舍弃分子文件中的H原子,因为H原子命名规则多,有的力场不认;-his 独个指定HIS残基的质子化位置。
------------------------------------------------------------------------其他的参数还不少,可以好好看一个pdb2gmx的帮助文件,一般的pdb2gmx 的执行格式如下(假设你的分子坐标文件为sen.pdb):pdb2gmx -f sen.pdb -o sen.gro -p sen.top -water tip4p -ignh -his该命令读入分子文件,使用tipp水模型,等等,然后pdb2gmx会让你选择力场文件。
然后它就很聪明的帮你建立初始模拟系统啦。
gromacs自带的力场有很多:------------------------------------------------------------------------0: GROMOS96 43a1 force field1: GROMOS96 43b1 vacuum force field2: GROMOS96 43a2 force field (improved alkane dihedrals)3: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)4: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)5: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)6: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)7: [DEPRECATED] Gromacs force field (see manual)8: [DEPRECATED] Gromacs force field with hydrogens for NMR9: Encad all-atom force field, using scaled-down vacuum charges 10: Encad all-atom force field, using full solvent charges------------------------------------------------------------------------本人建议使用OPLS力场和tip4p水模型。
tip4p水有四个粒子,分别是两个氢原子,一个氧原子和一个没有质量的电粒子。
这个电粒子在其他三个原子中间靠近氧原子。
tip4p水模型多了一个粒子,模拟代价高一点,但是结果要好一点。
但是最近有报道说使用spce水模型和GROMOS力场的计算结果最好,嘿嘿,好一个百家争鸣的学术氛围啊,我真的号感动哦。
Gromacs也可以使用其他力场,如AMBER力场等,使用方法请参考google。
pdb2gmx的输出基本可以做真空中模拟了,现在对MD模拟的要求高,一般都要有点水。
为分子系统添加水环境和离子环境需要其他命令,要慢慢来。
5、genion在给蛋白质添加水环境之后,一般要在水环境中添加金属离子,使模拟系统更加接近真实系统。
如果系统中蛋白质本身已经带了静电量,那么就更要给系统加几个带相反电量的金属离子,使系统处于电中性。
gromacs中添加金属离子的命令是genion,使用" genion -h "可以得到其使用的参数,其中有几个比较常用:---------------------------" -s: "指定系统tpr文件。
" -p: "指定系统拓扑文件,在往系统中添加金属离子时,genion会往拓扑文件最后的分子类型中写入添加的离子数,并修改拓扑文件中系统原子数。
" -o: "指定输出文件,genion的输出是pdb文件或者gro等结构文件。
也就是说你产生这个文件之后,还要再用这个文件产生tpr文件。
" -np/-nn: "带正/负电金属粒子的数目。
这个数目有一点讲究,一般需要看个人的应用。
假如想要得到" 0.1mol/L " 的离子浓度到底要加多少,可以自己算一下(很简单,方法很多,比如看课本)。