HPPD 酶及其抑制剂构效关系的研究进展
基于伏诺拉生方案根除幽门螺杆菌的研究进展

[基金项目] 云南省科技厅科技计划项目(2018FH 001-076;2018FH 001-080)。
△大理大学临床医学院2021级内科学在读硕士研究生▲通讯作者基于伏诺拉生方案根除幽门螺杆菌的研究进展刘 涛1△ 郑 盛2▲ 杨 涓21.大理大学临床医学院,云南大理 671000;2.云南省第三人民医院消化内科,云南昆明 650011[摘要] 伏诺拉生(VPZ)是新型抑酸药,其抑酸作用强大持久,优于传统质子泵抑制剂(PPI),可广泛应用于酸相关性疾病,目前在国内的适用证主要是反流性食管炎。
越来越多研究发现对比基于PPI 方案根除幽门螺杆菌(Hp),基于VPZ 的方案能提高Hp 根除成功率,而不良反应与之相当甚至更少,最新国内发布的Hp 治疗指南也提到VPZ 的应用,但并未明确用法。
本文就目前VPZ 用于Hp 治疗的作用机制、使用方案、疗效以及安全性等方面进行了总结,旨在为探索VPZ 用于治疗Hp 感染的提供参考。
[关键词] 伏诺拉生;幽门螺杆菌;质子泵抑制剂;疗效;安全性[中图分类号] R573 [文献标识码] A [文章编号] 2095-0616(2024)05-0046-04DOI:10.20116/j.issn2095-0616.2024.05.10Research progress on the eradication of Helicobacter pylori based on the Vonoprazan regimenLIU Tao 1 ZHENG Sheng 2 YANG Juan21. School of Clinical Medicine, Dali University, Yunnan, Dali 671000, China;2. Department of Gastroenterology, the Third People’s Hospital of Yunnan Province, Yunnan, Kunming 650011, China[Abstract] Vonoprazan (VPZ) is a new type of acid-suppressing drug, with a strong and long-lasting acid inhibitory effect, superior to traditional proton pump inhibitors (PPI), and can be widely used in acid-related diseases. Currently, its main indication in China is reflux esophagitis. More and more studies have found that compared to PPI-based regimens for eradicating Helicobacter pylori (Hp), the regimen based on VPZ can improve the success rate of Hp eradication, and the adverse reactions are equivalent or even fewer. The latest domestic guidelines for the treatment of Helicobacter pylori also mention the use of VPZ, but the usage is not clear. This article summarizes the mechanism of action, usage regimen, efficacy, and safety of VPZ in the treatment of Hp infection, aiming to provide a reference for exploring the use of VPZ in the treatment of Hp infection.[Key words] Vonoprazan; Helicobacter pylori ; Proton pump inhibitors; Efficacy; Safety1983年Marshall 和Warren 发现了幽门螺杆菌(Helicobacter pylori ,Hp),研究表明Hp 与胃癌等多种消化道疾病的发病密切相关[1]。
GLP_1受体激动剂及DPP_抑制剂的研究进展

Journal of China Pharm aceutical U n iversity 2008, 39( 5): 385- 391
38 5
药学前沿
GLP 1受体激动剂及 DPP 抑制剂的研究进展
周映红, 黄文龙* , 张惠斌, 对糖尿 病治疗药胰高血糖素样肽 1( G LP 1)受体激动剂和二肽基肽酶 ( DPP ) 抑制剂的研究进展进 行了 综述。介绍了 G LP 1的血糖调控机制, 还对 GLP 1受体激动剂 ( 如 Ex endin 4, Exentide LAR, L irag lutide, C JC 1131, 非 肽类 GL P 1受体激动剂 ) 和 DPP 抑制剂 ( 如 Sitag liptin, V ildag liptin, Saxag liptin, A log liptin) 进 行了详 细的介 绍, 为 2型糖 尿病 治疗药物的研发提供 参考。
Buse等 [ 11] 进行 了一项长达两 年的临床跟踪 调查, 研究对象为 974名患者, 观察的指标包括糖 化血 红 蛋 白 ( H bA1c )、空 腹 血 浆 葡 萄 糖 水 平 ( FPG )、体重、血压、胰岛 细胞的分泌功能 ( HO MA B) 、胰岛素敏感性 ( HOMA S) 以及对肝脏功能 ( 丙 氨酸 转 氨 酶, ALT ) 的影 响。统 计 结 果显 示 H bA1c下降 ( 1 1 ! 0 1) % , FPG 下降 ( 25 2 ! 2 8) m g /dL, 这两个指标在第 12 周观察到的数据分别 是下降 ( 1 1 ! 0 1) % 和 ( 25 1 ! 2 4) m g / dL。表 明 Exendin 4可将 H bA 1c维持在一个正常或相对 较低的水平。体重下降 ( 4 7 ! 0 3) % , 其中 81% 的患者伴有体重下降的症状。HOMA B 和 HOMA S与治疗前相比都有明显改善, 表明其对胰岛细胞 具有保护功能。对于 原来 ALT 水 平正常 的患者 ALT 值没有明显变化, 而 ALT 较高的患者则明显 的下降, 幅度为 ( 11 ! 1) IU /L ( P < 0 05) , 说明对肝 功能有所改善。血压平均下降 4 4 mmH g ( 舒张压 和收缩压都下降 ) , 高密度脂蛋白胆固醇 ( HDL C ) 平均上升 7 3 m g / dL; 甘油三酯 ( TG) 平均下降 9 3 m g /dL。血压、HDL C和 TG是 2型糖尿病治疗中的 3个心血管危险因子, 一般的药物治疗会引起血压 上升, HDL C下降和 TG水平上升, 增加了心血管危 险性, 相比之下, Exendin 4不存在这种问题 。 [ 11]
反义寡核苷酸化学修饰酶类药物的研究进展

反义寡核苷酸化学修饰酶类药物的研究进展[关键词]:化学修饰,靶向技术,序列选择,靶向转运摘要:反义寡核苷酸(antisense oligodeoxynucletide,as ODN)类药物是人工合成并经化学修饰的寡核苷酸(ODN)片段,能通过自身设计的特定序列与靶 mRNA 结合,在基因水平干扰致病蛋白的产生。
由于其高度的选择性和较低的副作用,as-ODN类药物已成为近年来药物研究和开发的热点。
最近,as -ODN类药物福米韦生(fomivirsen,Vitravene)通过美国FDA批准为第一个进入市场的反义药物。
其他asODN类药物ISIS2302,ISIS3521/CGP64128A和 G3139等在临床试验中也表现出良好的疗效。
as-ODN作为基因表达的反向抑制剂,首先必须具备三个主要条件:即它应有足够的稳定性、对目的基因的选择性以及对细胞的通透性和靶向性。
满足三个首要条件的方法主要是针对ODN在化学修饰、序列选择、靶向转运等方面加以改善。
反义寡核苷酸的化学修饰不经修饰的ODN不论在体液内还是细胞中都极易被降解,不能发挥其反义作用。
因而采用经化学修饰的ODN,以减少核酸酶对ODN的降解。
对ODN化学修饰的方法主要针对三方面,即碱基修饰、核糖修饰和磷酸二酯键修饰。
碱基修饰主要为杂环修饰、5-甲基胞嘧啶和二氨基嘌呤;核糖修饰主要为己糖。
2’-O-甲基取代核糖、环戊烷、α构象核糖;磷酸二酯键修饰主要为硫代和甲基代修饰等。
其中硫代寡核苷酸(phosphorothioate,PS-ODN)、混合骨架寡核苷酸(mixed backbone oligonucleic acid, MBO)和多肽核酸(peptide nucleic acid,PNA)应用广泛,成为具有代表性的第一、二、三代ODN。
硫代寡核苷酸由于磷酸二酯键是核酶的主要靶点,因此采用硫化试剂将ODN磷酸二酯键硫化成为PS-ODN类结构,是增强ODN稳定性的有效途忡。
微生态制剂及其临床应用研究进展_黄灿

广泛。 2. 1 预防抗生素相关性腹泻
抗生素的使用会破坏人体胃肠道内正常的菌群及其分布, 导致菌群失调,当肠道内的球 / 杆菌比例失调时,就会引起以腹 泻为主的临床症状,尤其是现今抗生素滥用的时代,预防抗生 素相关性腹泻就显得尤其重要。使用微生态制剂是目前预防 抗生素相关性腹泻的方法之一,马春英、姚晶晶[4]对酪酸梭菌 活菌预防抗生素相关性腹泻的临床疗效进行了研究,将 101 例 肺炎患者分为两组,其中对照组 53 例患者给予抗菌药以及对 症治疗,预防组 48 例患者在此基础上加服酪酸梭菌活菌,观察 两组患者出现腹泻情况、大便性状恢复正常时间及伴随症状发 生情况。结果预防组患儿腹泻发生率明显低于对照组( 分别 为 12. 5% 、39. 6% ,P < 0. 05) ,且腹泻发生时间明显晚于对照 组,伴随症状发生率也明显低于对照组,表明微生态制剂在预 防抗生素相关性腹泻的效果显著。从现有的临床证据来看,微 生态制剂可降低 抗 生 素 相 关 性 腹 泻 的 发 生 率[4],但 仍 需 大 样 本的随机双盲对照试验来进一步验证和支持[5]。 2. 2 肠易激综合征( irritable bowel syndrome,IBS)
组蛋白去乙酰化酶及去甲基化酶抑制剂在胃肠道肿瘤的研究进展

现代消化及介入诊疗 2021年第26卷第1期ModernDigestion&Intervention2021牞Vol.26牞No.1 ·综述·组蛋白去乙酰化酶及去甲基化酶抑制剂在胃肠道肿瘤的研究进展陈俊豪1,2,丁杰1,2,李显2,岑祥莹2,张林2,吴明2,樊斐2,曾家兴2 【提要】 组蛋白甲基化及乙酰化修饰的平衡失调与多种肿瘤的发生、发展、侵袭、转移密切相关,多种胃肠道肿瘤中发现组蛋白去乙酰化酶(HDAC)及组蛋白赖氨酸特异性去甲基酶1(LSD1)异常增高。
相应的,一些组蛋白去乙酰化酶抑制剂(HDACi)和LSD1抑制剂已在胃肠道肿瘤的研究中取得进展,如异羟肟酸类HDACi在胃肠道抗肿瘤研究中取得良好疗效,但因其特异选择性低,易产生耐药性和严重副作用,在临床的进一步研究中受到限制;苯甲酰胺类HDACi在特异选择性有所提高,并且能够通过抑制肿瘤细胞分化、诱导免疫自噬、抑制细胞周期蛋白产生抑瘤作用,但其由于活性低而受到限制;环肽类HDACi特异性进一步增加,但只是出于研究的基础阶段。
相应的,LSD1抑制剂,如苯环丙胺类、多肽类、小分子化合物抑制剂均在细胞层面有着良好的抑瘤作用,也在后期的整体实验均显示出耐药性和严重副作用。
这提示着单一的HDACi和LSD1抑制剂的抑瘤效应均不佳,由于HDAC常和LSD1形成复合体发挥转录调节作用,因此,双靶点抑制剂可能是有效的,后期的双靶点抑制剂,比如DuanYC等人报道了TCP和SAHA的组合产生的环戊二烯衍生物,AnastasJN报道的Corin,的确呈现出更加显著的抑瘤成效,本文就HDAC、LSD1抑制剂及二者的双重抑制剂在胃肠道肿瘤的研究进展进行综述。
【关键词】 组蛋白去甲基化酶;组蛋白去乙酰化酶;组蛋白去甲基化酶抑制剂;组蛋白去乙酰化酶抑制剂;双重抑制剂;胃癌与结肠癌中图分类号:R735.2;R57 文献标志码:A DOI:10.3969/j.issn.1672-2159.2021.01.028作者单位:1563003遵义医科大学研究生院;2550000贵州省人民医院胃肠外科通信作者:丁杰,E mail:dingjieboy@126.com基金项目:国家自然科学基金(81360366,81302169);贵州省社会发展攻关项目(黔科合SY字[2014]3023号);贵州省优秀青年科技人才培养对象(黔科合平台人才[2017]5602);贵州省高层次创新型人才培养对象(GZSYQCC[2014]001);贵州省科技计划项目(黔科合基础[2019]1198号,黔科合基础[2020]1Z064);贵州省高层次留学人才创新创业项目(留学人才择优资助合同[2018]04号) 胃癌新发病例排在恶性肿瘤的第5位[1],而结直肠癌是世界第三大恶性肿瘤和第四大癌症死亡原因,且无论是发病例数还是死亡率均呈上升趋势[2]。
农药化学的期末考试

农药:用于防治为害农作物及农副产品的病虫害、杂草及其它有害生物的化学药剂的统称。
急性毒性:药剂一次进入人体后短时间引起的中毒现象。
慢性毒性:药剂长时间作用于有机体后,引起药剂在体内的积蓄,或者造成有机体机能损害的积累而引起的中毒现象。
LD 50 :致死中量,或半致死量。
经口LD 50 :一次口服急性中毒死亡死亡半数的剂量经皮LD 50 :通过皮肤摄入极性中毒死亡半数的剂量。
农药残留:在农业生产中施用农药后一部分农药直接或间接残存于谷物、蔬菜、果品、畜产品、水产品以及土壤和水体中的现象。
农药代谢:农药的代谢是指作为农药进人生物体后,生物体利用自身的多种酶,对这些外源化合特产生化学作用,以达到排泄目的的过程,这类作用也称为生物转化。
初级代谢:一般将微生物从外界吸收各种营养物质,通过分解代谢和合成代谢生成维持生命活动的物质和能量的过程,称为初级代谢农药选择性:是指仅对某种或某几种病、虫、草害有防治效果的农药。
杀虫剂的主要类型:按作用方式可分类为:①胃毒剂。
②触杀剂。
③熏蒸剂。
④内吸杀虫剂。
按毒理作用可分为:①神经毒剂。
②呼吸毒剂。
③物理性毒剂。
④特异性杀虫剂。
制备反应:有机磷杀虫剂合成,吡虫啉合成化学除草剂的发展过程:19 世纪末:无机除草剂;1932 年:有机除草剂二硝酚;1942 年:第一个内吸性的有机除草剂2,4-D ;1980s :磺酰脲类除草剂的发现,掀起了超高效除草剂研究的热潮。
这是除草剂发展史上新的里程碑。
抑制植物氨基酸生物合成的除草剂。
目前,主要有两类氨基酸的生物合成过程已经被开发为除草剂的作用靶标:(1)支链氨基酸的生物合成:缬氨酸、亮氨酸、异亮氨酸(2)芳香氨基酸的生物合成:苯丙氨酸、色氨酸、酪氨酸为什么杀虫剂马拉硫磷会具有高效低毒的特点?杀虫剂马拉硫磷具有选择性, 马拉硫磷在昆虫体内转变为更毒代谢产物, 温血动物体内的转变为无毒代谢产物3说明杀虫剂马拉硫磷选择性原理 ?马拉硫磷具有生理选择性 , 具有在害虫和温血动物(人体)之间代谢的差异 干扰植物激素型除草剂 :干扰植物激素型除草剂就是破坏了植物体内激素的平衡,从而造成植物形态畸型,如茎加长、次生根及愈伤组织生长但却阻碍叶 片的发育等,严重破坏了植物的正常生理过程,最终导致植物死亡 赤霉素、脱落酸、细胞分裂素、吲哚乙酸、乙烯。
抗真菌药物靶标及其抑制剂的研究进展

抗真菌药物靶标及其抑制剂的研究进展徐波;蒋琰;张万年;盛春泉【摘要】目的综述抗真菌药物靶标及其抑制剂的研究进展.方法本文结合自身研究工作,分析近5年文献,总结抗真菌药物靶标及其抑制剂的最新进展.结果β-1,3-葡聚糖合成酶、羊毛甾醇14α-去甲基化酶、N-肉豆蔻酰基转移酶和分泌型天冬氨酸蛋白酶是目前研究最为集中的抗真菌药靶,其抑制剂显示了良好的新药开发前景.结论优化现有药物化学结构和发现全新作用机制的先导化合物,对研发新一代抗真菌药物具有重要意义.【期刊名称】《药学实践杂志》【年(卷),期】2013(031)005【总页数】6页(P321-325,379)【关键词】抗真菌药物靶标;β-1,3-葡聚糖合成酶;羊毛甾醇14α-去甲基化酶;N-肉豆蔻酰基转移酶;分泌型天冬氨酸蛋白酶【作者】徐波;蒋琰;张万年;盛春泉【作者单位】第二军医大学药学院,上海200433;海市食品药品监督管理局崇明分局,上海202150;第二军医大学药学院,上海200433;第二军医大学药学院,上海200433;第二军医大学药学院,上海200433【正文语种】中文【中图分类】R914近年来,真菌感染尤其是深部真菌感染(如侵入性白色念珠菌病,隐球菌病和曲霉菌病等)大幅上升,已成为一种严重威胁人类健康的疾病[1,2]。
这主要与免疫缺陷患者的急速增加有关,导管插管技术的普遍应用,广谱抗菌素的滥用,骨髓、器官移植和肿瘤放化疗,以及艾滋病患者的增加等因素引起免疫抑制,进而导致机会性深部真菌感染的发生。
目前临床上最常见的致病真菌主要有:白色念珠菌(Candida albicans)、烟曲霉菌(Aspergillus fumigatus)和新型隐球菌(Cryptococcus neoformans),其中白色念珠菌和烟曲霉菌感染分别占深部真菌感染的70%~90%和10%~20%[3]。
另一方面,一些新型致病真菌(如毛霉菌属、镰孢菌属、结合菌属和足放线病菌属等)不断出现。
乳腺癌放疗抵抗机制的研究进展

- 172 -*基金项目:国家自然科学基金青年项目(81903119);中山大学高校基本科研业务费青年教师培育项目(20ykpy52)①中山大学附属第一医院 广东 广州 510080通信作者:毕月乳腺癌放疗抵抗机制的研究进展*毕月① 【摘要】 放疗是鼻咽癌、乳腺癌及直肠癌等多种恶性肿瘤的重要治疗手段,不过部分患者会出现放疗抵抗的现象,造成转移及复发,从而降低患者生存率。
乳腺癌患者放疗抵抗的发生与多种因素有关,如肿瘤干细胞及肿瘤微环境等。
乳腺癌放疗患者的治疗效果与放疗抵抗密切相关,对放疗抵抗的产生机制进行研究有助于减少或者抑制放疗抵抗,提高放疗效果,改善患者预后。
本文从肿瘤干细胞、自噬性调节及DNA 损伤修复等多个方面做一综述,旨在为随后研究提供思路。
【关键词】 乳腺癌 放疗抵抗 肿瘤干细胞 自噬性调节 细胞周期调控 上皮间充质转化 DNA 损伤修复 Research Progress on the Mechanism of Radiotherapy Resistance in Breast Cancer/BI Yue. //Medical Innovation of China, 2023, 20(30): 172-176 [Abstract] Radiotherapy is an important treatment method for nasopharyngeal carcinoma, breast cancer, rectal cancer and other malignant tumors. However, some patients may experience resistance to radiotherapy, which may cause metastasis and recurrence, thus reducing the survival rate of patients. The occurrence of radiotherapy resistance in breast cancer patients is related to many factors, such as cancer stem cell and tumor microenvironment. The therapeutic effect of breast cancer patients undergoing radiotherapy is closely related to their resistance to radiotherapy. Studying the mechanism of resistance to radiotherapy can help reduce or inhibit resistance to radiotherapy, improve the effect of radiotherapy, and improve the prognosis of patients. This article reviews cancer stem cell, autophagy regulation, DNA damage repair and other aspects in order to provide ideas for subsequent research. [Key words] Breast cancer Radiotherapy resistance Cancer stem cell Autophagy regulation Cell cycle regulation Epithelial mesenchymal transformation DNA damage repair First-author's address: First Affiliated Hospital of Sun Yat-sen University, Guangzhou 510080, China doi:10.3969/j.issn.1674-4985.2023.30.040 乳腺癌是严重危害女性身心健康的一种恶性肿瘤。
组蛋白去乙酰化酶(SIRT1)抑制剂的研究进展

2019年第11期广东化工第46卷总第397期·107·组蛋白去乙酰化酶(SIRT1)抑制剂的研究进展杨利生,舒志豪,张齐玉,王德传*(中国药科大学理学院,江苏南京211198)[摘要]SIRT1是一种依赖NAD+辅酶的组蛋白去乙酰化酶,通过对组蛋白进行去乙酰化修饰,调控基因的的表达。
SIRT1与代谢,炎症和肿瘤等多种疾病有关,有望成为肿瘤治疗的新靶点。
本文对近些年SIRT1抑制剂的发展,以及它们存在的问题作了概述,并对SIRT1抑制剂的未来进行展望,将为以后SIRT1抑制剂的开发提供一些参考。
[关键词]SIRT1;组蛋白去乙酰化酶;SIRT1抑制剂[中图分类号]TQ[文献标识码]A[文章编号]1007-1865(2019)11-0107-02Advances in Research of Histone Deacetylase InhibitorsYang Lisheng,Shu Zhihao,Zhang Qiyu,Wang Dechuan*(College of Science,China Pharmaceutical University,Nanjing211198,China)Abstract:SIRT1are coenzyme NAD+-dependent histone deacetylases.It regulates gene expression by deacetylating histone.SIRT1is associated with many diseases,such as metabolism,inflammation and cancer,and is expected to become a new target for cancer treatment.In this paper,the development of SIRT1 inhibitors in recent years and their problems are summarized,which will provide some references for the future development of SIRT1inhibitors Keywords:SIRT1;Histone deacetylases;SIRT1inhibitorsSirtuin蛋白是酵母菌中的沉默信息调节因子2(Sir2)的同系物[1],在人类中一共有七种sirtuin蛋白,它们广泛分布于细胞的不同亚结构中[2]。
Src蛋白激酶的研究进展

㊀收稿日期:2022-03-30作者简介:陈烨(1965-)ꎬ男ꎬ辽宁沈阳人ꎬ研究员ꎬ博士生导师ꎬ研究方向:创新药物研发.㊀∗通讯作者:陈烨ꎬE ̄mail:chenye@163.com.㊀㊀辽宁大学学报㊀㊀㊀自然科学版第50卷㊀第4期㊀2023年JOURNALOFLIAONINGUNIVERSITYNaturalSciencesEditionVol.50㊀No.4㊀2023Src蛋白激酶的研究进展陈㊀烨∗ꎬ王㊀智ꎬ傅浩栋ꎬ车㊀晋(辽宁大学药学院ꎬ辽宁沈阳110036)摘㊀要:类固醇受体辅激活因子(SteroidreceptorcoactivatorꎬSrc)是一种由Src原癌基因编码的非受体型酪氨酸激酶ꎬ属于Src家族蛋白激酶(Src ̄familykinasesꎬSFKs)的核心成员.Src广泛存在于人体细胞中ꎬ可调节细胞分裂㊁运动㊁黏附㊁血管生成和存活等多种过程ꎬ对维持机体的正常生理功能活动具有重要作用.Src诱导各种恶性细胞的转化ꎬ在多种肿瘤细胞中都有发现ꎬ可以参与肿瘤的产生㊁生长㊁转移等多方面.与Src相关的信号通路异常激活或过表达会导致机体异常ꎬ进而导致癌症的产生.本文主要综述了Src的结构㊁Src的信号通路㊁Src对癌症治疗的作用及其抑制剂等.关键词:SrcꎻSrc信号通路ꎻ癌症ꎻ抑制剂中图分类号:R73㊀㊀㊀文献标志码:A㊀㊀㊀文章编号:1000-5846(2023)04-0359-07ResearchProgressofSrcProteinKinaseCHENYe∗ꎬWANGZhiꎬFUHao ̄dongꎬCHEJin(SchoolofPharmaceuticalSciencesꎬLiaoningUniversityꎬShenyang110036ꎬChina)Abstract:㊀Steroidreceptorcoactivator(Src)isakindofnon ̄receptortyrosinekinasesencodedbySrcproto ̄oncogenesꎬwhichisacorememberofSrc ̄familykinases(SFKs).Srciswidelypresentinhumancellsandplaysanimportantroleinthemaintainingnormalphysiologicalfunctionsofthebodybyregulatingvariousprocessessuchascelldivisionꎬmovementꎬadhesionꎬangiogenesisandsurvival.Srcinducesthetransformationofvariousmalignantcellsꎬwhichhasbeenfoundinavarietyoftumorcellsandcanbeinvolvedintheoccurrenceꎬgrowthandmetastasisoftumors.AbnormalactivationoroverexpressionofSrc ̄relatedsignalingpathwayscanleadtoabnormalitiesinthebodythatleadtocancer.InthispaperꎬthestructureꎬsignalingpathwayꎬroleofSrcincancertreatmentanditsinhibitorsarediscussed.Keywords:㊀steroidreceptorcoactivator(Src)ꎻSrcsignalingpathwayꎻcancerꎻinhibitors㊀㊀0㊀引言全球癌症死亡例数和发病例数持续上升[1]ꎬ癌症已经成为威胁人类健康的最大敌人.酪氨酸激酶(TyrosinekinaseꎬTKs)作为抗肿瘤药物研究的重要靶点ꎬ起到将细胞外环境中的信号传递到细胞内部的作用[2].根据是否具有细胞外配体结合和跨膜结构域的受体样特征ꎬTKs可以分为受体酪氨酸激酶(ReceptortyrosinekinasesꎬRTKs)和非受体酪氨酸激酶(NonreceptortyrosinekinaseꎬNRTKs).类固醇受体辅激活因子(SteroidreceptorcoactivatorꎬSrc)属于NRTKsꎬ能够参与细胞内信号转导并调节生命活动的生化反应ꎬ对维持细胞㊁组织和器官的稳态具有十分重要的意义[3].临床研究表明ꎬSrc在肺癌[4]㊁乳腺癌[5]等肿瘤细胞的产生㊁转移中有重要作用.1㊀Src的结构Src约为60kuꎬSrc与Blk(B淋巴酪氨酸激酶)㊁Fgr(猫肉瘤病毒原癌基因同系物)㊁Fyn(致密物酪氨酸激酶)㊁Hck(造血细胞激酶)㊁Lyn(一种酪氨酸蛋白激酶)㊁Lck(淋巴细胞特异性激酶)㊁Yes(一种酪氨酸蛋白激酶)㊁Yrk(一种酪氨酸蛋白激酶)共同构成Src家族蛋白激酶(SFKs)[6].基于它们的氨基酸序列差异ꎬSrc分为两个亚家族ꎬ第一类包括Src㊁Fyn㊁Yes和Yrkꎬ第二类包括Blk㊁Fgr㊁Hck㊁Lck和Lynꎬ主要存在于造血细胞中.Src结构由SH1㊁SH2㊁SH3㊁SH4组成[7]ꎬ其中SH4是膜附着所必需的ꎻSH2和SH3结构域不但可以将Src定位到合适的细胞位置ꎬ而且参与调节Src的催化活性ꎻSH1含有自身磷酸化位点酪氨酸416(Tyr416)ꎬ可以激活Src活性ꎬ而C端调节域的酪氨酸527(Tyr527)是磷酸化的调节位点和抑制因子ꎬ可以抑制Src的活性ꎬ在终止SFKs的功能中起着至关重要的作用[8].2㊀Src信号通路的调节2.1㊀Src与PI3K/Akt信号通路PI3K(Phosphatidylinositol ̄3 ̄kinasesꎬPI3K)是磷脂酰肌醇-3-激酶ꎬPI3K/Akt(蛋白激酶)信号通路广泛存在于肿瘤细胞中ꎬ影响着细胞的基本生命活动.研究表明ꎬ通过使用特异性Src抑制剂PP2(4-氨基-5-(4-氯苯基)-7-(t-丁基)吡唑[3ꎬ4 ̄d]嘧啶)处理肝癌细胞显著降低了Akt磷酸化水平ꎬ阻止PI3K/Akt信号通路的过表达或磷酸化ꎬ从而抑制恶性肿瘤细胞的异常增殖ꎻ另外ꎬPP2因进一步调节下游蛋白的功能而发挥生物抑制作用[9].Liu等[10]研究表明ꎬ乙型肝炎病毒表面大抗原(LargehepatitisBvirussurfaceantigenꎬLHBs)通过Src信号通路促进PI3K/Akt活化ꎬLHBs的表达可加速G1-S(DNA合成前期-DNA合成期)细胞周期进程并激活Src/PI3K/Akt信号通路ꎬ诱导肝癌发生.2.2㊀Src与FAK信号通路局部黏着斑激酶(FocaladhesionkinaseꎬFAK)是一种细胞质蛋白酪氨酸激酶ꎬFAK由一个N端的FERM(4.1 ̄ezfin ̄radixin ̄moesin)结构域ꎬ一个中心激酶结构域和一个C端黏着斑靶向(FAT)组成.FAK的N端接受来自上游的整合素等信号分子ꎬ活化FAK并使其磷酸化ꎬFAK进而激活下游信号通路并亲自参与多条信号通路转导[11].Src激活FAK并启动其向细胞膜的转运ꎬ在细胞膜上FAK与整合素结合并调节整合素介导的黏附作用.Thamilselvan等[12]采用细胞外压力诱导Src激活ꎬ它们将PI3K㊁FAK和Akt1(蛋白激酶B)信号通路联动起来ꎬ使胞浆中的FAK㊁p85(PI3K的调节亚基)和Akt随后转移到细胞膜上ꎬ通过FAK与β1(转化生长因子-β1)整合素异源二聚体结合ꎬ能够调节β1整合素异源二聚体与基质蛋白的结合亲和性ꎬ整合素结合亲和性的改变可以促进结肠癌细胞的063㊀㊀㊀辽宁大学学报㊀㊀自然科学版2023年㊀㊀㊀㊀黏附[12].2.3㊀Src与STAT3信号通路信号转导和转录激活因子(SignaltransducersandactivatorsoftranscriptionꎬSTATs)是一类具有类似结构的细胞质转录因子家族ꎬ起到转导细胞外细胞因子和生长因子的功能.STAT3(信号转导和转录激活因子3)是STATs的重要成员ꎬ可直接或通过其他转录因子间接调节基因表达.STAT3除了是细胞因子受体的下游ꎬ还可以被生长因子受体和非受体酪氨酸激酶激活[13].STAT3信号通路常在恶性细胞中被激活ꎬ能诱导大量对癌症产生至关重要的基因ꎬ成为癌症的主要内在途径.Zhu等[14]研究表明ꎬAhR-Src-STAT3-IL-10信号通路是参与炎性巨噬细胞免疫调节的关键通路ꎬ芳烃受体(AhR)通过Src-STAT3信号通路促进炎症巨噬细胞中1L-10(白细胞介素10)的表达ꎬ从而限制过度炎症的不良后果.3㊀Src与癌症3.1㊀乳腺癌乳腺癌是全世界女性癌症死亡的最常见原因ꎬ近年来发病率一直呈上升趋势ꎬ严重危害了女性的身体健康.Djeungoue-Petga等[15]研究表明ꎬ位于线粒体内的Src在乳腺癌中具有特定的功能ꎬ可以使三阴性乳腺癌更具侵袭性ꎬ并改变线粒体代谢.在87例三阴性乳腺癌和93例非三阴性乳腺癌中检测Srcꎬ结果显示ꎬSrc都有表达ꎬ且在三阴性乳腺癌中的表达频率高于非三阴性乳腺癌ꎬ因此ꎬSrc可能是治疗乳腺癌的潜在靶点[16].Ngan等[17]发现Src介导的LPP(脂质瘤首选伴侣)酪氨酸磷酸化对乳腺癌细胞的侵袭和转移至关重要.Song等[18]研究表明ꎬSrc在有丝分裂刺激下直接与lipin-1(磷脂酸磷酸酶)相互作用并使其磷酸化ꎬ有助于通过加速磷脂和甘油三酯合成来维持乳腺癌细胞的增殖.3.2㊀肺癌肺癌是一种极其复杂的恶性肿瘤ꎬ它的死亡率在所有肿瘤中位居首位.在肺癌的病例中ꎬ非小细胞肺癌(NSCLC)占比较大ꎬ是其主要类型.Dong等[19]通过体内和体外实验ꎬ将NSCLC细胞经不同浓度的槲皮素(Quercetin)给药ꎬ发现该化合物通过抑制Src/Fn14/NF-κB信号转导发挥抗NSCLC细胞增殖和转移的作用.Zhao等[20]采用荧光定量PCR法检测64例肺恶性组织和40例肺良性病变样本中葡萄糖转运蛋白(Glucosetransportprotein ̄1ꎬGlut ̄1)的表达ꎬ发现肺恶性组织Glut-1归一化值显著高于肺良性病变样本ꎬ差异具有统计学意义(P<0.05)ꎬ综合数据证实ꎬGlut-1通过整合素β1/Src/FAK信号通路调控NSCLC细胞增殖㊁迁移㊁侵袭和凋亡ꎬ可作为肺癌治疗的全新靶点.区豪杰等[21]研究表明ꎬRITA(肿瘤凋亡和P53再生化合物)提升肺鳞癌H226(人肺鳞癌细胞NCI-H226)细胞内活性氧水平ꎬ细胞内动态平衡被打破ꎬ从而导致Src/STAT3信号通路水平下降ꎬ最终诱导肺鳞癌细胞凋亡.3.3㊀前列腺癌前列腺癌是发病率和死亡率相差较大的男性常见恶性肿瘤ꎬ它的发病率随着年龄的增长而快速上升.CXC趋化因子配体1-脂质运载蛋白2(CXCL1-LCN2)激活Src信号ꎬ触发上皮-间充质转换(Epithelial ̄mesenchymaltransitionꎬEMT)ꎬ从而促进前列腺癌细胞的迁移ꎬ导致肿瘤转移增强[22].Dai等[23]研究发现ꎬ在缺氧条件下Src可以促进细胞的转移ꎬ这也正是前列腺癌治疗失败的原因ꎬ而Src抑制剂在缺氧条件下能降低细胞的转移功能ꎬ这表明此类药物具有治疗前列腺癌的潜力.Teng等[24]发现ꎬ达沙替尼阻断Src信号通路可以增强CYT997(微管聚合抑制剂)在前列腺癌中的抗癌活性.163㊀第4期㊀㊀㊀㊀㊀㊀陈㊀烨ꎬ等:Src蛋白激酶的研究进展㊀㊀3.4㊀肝癌肝癌是一种预后不良㊁治疗选择有限的恶性肿瘤ꎬ其中肝细胞癌(HepatocellularcarcinomaꎬHCC)是其主要类型.Wang等[25]研究发现ꎬmicroRNA24-2是一种具有癌变功能的microRNAꎬ至少在人类肝癌中有所体现ꎬ在人类肝癌干细胞(LivercancerstemcellsꎬHLCSCs)的实验中发现ꎬmicroRNA24-2通过增强HLCSCs中的PKM1(Pyruvatekinasemuscleisozyme1)来促进Src的表达ꎬ而Src正向调节和控制microRNA24-2在HLCSCs中的致癌功能.Suresh等[26]研究表明ꎬSrc-2可能具有致癌或抑癌活性ꎬ这取决于在不同组织中表达的靶基因和核受体ꎻ在肝脏中Src-2与多个肿瘤抑制因子包括甲状腺受体(TR)㊁雌激素受体(ER)等共同激活一个特定的靶基因程序ꎬ从而抑制肿瘤.3.5㊀卵巢癌卵巢癌是最为致命的妇女恶性肿瘤ꎬ其中ꎬ上皮性卵巢癌(EpithelialovariancancerꎬEOC)是其主要类型.由于预兆不显著ꎬ一直到晚期才易被发现ꎬ因此往往错过最佳治疗阶段.Huang等[27]运用免疫组织化学法检测c-Src(Cell ̄steroidreceptorcoactivator)在82例EOC患者和25例良性卵巢病变患者中的表达ꎬ并用20个正常卵巢组织作为对照ꎬ结果显示ꎬEOC中c-Src表达阳性的比例显著高于对照组ꎬ该研究还表明ꎬ通过Tyr416的磷酸化激活c-Src可能在卵巢癌发展的早期阶段发挥作用.Cheng等[28]发现ꎬZIP13(Zrt ̄andIrt ̄likeprotein13)是卵巢癌转移的主要介质ꎬ可以调节细胞内锌的分布ꎬ激活Src/FAK通路并导致卵巢癌的转移ꎬ因此ꎬZIP13可能是预防和治疗卵巢癌转移的一个有价值的治疗靶点.近年来ꎬBley等[29]在EOC衍生细胞中发现ꎬ胰岛素样生长因子2mRNA结合蛋白1(Insulinlikegrowthfactor ̄2mRNA ̄bindingprotein1ꎬIGF2BP1)通过刺激Src/ERK(Extracellularsignal ̄regulatedkinase)信号转导来促进卵巢癌侵袭性生长.Qiu等[30]研究发现TRIM50(Tripartitemotif ̄containing50)通过靶向Src并降低其活性来抑制卵巢癌ꎬ这为通过正向调节TRIM50来治疗Src过度激活的癌症提供了一种新的思路.3.6㊀宫颈癌宫颈癌是影响中年妇女健康的主要公共卫生问题ꎬ宫颈鳞状细胞癌(CSCC)占宫颈癌的绝大比例.Hou等[31]采用免疫组织化学法检测20例正常宫颈组织㊁20例宫颈原位癌(CIS)和87例宫颈鳞状细胞癌(CSCC)中磷酸化c-Src的表达.结果显示ꎬ磷酸化c-Src在正常宫颈组织㊁CIS和CSCC中的表达逐渐升高ꎬ此外ꎬ磷酸化c-Src的表达与宫颈癌的总生存率和复发率相关.Du等[32]研究发现ꎬ整合素α3与c-Src相互作用并激活ERK/FAK信号通路ꎬ导致黏着斑形成受损ꎬ这种作用使宫颈癌细胞的迁移和侵袭能力增强ꎬ并通过分泌基质金属蛋白酶-9(Matrixmetalloproteinase ̄9ꎬMMP-9)诱导宫颈癌血管生成.Yang等[33]发现ꎬ膳食油酸诱导的CD36(Clusterofdifferentiation36)通过上调Src/ERK信号通路促进宫颈癌细胞生长和转移.3.7㊀胰腺癌胰腺癌是一种高度致命㊁转移较快的消化道肿瘤ꎬ大多数患者在胰腺癌晚期之前一直没有明显症状.Kuo等[34]发现ꎬ在K-ras(KirstenRatSarcomaVirus)突变和p53基因缺失的条件下ꎬβ-连环蛋白通过上调PDGF(Platelet ̄derivedgrowthfactor)/Src信号ꎬ加速了胰腺癌的发生.Li等[35]研究表明ꎬ天然化合物OblongifolinC(OC)在体内对胰腺肿瘤的生长发挥抑制作用ꎬ并通过泛素-蛋白酶体途径下调Src表达来提高吉西他滨(Gemcitabine)的敏感性ꎬ有效抑制胰腺癌细胞增殖.An等[36]证实ꎬOxialisobtriangulata甲醇提取物(OOE)对胰腺癌细胞BxPC3(Biopsyxenograftofpancreaticcarcinomaline ̄3)具有抗癌活性ꎬOOE调控ERK/Src/STAT3激活ꎬ并调节与肿瘤发展相关的STAT3下游基因ꎬ展现了OOE作为抗癌药物的可能性.263㊀㊀㊀辽宁大学学报㊀㊀自然科学版2023年㊀㊀㊀㊀3.8㊀胃癌尽管胃癌发病率有所下降ꎬ但胃癌仍然是全球癌症死亡的常见原因之一.刘江惠等[37]应用流式细胞术检测c-Src在50例胃癌组织和10例胃黏膜中的表达情况ꎬ结果显示ꎬSrc在胃癌组织的表达高于胃黏膜组织(P<0.01)ꎬ且在临床晚期蛋白表达水平高于临床早期ꎬ差异有统计学意义(P<0.05).Qi等[38]的研究结果发现ꎬ红景天苷(Salidroside)通过抑制活性氧(ROS)介导的Src相关信号通路蛋白磷酸化和热休克蛋白70(HSP70)的表达来阻止胃癌细胞的增殖和迁移.Nam等[39]发现ꎬ塞卡替尼单独或与其他药物联合使用抑制Src激酶活性可降低胃癌细胞的增殖和迁移.4㊀Src抑制剂4.1㊀达沙替尼达沙替尼是一种广泛而有效的多酪氨酸激酶抑制剂.它主要用于抑制Abl和Srcꎬ除此之外还能够抑制c-KIT(c ̄Kitproto ̄oncogeneprotein)㊁PDGFR-α(Platelet ̄derivedgrowthfactorreceptorα)㊁PDGFR-β(Platelet ̄derivedgrowthfactorreceptorβ)和肾上腺素受体激酶.聚糖结合蛋白(Syndecan ̄bindingproteinꎬSDCBP)与c-Src的相互作用ꎬ促进c-Src在残基419处的酪氨酸磷酸化ꎬ增强了三阴性乳腺癌的增殖ꎬ而达沙替尼在残基419处抑制c-Src的酪氨酸磷酸化ꎬ并阻断SDCBP诱导的细胞循环进展[40].Redin等[41]研究表明ꎬ达沙替尼在NSCLC中与抗PD-1免疫疗法协同作用ꎬ可导致肿瘤消退.4.2㊀博舒替尼博舒替尼也是一种小分子Abl/Src双效抑制剂ꎬ但它对PDGFR和KIT(Kitproto ̄oncogeneprotein)受体无活性.Rabbani等[42]研究发现ꎬ博舒替尼通过调节参与癌症生长和骨骼转移的基因ꎬ阻断前列腺癌的侵袭㊁生长和转移.Src和c-Ab1(Abelsontyrosinekinase)是神经母细胞瘤的潜在治疗靶点ꎬ博舒替尼单独或与其他化疗药物联合可能是治疗神经母细胞瘤一种有价值的选择[43].4.3㊀来那替尼来那替尼是一种新型㊁不可逆的人表皮生长因子受体2(Humanepidermalgrowthfactor2ꎬHER2)靶向酪氨酸激酶抑制剂.曲妥珠单抗(Trastuzumab)已经被证明可以作为HER2阳性乳腺癌患者的新型疗法ꎬ然而很大一部分HER2阳性乳腺癌患者对曲妥珠单抗会产生耐药性ꎬ而来那替尼可以抵消这种耐药性ꎬ从而降低三阴性乳腺癌复发[44].5㊀展望Src在多种细胞信号转导途径中发挥着关键作用ꎬ也是癌症治疗中研究较好的靶点之一.通过本文的论述ꎬSrc的致癌激活已被证明在癌症中发挥重要作用ꎬ可以促进肿瘤生长和转移.一些针对Src的抑制剂已经开发出来ꎬ其中许多药物已经成功地用于临床治疗ꎬ但在临床中会有无法预料的并发症ꎬ还需要进一步的探索和阐述.随着未来研究的深入ꎬ针对Src的认识会更加清晰ꎬSrc抑制剂与其他抑制剂的联合使用会对癌症治疗发挥巨大作用.参考文献:[1]㊀SoerjomataramIꎬBrayF.Planningfortomorrow:Globalcancerincidenceandtheroleofprevention2020-2070[J].NatureReviewsClinicalOncologyꎬ2021ꎬ18(10):663-672.[2]㊀MaoLMꎬGeoslingRꎬPenmanBꎬetal.Localsubstratesofnon ̄receptortyrosinekinasesatsynapticsitesinneurons[J].ActaPhysiologicaSinicaꎬ2017ꎬ69(5):657-665.[3]㊀LowellCA.Src ̄familyandSykkinasesinactivatingandinhibitorypathwaysininnateimmunecells:Signaling363㊀第4期㊀㊀㊀㊀㊀㊀陈㊀烨ꎬ等:Src蛋白激酶的研究进展㊀㊀crosstalk[J].ColdSpringHarborPerspectivesinBiologyꎬ2011ꎬ3(3):a002352.[4]㊀ZhangJꎬKalyankrishnaSꎬWislezMꎬetal.Src ̄familykinasesareactivatedinnon ̄smallcelllungcancerandpromotethesurvivalofepidermalgrowthfactorreceptor ̄dependentcelllines[J].TheAmericanJournalofPathologyꎬ2007ꎬ170(1):366-376.[5]㊀JallalHꎬValentinoMLꎬChenGPꎬetal.ASrc/AblkinaseinhibitorꎬSKI ̄606ꎬblocksbreastcancerinvasionꎬgrowthꎬandmetastasisinvitroandinvivo[J].CancerResearchꎬ2007ꎬ67(4):1580-1588.[6]㊀BoggonTJꎬEckMJ.StructureandregulationofSrcfamilykinases[J].Oncogeneꎬ2004ꎬ23(48):7918-7927.[7]㊀BrownMTꎬCooperJA.RegulationꎬsubstratesandfunctionsofSrc[J].BiochimicaetBiophysicaActa(BBA) ̄ReviewsonCancerꎬ1996ꎬ1287(2/3):121-149.[8]㊀XuWCꎬAllbrittonNꎬLawrenceDS.Srckinaseregulationinprogressivelyinvasivecancer[J].PLoSOneꎬ2012ꎬ7(11):e48867.[9]㊀GingerichSꎬKrukoffTL.ActivationofERβincreaseslevelsofphosphorylatednNOSandNOproductionthroughaSrc/PI3K/Akt ̄dependentpathwayinhypothalamicneurons[J].Neuropharmacologyꎬ2008ꎬ55(5):878-885.[10]㊀LiuHOꎬXuJJꎬZhouLꎬetal.HepatitisBviruslargesurfaceantigenpromoteslivercarcinogenesisbyactivatingtheSrc/PI3K/Aktpathway[J].CancerResearchꎬ2011ꎬ71(24):7547-7557.[11]㊀MurphyJMꎬJeongKꎬLimSTS.FAKfamilykinasesinvasculardiseases[J].InternationalJournalofMolecularSciencesꎬ2020ꎬ21(10):3630.[12]㊀ThamilselvanVꎬCraigDHꎬBassonMD.FAKassociationwithmultiplesignalproteinsmediatespressure ̄inducedcoloncancercelladhesionviaaSrc ̄dependentPI3K/Aktpathway[J].FASEBJournal:OfficialPublicationoftheFederationofAmericanSocietiesforExperimentalBiologyꎬ2007ꎬ21(8):1730-1741.[13]㊀YuHꎬPardollDꎬJoveR.STATsincancerinflammationandimmunity:AleadingroleforSTAT3[J].NatureReviewsCancerꎬ2009ꎬ9(11):798-809.[14]㊀ZhuJYꎬLuoLꎬTianLXꎬetal.ArylhydrocarbonreceptorpromotesIL ̄10expressionininflammatorymacrophagesthroughSrc ̄STAT3signalingpathway[J].FrontiersinImmunologyꎬ2018ꎬ9:2033.[15]㊀Djeungoue ̄PetgaMAꎬLuretteOꎬJeanSꎬetal.IntramitochondrialSrckinaselinksmitochondrialdysfunctionsandaggressivenessofbreastcancercells[J].CellDeath&Diseaseꎬ2019ꎬ10(12):940.[16]㊀TryfonopoulosDꎬWalshSꎬCollinsDMꎬetal.Src:Apotentialtargetforthetreatmentoftriple ̄negativebreastcancer[J].AnnalsofOncologyꎬ2011ꎬ22(10):2234-2240.[17]㊀NganEꎬStoletovKꎬSmithHWꎬetal.LPPisaSrcsubstraterequiredforinvadopodiaformationandefficientbreastcancerlungmetastasis[J].NatureCommunicationsꎬ2017ꎬ8:15059.[18]㊀SongLTꎬLiuZHꎬHuHHꎬetal.Proto ̄oncogeneSrclinkslipogenesisvialipin ̄1tobreastcancermalignancy[J].NatureCommunicationsꎬ2020ꎬ11:5842.[19]㊀DongYꎬYangJꎬYangLYꎬetal.Quercetininhibitstheproliferationandmetastasisofhumannon ̄smallcelllungcancercellline:ThekeyroleofSrc ̄mediatedfibroblastgrowthfactor ̄inducible14(Fn14)/nuclearfactorkappaB(NF ̄κB)pathway[J].MedicalScienceMonitorꎬ2020ꎬ26:e920537.[20]㊀ZhaoHYꎬSunJꎬShaoJSꎬetal.Glucosetransporter1promotesthemalignantphenotypeofnon ̄smallcelllungcancerthroughintegrinβ1/Src/FAKsignaling[J].JournalofCancerꎬ2019ꎬ10(20):4989-4997.[21]㊀区豪杰ꎬ孙嘉ꎬ李华宇ꎬ等.RITA通过ROS/Src/STAT3通路诱导肺鳞癌H226细胞凋亡[J].天津医药ꎬ2021ꎬ49(8):785-790.[22]㊀LuYNꎬDongBJꎬXuFꎬetal.CXCL1 ̄LCN2paracrineaxispromotesprogressionofprostatecancerviatheSrcactivationandepithelial ̄mesenchymaltransition[J].CellCommunicationandSignalingꎬ2019ꎬ17(1):118.[23]㊀DaiYꎬSiemannD.c ̄Srcisrequiredforhypoxia ̄inducedmetastasis ̄associatedfunctionsinprostatecancercells[J].OncoTargetsandTherapyꎬ2019ꎬ12:3519-3529.[24]㊀TengYꎬCaiYFꎬPiWHꎬetal.AugmentationoftheanticanceractivityofCYT997inhumanprostatecancerbyinhibitingSrcactivity[J].JournalofHematology&Oncologyꎬ2017ꎬ10(1):118.[25]㊀WangLYꎬLiXNꎬZhangWꎬetal.miR24 ̄2promotesmalignantprogressionofhumanlivercancerstemcellsbyenhancingtyrosinekinaseSrcepigenetically[J].MolecularTherapyꎬ2020ꎬ28(2):572-586.[26]㊀SureshSꎬDurakoglugilDꎬZhouXRꎬetal.Correction:Src ̄2 ̄mediatedcoactivationofanti ̄tumorigenictargetgenes463㊀㊀㊀辽宁大学学报㊀㊀自然科学版2023年㊀㊀㊀㊀suppressesMYC ̄inducedlivercancer[J].PLoSGeneticsꎬ2018ꎬ14(4):e1007344.[27]㊀HuangYWꎬChenCꎬXuMMꎬetal.Expressionofc ̄Srcandphospho ̄Srcinepithelialovariancarcinoma[J].MolecularandCellularBiochemistryꎬ2013ꎬ376(1):73-79.[28]㊀ChengXXꎬWangJꎬLiuCLꎬetal.ZinctransporterSLC39A13/ZIP13facilitatesthemetastasisofhumanovariancancercellsviaactivatingSrc/FAKsignalingpathway[J].JournalofExperimental&ClinicalCancerResearchꎬ2021ꎬ40(1):199.[29]㊀BleyNꎬSchottAꎬMüllerSꎬetal.IGF2BP1isatargetableSrc/MAPK ̄dependentdriverofinvasivegrowthinovariancancer[J].RNABiologyꎬ2021ꎬ18(3):391-403.[30]㊀QiuYMꎬLiuPSꎬMaXMꎬetal.TRIM50actsasanovelSrcsuppressorandinhibitsovariancancerprogression[J].BiochimicaetBiophysicaActaMolecularCellResearchꎬ2019ꎬ1866(9):1412-1420.[31]㊀HouTꎬXiaoJꎬZhangHTꎬetal.Phosphorylatedc ̄Srcisanovelpredictorforrecurrenceincervicalsquamouscellcancerpatients[J].InternationalJournalofClinicalandExperimentalPathologyꎬ2013ꎬ6(6):1121-1127.[32]㊀DuQQꎬWangWꎬLiuTYꎬetal.Highexpressionofintegrinα3predictspoorprognosisandpromotestumormetastasisandangiogenesisbyactivatingthec ̄Src/extracellularsignal ̄regulatedproteinkinase/focaladhesionkinasesignalingpathwayincervicalcancer[J].FrontiersinOncologyꎬ2020ꎬ10:36.[33]㊀YangPꎬSuCXꎬLuoXꎬetal.Dietaryoleicacid ̄inducedCD36promotescervicalcancercellgrowthandmetastasisviaup ̄regulationSrc/ERKpathway[J].CancerLettersꎬ2018ꎬ438:76-85.[34]㊀KuoTLꎬChengKHꎬShanYSꎬetal.β ̄catenin ̄activatedautocrinePDGF/Srcsignalingisatherapeutictargetinpancreaticcancer[J].Theranosticsꎬ2019ꎬ9(2):324-336.[35]㊀LiYꎬXiZCꎬChenXQꎬetal.NaturalcompoundOblongifolinCconfersgemcitabineresistanceinpancreaticcancerbydownregulatingSrc/MAPK/ERKpathways[J].CellDeath&Diseaseꎬ2018ꎬ9:538.[36]㊀AnEJꎬKimYꎬLeeSHꎬetal.Anti ̄cancerpotentialofOxialisobtriangulatainpancreaticcancercellthroughregulationoftheERK/Src/STAT3 ̄mediatedpathway[J].Moleculesꎬ2020ꎬ25(10):2301.[37]㊀刘江惠ꎬ姜忠彩ꎬ郭建文ꎬ等.c-Src在胃癌中的表达与侵袭转移机制的探讨[J].河北医科大学学报ꎬ2010ꎬ31(3):252-255.[38]㊀QiZLꎬTangTꎬShengLLꎬetal.SalidrosideinhibitstheproliferationandmigrationofgastriccancercellsviasuppressionofSrc ̄associatedsignalingpathwayactivationandheatshockprotein70expression[J].MolecularMedicineReportsꎬ2018ꎬ18(1):147-156.[39]㊀NamHJꎬImSAꎬOhDYꎬetal.Antitumoractivityofsaracatinib(AZD0530)ꎬac ̄Src/Ablkinaseinhibitorꎬaloneorincombinationwithchemotherapeuticagentsingastriccancer[J].MolecularCancerTherapeuticsꎬ2013ꎬ12(1):16-26.[40]㊀QianXLꎬZhangJꎬLiPZꎬetal.Dasatinibinhibitsc ̄SrcphosphorylationandpreventstheproliferationofTriple ̄NegativeBreastCancer(TNBC)cellswhichoverexpressSyndecan ̄BindingProtein(SDCBP)[J].PLoSOneꎬ2017ꎬ12(1):e0171169.[41]㊀RedinEꎬGarmendiaIꎬLozanoTꎬetal.Srcfamilykinase(SFK)inhibitordasatinibimprovestheantitumoractivityofanti ̄PD ̄1inNSCLCmodelsbyinhibitingTregcellconversionandproliferation[J].JournalforImmunotherapyofCancerꎬ2021ꎬ9(3):e001496.[42]㊀RabbaniSAꎬValentinoMLꎬArakelianAꎬetal.SKI ̄606(Bosutinib)blocksprostatecancerinvasionꎬgrowthꎬandmetastasisinvitroandinvivothroughregulationofgenesinvolvedincancergrowthandskeletalmetastasis[J].MolecularCancerTherapeuticsꎬ2010ꎬ9(5):1147-1157.[43]㊀BieerkehazhiSꎬChenZHꎬZhaoYLꎬetal.NovelSrc/abltyrosinekinaseinhibitorbosutinibsuppressesneuroblastomagrowthviainhibitingSrc/ablsignaling[J].Oncotargetꎬ2017ꎬ8(1):1469-1480.[44]㊀CanoniciAꎬGijsenMꎬMulloolyMꎬetal.NeratinibovercomestrastuzumabresistanceinHER2amplifiedbreastcancer[J].Oncotargetꎬ2013ꎬ4(10):1592-1605.(责任编辑㊀郭兴华)563㊀第4期㊀㊀㊀㊀㊀㊀陈㊀烨ꎬ等:Src蛋白激酶的研究进展。
蛋白酶抑制剂的研究进展

蛋白酶抑制剂的研究进展郭川微生物专业,200326031摘要:自然界共发现四大类蛋白酶抑制剂:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂、金属蛋白酶抑制剂和酸性蛋白酶抑制剂,本文就各大类蛋白酶抑制剂的结构特点,活性部位的研究概况及其在各领域应用的原理及进展。
关键词:蛋白酶抑制剂;结构;应用天然的蛋白酶抑制剂(PI)是对蛋白水解酶有抑制活性的一种小分子蛋白质,由于其分子量较小,所以在生物中普遍存在。
它能与蛋白酶的活性部位和变构部位结合,抑制酶的催化活性或阻止酶原转化有活性的酶。
在一系列重要的生理、病理过程中:如凝血、纤溶、补体活化、感染、细胞迁移等,PI发挥着关键性的调控作用,是生物体内免疫系统的重要组成部分。
从Kunitz等最早分离纯化出一种PI至今,已有多种PI被发现,根据其作用的蛋白酶主要分以下几类:抑制胰蛋白酶、胰凝乳蛋白酶等的丝氨酸蛋白酶抑制剂,抑制木瓜蛋白酶、菠萝蛋白酶等的巯基蛋白酶抑制剂,抑制胃蛋白酶、组织蛋白酶D等的羧基蛋白酶抑制剂、抑制胶原酶、氨肽酶等的金属蛋白酶抑制剂等。
而根据作用于酶的活性基团不同及其氨基酸序列的同源性,可将自然界发现的PI分为四大类:丝氨酸蛋白酶抑制剂、巯基蛋白酶抑制剂(半胱氨酸蛋白酶抑制剂)、金属蛋白酶抑制剂和酸性蛋白酶抑制剂[1]。
1 结构与功能1.1丝氨酸蛋白酶抑制剂(Serine Protease Inhibitor,Serpin)丝氨酸蛋白酶抑制剂是一族由古代抑制剂趋异进化5亿年演变而来的结构序列同源的蛋白酶抑制剂。
Sepin为单一肽链蛋白质。
各种serpin大约有30%的同源序列,疏水区同源性高达70%。
血浆中的serpin多被糖基化,糖链经天东酰胺的酰胺基与主链相连。
位于抑制性serpin表面、距C端30~40个氨基酸处的环状结构区RSL(reactive site loop)中,存在能被靶酶的底物识别位点识别的氨基酸P1[2];近C端与P1相邻的氨基酸为P1’,依此类推,即肽链结构表示为N端-P15~P9~P1-P1’~P9’~P15’-C端。
苯甲酰 -1,3- 环己二酮类化合物互变异构的理论计算

21卷6期 结 构 化 学(JIEGOU HUAXUE) V ol. 21, No. 6 2002. 11 Chinese J. Struct. Chem. 678~682 苯甲酰-1,3-环己二酮类化合物互变异构的理论计算黄美兰a商志才a 邹建卫a①杨定亚b俞庆森aa (浙江大学化学系, 杭州 310027)b (台湾东海大学化学系, 台中 40704)对苯甲酰-1,3-环己二酮的各异构体进行了HF/6-31G*水平的几何优化,然后对能量低的三酮式和2个顺式烯醇式异构体进行了更广泛的计算,探讨了计算方法、基组大小对异构体相对稳定性的影响。
在此基础上,对该类化合物的互变异构化平衡常数、能垒进行了计算,在平衡常数的计算中充分考虑了溶剂的影响并结合实验结果进行了分析。
最后,对有代表性的苯环2-位取代衍生物进行了计算,发现互变异构自由能与其HPPD酶抑制活性间存在着较好的相关关系。
关键词: 苯甲酰-1,3-环己二酮, 互变异构, 从头算, 构效关系苯甲酰-1,3-环己二酮类化合物有很好的生物活性,如2-(2-硝基-4-三氟甲基苯甲酰)-1,3-环己二酮(NTBC)不仅对于治疗人体铬氨酸过多症有显著疗效[1],而且是一种高效的除草剂[2]。
动力学研究结果表明这类化合物与底物对羟苯基丙酮酸(HPP)键合到靶标-对羟苯基丙酮酸双氧化酶(HPPD)的同一部位,是底物的竞争抑制剂[3, 4]。
我们最近合成了一系列2-(2-取代苯甲酰基)及2-烷基-1,3-环己二酮化合物并测定了其体外HPPD 酶抑制活性,结构活性关系(SAR)表明,苯甲酰-1,3-环己二酮的苯环2-位取代对其酶抑制活性有重要影响[5]。
由于酮式和烯醇式结构质子转移的互变异构化性质,苯甲酰-1,3-环己二酮化合物存在多种可能的异构形式。
确定其优势结构、探讨各异构形式间的相对稳定性并研究取代基对结构的影响对于阐明该类化合物的构效关系及其与酶的作用机理有着重要的意义。
PTP1B抑制剂研究进展资料

PTP1B抑制剂研究进展资料PTP1B(蛋白酪氨酸磷酸酶1B)是一种重要的调节酪氨酸磷酸化信号传导的酶,其过度活化或异常表达与多种疾病的发生和发展密切相关,包括2型糖尿病、肥胖症、肝纤维化、乳腺癌和结肠癌等。
因此,研发PTP1B抑制剂是一项具有重要意义的研究。
以下是PTP1B抑制剂研究进展的资料。
1.自然产物抑制剂:自然产物是从植物、动物、微生物中分离提取的天然化合物。
许多天然产物被发现具有抑制PTP1B活性的潜力。
例如,山楂中的山楂酸、甘草中的甘草素,以及丹参中的左旋丹参酮等天然产物都显示出抑制PTP1B的活性。
2.合成抑制剂:许多研究人员通过有机合成方法来合成PTP1B抑制剂。
这些化合物通常是小分子有机化合物,具有选择性地抑制PTP1B酶活性。
例如,IOMA (iodoacetamide O-methyl ester) 是一种广泛应用的PTP1B抑制剂,它通过与PTP1B的活性中心发生自发磷酸化的方式来抑制其活性。
3.生物制药抑制剂:生物制药抑制剂是指通过生物技术手段制备的具有抑制PTP1B活性的蛋白质药物。
目前,许多研究人员致力于开发和研究PTP1B的生物制药抑制剂。
例如,一种叫做SHP2的PTP1B同源蛋白被发现在PTP1B的结构和功能上起到抑制作用,因此其在抗癌治疗中的应用正在研究中。
4.单克隆抗体抑制剂:单克隆抗体是一种在现代生物技术中得到广泛应用的抗体类药物,可用于特异性选择性地抑制PTP1B酶活性。
该类抗体抑制剂具有高度的特异性和亲和力,可针对性地靶向PTP1B,并阻断其活性。
例如,Recentin是一种被发现具有抑制PTP1B酶活性的单克隆抗体。
总结起来,目前对PTP1B抑制剂的研究主要集中在天然产物、合成化合物、生物制药和单克隆抗体等领域。
这些研究为开发新的抑制PTP1B活性的药物提供了基础,并有望在相关疾病的治疗中产生重要的临床应用价值。
成纤维细胞活化蛋白抑制剂在肿瘤诊疗中的研究进展
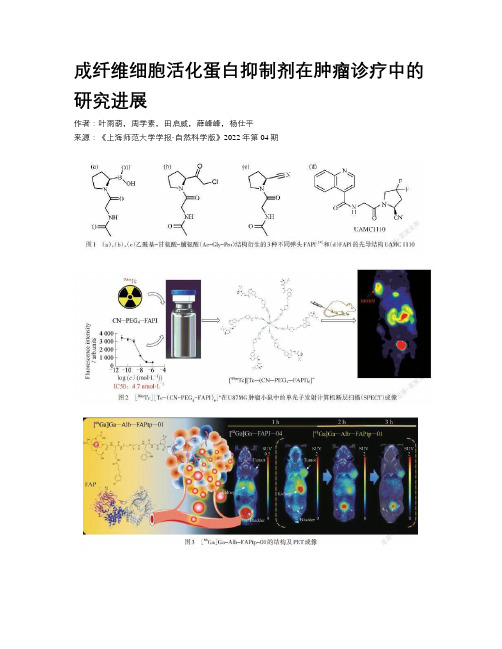
成纤维细胞活化蛋白抑制剂在肿瘤诊疗中的研究进展作者:叶雨萌,周学素,田启威,薛峰峰,杨仕平来源:《上海师范大学学报·自然科学版》2022年第04期摘要:成纤维细胞活化蛋白(FAP)在90%以上的上皮性癌间质中高表达,可以作为肿瘤成像和治疗的靶点.而一些已开发的成纤维细胞活化蛋白抑制剂(FAPI),由于对肿瘤的高亲和力和高肿瘤积聚,对肿瘤的诊断和治疗具有重大意义.文章综述了近年来FAPI在肿瘤诊疗方面的研究进展,重点阐述了新型FAPI在核医学上的诊疗应用,并且从构效关系上讨论了FAPI的靶向弹头结构,增强FAPI选择性及延长肿瘤保留时间的策略,进一步推动了FAPI向临床诊疗试剂转化.关键词:成纤维细胞活化蛋白抑制剂(FAPI); 核医学影像; 放射性治疗; 构效关系中图分类号: R 817.9 文献标志码: A 文章编号: 1000-5137(2022)04-0436-07Progress in fibroblast activation protein inhibitors for cancer diagnosis and treatmentYE Yumeng1, ZHOU Xuesu1, TIAN Qiwei1,2, XUE Fengfeng2, YANG Shiping1*(1. College of Chemistry and Materials Science, Shanghai Normal University, Shanghai 200234, China; 2. Shanghai Key Laboratory of Molecular Imaging, Shanghai University of Medicine and Health Sciences, Shanghai 201318, China)Abstract: Fibroblast activation protein(FAP) is highly expressed in more than 90% of epithelial carcinoma stroma and can be used as a target for tumor imaging and therapy. Some developed FAP inhibitors(FAPI) are of great significance in the diagnosis and treatment of tumors due to their high affinity for tumors and high tumor accumulation. Herein,the research progress of FAPI in tumor diagnosis and treatment in recent years was reviewed,with an emphasis on the clinical application of novel FAPI in nuclear medicine. In addition,FAPI targeting warhead structure and the strategies of enhancing FAPI selectivity and prolonging tumor retention time were discussed from the perspective of structure-activity relationship,which further promoted the transformation of FAPI into clinical diagnosis and treatment reagents.Key words: fibroblast activating protein inhibitors(FAPI); nuclear medical imaging; radiation therapy; structure-activity relationship0 引言癌相关成纤维细胞(CAFs)是一种异质性的成纤维细胞样细胞群,在肿瘤生长、迁移、转移、重构细胞外基质、治疗抵抗和免疫抑制中发挥关键作用,同时也是肿瘤微环境结构中最丰富的一类细胞[1-2].与癌细胞相比,CAFs的基因更稳定,更不易发生治疗耐药性[3-4],是癌症诊断和治疗的理想靶细胞.成纤维细胞活化蛋白(FAP)是一种II型膜结合的丝氨酸蛋白酶[5],在CAFs中过表达,而在健康成人组织中很少表达[6].有数据统计,FAP在90%以上的上皮性癌的间质中过表达[4].而且,在直肠癌、胰腺癌、卵巢癌等恶性肿瘤中,FAP的高表达与肿瘤局部浸润增加、淋巴结转移风险增加和患者生存期下降有关[7].从FAP与肿瘤组织的相关性、调节肿瘤行为的有效性可见,FAP是一个肿瘤靶向诊疗的理想靶点.因此,根据FAP在CAFs中的高表达及自身的蛋白酶特性,研究者们已经开发了一系列成纤维细胞活化蛋白抑制剂(FAPI).FAPI能够选择性地富集在肿瘤组织中,是一种有效的肿瘤靶向试剂,并且结合各种放射性同位素,展现出应用于癌症诊疗的巨大潜能.本文作者总结了近几年FAPI在肿瘤诊疗中的研究进展,重点介绍了新型FAPI在核医学领域肿瘤成像和治疗的应用,并从构效关系上讨论了增强FAPI选择性与延长保留时间的策略.1 FAPI的类型如图1所示,根据FAPI靶向弹头的伪肽结构,FAPI主要可分为下面几种类型:硼酸吡咯类、氯甲基酮类[8]和氰吡咯类.FAPI靶向弹头起抑制作用的机理是:FAPI中可分裂的肽键被不可分裂的亲电基团取代,引起FAP催化三联体中的丝氨酸羟基进行亲核攻击[9].硼酸吡咯类抑制剂由于对与FAP相关的多种脯氨酸肽酶有亲和力,对FAP的特异性受到了限制,并且还存在化学稳定性较低的缺点[10-11].而氰吡咯类抑制剂因为具有低纳摩尔FAP亲和性和高选择性等优异性质,已成为FAPI的主流构型.2014年,一种最有效的FAPI(简称:UAMC 1110)被开发出来,如图1(d)所示,它是一种典型的氰吡咯类抑制剂.目前,氰吡咯类抑制剂中具有代表性的是FAPI-02和FAPI-04,它们在临床实验中展现出高靶向性及高应用价值.另外,已有临床研究证明,相较于传统示踪剂氟代脱氧葡萄糖(FDG),FAPI-04在对各类肿瘤患者原發及转移灶的诊断上效果更优,尤其在肝转移瘤、腹膜癌、脑肿瘤的诊断上[12].FAPI-02和FAPI-04结构相似,两者的唯一区别在于FAPI-04的氰吡咯基团经二氟修饰,这增强了FAPI-04的疏水性,提高了抑制效力、配体效率和成纤维细胞活化蛋白与脯氨酰寡肽酶的比值(FAP/PREP)水平,提高了对FAP的选择性[13].2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各種癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 構建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各种癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性與延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设计了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫生组织(WHO)认定的分级为II级和III/IV级的异柠檬酸脱氢酶(IDH)突变星形细胞瘤的无创区分.虽然当前68Ga标记的FAPI在成像中得到了广泛关注,但由于68Ga的半衰期较短(半衰期(t1/2)为68 min),一次只能合成少量的放射性药物,并且不利于长距离的输送,这限制了68Ga-FAPI在实际医疗的应用.然而,放射性核素18F可以大量生产,并且它的发射器普遍可用,可以满足大量患者的需求.WANG等[19]开发了一种18F标记的铝与1,4,7-三氮杂环壬烷-N,N,N-三乙酸(NOTA)螯合的Al18F-NOTA-FAPI成像探针,可以在人工操作下制备,实现高放射性的批量生产.另外,如图4所示,HU等[20]通过临床研究证明一种18F标记的新型成纤维细胞活化蛋白抑制剂[18F]FAPI-42在各种癌症患者中表现出较高的病变检出率,可以成为68Ga-FAPI-04的替代品.在SPECT影像方面,由于SPECT具有低成本和广泛使用的特点,99 mTc标记的FAPI在实际患者的影像诊断中具有较大的应用潜能.LINDNER等[21]对一系列FAPI进行了99 mTc标记和临床研究,发现FAPI-34是一个优良的SPECT显像剂,可以通过快速肿瘤摄取和身体其他部位的快速清除获得高的对比度.TRUJILLO-BENITEZ等[22]还首次用99 mTc标记了硼酸吡咯类FAPI,结果显示:该示踪剂在人血清中的放射稳定性高,对FAP具有特异性识别,实现在肿瘤的高摄取和在肾脏中的快速清除.3.2 FAPI在肿瘤治疗中的应用目前,基于FAPI特异性靶向肿瘤的作用,许多对癌症具有靶向治疗效果的药物被开发出来.FAPI在肿瘤治疗上的应用大致可分为两类:一类是在FAPI上进行放射性核素标记用于放疗;另一类是通过化学合成在FAPI上偶联化疗药物进行化疗.在放疗上,WATABE等[23]使用半衰期较长的64Cu(t1/2为12.7 h)和225Ac(t1/2为10 d)标记FAPI,研究α-疗法治疗肿瘤的效果,结果表明:放射性铜和锕标记的FAPI-04(64Cu-FAPI-04和225Ac-FAPI-04)可用于治疗高表达FAP的胰腺癌.另一方面,如图5所示,为了研究短半衰期高能量同位素,如铼(188Re)、砹(21At)和铋(213Bi)等用于肿瘤放射治疗的效果,MA等[24]用与21At 化学性质相近的131I标记了FAPI-04,为后续21At放疗试剂的开发铺平道路.治疗结果表明:该种放射药物在胶质瘤近距离放疗中具有巨大的潜力.2 增强FAPI选择性与延长保留时间的策略许多已设计出来的FAPI对底物并非最佳特异性,易从体循环中被快速清除,在肿瘤中停留时间短,这限制了FAPI靶向诊断的精准性,而且不利于其在生物体内的长期跟踪.同时,较长的药物循环时间和肿瘤滞留时间是FAPI应用于放射性治疗的先决条件,所以这还阻碍了FAPI在肿瘤放疗上的应用.因此,增强FAPI对肿瘤的选择性与延长在肿瘤的保留时间就显得尤为重要.在此,总结了近年来几种常用的增强FAPI选择性与延长保留时间的策略.2.1 构建二聚体衍生物构建二聚体衍生物实质是在一个分子上同时连接2个相同的靶向药效基团,在相同的纳摩尔数上增加了药效团的数量,提高了FAPI的选择性,并且该策略易于在合成中实现,是一种增强FAPI肿瘤积聚和延长保留时间的有效策略.如EUY等[15]合成了2种方酰胺(SA)偶联的同型二聚体FAP抑制剂DOTA.(SA.FAPI)2和DOTAGA.(SA.FAPI)2,与传统的单体衍生物抑制剂相比,明显改善了肿瘤积聚和保留时间.2.2 连接强效配位基团与构建二聚体衍生物相似,在抑制剂结构上连接强配位基团也能增加药效基团的数量,从而提高FAPI的选择性.如图2所示,RUAN等[16]在抑制剂上连接强配位基团异氰酸,合成并用放射性锝标记了该含异氰化物的FAP抑制剂[99mTc][Tc-(CN-PEG4-FAPI)6]+.该络合物具有6个配位位点,在单个分子上携带了更多的药效基团,具有更高的肿瘤摄取及更好的肿瘤靶向性,是一种很有前景的肿瘤显像剂.2.3 合理选择连接基团设计合成一个有效的FAPI,不仅需要考虑分子靶向识别位点的数量,还需要考虑靶向位点作用的空间位阻.在螯合剂和药效基团之间选择更柔性的连接基团可以使药物更好地渗透到结合位点,增强FAPI的选择性.如SLANIA等[11]合成了2种新型FAPI:QCP01和[111In]QCP02,采用了灵活的线性酰胺烷基链替代原本支架结构上半刚性的哌嗪基团,结合时更好地渗透到结合位点,显示出肿瘤的高摄取率.2.4 增加体内保留基团在FAPI上增加体内保留基团,增强与生物组织的结合能力,从而获得较长的药物循环时间和延长FAPI的保留时间.如图3所示,LIN等[17]设計了以环螯合物四西坦(DOTA)为68Ga标记位点的FAP特异性体内正电子发射断层扫描(PET)示踪剂[68Ga]Ga-Alb-FAPtp-01,并在其上增加了与内源性白蛋白结合的基团4-对氯苯丁酸,以此延长药物的循环时间,增加在肿瘤病变的滞留时间,改善整体药代动力.并且这种与白蛋白的结合能力,使得PAPI能够通过内在血管或内部流体压力被稳定扩散并运输到肿瘤内.3 新型FAPI在肿瘤诊疗中的应用3.1 FAPI在肿瘤诊断中的应用在肿瘤诊断上,FAPI通过螯合不同的放射性核素,如68Ga,99mTc和18F等,进行核医学成像.目前基于FAPI的核医学成像主要包括PET影像和SPECT影像.在PET影像上,FAPI 除了在诊断原发及转移灶肿瘤上有优良的效果,在癌症的分级上也具有优势,如ROHRICH等[18]用68Ga标记的FAPI-02和FAPI-04对18例胶质瘤患者进行PET成像诊断,实现了世界卫。
_葡萄糖苷酶抑制剂的研究及应用

通讯作者 : 顾觉奋 ,教授 ; 研究方向 : 微生物制药教学与研究 ;
Tel: 025 2 86200377; E 2 ma il: yqyan1@ sina. com
2009, V o l . 33, N o. 2 63 P rog ress in P ha rm aceu tica l S c iences 改造 ,开发为降糖药米格列醇 。米格列醇安全有效 , 耐受性良好 ,现已成为治疗 2 型糖尿病的首选药物 。 此外 ,人们还发现合成米格列醇的关键中间体 N 2 取 代2 12 脱氧野尻霉素不仅具有降糖作用 ,还可抑制病 [2] 毒复制和调节激素分泌 , 因此加大了对该类化合 物的关注和研究力度 。
[5] [3]
α2 图 1 葡萄糖苷酶抑制剂的结构通式 Figure 1 The general structure ofα2glucosidase inhibitors
,如劳凤云等
[4]
从两株海洋放线菌
α2 葡萄糖苷酶抑制剂的作用及其机制
2. 1 降低血糖水平
Act 09和 Act 18 的发酵液中得到的乙酸乙酯提取物
・综述与专论 ・
2009年第 33卷 第 2期 第 63 页 α2 糖苷酶 ,如 α2 淀粉酶 、 葡萄糖淀粉酶和蔗糖酶等 , 其作用是将多糖 、 寡糖等水解成单糖 ,促进其被吸收 进入血液循环 。在胰岛素分泌正常的情况下 , 即使 食物中的淀粉等在小肠中被糖苷酶水解为大量葡萄 糖进入血液 ,血糖仍可维持在稳定 、 正常的水平 ; 但 糖尿病患者则因胰岛细胞功能障碍而导致胰岛素分 泌减少 ,使血液中的葡萄糖水平高于正常人 ,且伴有 脂蛋白浓度异常等现象 。 α2 葡萄糖苷酶抑制剂与寡糖结构相似 , 因此可 通过竞争性结合 α2 葡 萄糖苷酶上的碳水化合物结 合位点 ,使寡糖不能水解为单糖 , 阻止其被吸收 , 从 而使餐后血糖峰值渐变低平 、 波动减小 。 2. 2 抗 HI V 米格列醇的中间体 N 2 甲基 2 12 脱 氧野尻霉素和 N2 丁基 2 12 脱氧野尻霉素均可参与病毒的糖蛋白加 工过程 ,通过改变病毒上包膜糖蛋白 gp120 的糖侧 链结构 , 阻 断 H I V2 1 与宿主细胞结合 , 从而抑制
DPP-4抑制剂的降糖作用机制

DPP-4抑制剂的降糖作用机制何筱莹;李延兵【期刊名称】《药品评价》【年(卷),期】2012(000)034【总页数】4页(P17-19,23)【关键词】DPP-4抑制剂;2型糖尿病;GLP-1【作者】何筱莹;李延兵【作者单位】中山大学附属第一医院内分泌科;中山大学附属第一医院内分泌科【正文语种】中文【中图分类】587.1上个世纪三十年代,法国人La Barre首次提出了“肠促胰素”这一概念。
近年来的研究加深了人们对肠促胰素的认识,并且催生了一种新型的口服降糖药物——二肽基肽酶-4 (DPP-4)抑制剂,它可以提高血液中内源性GLP-1和GIP的浓度,并最终改善血糖控制。
目前已有数种DPP-4抑制剂在部分国家上市销售。
这些药物的化学结构和药效/代动力学或许各有差异,但它们都能有效地降低血糖(HbA1c降低0.5%~1%)并具有良好的耐受性。
本文将着重从A细胞和B细胞的双向调节方面阐述此类药物的作用机制。
肠促胰素效应与糖尿病“肠促胰素效应”指的是口服葡萄糖与静脉输注等量的葡萄糖相比,尽管后者可达到比前者更高的血糖水平,但前者能引起更强的胰岛素分泌反应。
人体在进食后,肠道受到饮食中脂肪和碳水化合物的刺激,以营养素依赖性的方式释放GLP-1和GIP等数种多肽,它们通过与胰岛B细胞表面的受体结合,增强胰岛素分泌囊泡的胞吐作用。
GLP-1由位于小肠远端(回肠)和结肠的L细胞分泌,而GIP则由位于十二指肠的K细胞分泌[1]。
正常人体在空腹状态下血浆中这两种激素的水平都很低,进餐后的数分钟内它们的血浆水平会迅速升高,并发挥葡萄糖依赖性的促胰岛素分泌作用,从而降低餐后血糖。
除了促胰岛素分泌作用之外,GLP-1还可以抑制胰岛A细胞释放胰高糖素,从而减少肝糖输出。
与促胰岛素释放作用相似,GLP-1对胰高糖素的抑制作用也依赖于血浆中的葡萄糖水平,在低于正常空腹血糖水平的状态下,GLP-1的抑制作用消失,从而降低了低血糖的发生风险。
药物化学中的抗艾滋病病药物研究

药物化学中的抗艾滋病病药物研究药物化学中的抗艾滋病药物研究艾滋病是由人体内感染人类免疫缺陷病毒(HIV)引起的一种严重的免疫缺陷疾病。
目前,艾滋病已成为全球范围内的重大公共卫生问题,给人类的生命和健康带来了严重威胁。
为了有效控制和治疗艾滋病,药物化学领域一直在致力于研究和开发抗艾滋病药物。
本文将介绍药物化学中的抗艾滋病药物研究的进展和挑战。
一、背景与意义随着艾滋病疫情的加剧,抗艾滋病病毒药物研究变得尤为重要。
抗艾滋病药物的研发可以有效控制病毒复制和进一步传播,提高患者的生活质量和预后。
因此,药物化学中对抗艾滋病病毒的研究具有重要的科学意义和社会意义。
二、抗艾滋病药物的研究进展1.核苷类逆转录酶抑制剂核苷类逆转录酶抑制剂是目前抗艾滋病药物研究的重点和主力。
这类药物通过干扰病毒的逆转录酶活性,阻断病毒的复制和扩散。
经过不断研究和进化,已经开发出多种有效的核苷类逆转录酶抑制剂,如拉米夫定、阿比卡韦等。
这些药物已广泛应用于临床治疗,并取得了良好的效果。
2.非核苷类逆转录酶抑制剂非核苷类逆转录酶抑制剂是另一类重要的抗艾滋病药物。
与核苷类逆转录酶抑制剂相比,非核苷类逆转录酶抑制剂通过与逆转录酶结合并抑制其活性,从而达到抗病毒的效果。
目前已经开发出一些有效的非核苷类逆转录酶抑制剂,如尼拉韦林、依非韦伦等。
这些药物可与核苷类逆转录酶抑制剂联合使用,提高治疗的效果。
3.蛋白酶抑制剂蛋白酶抑制剂是抗艾滋病药物研究中的新兴领域。
病毒的蛋白酶在病毒的复制过程中起到了关键作用,因此抑制蛋白酶的活性可以有效防止病毒的复制和传播。
目前,已经有一些蛋白酶抑制剂开始进入临床试验阶段,展现出潜在的应用前景。
三、抗艾滋病药物研究的挑战1.耐药性随着抗艾滋病药物的广泛应用,耐药性逐渐成为制约艾滋病治疗效果的重要因素。
病毒的变异性和复制速度快导致了药物的选择压力,从而促使病毒进化产生耐药突变。
因此,耐药性的监测和防控成为抗艾滋病药物研究中的重要课题。
HDAC抑制剂研究进展-樊后兴

第三页,共33页。
HDAC与肿瘤的发生
研究证实,HDAC的活性与癌症的发生有关。当HDAC过度表 达并被转录因子募集,就会导致特定基因的不正常抑制,从而导 致肿瘤,如急性粒细胞白血病被认为部分归因于早幼粒细胞白血 病蛋白/维A酸受体α融合蛋白对HDAC的非正常募集。
在乳腺癌中的SIRT7表达水平比良性乳腺组织中明显提高; 而乳腺癌的淋巴 结转移组和非转移组相比,SIRT3和SIRT7的表达水平差异也有显著性。
第八页,共33页。
组蛋白去乙酰酶类似物的结构特点及其功能
由于目前没有人的HDAC的晶体结构,所以有关HDAC的研究是通过与其有35.2%的氨基酸同一性 的超嗜热菌Aquifex aeolicus的组蛋白去乙酰酶类似物(HDLP)为基础来研究的。HDLP的晶体结构 显示,HDLP有两个明显的结构特征:a)一个深度为11A的管状通道,通向容纳TSA或SAHA的活性 部位,有催化活性的锌离子在通道的底部,TSA或SAHA的脂肪链从不同角度分析均利于与疏水通道 接近。b)一个长度为14A的内腔与锌离子的结合位点相邻。
第二页,共33页。
组蛋 白的乙酰化状态由两类酶来决定 ,即组蛋白乙酰转移酶(histone acetyltransferases, HAT)和组蛋白去乙酰化酶(histone de酰基转移到组蛋白N端赖氨酸残基,中和一个正电荷 ,使 DNA与组蛋白之间的静电引力减小 ,二者之间的相互作用减弱 ,染色质重塑 为转录活性结构 ,DNA易于解聚、 舒展 ,有利于转录因子、 调节因子复合物 和 RNA合成酶与 DNA模板相结合 ,激活基因转录。相反 , HDACs功能相反 ,抑制基因转录。
PTPB抑制剂研究进展

图3 PTP催化机理
通过对人类基因组的同源序列比对,发现人 类PTP家族共有112个成员,按其结构可以
分为两类:(1) 跨膜受体型,如LAR、PTPtx 和CD45等;(2) 胞内型,如PTP1B、TCPTP和 SHP2等。胞内型PTP-1B是最早被纯化和确
定生物学特性的蛋白酪氨酸磷酸酯酶,全
图11
图12 CoMFA 模型
图 13 CoMSIA 模型
表1 36个1,2-萘醌类化合物结构及活性数据
3.2 CADD方法对苯并三唑类化合物与PTP-1B
和TCPTP的相互作用方式进行比较研究
梁娜娜等利用计算机辅助药物分子设计 (Computer-Aided Drug Design, CADD) 采用定 量结构-活性(QSAR)和定量结构-选择性
图6 水杨酸类化合物
2.1.3 取代苯乙酮衍生物
α-卤代苯乙酮类衍生物是光致逆变共价型 PTP1B抑制剂[23]。Li等[12] 以苯乙酮为母 核,接合二氟亚甲基磷酸基团合成了化合 物(9)(见图 7) ,生物活性测试表明该7 取代苯乙酮衍生物
图1 PTP1B酶的活性位点结构示意
另一类研究较多的PTP1B抑制剂则是其变 构抑制剂 ,这一类化合物可以结合在α6 螺旋与α3螺旋之间的区域,使得 α7螺旋
不能再靠近酶的催化区域,从而阻止了 WPD 折叠形成具有催化作用的封闭构象 (见图2)。
图2 PTP1 B中变构位点结构示意图
1.2 PTP-1B的作用机制
国际上多个实验室大量的研究已经对PTP家族成员的
底物识别和作用机制有了很详尽的认识。由PTP介导的 催化过程一般分为两步,如图3所示。所有的PTP家族
成员都存在一个保守序列,即[I/V]HCXXGXXR[S/T],在
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
朱有全 男,38 岁,博士生,现从事高效 HPPD 酶抑制剂开发及构效关系研究。 *联系人 国家自然科学基金资助项目(29832050) 2003-04-22 收稿
化学通报 2004 年 第 67 卷
w018
有关 HPPD 提纯、晶体结构、在动植物体内的作用机制及其构效关系等方面的研究概况作一综述。
下,晶胞参数为 a=76.60 Å,b=142.75 Å,c=159.44 Å。
活性中心铁离子
图 2 晶体结构图 Fig.2 The active-site pocket of HPPd
图 3 模拟 HPPD-HPP 络合图 Fig.3 The binding model of HPPD-HPP
由图 1 可以看出,水平方向两个单体呈面背平行结合,上下通过 N-端结合,形成四聚体, 四个活性反应中心,当其中一个活性中心与底物结合时,相应会引起酶空间结构的变化,使水 平对称的另一活性中心的活性受到影响,活性降低,影响程度视抑制剂与酶结合的程度。这与 文献[9]所做的工作相符合:由于 DKN(diketonitrile)与 HPPD 二聚体为紧密结合,严重影响了另 一个的活性中心与 DKN 的结合反应,致使其与二聚体 HPPD 的化学计量比为 1/1。而与 HPPD
为 27.6%,R 因子为 21.9%,计算采用的衍射范围为 20.0~2.4Å,测定结果表明,具有非晶体 222
对称性,每个单体表面积为 2600Å2,晶包中包括 11119 个非氢原子、4 个铁原子、4 个二乙基
汞分子、4 个乙酸酯分子和 482 个水分子。晶体属正交晶系,空间群为 P212121;在 100K 条件
富马酸乙酰乙酯酶缺乏活性积累的琥珀酰基乙酰乙酸乙酯等体内物质,形成对肝肾有毒的代谢
物(即酪氨酸病产生的根本原因)。HPPD 抑制剂的使用,抑制了催化降解过程的发生,进而抑制
了有毒(副)产物的形成,从而达到了治疗疾病的目的[16]。
HPPD 在植物体内将 HPP 催化转化为 HGA 这一过程可能的反应机制,主要有:(1)Witkop
化学通报 2004 年 第 67 卷
HPPD 酶及其抑制剂构效关系的研究进展
朱有全 胡方中 杨华铮*
(南开大学元素元素有机化学研究所 有机化学国家重点实验室 天津 300071)
徐海珍
(天津师范大学化学与生命科学学院 天津 300074)
w018
摘 要 植物体中的对羟基苯基丙酮酸双氧化酶(HPPD)酶对动植物的生长发育起着至关重要的 作用。本文综述了 HPPD 酶的晶体结构、作用机制及其抑制剂的作用机制、构效关系。
关键词 对羟基苯基丙酮酸双氧化酶(HPPD) 抑制剂 构效关系
Reviews on 4-Hydroxyphenylpyruvate Dioxygenase Enzyme and the Structure-activity Relationships of its Inhibitors
Zhu Youquan, Hu Fangzhong, Yang Huazheng*
三酮部分
A
O
B
C
O
O R1 R2
R6
R3
环己二酮
R4
酮系统和其它等四部分,如右图所示。 4.1 苯环部分结构上的变化对化合物抑制能力的影响
R5
苯环部分
Lee 等[26]通过测定一系列的 2,X-二氯代化合物和 2,X,Y-三氯代化合物的活性发现,2,4-
二氯代、2,3,4-三氯代模式效果最好,2,4,5-三氯代效果最差。经进一步对一系列 2-氯(或硝基)-4-
1 HPPD 的提纯和活性分析
纯 HPPD 可以从动物肝、玉米、细菌等生物体中纯化来获得[9~11]。 HPPD 活性的分析通常采用的方法有:(1)测定 HPP 转化为 HGA 所消耗的氧气量[12,13];(2) 测定释放的 14CO2 的量[10];(3)分光光度法[11];(4)用高效液相法测定生成的 HGA 的量[7,14]。
化学通报 2004 年 第 67 卷
w018
4 HPPD 抑制剂的构效关系
HPPD 抑制剂和 HPP 在作用体系中,在氧气和铁离子存在下,竞争性地与 HPPD 活性中心
铁离子键合成配合物,抑制效果与抑制剂和酶的结合程度有关[18,23,24]。依据现有文献报道,HPPD
抑制剂主要有如下几类。
O
R1
O O
2 HPPD 的晶体结构
Laurence 等 [15] 采 用 SIRAS 方 法 测 定 了
Pseudomonas fluorenscens HPPD 的晶体结构。结果显
示,该酶以四聚体形式存在且通过两两 N-端相结合
形成上下对称的聚合结构,如图 1 所示。每一结构单
元都包含有两个重复的βαββα结构。正如图 2 所示,
深入的研究,有助于通过有效的抑制剂与酶结合的模拟,针对性地合成抑制剂目标分子,如图
3 所示。
O
O
SO 2CH 3
O
O
Cl
O
CH3
O
CN
DKN
H3C CF 3
O
S
CH3
RPA 206740 O
3 HPPD 及其抑制剂在动植物体内的作用机制
HPPD 在人及其它哺乳动物体内的主要作用是促进酪氨酸的代谢,但它还能够催化代谢因
2
化学通报 2004 年 第 67 卷
w018
二聚体结合较为松弛的 RPA206740 的实验结果显示,与二聚体的化学计量比为 1.2:0.5,即抑制
剂/活性中心的摩尔比为 1.2/1,这就是由于二者的结合能力差,导致蛋白质空间构型变化较小,
反应中心趋于全部参与,且抑制剂浓度的提高有利于平衡向络合物方向进行。因此,对酶结构
基化以及羧甲基的 1、2-迁移的反应过程机制。该反应六配位过渡态假设结构也获得 Laurence
等[15]报道的 HPPD 活性中心配位结构的佐证。
-O
O
O
H3C
H3C OH
Fe(II)
En
O
Fe
O
O
O
O
O
O-
-O
H+
OH
O
COO-
H
OH CO2
COO-
O
O O
En Fe(IV) O
OH
-O
O
En
Fe(IV)
Key words 4-Hydroxyphenylpyruvate Dioxygenase(HPPD), Structure-activity relationships, Inhibitor
对羟基苯基丙酮酸双氧化酶(4-hydroxyphenylpyruvate dioxygenase, HPPD)存在于各种生物体 中 并 被 提 取 出 来 [1] , 它 是 一 种 铁 - 酪 氨 酸 蛋 白 [2] , 在 植 物 体 内 可 将 对 羟 基 丙 酮 酸 (4hydroxyphenylpyruvate, HPP)催化转化为尿黑酸(2,5-dihydroxyphenylacetate, HGA)[3],进而转化为 光合作用中电子传递所需要的重要物质质体醌和生育酚[4,5],其中质体醌还是影响八氢番茄红素 去饱和酶催化的关键辅助因素[6],同时,该酶的抑制剂 NTBC(2-(2-nitro-4-trifloromethylbenzoyl)cyclohexane-1,3-dione)临床上已经应用于治疗酪氨酸病,对人体无不良作用。鉴于其上述重要作 用和特点,使之成为继 ALS、ACC 以及 Protox 之后的又一新的除草剂靶标酶和临床上治疗酪 氨酸病靶标酶[7]。由于该酶抑制剂用于除草方面时具有广谱、高效、残留低、环境相容性好、 使用安全的特点,且尚未发现有关其抗性的报道[8],这更加引起人们对其抑制剂及其构效关系 研究的重视,并进行了广泛的研究。为了使 HPPD 抑制剂的研制更具有针对性,本文对近年来
Abstract The crystal structure of 4-Hydroxyphenylpyruvate Dioxygenase(HPPD) enzyme, mechanism of action of HPPD enzyme and its inhibitors and structure-activity relationships of its inhibitors were reviewed.
三酮类、苯甲酰间苯二酚类在水中均能以烯醇形式存在且具有抑制 HPPD 和产生白化症状的作
用,在结构上和间苯二酚类具有极其相似的特点,通过合成相应的化合物证明苯甲酰乙烯醇结
构是 HPPD 抑制剂的基本结构,且需具有足够的酸性, PKa<6。
由于 HPPD 抑制剂起源于三酮类化合物,对 HPPD 抑制剂构效关系的研究也主要集中于三酮类。影响抑制 活性的结构部分可分为:苯环部分、环己二酮部分、三
O
O
En Fe(II) OH
O OH
O+
OH
OH
H3C
由于 HPPD 抑制剂在植物体内直接抑制了 HPP 转化为 HGA,间接抑制质体醌和生育酚甚
至类胡萝卜素的生成,最终导致植物的白化直至死亡。
从 HPPD 抑制剂在动植物体内作用机制可以看出,其对人畜是安全的,发展y Laboratory of Elemento-Organic Chemistry, Institute of Elemento- Organic Chemistry, Nankai University, Tianjin 300071)
Xu Haizhen
(College of Chemistry and Life,Tianjin Normal University, Tianjin 300074)