《结构化学》第九章 结构原理汇总
结构化学第九章
a 2
2
260 273 rS 2 184 pm rSe2 193 pm 2 2 Pauling的离子半径被广泛采用。
2. 有效离子半径 • 考虑价态、配位数、几何形状等条件对离 子半径的影响,经过修正的离子半径。 • Shannon(香农)以上千个氧化物和氟化物 的正负离子间距为基础(从F-133,O2-140pm 出发)拆分出的一套较完整的、经多次修 正的离子半径(参见表9.3.2)。 3. 离子半径的变化趋势——自学
(2)计算各个原子的价态,辨别原子的种类
• 硅酸盐中Si4+和Al3+由于核外电子数相同,配位情况相 似,互相易置换,不易区分,但通过键价计算可以辨 别。
(3)检验结构的正确性
• 若发现计算出的键价之和与原子价偏差太大,则 需考虑测定的结构是否正确,或结构中存在其它 因素,需重新审核。
(4)帮助确定晶体结构中轻原子的位置
Z Z e 2 AN m1 Z Z e 2 AN mB u B re m1 0 2 40 m 40 re re r r re Z Z e 2 AN 1 U NaCl 1 m可由晶体的压缩性因子求得, 40 re m
Z Z e A 40 r
2
A称为Madelung(马德隆)常数,收敛于1.7476。
同理, Cl
1 mol NaCl中,Cl-和Na+的数目均为N,每个离子 均计算了两次,所以,
N Z Z e2 EC Na Cl AN , ER Br m 2 40 r Z Z e2 总势能函数 u EC ER AN Br m 40 r 势能最低时,相邻Na+-Cl-间距即为平衡核间距re,
基础化学第九章分子结构精品文档

一、共价键的本质
而电子自旋方式相同的两个氢原子相互接近时,两 核间电子出现的概率密度降低,从而增大了两个原 子核的排斥力,使系统能量升高,所以无法形成化 学键。
由此可见,电子自旋相反的两个氢原子以核间距R0
相结合,比两个远离的氢原子能量低,可以形成稳 定的分子。而电子自旋相同的两个氢原子接近时, 系统能量比两个远离的氢原子能量还高,不能形成 稳定的分子。
第四节 轨道杂化理论
一 轨道杂化理论的基本要点 二 轨道杂化的类型与分子的空间构型
一、轨道杂化理论的基本要点
(1) 只有在形成分子中,能量相近的原子轨道才能进
行杂化。孤立原子不能杂化。常见杂化方式有nsnp杂化、ns-np-nd杂化和(n-1)d-ns-np杂化;
(2) 杂化轨道数等于参加杂化的原子轨道数; (3) 杂化轨道的成键能力比未杂化的原子轨道强,形
第二节 共价键的价键理论
一、共价键的本质 二、价键理论的基本要点 三、共价键的类型
引言:分子结构
分子是保持物质化学性质的最小微粒,而分子 的性质与分子结构密切相关。
分子内,相邻原子间存在强烈的相互作用—— 化学键——离子键、金属键和共价键。
分子间,存在着较弱的相互作用,包括分子间 作用力和氢键。
+-
±
双原子分子的极性
由相同原子组成的双原子分子,正、负电荷中心 重合,不具有极性,为非极性分子。
离子键
离子键的共价性
离子键形成的重要条件就是元素之间的电负性差值较大。
元素的电负性差值越大,形成的离子键越强。但离子化合物中
不存在100%的离子键,即使是 CsF 中的离子键,也有8%的
共价性。
>1.7,发生电子转移, 主要形成离子键;
九章分子结构-PPT精选

C2H 4 的构型为:
H 121o H
C = C 118 o
H
H
2020/8/21
§9.2 键参数
9.2.1 键级 9.2.2 键能 9.2.3 键长 9.2.4 键角 9.2.5 键矩与部分电荷
2020/8/21
9.2.1 键级
键级 B.O1(成键电子 反数键电子数)
2
N 2 ( 1 s ) 2 ( 1 * s ) 2 ( 2 s ) 2 ( 2 * s ) 2 ( π 2 p ) 4 ( 2 p ) 2
2020/8/21
第九章 分子结构
§9.1 价键理论 §9.2 键参数
2020/8/21
§9.1 价键理论
9.1.1 共价键的本质与特点 9.1.2 共价键的键型 9.1.3 杂化轨道
2020/8/21
9.1.1 共价键的本质与特点
化学键:
分子或晶体中相邻原子(或离子)之间 强烈的吸引作用。
化学键理论:
离子键理论
9.2.5 键矩与部分电荷
键矩是表示键的极性的物理量记作μ 。 μ= q ·l
式中 q 为电量,l 为核间距。μ为矢量,例如, 实验测得H-Cl
3.5 71 0 30 Cm HCl
2020/8/21
键参数小结: 键的强度 键级(B·O) 键能(E)
分子的空间构型
键角() 键长(l)
键的极性 ——键矩(μ)
E(H-H) =D(H-H) 多原子分子:原子化能 = 全部键能之和
Eatm(H2O) = 2E(O-H)
键焓与键能近似相等,实验测定中,常常得到 的是键焓数据。
2020/8/21
键能与标准摩尔反应焓变
2H2 (g) +
结构化学课后答案第9章晶体的结构习题解答

第9章 晶体结构和性质习题解答【9.1】若平面周期性结构系按下列单位并置重复堆砌而成,试画出它们的点阵结构,并指出结构基元。
●●●●●●●●●●●●●●●●●●●●○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○解:用虚线画出点阵结构如下图,各结构基元中圈和黑点数如下表:1234567○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○○●●●●●●●●●●●●●●●●●●●●图序号 1 2 3 4 5 6 7 结构基元数 1 1 1 1 1 1 1 黑点数 1 1 1 1 0 2 4 圈数1112313【评注】 从实际周期性结构中抽取出点阵的关键是理解点阵的含义,即抽取的点按连接其中任意两点的向量平移后必须能够复原。
如果不考虑格子单位的对称性,任何点阵均可划出素单位来,且素单位的形状并不是唯一的,但面积是确定不变的。
如果考虑到格子单位的对称形,必须选取正当单位,即在对称性尽量高的前提下,选取含点阵点数目尽量少的单位,也即保持格子形状不变的条件下,格子中点阵点数目要尽量少。
例如,对2号图像,如果原图是正方形,对应的正当格子单位应该与原图等价(并非现在的矩形素格子),此时结构基元包含两个黑点与两个圆圈。
【9.2】有一AB 型晶体,晶胞中A 和B 的坐标参数分别为(0,0,0)和(12,12,12)。
指明该晶体的空间点阵型式和结构基元。
解:晶胞中只有一个A 和一个B ,因此不论该晶体属于哪一个晶系,只能是简单点阵,结构基元为一个AB 。
【9.3】已知金刚石立方晶胞的晶胞参数a =356.7pm 。
请写出其中碳原子的分数坐标,并计算C —C 键的键长和晶胞密度。
解:金刚石立方晶胞中包含8个碳原子,其分数坐标为:(0,0,0),1(2,12,0),(12,0,1)2,(0,12,1)2,(14,14,1)4,3(4,34,1)4,(34,14,3)4,(14,34,3)4(0,0,0)与(14,14,14)两个原子间的距离即为C -C 键长,由两点间距离公式求得:C-C 356.7154.4pm r ====密度-13-10323-1812.0g mol 3.51 g cm (356.710cm)(6.022 10mol )A ZM D N V -⨯⋅==⋅⨯⨯⨯ 【9.4】立方晶系金属钨的粉末衍射线指标如下:110,200,211,220,310,222,321,400。
结构化学知识点汇总

结构化学知识点汇总关键信息项:1、原子结构原子轨道电子排布原子光谱2、分子结构化学键类型分子几何构型分子的极性3、晶体结构晶体类型晶格结构晶体的性质11 原子结构111 原子轨道原子轨道是描述原子中电子运动状态的数学函数。
主要包括s 轨道、p 轨道、d 轨道和 f 轨道。
s 轨道呈球形对称,p 轨道呈哑铃形,d 轨道和 f 轨道形状更为复杂。
112 电子排布遵循泡利不相容原理、能量最低原理和洪特规则。
电子按照一定的顺序填充在不同的原子轨道上,形成原子的电子构型。
113 原子光谱原子在不同能级间跃迁时吸收或发射的光子所形成的光谱。
包括发射光谱和吸收光谱,可用于分析原子的结构和成分。
12 分子结构121 化学键类型共价键:通过共用电子对形成,分为σ键和π键。
离子键:由正负离子之间的静电引力形成。
金属键:存在于金属晶体中,由自由电子和金属离子之间的相互作用形成。
氢键:一种特殊的分子间作用力,比一般的范德华力强。
122 分子几何构型通过价层电子对互斥理论(VSEPR)和杂化轨道理论来解释和预测。
常见的分子构型有直线型、平面三角形、四面体型、三角双锥型和八面体型等。
123 分子的极性取决于分子中正负电荷中心是否重合。
极性分子具有偶极矩,非极性分子则没有。
13 晶体结构131 晶体类型离子晶体:由离子键结合而成,具有较高的熔点和硬度。
原子晶体:通过共价键形成,硬度大、熔点高。
分子晶体:分子间以范德华力或氢键结合,熔点和硬度较低。
金属晶体:由金属键维系,具有良好的导电性和导热性。
132 晶格结构晶体中原子、离子或分子的排列方式。
常见的晶格有简单立方、体心立方、面心立方等。
133 晶体的性质各向异性:晶体在不同方向上的物理性质不同。
自范性:能够自发地呈现出多面体外形。
固定的熔点:在一定压力下,晶体具有固定的熔点。
21 量子力学基础211 薛定谔方程是描述微观粒子运动状态的基本方程,通过求解该方程可以得到粒子的能量和波函数。
最新结构化学重点掌握内容PPT课件

ms 自旋 磁量子数
n ,l,m ,m s( x ,y ,z ,m s ) n ,l,m ( x ,y ,z )( m s )
旋轨轨道或自旋轨道。
六:表示单电子原子状态的量子数
1. 主量子数n 2. 角量子数l
En
Z2 n2
R,
n1,2
Lll 1 , l 1 ,2n 1
3. 磁量子数m
lzm, m0,1 l
二、病因病机
(二)病机
3.病机转化: 较为复杂,既可由实转虚,
又可由虚转实,甚或虚中夹实;既可气滞及 血,又可血瘀阻气,但不外乎病在气,或病 在血,或气血同病。
4.预后:无论外感或内伤胁痛,只要治疗
将养得法,一般预后良好。
三、诊断要点
1.临床特征:一侧或两侧胁肋疼痛为
主要临床表现 ;疼痛性质可表现为刺痛、 胀痛、隐痛、闷痛或窜痛。
三、Zeeman效应 外磁场中原子光谱的分裂现象。
1四、、S多电子原s子i 的角S 动 量(S L(-S S耦1 合) )
i
S为原子的自旋量子数,
n, n 1,1
22
2
或
0(n:体系电子数)
Sz Ms M s S ,S 1 , , S
Ms有(2S+1)个取值
2、 L li LL(L1) i
多电子原子中的任何两个电子不可能具 有相同的4个量子数.
十、基态原子核外的电子排布遵循以下 三个原则: Pauli原理,能量最低原理, Hund规则。
第三章 原子光谱 一、谱项定义: 能级除以hc称为谱项,
T ~ 'E , T ~ E , ~T ~ T ~
hc hc
二、选择定则概念
两状态间发生跃迁,表示这些状态的 量子数之间需满足一定的条件,这些条件称 为选择定则。
结构化学知识点汇总

结构化学知识点汇总一、原子结构1、波粒二象性德布罗意波长公式:λ = h / p ,其中λ为波长,h 为普朗克常量,p 为动量。
海森堡不确定原理:ΔxΔp ≥ h /4π ,表明不能同时精确测定粒子的位置和动量。
2、原子轨道薛定谔方程:用于描述原子中电子的运动状态。
原子轨道的形状:s 轨道为球形,p 轨道为哑铃形。
原子轨道的能量:能层和能级的概念,以及能级交错现象。
3、电子自旋电子自旋量子数:取值为+1/2 和-1/2 。
泡利不相容原理:一个原子轨道最多只能容纳两个自旋相反的电子。
二、分子结构1、化学键离子键:由正负离子之间的静电引力形成。
共价键价键理论:包括原子轨道重叠、共价键的方向性和饱和性。
杂化轨道理论:解释分子的几何构型。
价层电子对互斥理论:预测分子的空间构型。
金属键:金属原子之间通过自由电子形成的化学键。
氢键:一种特殊的分子间作用力,具有方向性和饱和性。
2、分子的极性极性分子和非极性分子的判断依据:分子的正负电荷重心是否重合。
分子极性对物质性质的影响:如溶解性、熔沸点等。
3、分子间作用力范德华力:包括色散力、诱导力和取向力。
范德华力对物质物理性质的影响。
三、晶体结构1、晶体的特征有固定的熔点和规则的几何外形。
内部质点在三维空间呈周期性有序排列。
2、晶体的分类离子晶体:具有较高的熔点和硬度,如 NaCl 。
原子晶体:熔点和硬度很高,如金刚石。
分子晶体:熔点和硬度较低,如干冰。
金属晶体:具有良好的导电性和导热性,如铜。
3、晶胞晶胞的概念:晶体结构的基本重复单元。
晶胞中原子的占有率计算。
四、光谱学1、原子光谱发射光谱和吸收光谱。
原子光谱的应用:元素分析、测定原子结构。
2、分子光谱红外光谱:用于研究分子的化学键和官能团。
紫外可见光谱:反映分子中电子的跃迁。
五、量子化学计算方法1、从头算方法基于薛定谔方程的精确求解。
计算量较大,但结果较为准确。
2、半经验方法引入一些经验参数简化计算。
计算速度较快,但精度相对较低。
2015年《结构化学》电子课件 孙宏伟PPT Chap9 结构测定理论基础

h=2
A1
B1
A2 B2
A1B1 =/2
抵消
衍射强度与原子种类有关,即与原子的散射因子有 关,与各原子的分数坐标有关,与衍射方向有关。
《结构化学》第九章 分子、晶体结构测定方法理论基础 Nankai University
对空间点阵的劳埃方程有:
标量式 a(cos cos0) = h b(cos cos0) = k c(cos cos0) = l 矢量式 a· (S S0) = h b· ( S S0 ) = k c· ( S S0 ) = l h, k, l = 0, 1, 2, ... ...
《结构化学》第九章 分子、晶体结构测定方法理论基础 Nankai University
9.1.3 衍射强度与晶胞中原子的分布
1. 原子散射因子f 电子散射:
P 原生X射线 O r
O点放一个电子,距O为r的P点处的次生X射线的强度 设为 Ie 。若 O 点处 Z 个点电荷,则 P 点处的次生 X 射线 的强度为 I Ze=I e Z2
2. 晶胞散射因子
把O点放一个晶胞,则在衍射方向上散射次生X衍射的强度
Ic =Ie |F(hkl)|2 |F(hkl)|叫晶胞散射因子(叫结构振幅) Fhkl叫结构因子
分析晶胞内原子散射次生 X 射线的迭加情况,可以理 解晶胞的衍射强度即晶胞散射因子与什么有关。 设有一直线点阵:点阵的基本周期为a,一个结构基 元含2个原子A和B,B的坐标在a/4处
h k l为衍射指标,代衍射方向(与晶面指标不同, 不一定是互质的)
h=2 h=1 h=0 h=1 h=2 底片
h=1 h=0
h=1
《结构化学》第九章 分子、晶体结构测定方法理论基础 Nankai University
结构化学

1,泡利不相容原理:在一个多电子体系中,两个自旋相同的电子不能占据同一个轨道即同一原子中,两个电子的量子数不能完全相同。
2,泡利排斥原理,在一个多电子体系中,自旋相同的电子尽可能分开远离。
3,共价键的本质:当原子互相接近时,它们的原子轨道互相同号叠加,组合成成键分子轨道,当电子进入成键轨道,体系能量降低,形成稳定的分子。
此时原子间形成共价键。
4,原子轨道通过线性组合成分子轨道时,轨道数目不变,轨道能级改变,两个能级相近的原子轨道组合成分子轨道时,能级低于原子轨道能级的称为成键轨道,高于原子轨道能级的称为反键轨道,等于的称为非键轨道。
由两个原子轨道有效地组合成能级降低的分子轨道时,必须满足能级高低相近,轨道最大重叠以及对称性匹配三个条件。
5,价键理论认为电子隶属于原子,存在于特定的原子轨道上,原子轨道与原子轨道形成价键,原子间只有价键电子存在共用或偏移。
分子轨道理论认为电子隶属于整个分子,为原子所共有,存在于由原子轨道线性组合的分子轨道上。
6,价电子对互斥理论(VSEPR):原子周围各个价电子对之间由于相互排斥,在键长一定的条件下,互相间距离越远越稳定。
7,杂化轨道理论:原子在化合成分子的过程中,根据原子的成键要求,在周围原子的影响下,将原有的原子轨道进一步线性组合成新的原子轨道,这种在一个原子中不同原子轨道的线性组合称为原子轨道的杂化。
8,超共轭效应:指C-H等ε键轨道和相邻原子的π键轨道或其他轨道相互叠加,扩大ε电子的活动范围所产生的离域效应。
结构化学第9章

Z+
+ bX
Z−
→ M aXb +U
(气)
(气)
(晶体)
AN A Z + Z − e 2 1 U = (1 − ) 4 πε 0 re m
其中: 其中:
m 是晶体的压缩性因子 称为玻恩指数 它与离子的电子组态有关 是晶体的压缩性因子,称为玻恩指数 称为玻恩指数,它与离子的电子组态有关
电子组态
m
He 5
离 子 堆 积 描 述
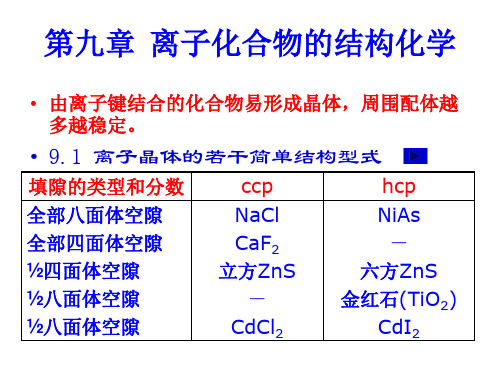
结构型式 化学组成比 n+/n负离子堆积方式 正负离子配位数比CN 正负离子配位数比 +/CN正离子所占空隙种类 正离子所占空隙分数
六方ZnS型 型 六方 1:1 六方最密堆积 4:4 正四面体 1/2
CaF2(荧石 型晶体结构 荧石)型晶体 荧石 型晶体结构
A: 0 0 0 0 1/2 1/2 1/2 0 1/2 1/2 1/2 0 分数坐标描述 B: 1/4 1/4 1/4 1/4 3/4 1/4 1/4 3/4 1/4 3/4 1/4 1/4 3/4 3/4 1/4 3/4
∆H1= -I
+
Cl-(g)
∆H3= E
∆H= U
NaCl(s)
Na(g)
∆H2= -S
Cl(g)
∆H4= -D/2
∆H5=∆Hf
Na(s)
+
(1/2)Cl2(g)
S为升华热,I为电离能,D为解离能,E为电子亲合能,∆Hf为生成热。 为升华热, 为电离能 为解离能, 为电子亲合能 为电子亲合能, 为生成热。 为升华热 为电离能, 为解离能 ∆H=∆H1+∆H2+∆H3+∆H4+∆H5 =-I-S+E-D/2+∆Hf=(-495.0-108.4+348.3-119.6-410.9)kJ·mol-1 =-785.6 kJ·mol-1 U= ∆H =-785.6 kJ·mol-1
九章分子结构-PPT精选

2020/7/21
直线形 三角形 四面体
BeCl2 BF 3 CH 4 HgCl2 BCl 3 SiCl 4
Be(ⅡA) B(ⅢA) C,Si
Hg(ⅡB)
(ⅣA)
三角锥
NH 3
PH 3
N,P
(ⅤA)
V型
H2O H 2S
O,S
(ⅥA)
思考题:解释CH4,C2H2,CO2的分子构型。
已知: C2H2,CO2均为直线型;
B.O = 1/2( 10 - 4 ) = 3
O 2 K ( 2 s ) 2 ( K 2 * s ) 2 ( 2 p ) 2 ( π 2 p ) 4 ( π * 2 p ) 2
B.O =1/2 ( 8 - 4 ) = 22Leabharlann 20/7/219.2.2 键能
键解离能(D) 在双原子分子中,于100kPa下将气态分
C2H 4 的构型为:
H 121o H
C = C 118 o
H
H
2020/7/21
§9.2 键参数
9.2.1 键级 9.2.2 键能 9.2.3 键长 9.2.4 键角 9.2.5 键矩与部分电荷
2020/7/21
9.2.1 键级
键级 B.O1(成键电子 反数键电子数)
2
N 2 ( 1 s ) 2 ( 1 * s ) 2 ( 2 s ) 2 ( 2 * s ) 2 ( π 2 p ) 4 ( 2 p ) 2
2020/7/21
sp3d2杂化
2020/7/21
小结:杂化轨道的类型与分子的空间构型
杂化轨道类型 sp sp2 sp3 不等性sp3
参加杂化的轨道s+p s+(2)p s+(3)p s+(3)p
结构化学知识点汇总

结构化学知识点汇总结构化学是一门研究原子、分子和晶体结构以及结构与性能之间关系的学科。
它为我们理解物质的性质和化学反应提供了基础。
以下是对结构化学中一些重要知识点的汇总。
一、原子结构1、玻尔模型玻尔提出了原子的行星模型,认为电子在特定的轨道上绕核运动,轨道具有固定的能量。
2、量子力学模型薛定谔方程是描述微观粒子运动状态的基本方程。
电子具有波动性和粒子性,其运动状态用波函数来描述。
3、原子轨道原子轨道是波函数的数学表达式,常见的有 s、p、d、f 轨道。
4、电子排布遵循能量最低原理、泡利不相容原理和洪特规则,电子依次填充不同的原子轨道。
二、分子结构1、价键理论认为原子通过共用电子对形成共价键,包括σ 键和π 键。
2、杂化轨道理论原子在形成分子时,轨道会杂化,形成等性杂化和不等性杂化。
3、价层电子对互斥理论用于预测分子的几何构型,根据中心原子的价层电子对数和孤电子对数来判断。
4、分子轨道理论将分子看作一个整体,电子在分子轨道中运动,分子轨道有成键轨道和反键轨道。
三、化学键1、离子键由正负离子之间的静电引力形成,通常在金属和非金属元素之间形成。
2、共价键原子间通过共用电子对形成,具有方向性和饱和性。
3、金属键金属原子通过自由电子形成的化学键,具有良好的导电性和导热性。
4、氢键一种特殊的分子间作用力,比范德华力强,但比化学键弱。
四、晶体结构1、晶体的分类根据晶体中粒子的排列方式,可分为离子晶体、原子晶体、分子晶体和金属晶体。
2、晶胞晶体的基本重复单元,通过晶胞可以研究晶体的结构和性质。
3、晶体的堆积方式如金属晶体的面心立方堆积、体心立方堆积等。
4、晶体的缺陷包括点缺陷、线缺陷和面缺陷,对晶体的性能有重要影响。
五、结构与性能的关系1、熔点和沸点与晶体类型和化学键的强度有关。
2、硬度和强度与晶体的结构和化学键的类型有关。
3、导电性和导热性金属晶体具有良好的导电性和导热性,而离子晶体在熔融或溶液状态下导电。
4、光学性质晶体的结构会影响其对光的折射、反射和吸收。
914704-结构化学-第9章

空隙位置 体心1个, 及数目 12条棱心 3个
占 有 位 置 体心1 ,棱心3
NaCl型
(111)方向正负离子堆积 s型:分数坐标描述(以负离子B为晶胞顶点,O点为坐标原点)
A(正离子)
B(负离子)
1/3 2/3 1/4
0
0
0
1/3 2/3 3/4 2/3 1/3 1/2
哥希密特指出:“晶体的结构型式主 要取决于组成晶体的原子、离子或原子团 的相对数量关系、相对大小关系及相互极 化性能三个因素。”
组成晶体的结构基元相对数量影响
晶体的结构一般可按化学式分类:例如, AB,AB2,AB3等,由于化学式不同,则晶体 结构一般不同,即组成者相对数量不同,结构 不同。
n+/n-=1 : 1
n+ : 1 +1/4 ×12
=4
n- : 1/8×8 +1/2×6
=4
NaCl型
2. 结构型式
结构型式是用一些 有代表性的晶体来命名的。 例如,MgO、SrS、LiF 等晶体的结构型式都属于 NaCl型,这只是说它们 的正、负离子空间排布方 式也采取NaCl晶体中那 种方式,而化学组成与 NaCl毫无共同之处。
NaCl型
CN+=6 CN-=6
NaCl型 5. 正离子所占空隙种类
正八面体
由CN+可知正离子所占空隙种类。
6. 正离子所占空隙分数
NaCl型
浅蓝色球代表的负离子(它们与绿色球是相同的负离子) 围成正四面体空隙, 但正离子并不去占据:
仔细观察一下: 是否有被占据的正四 面体空隙?
没有!
NaCl型
第9章 离子化合物的结构化学
离子化合物是指由正负离子结合在一起形 成的化合物,它一般由电负性较小的金属元素与 电负性较大的非金属元素构成。
结构化学的构造原理是

结构化学的构造原理是结构化学是化学领域的一个分支,它研究的是物质的化学结构及其与物质性质之间的关系。
结构化学主要包括两个方面的内容:化学键和化学功能团。
化学键是构成分子的原子之间的连接,它决定了分子的形状和稳定性,从而决定了分子的性质。
而化学功能团则是分子中具有特定性质和反应的基团,它们在分子中的位置和组合方式也会影响分子的性质。
结构化学的构造原理主要包括以下几个方面:1. 原子间的化学键:化学键是构成分子的原子之间的连接,它决定了分子的形状和稳定性。
根据化学键的类型,可以将化学键分为共价键、离子键和金属键。
共价键是通过原子之间共享电子而形成的,常见于非金属元素之间的连接。
离子键是由正负电荷之间的相互吸引力形成的,一般出现在金属元素与非金属元素之间的连接。
金属键是金属元素之间的连接方式,通过金属离子之间的电子云形成。
2. 分子的空间排列:化学键的连接方式决定了分子的形状。
空间排列对分子的性质具有重要影响。
空间排列可以分为线性、平面和立体排列。
线性排列的分子形状直线,平面排列的分子形状平面,而立体排列的分子则具有三维空间结构。
3. 化学功能团的种类和位置:化学功能团是分子中具有特定性质和反应的基团。
它们的存在使分子具有一定的化学活性和反应性。
不同的化学功能团会导致分子具有不同的性质和反应。
化学功能团的位置和组合方式也会影响分子的性质。
例如,醇基(OH)是一种常见的化学功能团,当它的位置发生变化时,分子的性质也会发生变化。
4. 电荷分布和分子极性:分子中电子的分布和分子极性对分子的性质具有重要影响。
根据电子云的分布,可以将分子分为极性分子和非极性分子。
极性分子具有正负电荷分布不均匀的特点,而非极性分子的电荷分布相对均匀。
分子的极性会影响分子的溶解性、沸点、熔点等性质。
总之,结构化学的构造原理是通过研究化学键、分子的空间排列、化学功能团的种类和位置、电荷分布和分子极性等因素,来理解分子的构造和性质之间的关系。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
傅里叶变换红外光谱仪框图
化学键的振动研究对光纤通讯材料也具有实际意义。光在光纤中传输时 的损耗与光纤的总体性质和原子尺度上的性质有关. 由玻璃密度、组成的随机涨落所引起的Rayleigh散射光强与波长的四次方 成反比,所以,与可见光和紫外光相比,使用红外光可以显著降低损耗。 从另一方面看,红外光可以激发玻璃中Si、O和杂质原子之间化学键的振 动跃迁. 尽管目前大量使用的高透明度SiO2玻璃已能满足远距离通信的要求, 但若要进一步降低光损耗,还需要减少振动激发. 使用原子量大于Si而化学键 更弱的材料,可以在更长的波长范围内实现红外透明,Rayleigh散射、振动激 发和电子激发都可以降至极低. 所以,近年来发展了性能更加优越的ZrF4、LaF4 和BaF2三元混合体氟玻璃,传输光信号上万千米不需任何中继站.
各类化学键振动波数
苯的红外光谱
红外分光光度计光路图
检测器 平衡马达
热电偶 滤光器 出射狭缝 光断续器 减光器 入射狭缝
减光器位置对应于纵坐标 参比槽
反射镜
光源
分光棱镜 描图马达通过波长 凸轮转动棱镜以改 变波长并带动记录 装置(横坐标)记录
立托夫镜
样品槽
傅里叶变换红外光谱
傅里叶变换红外光谱仪是非色散型光谱仪,其核心是 双光束干涉仪. 当动镜移动时,经过干涉仪的两束相干光 的光程差改变,到达探测器的光强随之改变,得到干涉图, 再经傅里叶变换得到入射光的光谱. 主要优点是:信噪比 和灵敏度高;以氦、氖激光波长为标准,波数值精确度可 达0.01cm-1 ; 增加动镜移动距离可提高分辨率;工作波段 从可见区可延伸到毫米区,实现远红外光谱测定.
4.拉曼光谱
分子振动也可能引起分子极化率的变化,产生拉曼 光谱. 拉曼光谱不是观察光的吸收, 而是观察光的非弹性 散射. 非弹性散射光很弱,过去较难观测. 激光拉曼光谱 的出现使灵敏度和分辨力大大提高,应用日益广泛.
激光拉曼光谱仪 (示意图)
拉曼现象可以在各种入射光频率下发生(例如用波长 为632.8 nm 的He-Ne 激光), 产生机理和红外光谱不同,选 律也不相同. 光子与分子碰撞时 , 少部分在侧向散射 . 散射光中又有 大部分是频率不变的弹性散射, 少部分是频率增大或减小的 非弹性散射. 频率增减是由于光子与分子交换能量引起,频 率减小和增大分别产生Stokes线与反Stokes线. 有对称中心的分子,任何一种振动方式不会在红外与拉 曼光谱上都观察到, 称为互斥规则. 研究分子结构时可互补. 没有对称中心的分子不存在互斥规则.
含 杂 原 子 不 饱 和 烃 分 子
E σ* π*
n π σ
这类分子中的杂原子可能直接形成双键,也可能用 孤对p电子与双键形成p-π共轭,前者属于生色基或发色 团,后者的含杂原子基团属于助色基. n π * ( 200~400 nm )和 n σ * ( ~200 nm )是这 类分子中新出现的跃迁. 没有p-π共轭时没有nπ*跃迁. n π * 跃迁是对紫外分析最有实际意义的另一种跃迁类 型.
第四章
结构分析原理
Chapter 9. Structural Analysis Theory
4.1 分子光谱
分子光谱与原子光谱有许多不同之处 , 谱线数目多且比 较密集. 一组吸收峰形成一个谱带, 各谱带之间有较大距离; 几个谱带又组成一组,成为一个谱带系,各谱带系之间的距 离更大. 这种特点与分子内部运动复杂性有关.分子中至少有两 个核,除电子相对于核的运动外,还有各核在平衡位置附近 的微小振动和分子整体绕质心的转动(分子平动能级间隔太 小, 可视为连续能级). 分子内部的转动、振动、电子的能级图如下:
双原子分子的转动光谱
将分子的转动与振动(及电子运动)近似分开,意 味着分子转动时核间距不变 . 这种模型称为刚性转子, 它大大简化了分子转动的数学处理. 设两原子的质量分别为m1、m2, 距质心O的距离分别 为r1、r2 ( r1与r2之和等于平衡核间距r, 即 r1+r2=r).
双 原 子 分 子 的 刚 性 转 子 模 型
注意: (1) 以下公式中振动波数以高能级振动量子数标 记, 因为低能级振动量子数已指定为0; (2) 非谐振子振动波 数公式中包含谐振子振动波数, 故分别加下标v和e区分.
2. 双原子分子振转光谱
振动激发可以同时伴随转动激发,用高分辨率红外光谱仪 可观察到振转光谱. 例如, HCl的振转光谱如下:
原子总动能
振动方程
ห้องสมุดไป่ตู้动能级
注意区别振动量子数 v (英文) 和经典振动频率ν (希文). 以下不再用颜色区分
振动跃迁选律: 整体选律:只有能够引起分子固有偶极矩变化的振动 方式才可能观察到红外光谱. 这对多原子分子也是适用的. 具体选律:才有Δv=±1的跃迁是允许的.
谐振子能级是等间隔hv. 具体选律Δv=±1, 即使分子分 布在多种振动能级上, 跃迁也只产生一条振动谱带. 实际上, 室温下大部分分子处于振动基态, 主要的振动跃迁是v=0到 v=1的跃迁. 实验表明这一推断基本正确, 但还发现了一些波数近似 等于基本谱带整数倍而强度迅速衰减的泛音谱带, 表明分子 不可能是完全的谐振子(否则分子永远不会离解), 而是非谐 振子模型:
分子的转动、振动、电子能级示意图
E
电子能级 ΔEe=1~20 eV
振动能级 ΔEv=0.05~1 eV 转动能级 ΔEt=10-4~10-2 eV
转动、振动、电子跃迁各有一定的能量,且相互影 响,但在一般近似情况下可忽略其相互影响,将分子的能 量视为这三部分能量之和:
E=Er+Ev+Ee
所以, 可以分别研究分子的转动、振动、电子光谱.
频率较高的振动不易受其他因素影响. 经验表明, 同 一种官能团的振动频率在不同化合物中大致相同, 据此可 鉴别不同类型的化合物. 频率较低的振动则易随环境而变, 反映每一种具体分子的结构信息, 出现的区域称为“指纹 区”. IR谱的扫描范围通常在4000~650 cm-1, 特征频率区与 指纹区大致以1500 cm-1为界.
含 杂 原 子 饱 和 烃 分 子
E
σ*
含杂原子饱和烃分 子中,杂原子的孤电子 对形成非键轨道n. 分子
9.2.1 转动光谱
转动能级间隔约为10-4~0.05 eV. 跃迁吸收或发射光的 波长处在远红外或微波区,故称远红外谱或微波谱. 远红外光谱光子能量低,测量易受干扰,应用不普 遍. 付里叶变换红外光谱和可调频率的激光红外光谱的出 现,使测量范围和精密度大大提高. 微波技术不需要分光 器,频率可连续改变,所以, 微波谱测量更加方便,研究 日益增多.
4.1.3 电子光谱
分子中价电子受到电磁辐 射作用 , 吸收辐射能在分子轨 道之间跃迁,产生的吸收光谱 称为电子光谱。价层分子轨道 之间的能级差对应于电磁辐射 的紫外或可见光区,因此也称 为紫外可见光谱。
1. 电子能级与跃迁类型
有机分子的价电子主要有三种类型: σ电子、 π 电子和非键电子n. 不同类型的分子可能包含不同类型的 价电子,具有不同类型的跃迁. 下面按分子类型分别讨论:
1. 双原子分子红外光谱
谐振子模型 双原子分子的振动可用谐振子模型作简化处理 . 下 图是两核在平衡距离 re 附近往复振动的示意图 ( 这是一 种经典图像 . 在量子力学中 , 振子在振动过程中位置按 概率分布, 而没有确切位置):
请单击按钮观看动画
谐振子势能曲线
设双原子分子振 动回复力服从虎克定 律, 则势能为
定义转动惯量I如下:
其中μ为折合质量. 这相当于将上述刚性转子等价地化成 了一个质量为μ的假想粒子在作半径为r的转动. 双质点问 题简化成了单质点问题.
将经典物理学中的平动与转动作一对比是有益的:
平动 质量 m 速度 v 动量 p=mv 动能 T=mv2/2=p2/(2m)
转动 转动惯量 I 角速度 ω 角动量 M=Iω 动能 T=Iω2/2=M2/(2I)
/cgi-bin/MolSpec/diperiodic.pl
4.1.2 振动光谱
分子振动能级间隔较大,约为0.05~l eV. 振动跃迁通常 伴有转动跃迁,称为振 - 转光谱 . 分子振动可能引起固有偶 极矩变化,也可能引起极化率变化. 分别产生红外光谱和拉 曼光谱. 下面先讨论红外光谱, 最后简单介绍拉曼光谱.
非谐振子
对于非谐振子模型提 出过几种势能函数, 例如 三方抛物线函数:
该函数曲线与左图 的实验势能曲线在高振 动能级仍不相符, 但振 动能级不太高时明显优 于谐振子的抛物线函数.
非谐振子的整体选律与谐振子相同, 但具体选律允许Δ v=±1, ± 2,±3,……. 考虑到室温下大部分分子处于振动基态, 可导出从v=0 跃迁到任一高能级v 的吸收波数公式(见下页).
刚性转子没有拉伸势能,总能量等于动能
Schrödinger方程为
回忆原子轨道角动量平方
仿照此式可写出刚性转子能量公式(转动量子数记作J, 并略去E的下标r):
B为转动常数. 能级差和波数差的下标都使用了较低能级 的量子数. 下图为转动能级及其一阶差分Δ EJ 、二阶差分Δ 2EJ 和 跃迁对应的谱线:
3. 多原子分子红外光谱
N原子分子有3N个自由度, 其中3个属于平动, 3个属于转 动(直线形分子为2个), 剩余3N-6个为振动自由度(直线形分子 为3N-5个). 每个振动自由度有一种正则振动. 原则上, 任何复 杂振动都可以分解为正则振动的叠加, 但实际上, 多原子分子 振动光谱主要由经验规律解析. 不过, 计算机辅助复杂分子结 构分析专家系统近年来也取得了引人瞩目的进展. 一般说来, 伸缩振动频率大于弯曲振动频率, 重键振动频 率大于单键振动频率, 连接较轻的原子(如H)的化学键振动频 率较高.
饱 和 烃 分 子
E
σ*
饱和烃分子只有σ电 子,只能产生σσ*跃 迁. 这种跃迁能级差很大 ,吸收波长很短,都在远 紫外区150 nm以下,所以 称这种基团为非生色基. 由于技术上的困难,研究 很少.