临床试验样本量的估算
临床研究中的样本量估算:临床试验

临床研究中的样本量估算:临床试验在临床研究领域,样本量估算无疑是临床试验设计中至关重要的一环。
它不仅关系到研究结果的可靠性和有效性,还直接影响到研究的成本、时间和可行性。
通俗来讲,样本量估算就是要回答“我们需要多少研究对象才能得出有意义的结论”这个关键问题。
为什么样本量估算如此重要呢?想象一下,如果样本量过小,就好像只从大海中舀了一小杯水来判断海水的成分,结果很可能因为随机性太大而不准确,甚至得出错误的结论。
相反,如果样本量过大,虽然能增加结果的可靠性,但会造成资源的浪费,研究时间延长,成本大幅增加。
所以,找到一个恰到好处的样本量,是每个临床试验设计者都必须面对的挑战。
那么,如何进行样本量估算呢?这可不是拍脑袋就能决定的,而是需要综合考虑多个因素。
首先要考虑的是研究的主要目的和假设。
比如,是要比较两种治疗方法的疗效差异,还是要评估某种药物的安全性?不同的研究目的和假设会对样本量产生不同的要求。
假设我们要比较一种新药物和传统药物治疗某种疾病的效果。
我们的假设是新药物的疗效优于传统药物。
为了能够有足够的把握检测出这种差异,我们需要根据预期的疗效差异大小来确定样本量。
如果预期的差异很小,那么就需要更多的样本才能检测到这种细微的差别;如果预期的差异较大,相对来说所需的样本量就会少一些。
其次,效应大小也是一个关键因素。
效应大小反映了研究中所关注的变量之间的差异程度。
比如,在药物疗效研究中,效应大小可以是新药物和传统药物在治愈率、症状改善程度等方面的差异。
一般来说,效应大小越大,所需的样本量就越小;效应大小越小,所需的样本量就越大。
再者,检验水准和检验效能也不能忽视。
检验水准通常设定为005,它表示在假设检验中,当原假设为真时,错误地拒绝原假设的概率。
而检验效能则是在给定的效应大小和样本量的情况下,正确拒绝原假设的概率,一般要求检验效能不低于 80%。
简单来说,检验水准和检验效能就像是天平的两端,我们需要在它们之间找到一个平衡,以确定合适的样本量。
临床试验样本量的估算30429

临床试验样本量的估算30429
1.实验目标:明确试验的主要目标是什么?是评估治疗的效果、副作用,还是探索新的治疗方法等?不同的目标可能需要不同的样本量。
2.效应大小:效应大小是指治疗或干预与对照组之间的差异有多大。
通常,需要根据预期的效应大小来估算样本量。
如果效应很小,那么需要
更大的样本量才能检测到显著的差异。
3.α和β错误:在样本量估算中,需要考虑到统计显著性水平(α)和统计功效(1-β)。
一般常用的显著性水平为0.05,统计功效为80%。
根据研究的特点和要求,也可以选择不同的显著性水平和统计功效。
4.可接受的误差:在样本量估算中,还需要考虑到可接受的误差范围。
例如,如果试验的目标是评估治疗效果,那么可接受的误差范围是多少?
一般来说,误差范围越小,需要的样本量也越大。
综合上述因素进行样本量估算的计算。
常用的样本量估算方法有多种,如假设检验的样本量估计、置信区间的样本量估计、生存分析中的样本量
估计等。
具体使用哪种方法取决于试验研究的设计和目标。
最后,需要注意的是样本量的估算只是一个大致的估计,并不代表最
终确定的样本量。
在实际进行试验之前,还需要进行实际情况的调查和分析,可能需要进行修正和调整。
临床试验样本量的估算

临床试验样本量的估算样本量的估计涉及诸多参数的确定,最难得到的就是预期的或者已知的效应大小(计数资料的率差、计量资料的均数差值),方差(计量资料)或合并的率(计数资料各组的合并率),一般需通过预试验或者查阅历史资料和文献获得,不过很多时候很难得到或者可靠性较差。
因此样本量估计有些时候不是想做就能做的。
SFDA的规定主要是从安全性的角度出发,保证能发现多少的不良反应率;统计的计算主要是从power出发,保证有多少把握能做出显著来。
但是中国的国情有多少厂家愿意多做建议方案里这么写:从安全性角度出发,按照SFDA××规定,完成100对有效病例,再考虑到脱落原因,再扩大20%,即120对,240例。
或者:本研究为随机双盲、安慰剂平行对照试验,只有显示试验药优于安慰剂时才可认为试验药有效,根据预试验结果,试验组和对照组的有效率分别为%和%,则每个治疗组中能接受评价的病人样本数必须达到114例(总共228例),这样才能在单侧显著性水平为5%、检验功效为90%的情况下证明试验组疗效优于对照组。
假设因调整意向性治疗人群而丢失病例达10%,则需要纳入病人的总样本例数为250例。
非劣性试验(α=,β=)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=× (S/δ)2等效性试验(α=,β=)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=× (S/δ)2上述公式的说明:1) 该公式源于郑青山教授发表的文献。
2) N 是每组的估算例数N1=N2,N1 和N2 分别为试验药和参比药的例数;3) P 是平均有效率,4) S 是估计的共同标准差,5) δ 是等效标准。
6) 通常都规定α=,β=(把握度80%)上述计算的例数若少于国家规定的例数,按规定为准;多于国家规定的则以计算值为准。
临床试验样本量的估算精编版

临床试验样本量的估算精编版临床试验样本量的估算是为了确保试验结果具有统计学意义和准确性而进行的,它直接关系到试验结果的可靠性和推广的可行性。
样本量的估算一般包括研究目的、研究设计、效应值、暴露率、有效α and β 水平以及研究变量等因素的考虑。
首先,研究目的是估算样本量的基础。
不同的研究目的需要不同的样本量。
例如,如果研究目的是描述性研究,那么样本量的估算就应该考虑到对总体特征参数的精确度要求,并按照这个要求选择样本量。
而如果研究目的是比较性研究,则需要估算出有效比较的样本量。
其次,研究设计也是影响样本量估算的重要因素。
常见的研究设计包括前瞻性队列研究、回顾性队列研究、前瞻性对照研究、回顾性对照研究等。
不同的研究设计需要不同的样本量估算方法。
一般而言,前瞻性研究需要相对较少的样本量,而回顾性研究需要相对较多的样本量。
此外,效应值也是影响样本量估算的重要因素。
效应值是指待研究变量之间的差异或相关关系的大小。
一般来说,如果关注的效应值较大,需要的样本量较小,反之则需要较大的样本量。
暴露率和有效α and β 水平也是样本量估算的重要考虑因素。
暴露率是指研究中具有待研究变量的人群的占比,它直接关系到样本量的多少。
一般而言,暴露率越高,需要的样本量越少。
有效α and β 水平是指接受两种处理的个体之间差异的显著性水平和检测到这种差异的能力,通常被设置为0.05和0.20,它们也会影响样本量的估算。
最后,研究变量的数量和类型也需要考虑。
当研究的变量较多时,往往需要更大的样本量来保证统计分析的有效性和可靠性。
总结起来,样本量的估算需要考虑研究目的、研究设计、效应值、暴露率、有效α和β水平以及研究变量等因素。
根据这些因素,可以选择合适的样本量估算方法,并计算出适当的样本量,以保证试验结果的准确性和可靠性。
临床试验样本量的估算介绍

临床试验样本量的估算介绍临床试验样本量的估算是一个关键性的步骤,它决定了试验结果的可靠性和统计学上的显著性。
样本量的估算需要考虑多个因素,如预期效应大小、研究设计、统计分析方法以及可接受的错误率等。
本文将详细介绍临床试验样本量估算的基本原理和常用方法。
一、样本量估算的目的和原则样本量估算的主要目的是确保试验具有足够的统计功效,能够检测到预期效应的存在或差异的显著性。
同时,也需要避免过大的样本量,以减少资源的浪费和人体试验的风险。
样本量估算的原则如下:1.统计功效要求:根据研究者的预期效应大小,确定试验能够达到的最小统计功效要求。
通常,我们希望试验能够有80%的功效检测到预期效应。
2.显著性水平:选择统计学上的显著性水平,通常为α=0.053.效应大小的估计:根据已有的文献或专家经验,估计预期效应的大小。
4.变异性的估计:估计所研究的指标的标准差或方差。
5.实用性考虑:考虑到资源和时间的限制,选择可实现的最小样本量。
二、常用的样本量估算方法1.基于效应大小的样本量估算方法这种方法根据研究者希望检测到的最小效应大小来估算样本量。
常用的方法有两组均数差异的估算、比例差异的估算以及相关系数的估算。
对于两组均数差异的估算,可使用t检验或方差分析来进行样本量估算。
对于比例差异的估算,可使用Z检验来进行样本量估算。
对于相关系数的估算,可使用相关系数检验来进行样本量估算。
2.基于统计功效的样本量估算方法这种方法以试验的统计功效为基础,确定对于预期效应的检测,需要多大样本量。
常用的方法有功效检验和样本量递推法。
对于功效检验法,可以通过计算给定样本量下的样本估计效应大小,并判断是否满足统计功效要求。
对于样本量递推法,可以根据初步样本量估计和实际效应大小来修正样本量,直到满足统计功效要求。
3.基于生存分析的样本量估算方法这种方法适用于生存分析或生存率的研究。
常用的方法有Kaplan-Meier方法和Cox比例风险模型。
临床试验样本量估算

临床试验样本量估算在估算样本量时,有几个关键要素需要考虑:1. 效应大小(Effect Size):效应大小是指在两个比较组之间预期的差异大小。
一般来说,效应大小越大,所需的样本量越小。
2. 置信度(Confidence Level):置信度是指研究者对样本调查结果的信任程度。
常用的置信度为95%或99%。
一般来说,置信度越高,所需的样本量越大。
3. 统计显著性(Statistical Significance):统计显著性是指试验结果的显著性水平。
常用的显著性水平为α=0.05或α=0.01、一般来说,显著性水平越低,所需的样本量越大。
4. 效应方向性(Directionality of Effect):效应方向性是指试验是否需要检测两组间的差异。
若只需检测差异是否存在,则样本量较小;若需检测差异的方向,则样本量较大。
5. 控制变量的数量(Number of Control Variables):增加控制变量的数量会增加结果解释的复杂度,从而需要更大的样本量。
6. 数据的可变性(Variability of Data):数据的可变性与样本量呈反比关系。
如果数据变异性大,所需的样本量就会相对较大。
7. 可行性和资源限制(Feasibility and Resource Constraints):实际操作中,样本量可能受到可行性和资源限制的影响。
研究者需要评估可行性因素,并根据实际情况确定样本量。
基于以上要素,常用的样本量估算方法有以下几种:1.参数估计法:通过统计分析来估计试验样本量。
研究者需要提供试验所需的显著性水平、效应大小以及控制变量的数量等参数。
常用的参数估计方法有t检验、方差分析、卡方检验等。
2. 非参数估计法:当样本不满足正态分布或总体参数未知时,可以采用非参数的方法进行样本量估算。
常用的非参数方法有Wilcoxon秩和检验、Mann-Whitney U检验、logistic回归等。
临床研究中的样本量估算_1_临床试验

临床研究中的样本量估算_1_临床试验临床研究中的样本量估算一、引言在临床研究设计的过程中,样本量估算是非常重要的一部分。
样本量的大小直接影响到研究结果的可信度和推广性,因此合理的样本量估算对于研究的有效性和准确性至关重要。
本文将介绍在临床试验中进行样本量估算的一般原则和方法。
二、样本量估算的原则1.研究目的和假设:在进行样本量估算之前,需要明确研究的目的和科学假设。
例如,研究目的是评价一种新药物的疗效,科学假设是该新药物相对于对照药物在治疗效果上具有显著差异。
2.效果大小和显著水平:样本量估算需要考虑研究的主要效果大小和研究结果的显著性水平。
主要效果大小通常由预期的效应量来衡量,显著性水平通常设定为0.05.3.统计方法和假设检验:样本量估算需要选择适当的统计方法和假设检验方法。
常用的统计方法包括t检验、χ2检验和生存分析等,根据研究设计和数据类型选择合适的方法。
4.统计功效和样本比例:根据效果大小、显著水平和统计方法,可以计算出研究的统计功效。
统计功效通常设定为80%或90%,根据研究的要求合理选择。
三、样本量估算的方法1.固定效应模型:适用于研究目的明确,效果大小可预期的情况。
根据预期的效应大小、显著水平和统计功效,可以计算出所需的最小样本量。
2.结果模拟法:适用于研究目的不明确,效果大小不确定的情况。
通过模拟多个样本量和效果大小的组合,评估不同样本量下的统计功效和效果大小的关系,从而选择合理的样本量范围。
3.临床合理性法:适用于研究目的和效果大小无法准确估计的情况。
根据研究领域的临床经验和专家意见,结合已有的临床试验结果,进行样本量估算。
四、样本量估算的考虑因素1.预计的失访率和样本无效率:由于各种原因,研究中可能存在部分受试者失访或数据无效的情况。
在样本量估算中需要考虑到这些因素的影响,增加适当的样本量。
2.子组分析和交互效应:如果研究中存在多个子组或交互效应的检验,需要进行适当的样本量调整,以保证对这些效应进行准确的评估。
临床试验样本量的估算

临床试验样本量的估算临床试验的样本量估算是研究计划中非常重要的一个环节。
样本量的大小会直接影响到试验结果的可靠性和推广性。
本文将介绍一些常用的方法和考虑因素,来进行临床试验样本量的估算。
一、统计学方法1.样本量估算的原则样本量估算的基本原则是保证试验结果的统计学意义和实际应用的可行性,同时控制样本量的大小。
在样本量估算时需要考虑的主要因素包括:研究目的、效应大小、α水平、β水平、检验类型和预估结果的方差。
2.效应大小效应大小(Effect Size)指的是一种观察、试验或实验中的两组之间的差异,并且是研究中最重要的指标之一、效应大小的选择需要基于研究目标和研究领域的实际情况。
常用的效应大小指标包括:风险比、比值比、均值差异等。
3.α水平和β水平α水平和β水平是两种错误假设的概率。
α水平(Type I错误)是拒绝了一个真假设。
通常是将p值设置在0.05以下。
β水平(TypeII错误)是接受了一个错误的假设。
常见的β值是0.2、0.1、0.05和0.01、α和β的选择需要根据实际情况和研究目的进行权衡。
4.检验类型根据研究目的和数据类型的不同,可以选择不同的检验类型。
常见的检验类型包括:t检验、方差分析、卡方检验等。
不同的检验类型需要不同的样本量估算方法。
5.预估结果的方差预估结果的方差是样本量估算的另一个重要因素。
方差的预估可以通过先前的研究结果或者基于临床经验来估算。
二、样本量估算方法1.均值差异的样本量估算方法均值差异的样本量估算方法适用于需要比较两个或多个组之间平均值差异的研究。
常用的方法有:Z检验样本量估算、t检验样本量估算和方差分析样本量估算。
2.分类变量的样本量估算方法分类变量的样本量估算方法适用于比较不同组之间的比率、风险比、比值比等。
常用的方法有:卡方检验样本量估算和Fishers精确检验样本量估算等。
3.生存分析的样本量估算方法生存分析的样本量估算方法适用于评估治疗或干预措施对患者生存时间或复发时间的影响。
临床试验样本量的估算

临床试验样本量得估算样本量得估计涉及诸多参数得确定,最难得到得就就是预期得或者已知得效应大小(计数资料得率差、计量资料得均数差值),方差(计量资料)或合并得率(计数资料各组得合并率),一般需通过预试验或者查阅历史资料与文献获得,不过很多时候很难得到或者可靠性较差。
因此样本量估计有些时候不就是想做就能做得。
SFDA得规定主要就是从安全性得角度出发,保证能发现多少得不良反应率;统计得计算主要就是从power出发,保证有多少把握能做出显著来。
但就是中国得国情?有多少厂家愿意多做?建议方案里这么写:从安全性角度出发,按照SFDA××规定,完成100对有效病例,再考虑到脱落原因,再扩大20%,即120对,240例。
或者:本研究为随机双盲、安慰剂平行对照试验,只有显示试验药优于安慰剂时才可认为试验药有效,根据预试验结果,试验组与对照组得有效率分别为65、0%与42、9%,则每个治疗组中能接受评价得病人样本数必须达到114例(总共228例),这样才能在单侧显著性水平为5%、检验功效为90%得情况下证明试验组疗效优于对照组。
假设因调整意向性治疗人群而丢失病例达10%,则需要纳入病人得总样本例数为250例。
非劣性试验(α=0、05,β=0、2)时:计数资料:平均有效率(P) 等效标准(δ)N=公式:N=12、365×P(1-P)/δ2计量资料:共同标准差(S) 等效标准(δ)N=公式:N=12、365× (S/δ)2等效性试验(α=0、05,β=0、2)时:计数资料:平均有效率(P) 等效标准(δ)N=公式:N=17、127×P(1-P)/δ2计量资料:共同标准差(S) 等效标准(δ)N=公式:N=17、127× (S/δ)2上述公式得说明:1) 该公式源于郑青山教授发表得文献。
2) N 就是每组得估算例数N1=N2,N1 与N2 分别为试验药与参比药得例数;3) P 就是平均有效率,4) S 就是估计得共同标准差,5) δ 就是等效标准。
临床试验样本量的估算精编版

临床试验样本量的估算精编版临床试验样本量估算是进行临床试验设计过程中极为重要的一环,其目的是通过合理的样本量确定试验的统计效力,确保试验结果的可靠性和可解释性。
样本量的大小直接关系到试验的结果,样本量过小容易导致试验结果的误差增大,而样本量过大则可能造成资源的浪费。
在进行临床试验样本量估算时,需要考虑以下几个因素:1.效应大小:效应大小指的是新型治疗与传统治疗之间的差异效应,通常通过之前的研究结果或临床经验作为参考。
效应大小越大,样本量可以相应减少。
2.类型I错误的控制:类型I错误也常被称为显著性水平或α错误。
它指的是在原假设为真的情况下,拒绝原假设的概率。
通常使用显著性水平α来控制类型I错误的概率,常见的α取值为0.053.类型II错误的控制:类型II错误也被称为β错误。
它指的是在备择假设为真的情况下,接受原假设的概率。
通常使用统计功效1-β来控制类型II错误的概率,常见的统计功效取值为0.8或0.94.变异性:变异性是指被试者在一些测量指标上的差异程度。
变异性越大,样本量需求相应增加。
5.置信区间:置信区间是指参数真值落在特定区间的概率。
通常使用95%的置信水平,即在95%的概率下,参数真值落在置信区间内。
1.样本量计算公式:根据试验的目标和研究假设,选择合适的样本量计算公式进行计算。
常用的样本量计算公式包括均数比较、比例比较、生存分析、相关性等。
2. 统计软件:利用统计软件进行样本量模拟和估算。
常用的统计软件包括PASS、G*Power和R软件等。
3.文献参考:根据类似的研究或文献中的样本量估算结果作为参考,结合本次研究的具体情况进行调整。
需要注意的是,样本量的估算是基于试验目标和预设的假设进行计算的,实际研究过程中可能会受到很多实际因素的影响,如实验设计的可行性、时间和资源限制等。
因此,在进行样本量估算时需要充分考虑这些实际因素,尽可能保证样本量的合理性和可行性。
总之,临床试验样本量估算是临床试验设计过程中重要的一环,合理的样本量估算可以确保试验结果的可靠性和可解释性。
临床试验样本量的估算

临床试验样本量的估算样本量的估计涉及诸多参数的确定,最难得到的就是预期的或者已知的效应大小(计数资料的率差、计量资料的均数差值),方差(计量资料)或合并的率(计数资料各组的合并率),一般需通过预试验或者查阅历史资料和文献获得,不过很多时候很难得到或者可靠性较差。
因此样本量估计有些时候不是想做就能做的。
SFDA的规定主要是从安全性的角度出发,保证能发现多少的不良反应率;统计的计算主要是从power出发,保证有多少把握能做出显著来。
但是中国的国情?有多少厂家愿意多做?建议方案里这么写:从安全性角度出发,按照SFDA××规定,完成100对有效病例,再考虑到脱落原因,再扩大20%,即120对,240例。
或者:本研究为随机双盲、安慰剂平行对照试验,只有显示试验药优于安慰剂时才可认为试验药有效,根据预试验结果,试验组和对照组的有效率分别为65.0%和42.9%,则每个治疗组中能接受评价的病人样本数必须达到114例(总共228例),这样才能在单侧显著性水平为5%、检验功效为90%的情况下证明试验组疗效优于对照组。
假设因调整意向性治疗人群而丢失病例达10%,则需要纳入病人的总样本例数为250例。
非劣性试验(α=0.05,β=0.2)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=12.365×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=12.365× (S/δ)2等效性试验(α=0.05,β=0.2)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=17.127×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=17.127× (S/δ)2上述公式的说明:1) 该公式源于郑青山教授发表的文献。
2) N 是每组的估算例数N1=N2,N1 和N2 分别为试验药和参比药的例数;3) P 是平均有效率,4) S 是估计的共同标准差,5) δ 是等效标准。
临床试验样本量估算

样本量估算(二):随机对照试验(两组均数)比较的样本量计算方法2020-07-16 18:54“样本量估算周一见”系列每周一呈现,敬请关注,本周展示的是医学研究最常见的两组均数比较样本量比较方法。
一、研究实例随机对照试验研究:探讨中西医结合治疗治疗女性膀胱过度活动症。
采用完全随机的方法将研究对象分为两组(中西医结合组和西医组),结局指标为排尿症状的评分预计西医对照组排尿症状评分的平均值为7.08±1.36分,中西医结合治疗组使用药物后预计降低1.2分,二者方差相似。
双侧检验,α为0.05,两组样本量比值1:1(即两组病例数相等),把握度(检验效能)1-β=90%,求需要多少样本量?二、样本量估算方法•案例解析:本案例比较的是某药物A治疗女性膀胱过度活动症,其结局指标为排尿症状评分,为定量数据,定量结局往往探讨的是2组或多组均数有无统计学差异。
本例为2组均数的比较。
•计算公式•n代表每组样本量。
•Zα和Zβ需要查表。
一般α为0.05,且Z值为双侧,则Z0.05=1.96;β为单侧,把握度(检验效能)为0.9时,Zβ=1.28,把握度(检验效能)为0.8时,Zβ=0.84,一般把握度0.9较多见,但需要更多样本量。
本例中Zα和Zβ分别等于1.96和1.28。
•σ代表标准差,本例中σ=1.36。
•δ代表差值,即治疗组与对照组平均值的差值,本例中δ=1.2。
三、直接利用公式计算样本量四、PASS操作计算样本量1. 打开PASS 15软件后,在左侧菜单栏中找到Means---TwoIndependence Means---T-Test(Inequality)---Two-Sample T-Tests Asuming EqualVariance (方差齐)、Two-Sample T-Tests AllowingUnequal Variance(方差不齐)。
2.这个研究中,把握度为90%,即Power=0.90;α为0.05,即Alpha=0.05;两组样本量比值1:1,即Group Allocation为Equal(N1=N2);μ1=5.88;μ2=7.08;标准差σ=1.36;其他为默认,点击Calculate。
临床试验样本量的估算
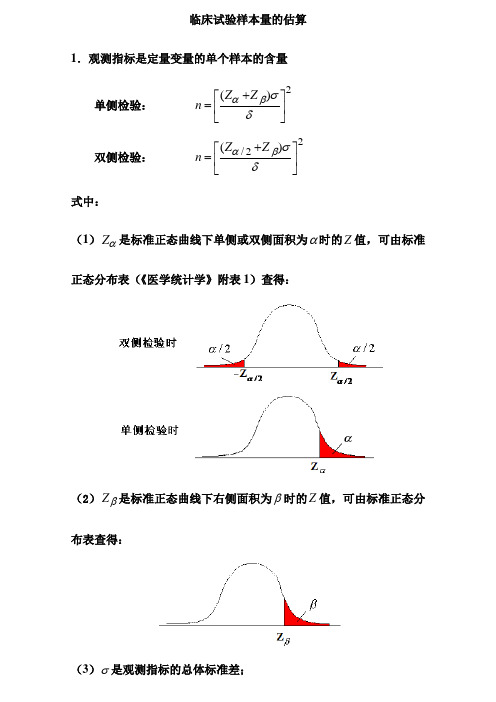
临床试验样本量的估算1.观测指标是定量变量的单个样本的含量单侧检验:2)(⎥⎦⎤⎢⎣⎡+=δσβαZZn双侧检验:22/)(⎥⎦⎤⎢⎣⎡+=δσβαZZn式中:(1)αZ是标准正态曲线下单侧或双侧面积为α时的Z值,可由标准正态分布表(《医学统计学》附表1)查得:(2)βZ是标准正态曲线下右侧面积为β时的Z值,可由标准正态分布表查得:(3)σ是观测指标的总体标准差;(4)δ是容许误差;2.观测指标是定量变量的实验组与对照组的样本含量双侧检验: 22/)(4⎥⎦⎤⎢⎣⎡+⨯=δσβαZ Z n 单侧检验: 2)(4⎥⎦⎤⎢⎣⎡+⨯=δσβαZ Z n 这是两个样本合计的例数,一般,可取21n n =,22n n =。
上式中, (1)αZ 是标准正态曲线下单侧或双侧面积为α时的Z 值,可由附表1查得;(2)βZ 是标准正态曲线下右侧面积为β时的Z 值,可由附表1查得 (3)σ是两个样本所取自的总体标准差的合并值:221σσσ+=;在实际研究时,σ一般是未知的,可以通过预试验或文献确定出1S 与2S ,用221S S S +=估计σ;(4)容许误差||21μμδ-=。
3.观测结果是二分类的单个样本的含量A 方法的阳性率:nb a +=1π B 方法的阳性率:n ca +=2π两种方法结果一致的例数:d a +,其中,阳性例数:a , 两种方法结果一致的阳性率:da a+=π 上述数据在实际中均是未知的,要从小规模的预试验的结果来估计。
样本量的估算公式:221212/)())((22⎥⎥⎥⎥⎦⎤⎢⎢⎢⎢⎣⎡---+=ππππππππβαZ Z n式中,2221ππππ-+=4.观测结果是二分类的实验组与对照组的样本含量双侧检验:22122112/))1()1((2)1(2⎥⎥⎦⎤⎢⎢⎣⎡--+-+-=ππππππππβαZ Z n c c单侧检验: 2212211))1()1((2)1(2⎥⎥⎦⎤⎢⎢⎣⎡--+-+-=ππππππππβαZ Z n c c式中, c π=221ππ+,1π是实验组的阳性率,2π是对照组的阳性率。
临床试验样本量的估算

临床试验样本量的估算样本量的估计涉及诸多参数的确定,最难得到的就是预期的或者已知的效应大小(计数资料的率差、计量资料的均数差值),方差(计量资料)或合并的率(计数资料各组的合并率),一般需通过预试验或者查阅历史资料和文献获得,不过很多时候很难得到或者可靠性较差。
因此样本量估计有些时候不是想做就能做的。
SFDA的规定主要是从安全性的角度出发,保证能发现多少的不良反应率;统计的计算主要是从power出发,保证有多少把握能做出显著来。
但是中国的国情?有多少厂家愿意多做?建议方案里这么写:从安全性角度出发,按照SFDAxx规定,完成100对有效病例,再考虑到脱落原因,再扩大20%,即120对,240例。
或者:本研究为随机双盲、安慰剂平行对照试验,只有显示试验药优于安慰剂时才可认为试验药有效,根据预试验结果,试验组和对照组的有效率分别为65.0%和42.9%,则每个治疗组中能接受评价的病人样本数必须达到114例(总共228例),这样才能在单侧显著性水平为5%、检验功效为90%的情况下证明试验组疗效优于对照组。
假设因调整意向性治疗人群而丢失病例达10%,则需要纳入病人的总样本例数为250例。
非劣性试验(a=0・05,3=0.2)时:计数资料:平均有效率(P)等效标准(&)N=公式:N=12・365xP(l-P)/&2计量资料:共同标准差(S)等效标准(&)N=公式:N=12・365x(S/6)2等效性试验(a=0.05,3=0.2)时:计数资料:平均有效率(P)等效标准(&)N=公式:N=17・127xP(l-P)/&2计量资料:共同标准差(S)等效标准(&)N=公式:N=17・127x(S/6)2上述公式的说明:1)该公式源于郑青山教授发表的文献。
2)N是每组的估算例数N1=N2,N1和N2分别为试验药和参比药的例数;3)P是平均有效率,4)S是估计的共同标准差,5)6是等效标准。
临床试验样本量的估算

临床试验样本量的估算首先,研究目的是样本量估算的基础,研究目的不同所需样本量也会不同。
例如,对于描述性研究,样本量的估算主要考虑数据的可行性,一般在50-100人左右即可;而对于验证性研究,需要进行统计检验或建立预测模型,则需要更多的样本量。
其次,研究设计也是样本量估算的重要因素。
常见的研究设计包括前瞻性队列研究、病例对照研究、随机对照试验等。
不同的研究设计对样本量的要求也不同。
例如,队列研究需要在较长的时间内追踪大量的受试者,样本量通常较大;而对照试验往往需要比较两组之间差异的显著性,样本量要求相对较少。
其次,统计学假设是影响样本量估算的主要因素之一、其中包括显著性水平(通常为0.05)、统计效应大小和统计检验的类型等。
较高的显著性水平(如0.01)和较小的统计效应大小都会增加样本量的要求。
此外,不同的统计检验方法也会影响样本量估算。
例如,判断两组均值是否有差异的双样本t检验需要较大的样本量,而判断两组比例是否有差异的卡方检验则需要较小的样本量。
最后,样本量的估算还需要考虑目标参数的合理估计。
目标参数包括所研究变量的均值、比例、相关系数等。
一般来说,样本量估计需要选取适当的目标参数,并根据目标参数的合理范围进行估计。
根据经验和实际情况,可以采用样本量估算公式来计算样本量。
n=(Zα/2*σ/δ)^2其中,n为所需样本量,Zα/2为给定显著性水平下的Z值,σ为总体标准差的估计值,δ为目标参数的边际误差。
综上所述,临床试验样本量的估算需要考虑研究目的、研究设计、统计学假设和目标参数等因素,并采用适当的样本量估算公式进行计算。
合理估算样本量可以确保研究结果的可靠性和统计分析的效力。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
临床试验样本量的估算 LG GROUP system office room 【LGA16H-LGYY-LGUA8Q8-LGA162】临床试验样本量的估算样本量的估计涉及诸多参数的确定,最难得到的就是预期的或者已知的效应大小(计数资料的率差、计量资料的均数差值),方差(计量资料)或合并的率(计数资料各组的合并率),一般需通过预试验或者查阅历史资料和文献获得,不过很多时候很难得到或者可靠性较差。
因此样本量估计有些时候不是想做就能做的。
SFDA的规定主要是从安全性的角度出发,保证能发现多少的不良反应率;统计的计算主要是从power出发,保证有多少把握能做出显着来。
但是中国的国情有多少厂家愿意多做建议方案里这么写:从安全性角度出发,按照SFDA××规定,完成100对有效病例,再考虑到脱落原因,再扩大20%,即120对,240例。
或者:本研究为随机双盲、安慰剂平行对照试验,只有显示试验药优于安慰剂时才可认为试验药有效,根据预试验结果,试验组和对照组的有效率分别为%和%,则每个治疗组中能接受评价的病人样本数必须达到114例(总共228例),这样才能在单侧显着性水平为5%、检验功效为90%的情况下证明试验组疗效优于对照组。
假设因调整意向性治疗人群而丢失病例达10%,则需要纳入病人的总样本例数为250例。
非劣性试验(α=,β=)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=× (S/δ)2等效性试验(α=,β=)时:计数资料:平均有效率(P)等效标准(δ)N=公式:N=×P(1-P)/δ2计量资料:共同标准差(S)等效标准(δ)N=公式:N=× (S/δ)2上述公式的说明:1) 该公式源于郑青山教授发表的文献。
2) N 是每组的估算例数N1=N2,N1 和N2 分别为试验药和参比药的例数;3) P 是平均有效率,4) S 是估计的共同标准差,5) δ是等效标准。
6) 通常都规定α=,β=(把握度80%)上述计算的例数若少于国家规定的例数,按规定为准;多于国家规定的则以计算值为准。
具体规定的最小样本量如下:II期,试验组100例;III 期,试验组300例;随机对照临床验证(如3类化药)试验组100例。
IV 期,2000例。
疫苗和避孕药与上述要求不同。
例1:某新药拟进行II 期临床试验,与阳性药按1:1 的比例安排例数,考察新药临床治愈率不差于阳性药。
根据以往的疗效和统计学的一般要求,取α=,β=,等效标准δ=,平均有效率P=,每组需要多少病例?由公式计算得,N=×/=88(例)以上88 例低于我国最低例数(100 例)的规定,故新药至少取100 例进行试验。
如上例作等效性分析,则得,N=×/=122(例)。
例2:某利尿新药拟进行II 期临床试验,与阳性药按1:1 的比例安排例数,考察24h 新药利尿量不差于阳性药。
根据以往的疗效和统计学的一般要求,取α=,β=,等效标准δ=60 ml,已知两组共同标准差S=180 ml,每组需要多少病例?由公式得,N=× (180/60)2=111 例。
故本次试验新药和阳性药的例数均不少于111 例。
如上例作等效性分析,则得,N=×(180/60)2=154(例)。
[s:11]临床试验研究中,无论是实验组还是对照组都需要有一定数量的受试对象。
这是因为同一种实验处理在不同的受试对象身上表现出的实验效应是存在着变异的。
仅凭一次实验观测结果或单个受试者所表现出来的实验效应说明不了什么问题。
必须通过一定数量的重复观测才能把研究总体真实的客观规律性显示出来,并且可以对抽样误差做出客观地估计。
一般说来重复观测次数越多,抽样误差越小,观测结果的可信度越高。
一定数量的重复还可起到部分抵消混杂因素影响的作用,增强组间的可比性。
但重复观测次数越多(即样本含量越大)试验所要消耗的人力、物力、财力和时间越多,可能会使试验研究成为不可能。
而且,样本含量过大还会增加控制试验观测条件的难度,有可能引入非随机误差,给观测结果带来偏性(bias)。
所以在实验设计中落实重复原则的一个重要问题就是如何科学合理确定样本含量。
由于在各对比组例数相等时进行统计推断效能最高,因此多数情况下都是按各组样本含量相等来估计。
但在个别情况下,也可能要求各组样本含量按一定比例来估计。
1 与样本含量估计有关的几个统计学参数在估计样本含量之前,首先要对以下几个统计学参数加以确定或作出估计。
规定有专业意义的差值δ,即所比较的两总体参数值相差多大以上才有专业意义。
δ是根据试验目的人为规定的,但必须有一定专业依据。
习惯上把δ称为分辨力或区分度。
δ值越小表示对二个总体参数差别的区分度越强,因而所需样本含量也越大。
确定作统计推断时允许犯Ⅰ类错误(“弃真”的错误)的概率α,即当对比的双方总体参数值没有差到δ。
但根据抽样观测结果错误地得出二者有差别的推断结论的可能性,α确定的越小,所需样本含量越大。
在确定α时还要注意明确是单侧检验的α,还是双侧检验的α。
在同样大小的α条件下;双侧检验要比单侧检验需要更大的样本含量。
提出所期望的检验效能power,用1-β表示。
β为允许犯Ⅱ类错误(“取伪”的错误)的概率。
检验效能就是推断结论不犯Ⅱ类错误的概率1-β称把握度。
即当对比双方总体参数值间差值确实达到δ以上时,根据抽样观测结果在规定的α水准上能正确地作出有差别的推断结论的可能性。
在科研设计中常把1-β定为或。
一般来说1-β不宜低于,否则可能出现非真实的阴性推断结论。
给出总体标准差σ或总体率π的估计值。
它们分别反映计量数据和计数数据的变异程度。
一般是根据前人经验或文献报道作出估计。
如果没有前人经验或文献报道作为依据,可通过预实验取得样本的标准差s或样本率P分别作为σ和π的估计值。
σ的估计值越大,π的估计值越接近,所需样本含量越大。
在对以上统计学参数作出规定或估计的前提下,就可以根据不同的推断内容选用相应的公式计算出所需样本含量。
由于在同样的要求和条件下完全随机设计(成组设计)所需样本含量最大,故一般都要按完全随机设计作出样本含量的估计。
2 常用的估计样本含量的方法两样本均数比较时样本含量估计方法(1)两样本例数要求相等时可按下列公式估算每组需观察的例数n。
n=2*[(α+β)σ/δ]^2 (公式1)式中δ为要求的区分度,σ为总体标准差或其估计值s,α、β分别是对应于α和β的u值,可由t界值表,自由度υ=∞-行查出来,α有单侧、双侧之分,β只取单侧值。
例1,某医师研究一种降低高血脂患者胆固醇药物的临床疗效,以安慰剂作对照。
事前规定试验组与对照组相比,平均多降低 mmol/L以上,才有推广应用价值。
而且由有关文献中查到高血脂患者胆固醇值的标准差为mmol/L,若要求犯Ⅰ类错误的的概率不超过5%,犯Ⅱ类错误的概率不超过10%,且要两组例数相等则每组各需观察多少例?本例δ= mmol/L,σ= mmol/L,α=,β=,1-β=,查t界值表自由度为∞一行得单侧=,=,代入公式(1)n=2*[+×]^2=44故要达到上述要求,两组至少各需观察44例。
(2)两样本例数要求呈一定比例(n2/n1=c)时,可按下列公式求出n1,再按比例求出n2=c*n1。
n1=[(α+β)σ/δ]^2*(1+C)/C (公式2)例2 对例1资料如一切要求都维持不变,但要求试验组与对照组的例数呈2∶1比例(即C=2),问两组各需观察多少例?n1=[+×]^2×(1+2)/2 =33(例)(对照组所需例数)n2=2×33=66(例)(试验组所需例数。
)两组共需观察99例多于两组例数相等时达到同样要求时两组所需观察的总例数2×44=88。
配对设计计量资料样本含量(对子数)估计方法配对设计包括异体配对、自身配对、自身前后配对及交叉设计的自身对照,均可按下列公式进行样本含量估计。
n=[(α+β)σd/δ]^2 (公式3)式中δ、α、β的含义同前,σd为每对差值的总体标准差或其估计值sd。
例3 某医院采用自身前后配对设计方案研究某治疗矽肺药物能否有效地增加矽肺患者的尿矽排出量。
事前规定服药后尿矽排出量平均增加mmol/L以上方能认为有效,根据预试验得到矽肺患者服药后尿矽排出量增加值的标准差 sd= mmol/L,现在要求推断时犯Ⅰ类错误的概率控制在以下(单侧),犯Ⅱ类错误的概率控制在以下,问需观察多少例矽肺病人?本例δ= mmol/L, sd= mmol/L,α=,β=。
1-β=,单侧=,=,代入公式(3)得到。
n=[+×89/]^2=54(例)故可认为如该药确实能达到平均增加尿矽排出量在 mmol/L以上,则只需观察54例病人就能有90%的把握,按照α=的检验水准得出该药有增加矽肺病人尿矽作用的正确结论。
样本均数与总体均数比较时样本含量估计方法可按下式估算所需样本含量n。
n=[(α+β)σ/δ]^2 (公式4)例4已知血吸虫病人血红蛋白平均含量为90g/L,标准差为25g/L,现欲观察呋喃丙胺治疗后能否使血红蛋白增加,事先规定血红蛋白增加10g/L 以上才能认为有效,推断结论犯Ⅰ类错误的概率α(双侧)不得超过,犯Ⅱ类错误的概率β不得超过,问需观察多少例病人?本例δ=10g/L,σ=25g/L,=(双侧),=代入公式(4)得:n=[+×25/10]^2=66(例)故如果呋喃丙胺确实能使血吸虫病人血红蛋白平均含量增加10g/L以上,则只需观察66例就可以有90%的把握在α=检验水准上得出有增加血吸虫病人血红蛋白平均含量的结论。