白凡士林检验标准操作规程
S HT0115凡士林检测方法

S HT0115凡士林检测方法拉丝性能拉丝性能其实并不是科学的名称,它主要是指用食指与拇指捏等量凡士林进行挤压、拉伸,拉出的丝越长、越细腻光亮,同时没有熔化感、不卷曲,则该凡士林的“拉丝性”越好。
但是拉丝性能的好坏在消费者和采购商眼里却是衡量一款凡士林好坏的最直观的检测指标。
影响拉丝性能的检测指标为脱壳性能、锥入度、运动粘度检测指标。
光稳定性凡士林光稳定性试验也就是紫外线吸收度的测定实验。
具体测试方法就是把各凡士林试样放在同一时间内进行紫外灯照射,然后对比色泽变化大小,色泽变化越大,越不稳定。
光稳定的测定主要是考虑凡士林保存问题。
包括可见光及紫外光都会加速凡士林的氧化反应,从而影响色泽及其质量。
脱壳性能及龟裂性能测定脱壳性能及龟裂性能测定方法主要是将凡士林样品加温熔化,同时把一个100mL量杯在100摄氏度烘箱中预热,将熔化好的凡士林倒入量杯5cm高度处,然后把量杯用棉衣包裹,在室温下缓慢冷却4小时以上,之后再将样品放在±1℃温度的冰箱里仿制6小时以上。
取出,观察凡士林表面及四周。
四周与量杯壁未出现脱离,则定性为“未脱”;周边50%以下脱离,则定性为“轻脱”;周边50%以上脱离,则定性为“脱壳”。
表面出现裂缝,则定性为“龟裂”;表面未出现裂缝,则定性为“未裂”。
异味及有毒有害物质异味的产生是因为凡士林在加工过程中会产生裂解物,而这些裂解物中可能还有硫醇、硫醚等物质。
这些物质挥发出来就会发出臭味,煤油味。
硫化物的测定可以帮我们很好的控制异味的产生。
凡士林中有毒有害物质主要是总金属,这里主要推荐检测铅含量及砷含量这两个指标。
所以,综上所述,凡士林检测一定要抓住重点,如果是企业研发、产品配方研究,工艺改进。
可以专注在某些性能的测定。
对影响这些性能的指标的来检测,从而得以掌控和改进。
企业采购,质检部分监督检测,则应该按照检测标准的所以规定项目进行全检。
拜恩检测可根据凡士林检测的各种需求,提供专业的第三方检测服务。
白凡士林检验操作规程

白凡士林检验操作规程1 目的:建立白凡士林检验操作规程。
2 适用范围:适用于白凡士林的检验操作。
3 职责:检验人员对本规程的实施负责。
4 规程:4.1 编制依据:《中国药典》2010年版二部P1192。
4.2 质量指标:见《白凡士林质量标准》。
4.3 仪器与用具:分光光度计、金属制圆筒状容器、水浴锅、烘箱。
4.4 试药与试液:重铬酸钾液、比色用硫酸铜液、三甲基戌烷、酚酞指示剂、甲基橙指示剂。
4.5 操作方法4.5.1 性状:本品为白色至微黄色均匀的软膏状物,无臭或几乎无臭,与皮肤接触有滑腻感,具有一定的拉丝性,本品在约35℃的苯中易溶,在约35℃的氯仿中溶解,在乙醚中微溶,在乙醇中或水中几乎不溶。
熔点:取本品,按《熔点检验操作规程》第三法检验,为45-60℃。
4.5.2 检查4.5.2.1 颜色:取本品10.0g,置烧杯中,在水浴上加热使熔融,移入比色管中,与同体积的对照液(取比色用重铬酸钾液7.8ml与比色用硫酸铜液0.2ml,混匀,取2.5ml,加水至25ml)比较,颜色不得更深。
4.5.2.2 杂质吸光度:取本品,加三甲基戊烷制成每1ml 中含0.50mg的溶液,照《紫外-可见分光光度法检验操作规程》检验,在290nm波长处的吸光度不得过0.50。
4.5.2.3 锥入度:取本品适量,在85℃±2℃熔融,倾入直径为100mm±5mm、高度不小于65mm的金属制圆筒状容器中,装至离容器上端6mm以内,于25℃±2℃放置16-18小时,并在试验前将容器移至25.0℃±0.5℃水浴中放置2小时;另将标准锥体(总重102.50g±0.05g,由表面光滑的圆锥体和可拆卸的锥尖以适当方式连接组成:锥尖部分呈30°角,尖端截面直径为0.38mm±0.02mm,基底直径为8.40mm±0.02mm,高15.00mm±0.05mm;圆锥体部分的夹角为90°0′±15 ′,高32.1mm±0.2mm,底部最大直径为69.3mm±0.1mm)置水浴中调节到25.0℃±0.5℃。
医药白凡士林执行标准

医药白凡士林执行标准项目执行标准(BP-98)
外观白色或淡黄色均匀的软膏状物,无臭无味,与皮肤接触有滑腻感,具有一定的拉丝性
滴熔点℃42—60
锥入度(0.1mm)130—210
稠环芳烃(265-420nm) 不大于 1.08
紫外线吸光度(290nm) 不大于0.5
硫酸盐灰分不大于0.1%
酸碱度BP-98
●已通过国家石蜡检测中心检测,本品为白色软膏状,有一定拉丝性,无臭,
有良好的化学稳定性及滑腻感,适用于作配制医药药膏及皮肤保护油膏的辅料,精密仪器和医疗器械等高级制品的防腐,以及高档化妆品及其他日用品的配料。
●质量标准:符合中国药典2000版要求和英国药典BP-98标准。
医药级白凡士林。
英国药典 白凡士林

DEFINITIONPurified and wholly or nearly decolorised mixture of semi-solid hydrocarbons, obtained from petroleum. It may contain a suitable antioxidant. White soft paraffin described in this monograph is not suitable for oral use.CHARACTERSAppearanceWhite or almost white, translucent, soft unctuous mass, slightly fluorescent in daylight when melted.SolubilityPractically insoluble in water, soluble in methylene chloride, practically insoluble in alcohol and in glycerol.IDENTIFICATIONFirst identification A, B, D.Second identification A, C, D.A. The drop point is between 35 °C and 70 °C and does not differ by more than 5 °C from the value stated on the label, according to method (2.2.17) with the following modification to fill the cup: heat the substance to be examined at a temperature not exceeding 80 °C, with stirring to ensure uniformity. Warm the metal cup at a temperature not exceeding 80 °C in an oven, remove it from the oven, place on a clean plate or ceramic tile and pour a sufficient quantity of the melted sample into the cup to fill it completely. Allow the filled cup to cool for 30 min on the plate or the ceramic tile and place it in a water bath at 24-26 °C for 30-40 min. Level the surface of the sample with a single stroke of a knife or razor blade, avoiding compression of the sample.B. Infrared absorption spectrophotometry (2.2.24).Comparison Ph. Eur. reference spectrum of white soft paraffin.C. Melt 2 g and when a homogeneous phase is obtained, add 2 ml of water R and 0.2 ml of0.05 M iodine. Shake. Allow to cool. The solid upper layer is violet-pink.D. It complies with the test for appearance (see Tests).TESTSAppearanceThe substance is white. Melt 12 g on a water-bath. The melted mass is not more intensely coloured than a mixture of 1 volume of yellow primary solution and 9 volumes of a 1 per cent m/V solution of hydrochloric acid R(2.2.2, Method II).Acidity or alkalinityTo 10 g add 20 ml of boiling water R and shake vigorously for 1 min. Allow to cool and decant. To 10 ml of the aqueous layer add 0.1 ml of phenolphthalein solution R. The solution is colourless. Not more than 0.5 ml of 0.01 M sodium hydroxide is required to change the colour of the indicator to red.Consistency (2.9.9)60 to 300.Polycyclic aromatic hydrocarbonsMaximum 300 ppm.Use reagents for ultraviolet spectrophotometry. Dissolve 1.0 g in 50 ml of hexane R which has been previously shaken twice with 10 ml of dimethyl sulphoxide R. Transfer the solution to a 125 ml separating funnel with unlubricated ground-glass parts (stopper, stopcock). Add 20 ml of dimethyl sulphoxide R. Shake vigorously for 1 min and allow to stand until 2 clear layers are formed. Transfer the lower layer to a second separating funnel. Repeat the extraction with a further 20 ml of dimethyl sulphoxide R. Shake vigorously the combined lower layers with 20 ml of hexane R for 1 min. Allow to stand until 2 clear layers are formed. Separate the lower layer and dilute to 50.0 ml with dimethyl sulphoxide R. Measure the absorbance (2.2.25) over the range 260 nm to 420 nm using a path length of 4 cm and as compensation liquid the clear lower layer obtained by vigorously shaking 10 ml of dimethyl sulphoxide R with 25 ml of hexane R for 1 min. Prepare a reference solution in dimethyl sulphoxide R containing 6.0 mg of naphthalene R per litre and measure the absorbance of the solution at the maximum at 278 nm using a path length of 4 cm and dimethyl sulphoxide R as compensation liquid. At no wavelength in the range 260 nm to 420 nm does the absorbance of the test solution exceed that of the reference solution at 278 nm.Sulphated ash (2.4.14)Maximum 0.05 per cent, determined on 2.0 g.STORAGEProtected from light.LABELLINGThe label states:—the nominal drop point,—where applicable, the name and concentrationDroping pointethod I(Ph. Eur. method 2.2.14)The melting point determined by the capillary method is the temperature at which the last solid particle of a compact column of a substance in a tube passes into the liquid phase.When prescribed in the monograph, the same apparatus and method are used for the determination of other factors, such as meniscus formation or melting range, that characterise the melting behaviour of a substance.Apparatus The apparatus consists of:—a suitable glass vessel containing a liquid bath (for example, water, liquid paraffin or silicone oil) and fitted with a suitable means of heating,—a suitable means of stirring, ensuring uniformity of temperature within the bath,—a suitable thermometer with graduation at not more than 0.5 °C intervals and provided with an immersion mark. The range of the thermometer is not more than 100 °C,—alkali-free hard-glass capillary tubes of internal diameter 0.9 mm to 1.1 mm with a wall 0.10 mm to 0.15 mm thick and sealed at one end.Method Unless otherwise prescribed, dry the finely powdered substance in vacuo and over anhydrous silica gel R for 24 h. Introduce a sufficient quantity into a capillary tube to give a compact column 4 mm to 6 mm in height. Raise the temperature of the bath to about10 °C below the presumed melting point and then adjust the rate of heating to about 1 °C/min. When the temperature is 5 °C below the presumed melting point, correctly introduce the capillary tube into the instrument. For the apparatus described above, immerse the capillary tube so that the closed end is near the centre of the bulb of the thermometer, the immersion mark of which is at the level of the surface of the liquid. Record the temperature at which the last particle passes into the liquid phase.Calibration of the apparatus The apparatus may be calibrated using melting point reference substances such as those of the World Health Organisation or other appropriate substances.Method II(No Ph. Eur. equivalent method)Apparatus(a) A glass heating vessel of suitable construction and capacity containing one of the following, or another suitable liquid, to a height of not less than 14 cm.(i) A liquid paraffin of sufficiently high boiling point.(ii) A silicone fluid of sufficiently high boiling point.(iii) Water.(b) A suitable stirring device capable of rapidly mixing the liquid.(c) An accurately standardised thermometer suitable for the substance being examined complying with the requirements of British Standard 1365:1990 (Specification for short-range short-stem thermometers) for thermometers designated by one of the following Schedule Marks.(d) Thin-walled capillary glass tubes of hard glass, closed at one end, with a wall thickness of 0.10 to 0.15 mm, at least 12 cm in length and of internal diameter 0.9 to 1.1 mm. The tubes should preferably be kept sealed at both ends and cut as required.Method Dry a small quantity of the finely powdered substance at a temperature considerably below its melting point or at a pressure of 2 kPa over a suitable desiccant, unless otherwise directed. Transfer a portion to a dry capillary tube and pack the powder by tapping on a hard surface so as to form a tightly packed column 4 to 6 mm in height. Heat a suitable liquid in the heating vessel and regulate the rate of rise of temperature, prior to the introduction of the capillary tube, to 3° per minute, unless otherwise directed, stirring constantly. When the temperature reaches 10°below the lowest figure of the range for the substance being tested, adjust the height of the thermometer so that the immersion mark is at the level of the surface of the liquid and insert the capillary tube so that the closed end is near the middle of the bulb of the thermometer. Note the temperature at which the liquefaction of the substance occurs, which is indicated by the formation of a definite meniscus or, for substances that decompose, the temperature at which frothing begins. Correct the observed temperature for any error in the calibration of the thermometer and for the difference, if any, between the temperature of the emergent stem of the thermometer and the temperature of the emergent stem under the conditions of standardisation of the thermometer. The temperature of the emergent stem is determined by placing the bulb of asecond thermometer in contact with the emergent stem at a point approximately midway along the mercury thread in the emergent stem.The correction to be applied is given by the following equation:t c = 0.00016n(t s– t d)wheret c=correction to be added to the observed temperature of the melting point,t s=mean temperature of the emergent column when standardised,t d=mean temperature of the emergent column at the observed melting point,n=number of °C over which the exposed column extends.The corrected temperature is regarded as the melting point of the substance. When the melting point in the monograph is expressed as a range, the melting point of the substance being tested must fall within that range.Method III(Ph. Eur. method 2.2.17)The drop point is the temperature at which the first drop of the melting substance to be examined falls from a cup under defined conditions.Apparatus The apparatus (see Figure 2.2.17.-1) consists of 2 metal sheaths (A) and (B) screwed together. Sheath (A) is fixed to a mercury thermometer. A metal cup (F) is loosely fixed to the lower part of sheath (B) by means of 2 tightening bands (E). Fixed supports (D) 2 mm long determine the exact position of the cup in addition to which they are used to centre the thermometer. A hole (C) pierced in the wall of sheath (B) is used to balance the pressure. The draining surface of the cup must be flat and the edges of the outflow orifice must be at right angles to it. The lower part of the mercury thermometer has the form and size shown in the Figure; it covers a range from 0 °C to 110 °C and on its scale a distance of 1 mm represents a difference of 1 °C. The mercury reservoir of the thermometer has a diameter of 3.5 ±0.2 mm and a height of 6.0 ± 0.3 mm. The apparatus is placed in the axis of a tube about 200 mm long and with an external diameter of about 40 mm. It is fixed to the test-tube by means of a stopper through which the thermometer passes, and is provided with a side groove. The opening of the cup is placed about 15 mm from the bottom of the test-tube. The whole device is immersed in a beaker with a capacity of about 1 litre, filled with water. The bottom of the test-tube is placed about 25 mm from the bottom of the beaker. The water level reaches the upper part of sheath (A). A stirrer is used to ensure that the temperature of the water remains uniform.Method Fill the cup to the brim with the substance to be examined, without melting it, unless otherwise prescribed. Remove the excess substance at the 2 ends of the cup with a spatula. When sheaths (A) and (B) have been assembled press the cup into its housing insheath (B) until it touches the supports. Remove with a spatula the substance pushed out by the thermometer. Place the apparatus in the water-bath as described above. Heat thewater-bath and when the temperature is at about 10 °C below the presumed drop point, adjust the heating rate to about 1 °C/min. Note the temperature at the fall of the first drop. Carry out at least 3 determinations, each time with a fresh sample of the substance. The difference between the readings must not exceed 3 °C. The mean of three readings is the drop point of the substance.Method IV(Ph. Eur. method 2.2.15)For certain substances, the following method is used to determine the melting point (also referred to as slip point and rising melting point when determined by this method).Use glass capillary tubes open at both ends, about 80 mm long, having an external diameter of 1.4 mm to 1.5 mm and an internal diameter of 1.0 mm to 1.2 mm.Introduce into each of 5 capillary tubes a sufficient amount of the substance, previously treated as described, to form in each tube a column about 10 mm high and allow the tubes to stand for the appropriate time and at the prescribed temperature.Unless otherwise prescribed, substances with a waxy consistency are carefully and completely melted on a water-bath before introduction into the capillary tubes. Allow the tubes to stand at 2-8 °C for 2 h.Attach one of the tubes to a thermometer graduated in 0.5 °C so that the substance is close to the bulb of the thermometer. Introduce the thermometer with the attached tube into a beaker so that the distance between the bottom of the beaker and the lower part of the bulb of the thermometer is 1 cm. Fill the beaker with water to a depth of 5 cm. Increase the temperature of the water gradually at a rate of 1 °C/min.The temperature at which the substance begins to rise in the capillary tube is regarded as the melting point.Repeat the operation with the other 4 capillary tubes and calculate the result as the mean of the 5 readings.Method V(Ph. Eur. method 2.2.16)The instantaneous melting point is calculated using the expression:in which t1 is the first temperature and t2 the second temperature read under the conditions stated below.Apparatus The apparatus consists of a metal block resistant to the substance to be examined, of good heat-conducting capacity, such as brass, with a carefully polished plane upper surface. The block is uniformly heated throughout its mass by means of amicro-adjustable gas heater or an electric heating device with fine adjustment. The block has a cylindrical cavity, wide enough to accomodate a thermometer, which should be maintained with the mercury column in the same position during the calibration of the apparatus and the determination of the melting point of the substance to be examined. The cylindrical cavity is parallel to the upper polished surface of the block and about 3 mm from it. The apparatus is calibrated using appropriate substances of known melting point.Method Heat the block at a suitably rapid rate to a temperature about 10 °C below the presumed melting temperature, then adjust the heating rate to about 1 °C/min. At regular intervals drop a few particles of powdered and, where appropriate, dried substance, prepared as for the capillary tube method, onto the block in the vicinity of the thermometer bulb, cleaning the surface after each test. Record the temperature t1 at which the substance melts instantaneously for the first time in contact with the metal. Stop the heating. During cooling drop a few particles of the substance at regular intervals on the block, cleaning the surface after each test. Record the temperature t2 at which the substance ceases to melt instantaneously when it comes in contact with the metalCalibration of the apparatus The apparatus may be calibrated using melting point reference substances such as those of the World Health Organisation or other appropriate substances.ethod III(Ph. Eur. method 2.2.17)The drop point is the temperature at which the first drop of the melting substance to be examined falls from a cup under defined conditions.Apparatus The apparatus (see Figure 2.2.17.-1) consists of 2 metal sheaths (A) and (B) screwed together. Sheath (A) is fixed to a mercury thermometer. A metal cup (F) is loosely fixed to the lower part of sheath (B) by means of 2 tightening bands (E). Fixed supports (D) 2 mm long determine the exact position of the cup in addition to which they are used to centre the thermometer. A hole (C) pierced in the wall of sheath (B) is used to balance the pressure. The draining surface of the cup must be flat and the edges of the outflow orifice must be at right angles to it. The lower part of the mercury thermometer has the form and size shown in the Figure; it covers a range from 0 °C to 110 °C and on its scale a distance of 1 mm represents a difference of 1 °C. The mercury reservoir of the thermometer has a diameter of 3.5 ±0.2 mm and a height of 6.0 ± 0.3 mm. The apparatus is placed in the axis of a tube about 200 mm long and with an external diameter of about 40 mm. It is fixed to the test-tube by means of a stopper through which the thermometer passes, and is provided with a side groove. The opening of the cup is placed about 15 mm from the bottom of the test-tube. The whole device is immersed in a beaker with a capacity of about 1 litre, filled with water. The bottom of the test-tube is placed about 25 mm from the bottom of the beaker. The water level reaches the upper part of sheath (A). A stirrer is used to ensure that the temperature of the water remains uniform.Method Fill the cup to the brim with the substance to be examined, without melting it, unless otherwise prescribed. Remove the excess substance at the 2 ends of the cup with a spatula. When sheaths (A) and (B) have been assembled press the cup into its housing in sheath (B) until it touches the supports. Remove with a spatula the substance pushed out by the thermometer. Place the apparatus in the water-bath as described above. Heat thewater-bath and when the temperature is at about 10 °C below the presumed drop point, adjust the heating rate to about 1 °C/min. Note the temperature at the fall of the first drop. Carry out at least 3 determinations, each time with a fresh sample of the substance. The difference between the readings must not exceed 3 °C. The mean of three readings is the drop point of the substance.Method IV(Ph. Eur. method 2.2.15)For certain substances, the following method is used to determine the melting point (also referred to as slip point and rising melting point when determined by this method).Use glass capillary tubes open at both ends, about 80 mm long, having an external diameter of 1.4 mm to 1.5 mm and an internal diameter of 1.0 mm to 1.2 mm.Introduce into each of 5 capillary tubes a sufficient amount of the substance, previously treated as described, to form in each tube a column about 10 mm high and allow the tubes to stand for the appropriate time and at the prescribed temperature.Unless otherwise prescribed, substances with a waxy consistency are carefully and completely melted on a water-bath before introduction into the capillary tubes. Allow the tubes to stand at 2-8 °C for 2 h.Attach one of the tubes to a thermometer graduated in 0.5 °C so that the substance is close to the bulb of the thermometer. Introduce the thermometer with the attached tube into a beaker so that the distance between the bottom of the beaker and the lower part of the bulb of the thermometer is 1 cm. Fill the beaker with water to a depth of 5 cm. Increase the temperature of the water gradually at a rate of 1 °C/min.The temperature at which the substance begins to rise in the capillary tube is regarded as the melting point.Repeat the operation with the other 4 capillary tubes and calculate the result as the mean of the 5 readings.Method V(Ph. Eur. method 2.2.16)The instantaneous melting point is calculated using the expression:in which t1 is the first temperature and t2 the second temperature read under the conditions stated below.Apparatus The apparatus consists of a metal block resistant to the substance to be examined, of good heat-conducting capacity, such as brass, with a carefully polished plane upper surface. The block is uniformly heated throughout its mass by means of amicro-adjustable gas heater or an electric heating device with fine adjustment. The block has a cylindrical cavity, wide enough to accomodate a thermometer, which should be maintainedwith the mercury column in the same position during the calibration of the apparatus and the determination of the melting point of the substance to be examined. The cylindrical cavity is parallel to the upper polished surface of the block and about 3 mm from it. The apparatus is calibrated using appropriate substances of known melting point.Method Heat the block at a suitably rapid rate to a temperature about 10 °C below the presumed melting temperature, then adjust the heating rate to about 1 °C/min. At regular intervals drop a few particles of powdered and, where appropriate, dried substance, prepared as for the capillary tube method, onto the block in the vicinity of the thermometer bulb, cleaning the surface after each test. Record the temperature t1 at which the substance melts instantaneously for the first time in contact with the metal. Stop the heating. During cooling drop a few particles of the substance at regular intervals on the block, cleaning the surface after each test. Record the temperature t2 at which the substance ceases to melt instantaneously when it comes in contact with the metalCalibration of the apparatus The apparatus may be calibrated using melting point reference substances such as those of the World Health Organisation or other appropriate substances.h. Eur. method 2.2.24)Infrared spectrophotometers are used for recording spectra in the region of 4000-650 cm-1 (2.5-15.4 µm) or in some cases down to 200 cm-1 (50 µm).ApparatusSpectrophotometers for recording spectra consist of a suitable light source, monochromator or interferometer and detector.Fourier transform spectrophotometers use polychromatic radiation and calculate the spectrum in the frequency domain from the original data by Fourier transformation. Spectrophotometers fitted with an optical system capable of producing monochromatic radiation in the measurement region may also be used. Normally the spectrum is given as a function of transmittance, the quotient of the intensity of the transmitted radiation and the incident radiation. It may also be given in absorbance.The absorbance (A) is defined as the logarithm to base 10 of the reciprocal of the transmittance (T):T=I0=intensity of incident radiation,I=intensity of transmitted radiation.Preparation of the sampleFor recording by transmission or absorptionPrepare the substance by one of the following methods.Liquids Examine a liquid either in the form of a film between 2 plates transparent to infrared radiation, or in a cell of suitable path length, also transparent to infrared radiation.Liquids or solids in solution Prepare a solution in a suitable solvent. Choose a concentration and a path length of the cell which give a satisfactory spectrum. Generally, good results are obtained with concentrations of 10-100 g/l for a path length of 0.5-0.1 mm. Absorption due to the solvent is compensated by placing in the reference beam a similar cell containing the solvent used. If an FT-IR instrument is used, the absorption is compensated by recording the spectra for the solvent and the sample successively. The solvent absorbance, corrected by a compensation factor, is subtracted using calculation software.Solids Examine solids dispersed in a suitable liquid (mull) or in a solid (halide disc), as appropriate. If prescribed in the monograph, make a film of a molten mass between 2 plates transparent to infrared radiation.A. MullTriturate a small quantity of the substance to be examined with the minimum quantity of liquid paraffin R or other suitable liquid; 5-10 mg of the substance to be examined is usually sufficient to make an adequate mull using one drop of liquid paraffin R. Compress the mull between 2 plates transparent to infrared radiation.B. DiscTriturate 1-2 mg of the substance to be examined with 300-400 mg, unless otherwise specified, of finely powdered and dried potassium bromide R or potassium chloride R. These quantities are usually sufficient to give a disc of 10-15 mm diameter and a spectrum of suitable intensity. If the substance is a hydrochloride, it is recommended to use potassium chloride R. Carefully grind the mixture, spread it uniformly in a suitable die, and submit it to a pressure of about 800 MPa (8 t·cm-2). For substances that are unstable under normal atmospheric conditions or are hygroscopic, the disc is pressed in vacuo. Several factors may cause the formation of faulty discs, such as insufficient or excessive grinding, humidity or other impurities in the dispersion medium or an insufficient reduction of particle size. A disc is rejected if visual examination shows lack of uniform transparency or when transmittance at about 2000 cm-1 (5 µm) in the absence of a specific absorption band is less than 60 per cent without compensation, unless otherwise prescribed.Gases Examine gases in a cell transparent to infrared radiation and having an optical path length of about 100 mm. Evacuate the cell and fill to the desired pressure through a stopcock or needle valve using a suitable gas transfer line between the cell and the container of the gas to be examined.If necessary adjust the pressure in the cell to atmospheric pressure using a gas transparentto infrared radiation (for example nitrogen R and argon R). To avoid absorption interferences due to water, carbon dioxide or other atmospheric gases, place in the reference beam, if possible, an identical cell that is either evacuated or filled with the gas transparent to infrared radiation.For recording by diffuse reflectanceSolids Triturate a mixture of the substance to be examined with finely powdered and dried potassium bromide R or potassium chloride R. Use a mixture containing approximately 5 per cent of the substance, unless otherwise specified. Grind the mixture, place it in a sample cup and examine the reflectance spectrum.The spectrum of the sample in absorbance mode may be obtained after mathematical treatment of the spectra by the Kubelka-Munk function.For recording by attenuated total reflectionAttenuated total reflection (including multiple reflection) involves light being reflected internally by a transmitting medium, typically for a number of reflections. However, several accessories exist where only one reflection occurs.Prepare the substance as follows. Place the substance to be examined in close contact with an internal reflection element (IRE) such as diamond, germanium, zinc selenide, thallium bromide-thallium iodide (KRS-5) or another suitable material of high refractive index. Ensure close and uniform contact between the substance and the whole crystal surface of the internal reflection element, either by applying pressure or by dissolving the substance in an appropriate solvent, then covering the IRE with the obtained solution and evaporating to dryness. Examine the attenuated total reflectance (ATR) spectrum.Identification using reference substancesPrepare the substance to be examined and the reference substance by the same procedure and record the spectra between 4000-650 cm-1 (2.5-15.4 µm) under the same operational conditions. The transmission minima (absorption maxima) in the spectrum obtained with the substance to be examined correspond in position and relative size to those in the spectrum obtained with the reference substance (CRS).When the spectra recorded in the solid state show differences in the positions of the transmission minima (absorption maxima), treat the substance to be examined and the reference substance in the same manner so that they crystallise or are produced in the same form, or proceed as prescribed in the monograph, then record the spectra.Identification using reference spectraControl of resolution performance For instruments having a monochromator, record the spectrum of a polystyrene film approximately 35 µm in thickness. The difference x (see Figure 2.2.24.-1) between the percentage transmittance at the transmission maximum A at 2870 cm-1 (3.48 µm) and that at the transmission minimum B at 2849.5 cm-1 (3.51 µm) must be greater than 18. The difference y between the percentage transmittance at the transmission maximum C at 1589 cm-1(6.29 µm) and that at the transmission minimum D at 1583 cm-1 (6.32 µm) must be greater than 10.。
凡士林纱布产品技术标准2023年

凡士林纱布1本标准规定了凡士林纱布的分类、要求、试验方法、检验规则、标志标签、使用说明书和包装、运输、贮存等要求。
本标准适用于凡士林纱布。
主要用于外科、烧伤科、皮肤感染及手术后贴敷伤口、疮面、及填充瘘道用。
2下列文件中的条款通过本标准的引用而成为本标准的条款,凡是注日期的引用的引用文件,其随后所有的修改单 (不包括勘误内容) 或修订版本均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。
凡是不注明日期的引用文件,其最新版本适用于本标准。
GB/T6682 分析实验室用水规格和实验方法GB191-2008 包装储运图示标志GB/T2828.1-2003 计数抽样检验程序第一部分,按接收质量限(AQL)检索的逐批检验抽样计划GB/2829-2002 周期检验计数抽样程序及表(适用于对过程稳定的检验)GB/T14233.2-2005 法GB/T16886.5-2003 GB/T16886.10-2005 试验医用输液、输血、注射器具检验方法第二部分:生物试验方医疗器械生物学评价第 5 部分:体外细胞毒性试验医疗器械生物学评价第10 部分: 刺激与迟发型超繁反应《中华人民共和国药典》(2015 年版) 第二部33.1 产品型号分为:轻载型(S) 、中载型(M)和普通型(P) 3.2型号按醚中可溶物含量划分:对于非单片包装的凡士林纱布,轻载型凡士林纱布的醚中可溶物为90g/m ~1302 g/m2 ;中载型凡士林纱布的醚中可溶物应大于130 g/m2 ;普通型凡士林纱布的醚中可溶物不少于200 g/m2 。
对于单片包装的凡士林纱布,轻载型凡士林纱布的醚中可溶物不得少于80g/m2 ;中载型凡士林纱布的醚中可溶物不得少于110 g/m2 ;普通型凡士林纱布的醚中可溶物不得少于175g/m2 。
44.1 凡士林纱布的外观要求:4.1.1 凡士林纱布应呈湿润状、无异味。
4.1.2 凡士林纱布外观应平整、无杂质、无污点、无破损、无发霉等缺陷,色泽均匀、无明显可见的外来物质和纱线脱落。
WHITE PETROLEUM JELLY白凡士林质检报告BP2012批号[1]..
![WHITE PETROLEUM JELLY白凡士林质检报告BP2012批号[1]..](https://img.taocdn.com/s3/m/97b62216964bcf84b8d57b0f.png)
检验结果
TESTRESULT
1.外观
Appearance
Whiteor almost white, translucent, soft unctuous mass, slightly fluorescent in daylight when melted.
符合
Complies
2.溶解性
Density
0.81-0.88
0.85
4.鉴别
Identification
First identification A, B, D
Second identification A, C, D
A.Drop point: Between35℃and70℃and doesn’t differ by more than5℃from the value stated on the label.
包头市华海化工有限责任公司
白凡士林质检报告
CERTIFICATEOFANALYSISOFWHITEPETROLEUM JELLY
BATCH NO.:20141210MFG . DATE:DEC 10,2014
EXPIRY DATE:DEC 10,2016
检验项目
TEST ITEMS
检验方法(BP-2012)
符合
s
10.异性有机物
Foreign Organic Matter
Heat1 guntil fumes appear, No acrid odor is evolved.
符合
Complies
11.硫酸盐灰分
Sulphated Ash
Not more than 0.05%
0.04%
凡士林检测报告
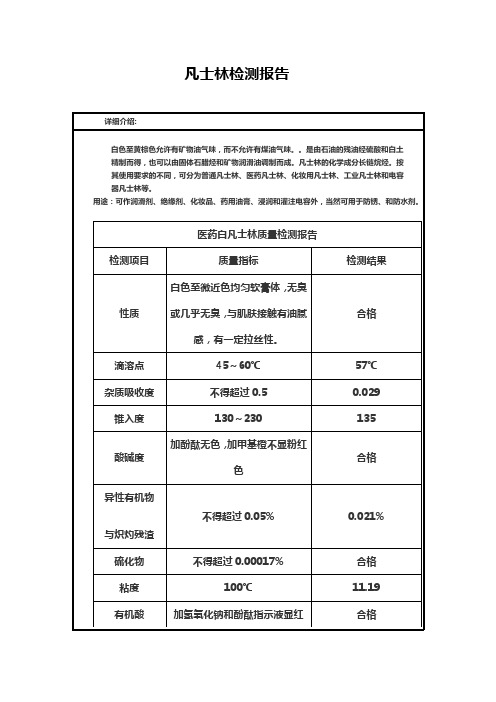
检测项目
质量指标
检测结果
性质
白色至微近色均匀软膏体,无臭或几乎无臭,与肌肤接触有油腻感,有一定拉丝性。
合格
滴溶点
45~60℃
57℃
杂质吸收度
不得超过0.5
0.029
锥入度
130~230
135
酸碱度
加酚酞无色,加甲基橙不显粉红色
合格
异性有机物
与炽灼残渣
不得超过0.05%
0.021%
凡士林检测报告
详细介绍:
白色至黄棕色允许有矿物油气味,而不允许有煤油气味。。是由石油的残油经硫酸和白土
精制而得,也可以由固体石腊烃和矿物润滑调制而成。凡士林的化学成分长链烷烃。按
其使用要求的不同,可分为普通凡士林、医药凡士林、化妆用凡士林、工业凡士林和电容
器凡士林等。
用途:可作润滑剂、绝缘剂、化妆品、药用油膏、浸润和灌注电容外,当然可用于防锈、和防水剂。
硫化物
不得超过0.00017%
合格
粘度
100℃
11.19
有机酸
加氢氧化钠和酚酞指示液显红色
合格
检测依据
中国药典2005版二部
凡士林说明书

篇一:凡士林安全技术说明书凡士林安全技术说明书第一部分化学品化学品中文名称:凡士林化学品英文名称:vas中文名称2:英文名称2:技术说明书编码: cas no.:8009-03-8分子式:分子量:第二部分成分/组成信息有害物成分含量cas no.:第三部分危险性概述危险性类别:侵入途径:通过食入吸收到体内健康危害:环境危害:燃爆危险:第四部分急救措施皮肤接触:用清水冲洗眼睛接触:先用大量清水冲洗几分钟,然后就医吸入:迅速脱离现场至空气新鲜处食入:饮足量温水,催吐。
就医。
第五部分消防措施危险特性:有害燃烧产物:灭火方法:使用泡沫、干粉、二氧化碳和雾状水第六部分泄露应急处理应急处理:第七部分操作处置与储存操作注意事项:1.刚刚烧伤时最好不用,否则热量散不出去,反而会影响伤口愈合。
2.鼻子阻塞时不要使用,因为凡士林会影响鼻毛对脏空气的清洁能力。
储存注意事项:阴凉处储存避免阳光直射第八部分接触控制/个体防护职业接触限值:中国mac(mg/m3):前苏联mac(mg/m3):tlvtn:tlvwn:监测方法:工程控制:禁止明火呼吸系统防护:眼睛防护:戴安全护目镜身体防护:穿普通工作服手防护:戴橡胶手套其他防护:工作时不得进食、饮水或吸烟第九部分理化特性主要成分:蜡膏,不同量的高、中黏度润滑油外观与性状:外观为白色的半透明软膏,有滑腻感,具有一定的拉丝性和粘附性。
擦在皮肤上无嗅味ph:熔点(℃):36-60沸点(℃):302相对密度(水=1):0.9相对蒸气密度(空气=1):无资料饱和蒸气压(kpa):20℃时1.3pa燃烧热(kj/mol):无资料临界温度(℃):无资料临界压力(mpa):无资料辛醇/水分配系数的对数值:6闪点(℃):182-221℃引燃温度(℃):290爆炸上限%(v/v):7%爆炸下限%(v/v):0.9%溶解性:基本不溶于水和乙醇(96%),溶于氯仿、乙醇和溶剂汽油主要用途:适用于配制医用药膏及皮肤保护油膏的原料第十部分稳定性和反应活性稳定性:常温下稳定禁配物:避免接触的条件:聚合危害:分解产物:第十一部分毒理学资料急性毒性:ld50:无资料;lc50:无资料亚急性和慢性毒性:刺激性:致敏性:致突变性:致畸性:致癌性:第十二部分生态学资料生态毒理毒性:生物降解性:非生物降解性:生物富集或生物积累性:其它有害作用:无资料。
白凡士林的粘度检验方法

白凡士林的粘度检验方法说实话白凡士林的粘度检验方法这件事,我一开始也是瞎摸索。
我就想嘛,这白凡士林粘粘乎乎的,得怎么检验它的粘度才好呢。
我试过好几个方法呢。
最开始我就想简单粗暴一点,拿个小棍子插到凡士林里面,然后再拔出来,看凡士林黏在棍子上的情况。
我寻思着如果凡士林在棍子上挂得厚厚的,那肯定粘度就大呗。
可是啊,这方法太不靠谱了。
因为我发现我每次插进去的深度不一样,拔出来的速度也不一样,结果每次凡士林挂在棍子上的量和状态都不太一样,完全没有个准头。
这可把我愁坏了。
后来我就想啊,能不能像测量蜂蜜的流动速度那样来测量白凡士林呢。
我找了个倾斜的板子,把凡士林薄薄地抹在上面,想看看它从上往下流的速度。
但是白凡士林根本不像蜂蜜那么容易流动啊,它半天就在原地不动,我等了好久,它就那么一小团呆着。
我这才意识到,这种适用于流动性比较好的液体的方法,对白凡士林这种半固体的东西是不适用的。
再后来呢,我做了个新的尝试。
我找了个小容器,把白凡士林装进去,然后又找了个小的砝码,把砝码轻轻放在凡士林的表面。
然后就观察砝码陷入凡士林的速度和深度。
如果砝码陷得快又深,就说明凡士林的粘度比较小。
这个方法好像有点靠谱了,但是我又有新的问题了。
我发现不同形状的砝码得到的结果不太一样,方的和圆的就有差异。
我就想啊,这肯定是因为接触面的问题。
所以我之后就固定使用一种形状的砝码。
我感觉这差不多就是个可行的办法了,但是我还不太确定是不是特别准确。
不过对于我们平时大概判断一下白凡士林的粘度来说,已经足够用了。
我觉得要是想更准确一点的话,可以多做几次实验,取个平均值之类的。
我还想啊,如果有那种很精密的仪器就好了,就像那些科学家测量别的东西粘度用的那种高级仪器,可那咱也没有啊。
反正就通过这种土方法,也算是能对付着检验白凡士林的粘度了。
我还想说,其实在这个摸索的过程中,我发现任何测试都得保持一定的条件不变,这样得到的结果才会相对准确。
就像我用砝码测试的时候,你要是每次放置砝码的角度不一样,都可能影响结果。
凡士林标准

凡士林标准
凡士林是一种烷系烃或饱和烃类半液态的混合物,也叫矿脂,由石油分馏后制得。
其状态在常温时介于固体及液体之间,因不同用途而有棕、黄、白三种颜色。
凡士林根据使用范围、理化性质和生产要求的不同,采用国际通用的产品标准分为三类,而在中国增加制订了普通凡士林国家标准,四个分别为:医药凡士林(GB1790—86)、工业凡士林(GB6731—86)、普通凡士林(GB6732—86)、电容器凡士林(GB6733—86)。
1、医用凡士林:是具有一定的拉丝性、拉丝甚短的均匀软膏状物质,无臭味,其中按精制深度又分为医药白凡士林和医药黄凡士林,分别为白色、淡黄色至浅黄色[7];适用于医药软膏、乳膏的原料和护肤油膏;用石蜡脂与矿物油混合或是用香料油与熔化的白色石蜡或地蜡相混合制得,可用以制造医用油膏,在纺织工业上用以制造乳化液,也可作为测量器械及外科器械抗腐蚀润滑脂用。
经过高度精制达到药用级时可用作油膏,用于医药的矿脂冻可能配有其他物质。
2、工业凡士林:是一种固体烃基润滑脂,用来保护金属制品;也可在温度不高于45℃和负荷不大的条件下作为减摩润滑脂使用[8]。
适用于金属物品,金属零件及机械防蚀和机械的减摩润滑。
凡士林(矿脂)用于润滑和橡胶及树脂配方中,用作润滑时,只应经过过滤精制而不能用酸处理,且也不应掺石蜡,其熔点应在46-54℃之间;用作橡胶柔顺剂的O级矿脂是比重为0.84的浅黄色无味半固体物,熔点为46-48℃。
3、普通凡士林:分为普通白凡士林、普通黄凡士林两种;适用于润滑橡胶制品软化剂,玻璃纤维拉丝成型乳液等。
4、电容器凡士林:是白色以至于浅黄色均匀油膏;适用于浸渍电容器绝缘材料和浇注电容器。
白矾检验标准操作规程
白矾检验标准操作规程目的:建立饮片用辅料检验标准操作规程,以规范操作过程,保证检验准确无误。
责任人:文件制订人及所有相关人员。
内容:1、性状取本品适量,放入白瓷盘中,用眼观察,可见以下性状特征:本品呈不规则的块状或粒状,无色或淡黄白色,透明或半透明。
表面略平滑或凹凸不平,具细密纵棱,有玻璃样光泽。
质硬而脆。
气微,味酸、微甘而极涩。
2、鉴别2.1理化鉴别2.1.1铝盐2.1.1.1取供试品溶液,滴加氢氧化钠试液,即生成白色胶状沉淀;分离,沉淀能在过量的氢氧化钠试液中溶解。
2.1.1.2取供试品溶液,加氨试液至生成白色胶状沉淀,滴加茜素磺酸钠指示液数滴,沉淀即显樱红色。
2.1.2钾盐2.1.2.1取铂丝,用盐酸湿润后,蘸取供试品,在无色火焰中燃烧,火焰即显紫色;但有少量的钠盐混存时,须隔蓝色玻璃透视,方能辩认。
2.1.2.2取供试品,加热炽灼除去可能杂有的铵盐,放冷后,加水溶解,再加0.1%四苯硼钠溶液与醋酸,即生成白色沉淀。
2.1.3硫酸盐2.1.3.1取供试品溶液,滴加氯化钡试液,即生成白色沉淀;分离,沉淀在盐酸或硝酸中均不溶解。
2.1.3.2取供试品溶液,滴加醋酸铅试液,即生成白色沉淀;分离,沉淀在醋酸铵试液或氢氧化钠试液中溶解。
2.1.3.3取供试品溶液,加盐酸,不生成白色沉淀( 与硫代硫酸盐区别)。
3、检查主要使用仪器:电子分析天平、量筒等。
3.1 铵盐取本品0.1g,加无氨蒸馏水100ml使溶解,取10 ml,置比色管中,加无氨水40ml 与碱性碘化汞钾试液2ml,如显色,与氯化铵溶液(取氯化铵31.5mg,加无氨蒸馏水使成1000ml)1ml、碱性碘化汞钾试液2ml及无氨蒸馏水49ml的混合液比较,不得更深。
3.2 铜盐与锌盐取本品1g,加水100ml与稍过量的氨试液,煮沸,滤过,滤液不得显蓝色,滤液中加醋酸使成酸性后,再加硫化氢试液,不得发生浑浊。
3.3 铁盐取本品0.35g,加水20ml溶解后,加硝酸2滴,煮沸5分钟,滴加氢氧化钠试液中和至微显浑浊,加稀盐酸1ml、亚铁氰化钾试液1ml与水适量使成50ml ,摇匀,1小时内不得显蓝色。
ZL-TS-10-F004-00白凡士林质量标准
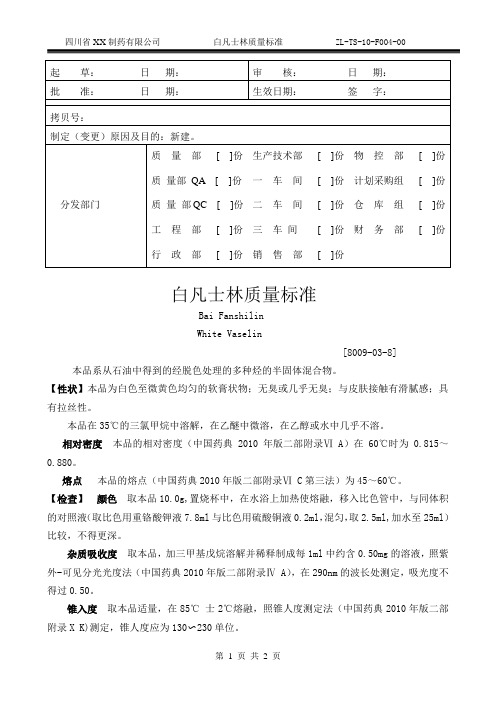
白凡士林质量标准Bai FanshilinWhite Vaselin[8009-03-8] 本品系从石油中得到的经脱色处理的多种烃的半固体混合物。
【性状】本品为白色至微黄色均匀的软膏状物;无臭或几乎无臭;与皮肤接触有滑腻感;具有拉丝性。
本品在35℃的三氯甲烷中溶解,在乙醚中微溶,在乙醇或水中几乎不溶。
相对密度本品的相对密度(中国药典2010年版二部附录Ⅵ A)在60℃时为0.815~0.880。
熔点本品的熔点(中国药典2010年版二部附录Ⅵ C第三法)为45~60℃。
【检查】颜色取本品10.0g,置烧杯中,在水浴上加热使熔融,移入比色管中,与同体积的对照液(取比色用重铬酸钾液7.8ml与比色用硫酸铜液0.2ml,混匀,取2.5ml,加水至25ml)比较,不得更深。
杂质吸收度取本品,加三甲基戊烷溶解并稀释制成每1ml中约含0.50mg的溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A),在290nm的波长处测定,吸光度不得过0.50。
锥入度取本品适量,在85℃士2℃熔融,照锥人度测定法(中国药典2010年版二部附录X K)测定,锥人度应为130〜230单位。
酸碱度取本品35.Og,置250ml烧杯中,加水100ml,加热至微沸,搅拌5分钟,静置放冷,分取水层,加酚酞指示液1滴,应无色;再加甲基橙指示液0.10ml,不得显粉红色。
硫化物取本品3.0g,依法检査(中国药典2010年版二部附录Ⅷ C),应符合规定 (0. 000 17%)。
有机酸取本品20.0g,加中性稀乙醇100ml,搅拌并加热至沸,加酚酞指示液lml与氢氧化钠滴定液(0.1mol/L)0.40ml,强力搅拌,应显红色。
异性有机物与炽灼残渣取本品2.0g,用直火加热,应无辛臭;再炽灼,遗留残渣不得过lmg(0.05%)。
微生物限度照微生物限度检查法(中国药典2010年版二部附录XI J)常规方法检查:细菌数应≤80cfu/g,霉菌和酵母菌数应≤80cfu/g,金黄色葡萄球菌、铜绿假单胞菌每lg不得检出。
凡士林锥入度测定仪安全操作及保养规程

凡士林锥入度测定仪安全操作及保养规程随着科技的不断发展,各种测量仪器在工业生产、实验室、医疗保健等领域都得到了广泛的应用。
作为一种常用的粘度测量仪器,凡士林锥入度测定仪在化工、食品、医药等领域扮演着重要角色。
然而,由于该仪器的使用需要较高的技能水平和严格的操作规程,一旦使用不当会对人身安全和仪器的使用寿命产生严重影响。
因此,本文将针对凡士林锥入度测定仪的安全操作及保养规程进行详细介绍,以确保您的工作安全和测量结果的准确性。
一、安全操作1. 操作前检查在使用凡士林锥入度测定仪前,应该进行以下基本检查,以确保操作环境和仪器本身的安全性。
•检查周围环境是否具备安全条件,如够大的操作空间、不易引起外力干扰的稳定位置等;•检查本机工作电源是否正常并已正确接地;•检查防护罩是否安装牢固,不能存在松动或损坏的情况;•确认测试样品的温度与室温相同。
2. 操作流程凡士林锥入度测定仪的操作流程较为复杂,顺序不能颠倒,下面将分步骤进行详细介绍。
2.1 仪器预热打开仪器电源,选择温度设定条件,将样品加热到所需测试温度。
在设定温度下进行预热,直至仪器稳定工作,提示可以进行下一步操作。
注意事项:•在加热过程中,应观察仪器的工作状态,如有异常或热线颜色不正常,应停止使用并联系维修人员。
•避免高温空转情况发生,以延长仪器使用寿命。
2.2 样品准备使用准确的天平将适量的测试样品称入样品容器中,并严格控制称量误差,保持实验结果的精度。
注意事项:•在称量样品前,应先进行容器的校准,以避免误差发生;•对于不同种类的样品,应严格按照规定的称量要求进行操作。
2.3 操作流程将预热好的凡士林锥入度测定仪放置在水平的桌面上,并按照以下步骤进行操作。
1.清洗仪器:用反应用的溶剂和棉纱球对仪器进行彻底的清洗。
清洗时,注意不能将溶剂加入样品容器中,在清洗后,用干净纸巾或棉纱球将仪器擦干。
2.测定准备:将样品容器放入仪器中,打开进料阀门,在空气压力控制系统的作用下,将样品缓慢加入仪器中。
白凡士林 质量标准

白凡士林质量标准
白凡士林是一种常见的皮肤护理产品,通常用于保湿和保护皮肤。
它的质量标准通常由国家药品监督管理部门或相关标准化组织
制定和监管。
一般来说,白凡士林的质量标准包括以下几个方面:
1. 成分标准,白凡士林通常由石蜡和凡士林油等成分制成,其
质量标准应包括原材料的纯度、生产工艺、添加剂使用等方面的要求。
2. 产品性能标准,白凡士林的产品性能标准通常包括外观要求、质地、透明度、pH值、水含量等指标,以确保产品符合相关的安全
和使用要求。
3. 医药标准,如果白凡士林被归类为医药产品,那么其质量标
准还需要符合相关的药典要求,包括药物的纯度、稳定性、微生物
限度等指标。
4. 包装标准,白凡士林的包装标准通常包括包装材料的要求、
标签标识、包装规格等,以确保产品在运输和存储过程中不受污染
或损坏。
总的来说,白凡士林作为一种常见的皮肤护理产品,其质量标准应当涵盖原材料、生产工艺、产品性能、医药要求和包装标准等多个方面,以确保产品的质量和安全性。
消费者在购买和使用白凡士林时,可以关注产品的生产许可证、相关认证标识和质量检测报告,以确保产品符合相关的质量标准。
白凡士林成品质量标准
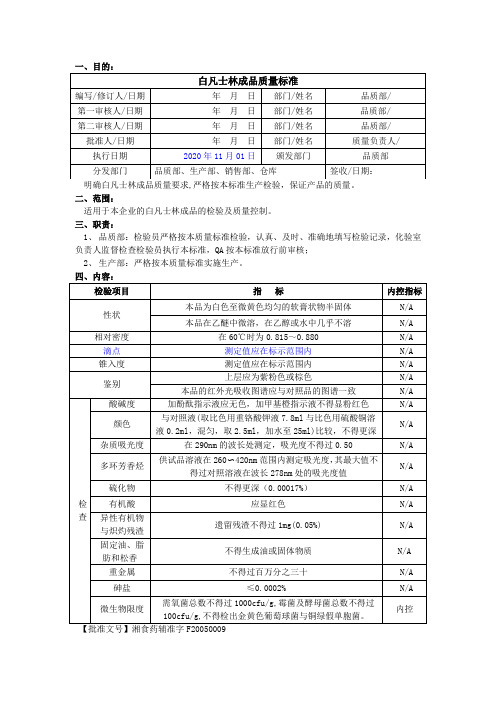
一、目的:
二、范围:
适用于本企业的白凡士林成品的检验及质量控制。
三、职责:
1、品质部:检验员严格按本质量标准检验,认真、及时、准确地填写检验记录,化验室负责人监督检查检验员执行本标准,QA按本标准放行前审核;
2、生产部:严格按本质量标准实施生产。
四、内容:
【批准文号】湘食药辅准字F20050009
【代码】CF001
【规格】165kg/桶(药用钢桶),20kg/袋(药用低密度聚乙烯袋),2kg/袋(药用低密度聚乙烯袋),500g/瓶(药用高密度聚乙烯瓶)
【类别】药用辅料,软膏基质和润滑剂等。
【贮藏】避光,密闭保存。
【标示】应标明滴点、锥入度的标示范围。
如加入抗氧剂或稳定剂,应标明名称和含量。
【有效期】三年
【注意事项】N/A
五、参考文献:
《中国药典》2020年版四部
六、相关文件:
EK/SMP-QC0005 取样管理规程
EK/SOP-QC3002 白凡士林成品检验操作规程
EK/SOP-QC300201 白凡士林成品检验原始记录
七、相关记录:
N/A。
GC-YL-70030白矾原料检验操作规程

【鉴别】
(1)理化鉴别
本品水溶液显铝盐(通则0301)、钾盐(通则0301)与硫酸盐(通则0301)的鉴别反应。
【检查】
铵盐
仪器与试剂:分析天平、无氨蒸馏水、比色管、碱密称定,照氮测定法(通则0704第二法或第三法,无需消解)测定,含鞍盐以总氮(N)计,不得过0.3%
分发部门: 质量部、化验室、生产部
标 题
正 文
1
2
2.1
3
3.1
4
4.1
4.1.1
4.1.2
4.2
4.2.1
4.2.2
4.3
4.3.1
4.3.2
4.4
4.4.1
4.4.2
5
5.1
5.1.1
5.1.2
标准依据:《中华人民共和国药典》2020年版一部及四部
【性状】
方法:取本品,置日光下观察:本品呈不规则的块状或粒状。无色或淡黄白色,透明或半透明。表面略平滑或凹凸不平,具细密纵棱,有玻璃样光泽。质硬而脆。气微,味酸、微甘而极涩。
重金属
仪器与试剂:分析天平、稀醋酸等。
方法:取本品1g,加稀醋酸2ml与水适量使溶解成25ml,依法检查(通则0821第一法),含重金属不得过20mg/kg。
GC-YL-10390白及检验操作规程
【检查】
水分不得过15.0%(通则0832第二法)。
仪器:电热鼓风干燥箱、分析天平、粉碎机、药筛等。
方法:取供试品2~5g,平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm,疏松供试品不超过10mm,精密称定,开启瓶盖在100~105℃干燥5小时,将瓶盖盖好,移置干燥器中,放冷30分钟,精密称定,再在上述温度干燥1小时,放冷,称重,至连续两次称重的差异不超过5mg为止。根据减失的重量,计算供试品中含水量(%)。
仪器:粉碎机、药筛、分析天平、坩埚、箱式电阻炉等。
方法:取供试品2~3g,过二号筛混合均匀后,置炽灼至恒重的坩埚中称定重量,缓缓炽热,注意避免燃烧,至完全炭化时,逐渐升高温度至500~600℃,使完全灰化至恒重。根据残渣重量,计算供试品中总灰分的含量(%)。
计算公式:
W1-W0
总灰分% =×100%
计算公式:
W样+W0-W1
样品含水量% =×100%
式中:
W0----------------------------称量瓶的重量(g)。
W1---------------------------称量瓶与样品的重量(g)。
W样---------------------------样品的重量(g)。
总灰分不得过5.0%(通则2302)。
【含量测定】 照高效液相色谱法(通则0512)测定。
仪器与试剂:高效液相色谱仪、数显恒温水浴锅等。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸溶液(22:78)为流动相,检测波长为223nmo理论板数按1,4-二[4-(葡萄糖氧)节基]-2-异丁基苹果酸酯峰计算应不低于2000.
凡士林检测 凡士林测试

凡士林检测一:凡士林(003)凡士林是一种油脂状的石油产品。
白色至黄棕色允许有矿物油气味,而不允许有煤油气味。
滴点约37-54度。
是由石油的残油经硫酸和白土精制而得,也可以由固体石腊烃和矿物润滑油调制而成。
凡士林的化学成分主要是长链烷烃。
凡士林的学名叫石油脂,它的主要原料是从原油经过常压和减压蒸馏后留下的渣油中脱出的蜡膏,同时还需按照要求掺和不同量的高、中黏度润滑油。
二:凡士林的分类按其使用要求的不同,可分为普通凡士林、医药凡士林、化妆用凡士林、工业凡士林和电容器凡士林等。
除去可作润滑剂、绝缘剂、化妆品、药用油膏、浸润和灌注电容外,当然可用于防锈、和防水剂。
在做防水薄膜时,建议用凡士林和桐油调和好擦在要防水的物件的表层,干了后反复涂2-3次干后就会生成一层防水膜了。
三:凡士林的主要检测项目外观,颜色,密度,闪点,粘度,倾点,灰分,硅含量,燃点,酸度,酸值,添加元素(钡、钙、锂、磷、硫、镁、硼、锌)含量,含硫量,水分,残碳,灰分,热值,铜片腐蚀,馏程,十六烷值,博士试验,凝点,冷滤点,润滑性,锰含量,铅含量,苯含量,芳烃含量,烯烃含量,胶质,甲醇含量,辛烷值,蒸气压等检测。
四:凡士林的部分检测标准GB 1790-2003 医药凡士林GB/T 269-1991 润滑脂和石油脂锥入度测定法GB/T 6733-1986 电容器凡士林GB 8025-1987 石油蜡和石油脂微量硫测定法(微库仑法)GB 8026-1987 石油蜡和石油脂滴熔点测定法SH 0008-1990 化妆用凡士林SH 0039-1990 工业凡士林SH/T 0405-1996 凡士林重金属限量试验法SH/T 0406-1992 凡士林紫外吸光度测定法SH/T 0655-1998 凡士林稠环芳烃试验法SH/T 0678-1999 凡士林滴点测定法SH/T 0767-2005 食品级凡士林科标能源检测检测中心,专业从事凡士林检测、凡士林测试、凡士林性能检测、化工材料与制品老化测试、可靠性检测、阻燃检测、有害物质检测、性能测试、成分分析、配方研究的分析测试研发机构。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
的对照液(取比色用重铬酸钾液7.8ml与比色用硫酸铜液0.2ml,摇匀,取2.5ml,加水至25ml)比较,颜色不得更深(比色时应将比色管靠近白色背景成一无荧光的角度,在反射光下进行观察)为符合规定。
2.3.2杂质吸收度
取本品,加三甲基戊烷制成每1ml中含0.50mg的溶液,照紫外-可见分光光度法(《中国药典》2005版二部附录ⅣA),在290nm的波长处测定,吸收度不得过0.50为符合规定。
题目:白凡士林检验标准操作规程
编码:SOP-QC-202-A
起草:
日期:
审核:
日期:
批准:
日期:
生效日期:
颁发部门:质控部
分发部门:检验室
变更记载修改号
批准日期执行日期
变更原因及目的
标准依据:《中国药典》2005年版二部
目的:建立白凡士林检验操作规程。
范围:本规程适用于白凡士林的检验。
职责:检验员、质控部经理对本规程实施负责。
2.3.6异性有机物与炽灼残渣
取本品2.0g,置炽灼恒重的坩埚中,用直火加热(注意勿使供试品着火),应无辛臭;再强热炽灼,遗留残渣不得过1mg为符合规定(0.05%)。
2ห้องสมุดไป่ตู้3.7硫化物
取本品3.0g,按硫化物检查法(《中国药典》2005版二部附录ⅧC)检查,应符合规定(0.00017%)。
2.3.8微生物限度
2.3.4酸碱度
取本品35.0g,置250ml烧杯中,加水100ml,加热微沸,搅拌5分钟,静置放冷,分取水层,加酚酞指示液1滴,应无色;再加甲基橙指示液0.10ml,不得显粉红色为符合规定。
2.3.5有机酸
取本品20.0g,加中性稀乙醇100ml,搅拌并加热至沸,加酚酞指示液1ml与氢氧化钠滴定液(0.1mol/L)0.40ml,强力搅拌,应显红色为符合规定。
规程:
1.[性状]
取本品适量,置表面皿中,在自然光下观察,为白色至微黄色均匀的软膏状物;无臭或几乎无臭;与皮肤接触有滑腻感;具有一定的拉丝性。
本品在35℃的苯中易溶,在约35℃的氯仿中溶解,在乙醚中微溶,在乙醇或水中几乎不溶。
2.[检查]
2.1仪器与用具
扭力天平刻度吸管1ml、5ml、10ml恒温水浴分光光度计锥入度计
取本品10g加至含溶化的(温度不超过45℃)5g司盘80,3g 单硬脂酸甘油酯,10g聚山梨酯80的无菌混合物的烧杯中,用无菌玻棒搅拌成团后,加入45℃的PH7.0的无菌氯化钠-蛋白胨缓冲液至100ml,混匀使充分乳化,作为1:10的供试液。取适量供试液,照微生物限度检查平皿法(《中国药典》2005版二部附录Ⅺ J)检查,细菌数、霉菌和酵母菌数分别不得过100个/g,金黄色葡萄球菌、铜绿假单胞菌不得检出为符合规定。
2.3.3锥入度
取本品适量,在85℃±2℃熔融,倾入直径为100mm±5mm、高度不小于65mm的金属制圆筒状容器中,装置离容器上端6mm以内,于25℃±2℃放置16~18小时,并在试验前将容器移至25℃±0.5℃水浴中放置2小时;另将标准锥体(总重102.50g±0.05g,由表面光滑的圆锥体和可拆卸的锥尖以适当方式连接组成:锥尖部分呈30°角,尖端载面直径为0.38mm±0.02mm,基底直径为8.40mm±0.02mm,高15.00mm±0.05mm,圆锥体部分的夹角为90°±15′,高32.1mm±0.2mm,底部最大直径为69.3mm±0.1mm)置水浴中调节到25.0℃±0.5℃。将上述容器从水浴中取出,立即置于预先调至水平的针入度计平台上,调节仪器使锥尖刚好与距容器壁25~38mm处的本品表面相接触,调刻度盘指针至零位,迅即按下“启动”钮使锥体自由下落5.0秒±0.1秒,使锥体停止沉入,轻轻调节指示器至恰被锥杆挡住为止。从指示器刻度盘上读取锥入度。若测得的锥入度小于200单位,可在同一容器中连续进行三次测定,测定部位应有一定距离,以保证扰动区域不相重叠;当测得的锥入度超过200单位时,应另取本品置另一容器中同法试验,测定点应在容器中心位置。三次读数的相对偏差应在±3%之内,取三次测定平均值为本品的锥入度(系指标准锥体在5秒钟内沉入凡士林中所达到的深度,其单位为0.1mm),应为130~230单位为符合规定。
电炉酒精灯量筒100ml烧杯250ml比色管25ml坩埚坩埚钳
2.2试药与试液
酚酞指示液取酚酞1g,加乙醇100ml使溶解,即得。
甲基橙指示液取甲基橙0.1g,加水100ml使溶解,即得。
氢氧化钠滴定液(0.1mol/L)
三甲基戊烷乙醇
2.3操作方法
2.3.1颜色
取本品10g,置烧杯中,在水浴上加热使熔融,倾注入25ml比色管中与同体积