α-2受体激动剂作用机制及应用
探讨α受体激动对支气管平滑肌的舒张作用

探讨α受体激动对支气管平滑肌的舒张作用中山医学院临床医学02级14班第四组陆文霞吴盛喜吴健彬李嘉欣林树洪李雪梅摘要:目前在临床上治疗支气管哮喘的药物主要分为四大类:β2-受体激动剂和α2-受体拮抗剂,抗胆碱药物,钾通道激活剂及磷酸二酯酶抑制剂。
而又以β2-受体激动剂和α2-受体拮抗剂的联合用药最为常用。
但有某些文献资料显示,支气管平滑肌上还存在一定量的α-肾上腺受体,其激动对支气管平滑肌的舒缩可能产生影响,但鲜有实验支持。
为了探讨该观点的正确性,本组设计了这个实验,以观察α受体激动药与β受体激动药对豚鼠支气管平滑肌舒缩的调节作用,着重研究α受体激动药是否存在对支气管哮喘的解痉作用。
关键词:支气管α受体舒张1.实验对象:豚鼠4只,体重300g左右。
2.实验器材和药品恒温浴槽装置一套、二道生理记录仪(或计算机实时分析系统)、张力换能器、恒温水浴箱、豚鼠手术器械一套、培养皿、注射器(1ml)、烧杯、铁支架、双凹夹、木锥、丝线等。
2×10-3乙酰胆碱、10-3间羟胺、10-3普洛萘尔、10-3肾上腺素、10-3酚妥拉明、10-3异丙肾上腺素、氧气、Krebs-Henseleit(K-H)液含(mol/L:NaCl 118,KCl 4.7,MgSO4 1.2,KH2PO4 1.2,NaHCO3 25,CaCl2 2.5和葡萄糖11.1×10-3)。
3.标本制备豚鼠击昏后切开颈正中皮肤,迅速取出喉头与隆突之间的气管,浸入通5%CO2和95%O2混合气的Krebs—Henseleit液中。
细心去除气管周围的疏松结缔组织后,按参考文献方法,剪成宽约3mm,长约20mm的螺旋条,放入含30mlK—H液的37℃恒温浴槽中, 在气管螺旋条标本两端穿线,下端固定在浴槽内,上端固定于拉力换能器,离体气管螺旋条静止负荷为2.0g,用平衡记录仪记录收缩曲线。
每15min换1次营养液,稳定30min待收缩基线平稳后进行试验。
α2受体激动剂应用于紧张型头痛

-52.3
-60
Bettucci D,et al. Combination of tizanidine and amitriptyline in the prophylaxis of chronic tension-type headache: evaluation of efficacy and impact on quality of life. J Headache Pain. 2006 Feb;7(1):34-6
40
32.3
30
28.8
患者比例(%)
20.9 20
14.5
12.8
10
11.3 10.2 9.6 7.6
0
不良情绪 睡眠障碍 阳光
刮风
疲劳
寒冷
炎热 天气变化 饮酒
4.4 4.4 2.3
气味 鼻部疾病 月经周期
Wang J,et al. Triggers of migraine and tension-type headache in China: a clinic-based survey. Eur J Neurol. 2013 Apr;20(4):689-96
50 42
40
• 中国一项入户调查研究结果显示:TTH发病率 占10.8%3
25 23.8
20
发病率(%) 发病率(%)
30
20
10
0 TTH
11 偏头痛
3 CDH
15 10.8
10
5
0 原发性头痛 TTH
9.3 偏头痛
1.0 CDH
1. Stovner L,et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193– 210. 2. Bendtsen L, et al. EFNS guideline on the treatment of tension-type headache - report of an EFNS task force. Eur J Neurol. 2010 Nov;17(11):1318-25. 3. Yu S,et al. The Prevalence and Burden of Primary Headaches in China: A Population-Based Door-to-Door Survey. Headache. 2012 Apr;52(4):58291.
第四节α2激动剂

第四节α2激动剂第四节α-肾上腺素受体激动剂(Alpha-Adrenergic Agonists)⼀、盐酸可乐定(clonidine Hydrochloride)可乐定是中枢α2-去甲肾上腺素受体激动剂,可以优先激动下丘脑及延脑的中枢突触前α2受体,抑制⼤脑的内源性去甲肾上腺素释放。
减少中枢交感神经冲动传出,从⽽抑制外周交感神经活动。
近年来的⼀些研究认为脑内去甲肾上腺素系统在注意缺陷多动障碍的病理机制中具有重要作⽤。
(⼀)药代动⼒学本品⼝服后70%~80%吸收,并很快分布到各器官,组织内药物浓度⽐⾎浆中⾼,能通过⾎脑屏障蓄积于脑组织。
蛋⽩结合率为20%~40%。
⼝服本品后半⼩时到1⼩时发挥作⽤,3~5⼩时⾎药浓度达峰值,⼀般为1.35ng/ml,作⽤持续时间6~8⼩时。
消除半衰期为12.7(6~23)⼩时,肾功能不全时延长。
在肝脏代谢,约50%吸收的剂量经肝内转化。
40%~60%以原形于24⼩时内经肾排泄,20%经肝肠循环由胆汁排出。
(⼆)禁忌症对可乐定过敏者禁⽤。
有⼼⾎管疾病者是相对禁忌症,使⽤时需要进⾏密切监测。
不能⽤于有抑郁症状或抑郁症病史、抑郁症家族史或双向情感障碍家族史的⼉童少年。
(三)药物相互作⽤1.与⼄醇、巴⽐妥类或镇静药等中枢神经抑制药合⽤,可加强中枢抑制作⽤。
2.与其他降压药合⽤可加强降压作⽤。
3.与β受体阻滞剂合⽤后停药,可增加可乐定的撤药综合征危象,故宜先停⽤β受体阻滞剂,再停可乐定。
4.与三环类抗抑郁药合⽤,减弱可乐定的降压作⽤。
可乐定须加量。
5.与⾮甾体类抗炎药合⽤,减弱可乐定的降压作⽤。
可乐定与哌甲酯合⽤1995年美国媒体报道了3名⼉童服⽤哌甲酯与可乐定后死亡,但美国⾷品药品管理局并未将此事通知临床医师,Popper认为这两者间的关系⾮常不能确定。
之后有许多报道,均认为没有令⼈信服的证据说明两者间的关系。
Popper (1995)和Swanson(1995)都认为可乐定与哌甲酯联合⽤于治疗ADHD是安全的,但还缺乏系统的研究。
右美托咪定临床经验分享

Can J Physiol Pharmacol, 2004,82(5):359-62.
➢稳定患者血压、心率及术中血流动力学,提高缺血区/非缺血区
血流比例,降低心肌需氧,减少心肌缺血的发生
➢ 降低围术期心肌缺血和心肌梗塞的发生,降低围术期死亡率
Can J Physiol Pharmacol, 2004,82(5):359-62.
12
• 右美托咪定组平均拔管时间 显著较安慰剂组缩短
• 安慰剂组5.8±1.2 (3~27) min Dex 0.2μg/L组3.6±1.5 (0~13) min Dex 0.4μg/L组2.7±1.3 (0~20) min (P<0.05)
Br J Anaesth, 2006,97(5):658-65.
不良反应及注意事项
•暂时性高血压 •低血压 •心动过缓及窦性停搏 •治疗可能包括减少或停止本品输注,增加静脉液体的流速,抬高下肢,以及使 用升高血压的药物。(静脉给予抗胆碱能药物。例如,格隆溴铵、阿托品) •患有严重心脏传导阻滞和/或严重的心室功能不全的患者 当给予其他血管扩张剂 或负性频率作用药物时,同时给予本品可能有附加的药效影响,应该谨慎给药。
右美——畅快呼吸,拔管无忧
➢ 右美托咪定对自主呼吸无抑制,可安全用于拔管期间患者的
镇静
➢ 机械通气拔管失败的患者,给予右美托咪定,都可成功拔管,且在
拔管期间和拔管后, 患者血流动力学稳定,无躁动
Anesth Analg, 2000,90(4):834-9.
13
Middle East J Anesthesiol, 2002,16(6):597-606.
注0.15~1.2 μg/min右美托咪定(Dex)或安慰剂,至手术结束,评估两组
肾上腺素受体激动

THANKS FOR WATCHING
感谢您的观看
02 肾上腺素受体激动剂
肾上腺素受体激动剂的种类
选择性肾上腺素受体激动剂
只作用于特定的肾上腺素受体亚型,对其他受体亚型影响较小。
非选择性肾上腺素受体激动剂
作用于多种肾上腺素受体亚型,产生广泛的药理作用。
肾上腺素受体激动剂的作用机制
兴奋心血管系统
01
通过激动心肌和血管平滑肌上的肾上腺素受体,增加心肌收缩
开发新型肾上腺素受体激动剂
针对不同亚型肾上腺素受体,开发出具有创新性的肾 上腺素受体激动剂,为疾病治疗提供更多选择。
05 安全使用肾上腺素受体激 动剂
肾上腺素受体激动剂的副作用
心跳加速
肾上腺素受体激动剂可能导致心跳加速,引 发心悸和心律不齐。
血压升高
使用肾上腺素受体激动剂可能导致血压升高, 增加心血管疾病的风险。
心血管疾病
研究肾上腺素受体激动剂在高血压、冠心病 等心血管疾病治疗中的应用,探索其对心血 管系统的保护作用。
神经系统疾病
研究肾上腺素受体激动剂在焦虑、抑郁、帕 金森病等神经系统疾病治疗中的应用,探索 其对神经系统的调节作用。
肾上腺素受体激动的研究前景
深入探究肾上腺素受体激动剂的作用 机制
通过深入研究肾上腺素受体激动剂的作用机制,为新 药研发提供理论急情况
在应激状态下,肾上腺素受体激动可引发一系列生理反应, 如心跳加速、血压升高和血糖升高,以应对紧急情况。
调节代谢
肾上腺素受体在代谢调节中发挥重要作用,如β₂受体激动 促进脂肪和葡萄糖代谢,而β₃受体激动则抑制脂肪合成和 促进脂肪分解。
中枢神经系统作用
肾上腺素受体在中枢神经系统中也有分布,参与神经递质 释放和突触传递等过程,对学习和记忆等认知功能产生影 响。
α2受体激动剂在围术期的应用进展

α2受体激动剂在围术期的应用进展α2受体激动剂与中枢及外周神经系统α2受体特异性结合后产生多种生理效应,以可乐定(clonidine)或美托咪啶(medetomidine)作为代表药物,其具有降压、镇静、镇痛、抗焦虑等多重作用,临床围术期的应用越来越广泛。
现就α2受体激动剂在围术期的应用进展做一综述。
一、α2受体及其亚型α2受体属于α受体亚型,广泛分布于中枢神经系统和外周组织【1】,通过药理学和分子生物学的进一步研究发现,α2受体又可以分成多种亚型,如α2A、α2B 、α2C和α2D等,这些亚型广泛分布在中枢神经系统。
由于α2受体的多样性,以及在动物和人体内各个组织器官的分布多样性,使药物作用于受体产生不同的结果。
临床上最重要的α2受体亚型是α2A和α2B亚型,α2A主要分布于脑干,参与调解知觉、觉醒、和警觉的状态。
α2B亚型主要调解外周血管的紧张度。
物种多样性决定了α2受体亚型在脑干的作用特性。
在犬和鼠的脑干以α2A亚型分布为主,而在绵羊的脑干以α2D 亚型为主【2】。
α2受体激动可以产生镇静和镇痛效应,这种效应不仅和受体所在部位、分布密度以及亚型有关,还与α1、α2和药物的亲和力有关。
目前临床使用的α2受体激动剂均有不同程度的激动α1的作用,因此均有不同程度的α1激动效应【3】。
中枢α1刺激,可以拮抗强效α2受体激动剂产生的催眠作用【4】,而且增加α2受体激动剂药物剂量或达到中毒剂量时,α1激动效应将占优势。
临床使用的α2受体激动剂,其受体选择性越高,其作用效应越显著,因此使用较低的剂量,或者增加药物使用剂量时只需要增加较低的剂量,即可达到预期效应。
下列药物α2/α1受体的选择性如下:美托咪啶(1620:1);地托咪啶(detomidine)(260:1);可乐定(220:1);赛拉嗪(xylazine)(160:1)【5】。
二、α2受体激动剂生理效应α2受体激动可以产生多种效应,包括对意识、循环、呼吸、骨骼肌的影响等等。
α2受体激动剂作用机制及应用资料课件

18
VLPO
组胺能
甘丙肽和γ氨基丁酸能
去甲肾上腺素能
非快动眼相睡眠
18
脑侧室视前核损害可消除右美托咪定的催眠作用
VLPO
组胺能
甘丙肽和γ氨基丁酸能
去甲肾上腺素能
右美托咪定诱导睡眠Leabharlann 22安慰剂-右美托咪定
22
图片
GABA类似物不产生非快速动眼型改变
Nelson et al,Nature NS 220402
ITU催眠和镇痛的另一选择
图片
ITU催眠存在的问题
增加机械通气增加ICU住院时间无法进行神经学检查患谵妄的危险增加
ITU采用可有效唤醒的镇静方式的益处
39
合作对医务人员进行应答镇痛需求物理疗法系统功能的评估呼吸神经系统减少隔离在维持镇静的同时允许唤醒
ICU谵妄诊断治疗的重要性
39
Pun BT, Ely EW Chest 132:624-36,2007
MENDS研究随机双盲对照试验
Pandharipande et al,JAMA204507
MICU/SICU机械通气患者,对镇静剂知情同意
对照组劳拉西泮(GABA)+/-芬太尼
干预组右美托咪定(α2)+/-芬太尼
双盲镇静方案
5cc剂量
1-2cc/小时输注
滴定至靶RASS (最大10cc/小时)
主要终点的评估
右美托咪定长期治疗研究
MICU 机械通气并使用镇静剂2(右美托咪定):1(咪达唑仑)随机分组
对照组咪达唑仑(GABA)+/-芬太尼
干预组 右美托咪定+/-芬太尼
64
右美托咪定长期治疗研究结果
Riker R.The Precedex ICU long term sedation trial SCCM,Feb 3rd,200866
ICU常用的镇静镇痛药物特点及应用

吗啡
副作用:
①呼吸抑制:在低剂量下的使用下亦会产生呼吸次数及呼吸 深度改
第15页/共20页
瑞芬太尼
瑞芬太尼是一种新的短效镇痛药
适应症:可用于短时间镇痛或持续输注的病人,也可用
于肝肾功能不全病人。
给药途径:只能用于静脉给药,特别适用于输液泵静脉
持续滴注
给药速度:
1、成人按的输注速率持续静滴。 2 、 先 给 予 的 初 始 剂第1量6页静/共推20(页 时 间 应 大 于 6 0 秒 ) , 再
成。
变,主要是吗啡会影响到脑干的呼吸中枢所造
②耐药、成瘾
③低血压:吗啡会造成周边血管扩张
④便秘:大多数的病患皆会发生,因吗啡会降低肠胃道的蠕 动并响
中枢神经的排便反射,因而造成便秘。
⑤排尿困难:主要为吗啡会抑制排尿反射,尿液潴留。
⑥恶心、呕吐
⑦皮肤发痒:吗啡会使表皮血管扩张,皮肤发红、发痒。可
用抗织
(B级)。 4、急性疼痛病人的短期镇痛可选用芬太尼。(C级)
第17页/共20页
ICU病人镇痛镇静指南推荐(中国)
7、对急性躁动病人可以使用咪唑安定、安定或丙泊酚来获得快 速的镇静。 (C级)
8、需要快速苏醒的镇静,可选择丙泊酚。 (B级) 9、短期的镇静可选用咪唑安定或丙泊酚。(A级) 10、长期镇静治疗如使用丙泊酚,应监测血甘油三酯水平,并
肌注吸收慢而不规则,20minh达高峰。静脉给药1 -3min起效,15min达高峰,4-10天血药浓度达稳 态。
α受体激动剂

药理学
15
2024/1/5
【禁忌证】
高血压、动脉硬化、器质性心脏病、少 尿或无尿者应慎用或禁用。
药理学
16
2024/1/5
间羟胺 (metaraminol;阿拉明,aramine) 为人工合成品 作用机制: 1.直接作用于1受体和1受体。 对1受体作用较 弱。 2.间羟胺可被肾上腺素能神经末梢摄取,进入囊 泡,置换囊泡中的NA,促进NA释放。
视力。比阿托品作用弱,不升高眼内压.
药理学
20
2024/1/5
Clonidine可乐定
中枢性α2受体激动剂,通过激活抑制性神经元,降 低血管运动中枢的紧张性,使外周交感神经的功能 降低而引起降压作用
适用治疗中度高血压,常用于其他药无效时
适用治疗中度高血压,常用于其他药无效时。降压作用中等 偏强,不显著影响肾血流量和肾小球滤过率,一般用于高 血压的长期治疗。高血压急症用静脉注射,也可静脉滴注。 口服也用于预防偏头痛,或作为治疗吗啡类镇痛药或瘾者 的戒毒药。点眼用于治疗开角型青光眼。
作用机制:只激动α1受体
药理学
19
2024/1/5
[临床应用]
激动血管平滑肌细胞膜上的α1受体,使血管收 缩,血压升高,可用于防治低血压;如果血压 正常,用药后血压超过生理水平,反射性兴奋 迷走神经,使心率减慢,用于治疗阵发性室上 性心动过速;去氧肾上腺素滴眼后可激动瞳孔 开大肌细胞膜上的α1受体,使其收缩,瞳孔扩 大,用于检查眼底,既作用时间短,又不影响
药理学
11
2024/1/5
2.上消化道出血 对食管下端静脉曲张出 血以及胃出血等,取本品1~3mg,适 当稀释后口服,使上消化道粘膜血管剧 烈收缩,发挥局部作用,有助于控制上 消化道大出血。
血管活性药物的临床应用

常用药物
1.肾上腺素 α、β受体激动剂 药理作用: α受体兴奋:血管平滑肌收缩,血压上升。 β1受体兴奋:心肌收缩力增强、心率增快、传导性增
加、心肌耗氧量增加。 β2-受体兴奋:舒张冠脉、支气管平滑肌舒张 ,并抑
制肥大细胞释放过敏性物质
心脏骤停
CPR首选药物,适用于各种原因导致的心脏骤停
主要作用机制:降低左右心室充盈压和全身血管阻力,从而减轻心脏负荷
多巴酚丁胺 短期应用可增加心输出量,改善外周灌注,缓解症状,2-20ug/kg▪min静滴,使用时监测血压
肾上腺素受体阻滞剂 2、多巴酚丁胺:充分体液复苏后仍然存在低心排出量,可以使用多巴酚丁胺增加心排出量,若同时存在低血压可以考虑联合使用升压
胆碱能系统 β1受体兴奋:心肌收缩力增强、心率增快、传导性增加、心肌耗氧量增加。
一、急性心衰、心源性休克
如果出现低血压或对起始的肾上腺素剂量无反应,静脉给予1:10000肾上腺素。
钙通道阻滞剂 5-单硝酸异山梨酯静脉滴注的起效、达峰、达稳态的时间明显延迟于同等剂量的口服片,弹丸式静脉推注虽可明显加快起效时间,但
2、去甲肾上腺素 过敏性休克
广义:血管活性药物通过作用于血管上受体或直接改变血管平滑肌张力,影响心脏前负荷、后负荷,或通过影响心脏的变力、变时效
应来产生血管收缩、舒张效应。
α受体激动药。α受体兴奋作用强于肾上腺素,β1受体 当其中任何一因素的改变,超出了人体的代偿限度时,即可导致有效循环血量的急剧下降
3.多巴胺
药理作用:
小剂量(1-2μg/kg/min)主要兴奋外周多巴胺受体, 有肾血管扩张作用,尿量可能增加;同时兴奋心脏β1受体,有轻度正性肌力作用。
中等剂量(2-5μg/kg/min)β1,β2受体激动作用, 表 现为心肌收缩力增强,心率增快不明显,能显著改善 心力衰竭的血流动力学异常。
α2肾上腺素能受体激动剂在疼痛治疗中的使用【内容详细】

α2肾上腺素能受体激动剂在疼痛治疗中的使用从1970年开始,α2肾上腺素能受体激动剂在临床上被用来治疗高血压和药物及乙醇的戒断症状。
这类药物能产生抗焦虑、镇静、抗交感及镇痛等多种作用,因此可以用于手术期间以满足不同的需要。
目前在西方国家中有3种α2肾上腺素能受体激动剂在临床中使用,它们分是可乐定、右美托咪啶和替扎尼定,但在中国右美托咪啶尚未上市。
因此还是有必要就这类药物向中国的疼痛学专家作个简要介绍。
α2肾上腺素能受体在体内分布广泛,当α2肾上腺素能受体激动剂与其结合后就能产生临床效应。
α2肾上腺素能受体有3种亚型,分别是α2a, α2b andα2c,α2肾上腺素能受体激动剂结合每种不同的亚型都能产生独特的效应,例如α2a受体能产生麻醉、镇痛及抗交感作用(低血压和心动过缓),α2b受体有间接升高血压的作用(血管收缩),α2c受体与感觉与运动门控欠缺有关,如精神分裂症, 注意力缺乏及过动症,创伤后功能障碍和停药反应(调节多巴胺的活性)。
在中枢神经系统中α2受体亚型有不均匀的分布,3种受体中α2a受体最普遍且到处存在,α2b 受体仅存在于少数部位。
所有的α2肾上腺素能受体激动剂都是不同程度地作用于各受体亚型,所有的受体亚型都是通过结合G蛋白而产生细胞效应,尤其是对百日咳-毒素易感的G蛋白:Go和G1。
因为没有选择性亚型受体激动剂可供使用,所以想只产生单一所需要的α2肾上腺素能效应可能是不行的,如只是产生镇痛作用,而不会产生其他不利作用如低血压等。
激活α2肾上腺素能受体可抑制腺苷酸环化酶,导致cAMP生成减少,cAMP是许多细胞作用的重要调节剂,它能通过cAMP 依赖的蛋白激酶而控制调节蛋白的磷酸化状态。
另外α2肾上腺素能受体兴奋导致了神经递质释放受到抑制,这是通过在电压门控钙离子通道中钙离子的减少而介导的,这个过程需要结合一个Go蛋白。
激活α2肾上腺素能受体还可加速Na+-H+的交换,引起血小板内部碱化,刺激磷脂酶A2活性的增加,最终导致血栓素A2的生成增多。
右美托咪定药理特点及对脏器保护作用的研究进展殷国英吴慢慢张晨许国庆李艳美(通讯作者)

右美托咪定药理特点及对脏器保护作用的研究进展殷国英吴慢慢张晨许国庆李艳美 *( 通讯作者 )发布时间:2023-06-09T07:17:10.733Z 来源:《医师在线》2023年5期作者:殷国英吴慢慢张晨许国庆李艳美 *( 通讯作者 )[导读] 右美托咪定(dexmedetomidine,DEX)作为一种高度选择性 α2 受体激动剂,在临床上不仅可用于镇静和减轻焦虑,还能抑制交感神经、稳定血流动力学、减轻应激反应和发挥抗炎作用,因此,右美托咪定在临床上被广泛应用。佳木斯大学黑龙江佳木斯 154000摘要:右美托咪定(dexmedetomidine,DEX)作为一种高度选择性α2受体激动剂,在临床上不仅可用于镇静和减轻焦虑,还能抑制交感神经、稳定血流动力学、减轻应激反应和发挥抗炎作用,因此,右美托咪定在临床上被广泛应用。此外,越来越多的研究表明右美托咪定具有保护多个器官的作用。本文就右美托咪定的药理特点及其脏器保护作用和相关机制进行综述,为其在临床应用提供参考。关键词:右美托咪定;α2肾上腺素受体;器官保护作用1右美托咪定的药理特点药代动力学:DEX是一种咪唑类衍生物,具有高度选择性地激活α2肾上腺素受体的特性。它的α2受体亲和力比可乐定高8倍(α2与α2受体亲和力之比为1600∶1)。DEX的分布半衰期约为6分钟,消除半衰期大约为2小时。经由颊黏膜给药时,它的吸收效果良好。肌肉注射DEX2μg/kg后,平均12分钟达到血浆峰值浓度,终末半衰期为(281±177)分钟[1]。DEX还表现出很高的亲脂性,几乎完全被生物转化,随后通过粪便和尿液排出。对于那些肝脏或肾脏功能受损者而言,其药物清除率低于正常人,因此DEX应用的剂量应减少[2]。药效动力学:机体的α2肾上腺素受体主要分布于交感神经末梢和中枢神经系统的肾上腺素神经细胞中。这些受体有三种亚型(α2 A、α2 B和α2 C),各自发挥不同的作用。据认为,α2 A受体亚型介导了非选择性α2肾上腺素受体激动剂的镇静和抗交感神经效应,α2 C受体亚型介导了其激动剂的抗焦虑效应。激动剂与α2 B受体亚型结合后可引起短暂的血压升高反应。DEX发挥镇静和催眠效应的机制是作用于蓝斑核,其抗伤害性感受效应则主要是通过对脊髓后角的作用而实现的。此外,DEX还能够通过对外周和中枢的作用来发挥抗交感活性的效应[3]。2 DEX对器官的保护作用2.1脑的保护作用:研究表明,脑缺血和缺氧会导致儿茶酚胺(CA)过度释放,可以导致神经细胞发生钙超载,进而生成大量神经毒性自由基,从而加重缺血和脑积水,增加神经细胞对谷氨酸的敏感性,最终导致神经细胞受损[4-5]。DEX可以有效降低CA的生成,减轻脑血管痉挛和其伴随的脑损伤[6]。此外,创伤性损伤、脑缺血、术后认知功能障碍等问题与神经细胞炎症反应及细胞凋亡密切相关。DEX可以通过抑制TLR4/NF-κB、JAK2-STAT3和NF-κB/COX-2通路抑制神经细胞炎症产生,从而发挥保护作用。DEX还能够通过减少神经细胞凋亡发挥保护效应。细胞程序性死亡被称为细胞凋亡,它是由多种基因介导的。关键的凋亡调节蛋白包括Bcl-2、BCL-XL和Bax。研究表明右美托咪定直接作用于α2 A受体,促进脑源性神经营养因子(BDNF)的表达。BDNF通过调节ERK1/2通路,激活PI3K/Akt信号通路和MAPK通路,同时抑制p38和c-Jun N-末端激酶(JNK)通路,从而促进了Bcl-2和BCL-XL的表达,减少神经细胞凋亡[7]。2.2心脏的保护作用:患者在手术过程中容易受到外科手术、气管插管等刺激,可能会引起机体交感神经兴奋,导致心脏并发症,如心动过速、血压升高、心肌缺血等。DEX通过降低交感神经的兴奋性,减弱应激反应,稳定血流动力学,有效预防围手术期心肌缺血。据临床观察证实,DEX可降低术后死亡率和心肌梗死概率[8-9]。据文献[10]检索发现,DEX的心脏保护机制主要有三个方面:(1)抑制蓝斑核去甲肾上腺素神经细胞活性,降低血液中CA水平、心脏负荷和心肌耗氧量,同时增加左室冠状动脉血流量,提高心肌对缺血缺氧的抵抗力;(2)去甲肾上腺素(NE)大量释放能导致心律失常的风险增高,DEX能通过减少NE的释放,降低高风险患者心律失常发生的概率;(3)当心脏缺血再灌注(IR)损伤之前应用DEX,可以激活PI3K/Akt、MEK1/2-ERK1/2等信号通路,减弱IR引起的细胞凋亡和炎症反应,从而缩小心肌梗死面积。但其下游分子机制仍有待于进一步研究发现。2.3肺脏的保护作用:肺是一个敏感的器官,易遭受到全身炎症反应综合征(SIRS)和远端器官IR损伤的影响。许多临床实践,如创伤、单肺通气、体外循环和肝移植,都可能引起肺损伤,主要通过炎症和细胞凋亡来实现。近年来的研究表明,DEX通过影响肺血管的收缩机制以及炎性因子的释放等作用,可以在许多肺损伤的情况下发挥肺保护作用。研究发现在肾IR损伤造成的急性肺损伤实验中,发现髓过氧化物酶、细胞间粘附分子-1及TNF-α mRNA的表达会显著升高。DEX能下调它们的表达水平,显著减弱炎症反应,从而减轻肺水肿[11-12]。Lameire等[13]在脓毒症大鼠模型中发现,DEX能通过TLR4/MyD88/NF-κB信号通路抑制肺部炎症反应。吴晓静[14]等在大鼠胸部创伤模型中发现,NF-κB的活性会增强,DEX可以抑制其活性,降低促炎因子的表达,进而对肺挫伤产生保护作用。此外实验[15]发现,DEX还可以减轻脂多糖(LPS)诱导的急性肺损伤中的线粒体氧化应激和细胞凋亡,并发现DEX对肺保护作用的强度与其浓度呈抛物线关系。2.4肾脏的保护作用:肾脏是一个重要的器官,占据了心输出量的20%,其中90%-95%分布在皮质。围手术期的休克、血管活性药物的应用都容易造成肾实质特别是肾皮质损伤。IR会导致交感神经活动增强,引起肾皮质血管收缩加剧。DEX可以通过局部舒张肾脏血管来改善肾外髓质的血流量,提高肾小球过滤率,减弱精氨酸加压素在集合管的作用,减少水通道蛋白的表达,促进尿液排泄,从而改善肾脏缺血损伤[16-17]。DEX还可以扩张机体血管、加快血液循环、减少红细胞聚集所造成血液瘀滞,从而减轻IR对肾脏的危害。此外,肾IR损伤会导致TLR4/NF-κB、JAK/STAT信号通路被激活,引起NF-κB的转录增多,而NF-κB是炎症相关酶诱导型一氧化氮合酶的启动子,NF-κB表达上调可促进一氧化氮释放,造成氧化应激水平上升,线粒体损伤加重。DEX可以抑制上述途径,从而减轻肾IR损害[10]。2.5肝脏的保护作用:IR会造成肝脏不同程度的损害,严重影响患者的预后。肝组织IR损害后可见肝窦淤血、炎性细胞浸润、局灶性坏死。肝组织损害的过程极其复杂,常常伴随细胞内信号通路、细胞和病理生理学的改变[18]。张世霞[19]等研究发现,肝IR之前应用DEX能显著提高BRL-3A细胞的超氧化物歧化酶和谷胱甘肽活性,同时降低了活性氧和丙二醛的水平,缓解了氧化应激,减轻了肝脏IR损害。另一项研究[20]显示,DEX可以减轻LPS引起的肝损伤。其是因为DEX可以引起乙酰胆碱、α7 烟碱型乙酰胆碱受体表达上升,TLR4、MyD88、NF-κB表达下降,降低肿瘤坏死因子-α、白介素-6等炎症因子的水平,减弱了炎症反应,最终减轻了LPS所造成的肝损害。同时该研究还发现DEX预处理后,可以调节肝细胞凋亡相关蛋白caspase-3和Bcl-2的表达,抑制了肝细胞凋亡,从而发挥保护肝脏作用。但α7nAChR与TLR4活化的关系有待进一步研究。3结语综述所述,DEX是一种常用的临床药物,由于其广泛的应用价值而值得推广。它能通过多种机制对大脑、心脏等多个器官产生保护作用,但仍有部分机制尚未未完全清楚,仍需更多的基础和临床实验来阐明。参考文献[1]张青,王竹梅.右美托咪啶的药理作用及临床应用进展[J].贵州医药, 2016, 40(5): 544-546.[2]赵秀洁, 王业文, 宋倩倩, 等. 右美托咪定药理作用及肺保护作用机制的研究进展[J]. 中国医学创新, 2022, 19(33): 185-188.[3]张燕, 郑利民. 右美托咪啶的药理作用及临床应用进展[J]. 国际麻醉学与复苏杂志, 2007, 28(6):544-547.[4] J. Lutz, L. A. Luong, M. Strobl et al., “The A20 gene protects kidneys from ischaemia/reperfusion injury by suppressing pro-inflammatory activation,” Journal of Molecular Medicine (Berlin, Germany), vol. 86, no. 12, article 405, pp. 1329–1339,2008.[5] C. G. da Silva, E. R. Maccariello, S. W. Wilson et al.,“Hepatocyte growth factor preferentially activates the anti-inflammatory arm of NF-κB signaling to induce A20 and protect renal proximal tubular epithelial cells from inflammation,” Journal of Cellular Physiology, vol. 227,no. 4, pp. 1382–1390, 2012.[6] U. Kunter, S. Daniel, M. B. Arvelo et al., “Combined expression of A1 and A20 achieves optimal protection of renal proximal tubular epithelial cells,” Kidney International,vol. 68, no. 4, pp. 1520–1532, 2005.[7] ZHAO Y, HE J, YU N, et al. Mechanisms of Dexmedetomidine in Neuropathic Pain[J/OL]. Frontiers in Neuroscience, 2020, 14: 330.[8] D. N. Wijeysundera, J. S. Naik, and W. S. Beattie, “Alpha-2 adrenergic agonists to prevent perioperative cardiovascular complications: a meta-analysis,” The American Journal of Medicine, vol. 114, no. 9, pp. 742–752, 2003.[9] B. M. Biccard, S. Goga, and J. de Beurs, “Dexmedetomidine and cardiac protection for non-cardiac surgery: a meta-analysis of randomised controlled trials,” Anaesthesia,vol. 63, no. 1, pp. 4–14, 2008.[10] BAO N, TANG B. Organ-Protective Effects and the Underlying Mechanism of Dexmedetomidine[J/OL]. Mediators of Inflammation, 2020, 2020: 6136105.[11] J. Gu, J. Chen, P. Xia, G. Tao, H. Zhao, and D. Ma, “Dexme-detomidine attenuates remote lung injury induced by renal ischemia-reperfusion in mice,” Acta Anaesthesiologica Scan-dinavica, vol. 55, no. 10, pp. 1272–1278, 2011.[12] N. Bao and D. Dai, “Dexmedetomidine protects against ischemia and reperfusion-induced kidney injury in rats,”Mediators of Inflammation, vol. 2020, Article ID 2120971,8 pages, 2020.[13] N. H. Lameire, A. Bagga, D. Cruz et al., “Acute kidney injury:an increasing global concern,” Lancet, vol. 382, no. 9887,pp. 170–179, 2013.[14] X. Wu, X. Song, N. Li, L. Zhan, Q. Meng, and Z. Xia, “Protec-tive effects of dexmedetomidine on blunt chest trauma-induced pulmonary contusion in rats,” Journal of Trauma and Acute Care Surgery, vol. 74, no. 2, pp. 524–530, 2013.[15] C. Fu, X. Dai, Y. Yang, M. Lin, Y. Cai, and S. Cai, “Dexmede-tomidine attenuates lipopolysaccharide-induced acute lung injury by inhibiting oxidative stress, mitochondrial dys function and apoptosis in rats,” Molecular Medicine Reports,vol. 15, no. 1, pp. 131–138, 2017.[16] F. T. Billings, S. W. C. Chen, M. Kim et al., “α2-Adrenergic agonists protect against radiocontrast-induced nephropathy in mice,” American Journal of Physiology-Renal Physiology,vol. 295, no. 3, pp. F741–F748, 2008.[17] A. J. Rouch, L. H. Kudo, and C. Hebert, “Dexmedetomidine inhibits osmotic water permeability in the rat cortical collecting duct,” The Journal of Pharmacology and Experimental Therapeutics, vol. 281, no. 1, pp. 62–69, 1997.[18] TÜFEK A, TOKGÖZ O, ALIOSMANOGLU I, elat.The protective effects of dexmedetomidine on the liver and remote organs against hepatic ischemia reperfusion injury in rats[J/OL]. International Journal of Surgery (London, England), 2013, 11(1): 96-100.[19] ZHANG S, TANG J, SUN C, et al. Dexmedetomidine attenuates hepatic ischemia-reperfusion injury-induced apoptosis via reducing oxidative stress and endoplasmic reticulum stress[J/OL]. International Immunopharmacology, 2023, 117: 109959.[20] ZI S F, LI J H, LIU L, et al. Dexmedetomidine-mediated protection against septic liver injury depends on TLR4/MyD88/NF-κB signaling downregulation partly via cholinergic anti-inflammatory mechanisms[J/OL]. International Immunopharmacology, 2019, 76: 105898.。
第二节_阿片类镇痛药
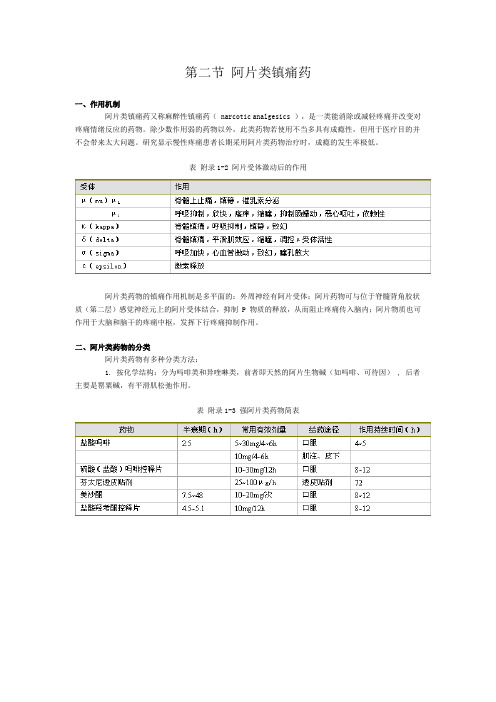
第二节阿片类镇痛药一、作用机制阿片类镇痛药又称麻醉性镇痛药( narcotic analgesics ),是一类能消除或减轻疼痛并改变对疼痛情绪反应的药物。
除少数作用弱的药物以外,此类药物若使用不当多具有成瘾性,但用于医疗目的并不会带来太大问题。
研究显示慢性疼痛患者长期采用阿片类药物治疗时,成瘾的发生率极低。
表附录1-2 阿片受体激动后的作用阿片类药物的镇痛作用机制是多平面的:外周神经有阿片受体;阿片药物可与位于脊髓背角胶状质(第二层)感觉神经元上的阿片受体结合,抑制 P 物质的释放,从而阻止疼痛传入脑内;阿片物质也可作用于大脑和脑干的疼痛中枢,发挥下行疼痛抑制作用。
二、阿片类药物的分类阿片类药物有多种分类方法:1. 按化学结构:分为吗啡类和异喹啉类,前者即天然的阿片生物碱(如吗啡、可待因) , 后者主要是罂粟碱,有平滑肌松弛作用。
表附录1-3 强阿片类药物简表表附录1-4 弱阿片类药物简表2. 按来源该类药物可分为天然阿片类、半合成衍生物 ( 如双氢可待因,二乙酰吗啡 ) 和合成的阿片类镇痛药。
合成药物又分为四类:①苯丙吗啡烷类 (phenylpiperidine derivatives) ,如哌替啶、芬太尼等;②吗啡喃类 (morphinenans) ,如左吗喃;③苯异吗啡烷类 (bengmorphans) ,如喷他佐辛;④二苯甲烷类 (diphenylmethanes) ,如美散酮。
3. 按受体类型可分为μ、κ、δ受体,该三种受体的分子结构已被确定,并被成功克隆。
从功能上还可能存在ε和δ受体,并可能进一步分为μ 1 、μ 2 、κ 1 、κ 2 、κ 3 和δ 1 、δ 2 等亚型。
表 3-2 为受体激动后的药理作用。
4. 按药理作用分,阿片类镇痛药又可分为激动药 ( 吗啡、芬太尼、哌替啶等 ) ,激动一拮抗药 ( 喷他佐辛、纳布啡等 ) ,部分激动药(丁丙诺啡)和拮抗药 (纳洛酮等) 。
a2肾上腺素能受体激动剂

0
10
20
30
B组
40
A组
50
心率(bpm)
60
70
80
90
心率变化
100
右美托咪啶Ⅱ期临床研究(国内)
右美托咪啶Ⅱ期临床研究(国内)
两组病人用药总剂量(mg)比较
药物
组别
x ±s
min M max
P
芬太尼
A组
0.16±0.07 0.10 0.15
0.30
0.0024
B组 0.14±0.07 0.10 0.10 0.40
副交感神经
运动神 经系统
M2
乙酰胆碱
肾上腺髓质 N1
M2
乙酰胆碱
N1
肾上腺素及去甲 肾上腺素入血
α2
肾上腺素 去甲肾上腺素
全身肾上腺素能受体
M2
乙酰胆碱
N1
M2
乙酰胆碱
M2
乙酰胆碱
胆碱能受体
N2
a2受体对神经递质释放的调节作用
突触囊泡
负反馈 (-)
去甲肾上腺素
a2受体 a1受体
a2受体的分布及其作用
睡眠时间(min)
右美托咪啶的剂量依赖性镇静作用
右美托咪啶(mg/kg)
右美托咪啶调节镇痛的部位
• 脊髓上的很多位点 • 脊髓a2受体也可能参与 右美托咪啶
• 最重要的作用位点可能
为脊髓
硬膜外或鞘内注射
• 外周确切的机制和通路
尚不清楚
初级传入纤维
皮层 丘脑 中脑 延髓 投射神经元
脊髓
右美托咪啶的镇痛作用
助用量。
– 无呼吸抑制产生。 – 心率减慢和血压轻度下降并可预见。 – 病人易于唤醒,给予刺激时有警觉性。
α2肾上腺素能受体激动剂在疼痛治疗中的使用

α2肾上腺素能受体激动剂在疼痛治疗中的使用从1970年开始,α2肾上腺素能受体激动剂在临床上被用来治疗高血压和药物及乙醇的戒断症状。
这类药物能产生抗焦虑、镇静、抗交感及镇痛等多种作用,因此可以用于手术期间以满足不同的需要。
目前在西方国家中有3种α2肾上腺素能受体激动剂在临床中使用,它们分是可乐定、右美托咪啶和替扎尼定,但在中国右美托咪啶尚未上市。
因此还是有必要就这类药物向中国的疼痛学专家作个简要介绍。
α2肾上腺素能受体在体内分布广泛,当α2肾上腺素能受体激动剂与其结合后就能产生临床效应。
α2肾上腺素能受体有3种亚型,分别是α2a, α2b andα2c,α2肾上腺素能受体激动剂结合每种不同的亚型都能产生独特的效应,例如2a受体能产生麻醉、镇痛及抗交感作用(低血压和心动过缓),α2b 受体有间接升高血压的作用(血管收缩),2c受体与感觉与运动门控欠缺有关,如精神分裂症, 注意力缺乏及过动症,创伤后功能障碍和停药反应(调节多巴胺的活性)。
在中枢神经系统中α2受体亚型有不均匀的分布,3种受体中2a受体最普遍且到处存在,2b受体仅存在于少数部位。
所有的α2肾上腺素能受体激动剂都是不同程度地作用于各受体亚型,所有的受体亚型都是通过结合G蛋白而产生细胞效应,尤其是对百日咳-毒素易感的G蛋白: Go和G1。
因为没有选择性亚型受体激动剂可供使用,所以想只产生单一所需要的α2肾上腺素能效应可能是不行的,如只是产生镇痛作用,而不会产生其他不利作用如低血压等。
激活α2肾上腺素能受体可抑制腺苷酸环化酶,导致cAMP生成减少,cAMP是许多细胞作用的重要调节剂,它能通过cAMP依赖的蛋白激酶而控制调节蛋白的磷酸化状态。
另外α2肾上腺素能受体兴奋导致了神经递质释放受到抑制,这是通过在电压门控钙离子通道中钙离子的减少而介导的,这个过程需要结合一个Go蛋白。
激活α2肾上腺素能受体还可加速Na+-H+的交换,引起血小板内部碱化,刺激磷脂酶 A2活性的增加,最终导致血栓素A2的生成增多。
α受体激动药

欢迎阅读拟肾上腺素药α受体激动药一、α1受体激动药1、去甲肾上腺素1)药理作用:是肾上腺素能神经末梢释放的递质,肾上腺髓质分泌少量。
上腺素虽肾血流低下但仍可迅速改善利尿状态。
因此,去甲肾上腺素近年来再次被临床重视。
体内过程:该药起效迅速,停药后1-2min失效,大部分被体内酶代谢。
2)临床应用:主要用于治疗低血压,特别对于感染性休克高排低阻者,在充分扩容后应用可显着见效;嗜铬细胞瘤切除后低血压应持续静脉注射一段时间;近年来常与扩血管药物并用,治疗低心排血量或难治性休克。
具体用法:2-4mg,加入500ml水中,每分钟1ml。
每小时应该是60ml。
一3452素,效应稍弱,但持续时间长。
主要作用α1受体,对β受体作用弱。
长时间滴注可耗尽体内储存的去甲肾上腺素而失效,应改用去甲肾上腺素。
3、甲氧明:只选择性激动α1受体,对其他受体无作用;只激动小动脉α1受体,很少兴奋小静脉α1受体,所以回心血量不多,血压升高不明显;可反射性促进迷走神经兴奋,故能治疗室上性心动过速。
成人剂量10-20mg静脉注射。
4、去氧肾上腺素:性质稳定,作用时间5-10min。
直接激动α1受体,收缩小动脉和小静脉,并稍有促去甲肾上腺素释放功能。
对β受体作用弱,升高血压可反射性减慢心率,对心肌应激性小。
按血压二、α21因不阻滞肾上腺素受体,可保持机体正常反射功能。
显效时间:30-60min,峰值时间2-4h。
临床应用:治疗高血压,成人0.1-1.2mg∕d,分次口服。
临床麻醉:麻醉前用药:口服0.2-0.3mg(5μg∕kg)可显着镇静,减少麻醉药及阿片类药物剂量40-50%,还有预防气管插管的心血管副反应等。
椎管内应用:有显着的镇痛效应,并延长镇痛时间,无恶心、呕吐、乏力及呼吸抑制现象。
(70-150μg)神经阻滞时应用:可强化镇痛效应,延长镇痛时间(150μg)。
注意事项:长期应用突然停药可出现反跳性高血压和心律失常。
可能有噩梦、嗜睡、不安、焦虑和压抑感。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.Maze.White paper;2000. 2.Khan et al.Anaesthesia.1999;54:146-155. 3.Kamibayashi,Maze.Anesthesiology.2000;93:1345-1349
甘丙肽和γ氨基丁酸能
VLPO
去甲肾上腺素能
TMN:结节乳头核
LC
VLPO:脑侧室视前核 LC:蓝斑
唤 醒
(+) HIS 组胺能 TMN
甘丙肽和γ氨基丁酸能
VLPO
(-) NE 去甲肾上腺素能 TMN:结节乳头核
LC
VLPO:脑侧室视前核 LC:蓝斑
非快动眼相睡眠
组胺能 TMN (-) 甘丙肽和γ氨基丁酸能
假说
以α2受体为作用靶点
(右美托咪定) 与以GABAA受体为靶点的镇静方案(劳拉西泮)相比, 对进行机械通气的内外科ICU患者更易达到镇静 目的,并缩短谵妄和昏迷时间.
Maximize Efficacy of targeted sedation and reduce Neurological Dysfunction 靶向镇静的最大效应及减少神经系统功能障碍
MENDS 研究 • 主要结果
– 急性脑功能障碍的持续时间和患病率(谵妄和昏迷) – 用右美托咪定或劳拉西泮获得理想的镇静水平
• 其他结果
– 不用机械通气的日数 – ICU和医院内入住日数 – 28天死亡率
Pandharipande et al,JAMA2007
MENDS研究 随机双盲对照试验
MICU/SICU机械通气患者,
Fischer大鼠结节乳头核应用GABA 对右美托咪定催眠作用的影响
右美托咪定诱导的LORR时间 250 NS
200
150 100 50 0
GBZ
Nelson et al, Anesthesiology 2003
非快动眼相睡眠
组胺能 TMN (-) 甘丙肽和γ氨基丁酸能
VLPO
(-)
去甲肾上腺素能
ITU采用可有效唤醒的镇静方式的益处
• 合作
– 对医务人员进行应答
• 镇痛需求 • 物理疗法
– 系统功能的评估
• 呼吸
• 神经系统
– 减少隔离
• 在维持镇静的同时允许唤醒
ICU谵妄 诊断治疗的重要性
Pun BT, Ely EW Chest 132:624-36,2007
ICU谵妄的危险因素
• 年龄 • 痴呆病史 • 基础疾病
500
睡眠时间(min)
250
0 结节乳头核双侧注射毒蝇母 Nelson et al,Nature NS 2002
GABA类似物对睡眠相关神经基质的作用
催眠 皮质 皮质下区
组胺能结节乳头体核 (-) 甘丙肽和γ氨基丁酸能 脑侧室视前核
(-)
TMN
毒蝇母 (-)
下丘脑前部
VLPO
NE
基底前脑
去甲肾上腺素能蓝斑核
VLPO
(-)
去甲肾上腺素能
TMN:结节乳头核
LC
VLPO:脑侧室视前核 LC:蓝斑
大鼠全身应用GABA 对右美托咪定催眠反应的影响
100 80 %LORR 60 40 20 0 对照 +GBZ
1
2
4
6
8
10
右美托咪定剂量(μg/0.2μl)
Nelson et al,Nat Neurosci 2003
“尽管接受插管或机械通气,接受右美托咪定镇
静的患者仍可轻松唤醒并配合诊疗操作(如 理疗,X线检查),无烦躁”
在右美托咪定和咪达唑仑镇静期间被 噪音唤醒并完成任务的能力比较
110 100
任务和噪音
任务
%采样数
90 80 70 60 50 安慰剂 右美托咪定 咪达唑仑
图片
穹隆周核食欲素能神经元 在唤醒过程中起关键作用
• Orion, Abbott提供咨询和实验室支持
• 右美托咪定未获准连续输注超过24小时
报告内容
• 右美托咪定和可乐定的药代动力学特性
• 右美托咪定催眠作用的药效学特性
• 右美托咪定催眠作用的分子机制
• 右美托咪定催眠作用的神经基质
• 与其他催眠剂的比较 • 右美托咪定在ITU的临床预后研究
α-2受体激动剂
ITU催眠和镇痛的另一选择
• 图片
ITU催眠存在的问题
• 增加机械通气
• 增加ICU住院时间
• 无法进行神经学检查
• 患谵妄的危险增加
Kollef,M et al.Chest,1998;114:541-548
Pandharipande et al.Anesthesiology 2006;124:21-6
LDGg/PPTg
清醒
PeF LC TMN Raphe
(ox)
“开”
VLPO eVLPO
睡眠
“关”
与催眠等剂量的全麻过程中 食欲素能神经元的活性
c-Fos-ir核食欲素能神经元%
60 50 40 30 20 10 0 SAL DEX ISO PTB PRO MUS
* * *
*
解
释
右美托咪定的催眠作用与自然睡眠 相似,且具有有效的唤醒系统
– 炎症 – 凝集(?凝血障碍?)
• 精神活性药物 (苯二氮卓类,阿片类) • 睡眠缺乏
• 代谢紊乱 • 血氧不足 • 遗传易患性(?)
Inouye, JAMA 1996;275:852-57 Dubois,Interns Care Med 2001;27:1297-1304 Inouye,NEJM 1999;340:669-676 Mibrandt,Crit Care Med.2005;33:116-9
150(0,922)
镇静效果适当的患者
P=0.04
右美托咪定
劳拉西泮
坐标轴标注看不清楚
Pandharipande et al,JAMA2007;298:2644-53
过度镇静的患者
右美托咪定
劳拉西泮
Pandharipande et al,JAMA2007;298:2644-53
镇静不足的患者
右美托咪定
0.69
镇静效果
人口统计学资料
劳拉西泮(n=51)
右美托咪定(n=51) P值 80(58,100) 67(50,85) 575(140,2206) 0.04 0.008 0.006
达到护理目标的日数% 67(48,83) 达到医生目标的日数% 55(8,67) 每日平均芬太尼
给药剂量: 右美托咪定 - 0.74(0.39-1.04)mcg/kg/hr 劳拉西泮 - 3(2.2,6)mg/hr
主要终点的评估
• 谵妄
– +ve ICU谵妄诊断的意识状态评估法 (CAM-ICU)
• 精神状态突然发生改变 • 注意力不集中 • 意识水平和思维无序性改变
• 昏迷
– RASS>-4(只对物理刺激有反应)
• 纳入超过12天时间 (5天镇静时间+之后7天停止镇静的时间)
基线特征
人口统计学资料 劳拉西泮(n=51) 年龄 男性 APACHE II SOFA评分 认可的诊断 败血症/ARDS COPD 肺及其他疾病 39% 4% 22% 37% 4% 23% 0.78 0.99 0.85 59(45,67) 45% 27(24,32) 9(7,11) 右美托咪定(n=51) 60(49,65) 58% 29(24,32) 10(8,2) P值 0.96 0.20 0.75 0.23
对镇静剂知情同意
对照组
干预组
劳拉西泮(GABA)
+/-芬太尼
右美托咪定(α2)
+/-芬太尼
Pandharipande et al,JAMA2007
双盲镇静方案
5cc剂量
1-2cc/小时 输注
滴定至靶RASS (最大10cc/小时)
Pandharipande et al,JAMA2007;298:2644-53
TMN:结节乳头核
LC
VLPO:脑侧室视前核 LC:蓝斑
右美托咪定可使参与非快动眼相睡眠的 神经元活性发生改变
Nelson et al, Anesthesiology 2003
非快动眼相睡眠
组胺能 TMN (-) 甘丙肽和γ氨基丁酸能
VLPO
(-)
去甲肾上腺素能
TMN:结节乳头核
LC
VLPO:脑侧室视前核 LC:蓝斑
脑侧室视前核损害 可消除右美托咪定的催眠作用
100
表现为慢波脑电图的时间%
生理盐水 右美托咪定
80 60 40 20 0
脑侧室视前核损害 右美托咪定+脑侧室视前核损害
Nelson et al, Anesthesiology 2003
右美托咪定诱导睡眠
组胺能 TMN (-) 甘丙肽和γ氨基丁酸能
VLPO
α-2受体激动剂作用机制及应用
Mervyn Maze
右美托咪啶
• 右美托咪定的催眠特性
– 1986年在斯坦福被发现 – 与 Mika Scheinin共用专利资料 – 恢复本人对Orion-Farmos的专利权 – 本人在斯坦福的实验室获得$250000
• Abbott实验室获得美国市场权,并将此权利归还 Hospira
自然睡眠的潜在益处
• 避免睡眠不足