高等有机化学第四章有机反应中间体解析
合集下载
高等有机化学第四部分4-4金属有机化合物的制备.ppt
(三)其它价态金属
4EtCl + 4NaPb 4EtBr + Mg2 Sn
Et4 Pb + 4NaCl + 3Pb E4t Sn + 2MgB2r
2C6H6 + Cr*
Cr(C6H6)2
PEt3
C6F5Br +Ni* + 2 Et3P
C6F5 _ Ni Br
PEt3
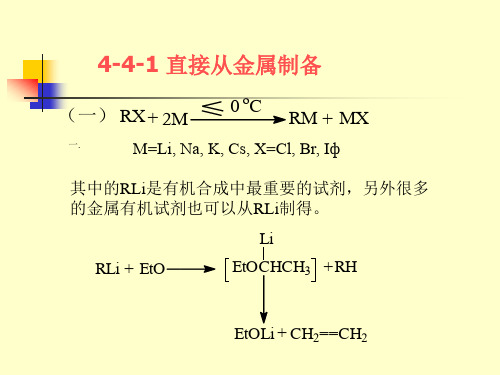
4-4-2 从金属有机试剂的 交换反应制备
尽管这些取代基有的推电子,有的吸电 子,但都得到邻位的Li化试剂,这主要是诱导 效应和取代基时Li的配位作用所至。
Li
MeO
R
H
MeO Li
当然这反应也不限于芳基金属试剂,如:
+ ( i Pr )2NU1 HMPT
N
CH2LI W
(四)金属化合物对双键的加成
1、 RLi + C C
R C C Li ( 增加两个碳 )
M=Fe,Cr,V
这类金属交换反应在有机合成中非常有用,
因为可以直接从金属本身制备的金属有机化 合物太活泼,反应的选择性差,而选择性高 的金属有机试剂活性低,难以直接从金属制 备,所以只好用间接方法。
(二)另一种交换方式
R M + R' X
R' M + RX
其中用得最多的是Li交换反应。
BuLi +
2、M(CO)6 + RLi [ Me3O]BF4
(CO)5M
C
OLi R
(CO)5M
C
OMe R
( Fisher Carbene)
3、B2H6 + 6MeCH2CH CH2
2Bu3
高等有机化学4

醛酮羰基的亲核加成
醛酮羰基与含碳亲核试剂的亲核加成 Wittig-Horner反应
(EtO)3P + RCH2Br
亚磷酸酯
EtO + O Et
-EtBr
P EtO CH2R
Br-
R'CHO R
H Mechanism:
H
O
+ (EtO)2P-O-Na+ R'
溶于水
OEt
O O P OEt + -C
R' H H R
(2) 立体选择性高, 以E-型产物为主, 副产物二乙基磷酸盐溶于水, 容易除去23.
醛酮羰基的亲核加成
醛酮羰基与含碳亲核试剂的亲核加成 Wittig-Horner反应的应用
1.
NaH
O + (EtO)2P(O)CH2CO2Et
CHCO2Et 67-77%
2.
NaOEt
O + (EtO)2P(O)CH2CO2Et
3
Ph Me
Ph
Ph Ph
Proposed mechanism: Cl4Sn
Cl4Sn O
O
-HCl
CH2 Me
H Ph C
H2
Cl3Sn O Ph
Ph O Me
Ph H
OSnCl4 Ph
Cl4Sn
SnCl4
H O H2C
Cl3Sn -HCl
O
CH2
O Me Ph
Ph
Ph
Ph
第四章 碳杂不饱和键的亲核加成
许家喜 北京化工大学
1. 醛酮C=O键的亲核加成 2. 亚胺C=N键的亲核加成 3. 腈CN键的亲核加成 4. 累积双键的亲核加成
南开大学高等有机化学课件第四章有机反应机理的研究和描述

Ea ln k ln A RT
R: 气体常数, A: 频率因数,
在不同温度下测速率常数, 可计算出 Ea: Arrhenius活化能
Ea ΔH RT ΔS Ea log k 10 .753 log T 4.576 4.576T
4.3.1 简单速率表达式的积分形式
正常情况下动力学数据用微分方程的积分形式来处理:
如简单的一级反应和二级反应:
1 C0 一级级反 : k ln( ) t C 1 b0(a) 二级反应 : k (a0 - b0)ln t a0(b)
a, b, c: 时间t时浓度
a0, b0, c0: 起始浓度
一些反应速率方程积分形式的推导:
4.2 动力学数据(Kinetic Data)
动力学数据使我们能更详细地洞察反应机理。用跟踪反 应物消失和产物出现的方法可以测定某一个反应的速度。波 谱技术提供了一个迅速又连续地监测浓度变化的方法,因而 往往被用来测量反应进行的程度。总之,任何与一种反应物 或产物的浓度有关而且能被测量的性质,都可利用来测定反 应速度。 动力学研究的目的是为了在反应物和催化剂的浓度以及 反应速度之间建立定量关系。
k1[A][B] [C] k -1
d[D] k1 k2[C] k2 [A][B] kobs.[A][B] dt k -1
大多数反应不止一步, 可以参考一些重要的多步反应例子来得出动 力学表达式, 例如在决速步之前可以有一个快速平衡:
ROH + H+
+ ROH2
快 k1 k -1
_
ROH2 RBr +H2O
计算出一个反应的自由能变化,就使反应平衡位置的计算有了 可能,也就指出了某一化学过程的可实现性。 有兴趣的反应大多数发生在溶液中,任何这种反应的焓、熵和 自由能都与溶剂介质有关。 但是,热力学数据并不能说明是否存在一个能量上有利的潜在 反应途径,即反应速度上的情报。因此,深入了解反应机理以及 有机反应进行是中间所经各步的速度和能量要求是极为重要的。
高等有机化学第四部分4-6金属有机化合物

O
四、反应还被用来合成高分子(特殊 的高分子,如导电子分子)
X Ar MgCl
Ni (Ar)n
Br Ar Br+ (HO)2B Ar B(OH)2 Pd
(Ar) (Ar')
*从机理看反应应用了 a)O.A. b)烷基化 c)R.E.
R'M'
RX
M(O) O.A
R MX
M'X R
M R'
R R' +M(o)
实验二:
CH3 CH = CH CH3
+
CO3 CO = CO CO3
CH3 CH = CO CO3
"W"
CH3 CH = CH CO3
CH3 CO = CO CH3
CH3 CH = CO CH3
只有这种产物所以证明反应中没有氢或烷基的转移。
最早认为机理:
R M ⅡⅡ
M
R
R
RR
R
R
到1972年.E.O.Fishec发现:
1、
(CO)5CR = C
Ar OCH3
+ CH2 = C
CO3Et H
Ar CH2 = C OMe
2、
Ar (CO)5W = C Ar +
CH2 = C
OCH3 Ar
(CO)5W = C
R.E.
O ?Ⅱ ?Ⅱ ?Ⅱ
X_ Pd
OH
R
X
O
Pd R
HO ?Ⅱ ?Ⅱ H
O
Ph R CH CH2 + CO
O
Ph
R CH CH2 CO
O
β_ ⅡⅡ
O CH2 Rh CHR
高等有机化学课件4-第四章 活性中间体

CH2
CH2
CH CH2
-
-
CH2
CH CH2
CH2
CH2
CH2
CH3CN
CH2(CN)2
CH(CN)3
pKa
25
CH3NO2
12
CH2(NO2)2
0
CH(NO2)3
pKa 10.2
O CH3CCH3
4
CH2(COOEt)2 COCH3 CH2 COOEt
0
COCH3 CH2 COCH3
pKa 20
CH3 H3C C OH CH3 CH3
HSO3F - SbF5 - SO2 -60℃
H3C C + H +O + SO F3 3 CH3 + SbF + SO
5 2
很多碳正离子的结构与稳定性的研究都是在超酸 介质中进行的。
(2)正离子或Lewis酸对中性分子加成
C CH2 H+
BF3
C CH3
H C CH2 BF3
乙、诱导效应
pKa
H3C H
43
F3C
H
(F3C)3C H
28
11
(F3C)3C > F3C > H3C
R3C < R2CH < RCH2 < CH3
R3N CH2 > R2N CH2
R3N CH3
+
+
-
R3N+ CH2D
DO-
R3N CH2
+
-
D2O
丙、共轭效应: 碳负离子sp2杂化
CH2 CH CH2 CH2 -
(CH3)3C > (CH3)2CH > CH3CH2 > H3C
高等有机第四章有机化学反应中间体

2014-12-28
18
5 .伯胺与 HNO2 作用,先生成重氮离子,然后失去 N2形成碳正离子。 R-NH2 → R-N2+ → R+ + N2↑ (二)亲电试剂与重键加成 1.烯烃酸催化水合
C C + H+ C CH
2.羧基化合物的氧质子化
R C R O + H
+
R R C OH
2014-12-28
31
(四)空间效应
不饱和键与碳负离子的电子对共轭,则碳负离子必 然成为平面构型,以利于 P 轨道的最大重叠;若结构上 或空间上因受阻碍达不到最大重叠,则相应碳负离子的 稳定性就小。
1 O H H O O H O
1,3-环己二酮 可与NaOH液反应
双环[2.2.2]辛-2,6-二酮 不能与NaOH水液反应,
(一)S特性效应(杂化效应)
S轨道比相应的 P 轨道离原子核较近,故原子核对S 轨 道中的电子吸引力比相应P轨道的大。这种差别也反映在 杂化轨道中。
轨道吸电子能力: SP > SP2 > SP3 碳负离子稳定性: CH≡C- > -CH=CH2 > -CH2-CH3 这种影响是由于碳原子杂化轨道中S成分不同造成的, 称S特性效应,又叫杂化效应。
CH3NO2
CH2NO2
2014-12-28
34
2 . 碱性条件下脱羧(C-C键异裂)
CN C2H5 C COOH C6H5 碱 CN C2H5 C C6H5
(二)亲核试剂与重键加成(略)
四.碳负离子的反应
(一)对重键的加成
2014-12-28
35
1. 对羰基的加成
C O + R-MgX R C OMgX H3O R C OH
有机活性中间体-苯炔

实验结果表明: (1)重排产品中引入的基团在脱掉的原子的邻位; (2)重排产品是从邻位脱掉原子(重氢)形成的。因而这些 反应是通过一个对称的中间体苯炔进行的。
环加成反应(Diels-Alder反应)
苯炔是具有高度反应活性的中间体,它的反应不受生成方法的影响, 即不论苯炔的来源如何,和同一作用物反应形成的产品是相同的。 苯炔的反应总是涉及到对“三键”的加成,从而在产品中恢复其芳香性。 苯块的反应可以是极性的。也可以是协同的环加成。
关于苯炔的结构,倾向性的意见为:
除了脱掉两个相邻的氢原子以外,苯环上基本没被扰乱。 在相邻的sp2杂化轨道之间必然有重叠,这样形成一个很弱 的键,这种情况和观察到的活泼性是一致的,红外光谱也 表明苯炔应具有这样的结构。
苯炔的反应
苯炔(或去氢苯)是从苯消除两个邻位取代基得到的具有高度反应活性的中间体。 芳香亲核取代反应过去存在着许多难以解释的现象。如用强碱处理芳香卤代物, 在某些情况下不仅形成正常的取代产品。而且同时也得到异构的化合物,其中 新的取代基在原来连接卤素的碳原子的体主要有: 碳正离子、碳负离子、自由基、 碳正离子、碳负离子、自由基、卡宾 碳烯)、乃春(氮烯)和苯炔等。 )、乃春 (碳烯)、乃春(氮烯)和苯炔等。
影响活性中间体稳定性的因素
诱导效应 共轭效应 空间效应 芳香性及其结构
苯炔的结构
苯炔的结构式可表示为:
有机反应活性中间体—苯炔
简介及意义
– 简介:所谓“活性中间体”就是指具有一定的反应活性 简介:所谓“活性中间体” 或不稳定性的中间体。 或不稳定性的中间体。有机反应的活性中间体不同于中 间过渡状态,它是真实存在的,且不少的活性中间体目 间过渡状态,它是真实存在的, 前已经被人们通过物理或化学的方法检测到或分离出 例如1900年Gowmberg首次发现的三苯甲基自由基 来.例如1900年Gowmberg首次发现的三苯甲基自由基 ((C6H5 )3一C·)就是一种中间体。 就是一种中间体。 就是一种中间体 – 意义:有机化学反应类型很多,反应过程也复杂。根据 意义:有机化学反应类型很多,反应过程也复杂。 过渡状态理论, 过渡状态理论,有机反应中反应物分子一般是通过一个 或几个能量最高的过渡状态形成产物,或者是经过某些 或几个能量最高的过渡状态形成产物, 中间体形成产物的。因此, 中间体形成产物的。因此,讨论反应活性中间体的稳定 性对研究有机化学反应的活性具有十分重要意义。 性对研究有机化学反应的活性具有十分重要意义。
高等有机化学-活性中间体

C
CH3
溶剂
高 等 有 机 化 学
7
Advanced Organic Chemistry
碳正离子的反应
1)与负离子或具有未成对电子的中性分子结合
R + Y
Ph CH + OHCH3
CH 3 CH3 C CH 3 CH 2 CH 3 C CH 3 + H C(CH3) 3 CH 3
R
Y
OH
Ph CH CH3
高 等 有 机 化 学
16
Advanced Organic Chemistry
4)消去反应 E1历程
RCH2 CHR' X RCH2 CHR' B RCH2 CHR' + BH -X slow RCH2 CHR'
单分子历程;一级反应;生成碳正离子中间体
E1 CH3 H3C C Cl CH3 slow H3C CH3 C CH3 (CH3)2C CH2 + H3O H2 O fast SN1 17%
Br Br Br 10-6 10-13
相对速度 1
10-3
桥头碳正离子的形成比较困难
高 等 有 机 化 学
Advanced Organic Chemistry
溶剂效应
1) 溶剂的诱导极化作用,利于底物的解离 2) 溶剂使正碳离子稳定 3) 极性溶剂:溶剂化作用 强,利于底物的解离 空的 p 轨道 易于溶剂化
结构 生成 性质 反应
高 等 有 机 化 学
1
Advanced Organic Chemistry
碳正离子(Carbocations)
结构
R R C R
H
sp2杂化
有机化学第4章 反应机理讲解

(fǎnyìng)。 以甲烷氯代反应(fǎnyìng)为例,整个反应(fǎnyìng)过程如图4.1所示。
链引发 Cl2
光
2Cl
a
链增长 CH4 + Cl
CH3 + HCl
b
CH3 + Cl2
CH3Cl + Cl
c
链终止 2 Cl
Cl2
Cl + CH3
CH3 + CH3
CH3Cl CH3CH3Fra bibliotek精品资料
精品资料
6
(1)链引发:这是反应(fǎnyìng)起始阶段,产生一个活性自由基中间体。 (2)链增长:链引发阶段产生的活性自由基中间体和稳定分子反应(fǎnyìng)形
成另一个活性自由基中间体,使反应(fǎnyìng)链不断传递下去。 (3)链终止:破坏活性自由基中间体及减慢或终止反应(fǎnyìng)的副反应
子中间体接受亲核试剂进攻时可以从平面两侧被进攻,而且几 率相等,结果得到外消旋产物(构型保持和构型转化各为 50%)。 4.2.3.3 影响亲核取代反应的因素 (1) 卤原子对亲核取代活性的影响 C-X键键能与极性大小次序相同(xiānɡ tónɡ): C-F>C-Cl>C-Br>C-I。 因此卤代烃亲核取代反应活性次序应该是: RI>RBr>RCl>RF。
A
A
精品资料
1
4.1.2 反应机理分类 有机(yǒujī)反应的实质就是化学键的旧键断裂和新键生成。
即在一定的条件下,有机(yǒujī)化合物分子中的成键电子 发生重新分布,原有的化学键断裂,新的化学键形成,从
而使原分子中原子间的组合发生了变化,新的分子产生。 4.1.2.1按化学键的断裂和生成可以将有机(yǒujī)化学反应
链引发 Cl2
光
2Cl
a
链增长 CH4 + Cl
CH3 + HCl
b
CH3 + Cl2
CH3Cl + Cl
c
链终止 2 Cl
Cl2
Cl + CH3
CH3 + CH3
CH3Cl CH3CH3Fra bibliotek精品资料
精品资料
6
(1)链引发:这是反应(fǎnyìng)起始阶段,产生一个活性自由基中间体。 (2)链增长:链引发阶段产生的活性自由基中间体和稳定分子反应(fǎnyìng)形
成另一个活性自由基中间体,使反应(fǎnyìng)链不断传递下去。 (3)链终止:破坏活性自由基中间体及减慢或终止反应(fǎnyìng)的副反应
子中间体接受亲核试剂进攻时可以从平面两侧被进攻,而且几 率相等,结果得到外消旋产物(构型保持和构型转化各为 50%)。 4.2.3.3 影响亲核取代反应的因素 (1) 卤原子对亲核取代活性的影响 C-X键键能与极性大小次序相同(xiānɡ tónɡ): C-F>C-Cl>C-Br>C-I。 因此卤代烃亲核取代反应活性次序应该是: RI>RBr>RCl>RF。
A
A
精品资料
1
4.1.2 反应机理分类 有机(yǒujī)反应的实质就是化学键的旧键断裂和新键生成。
即在一定的条件下,有机(yǒujī)化合物分子中的成键电子 发生重新分布,原有的化学键断裂,新的化学键形成,从
而使原分子中原子间的组合发生了变化,新的分子产生。 4.1.2.1按化学键的断裂和生成可以将有机(yǒujī)化学反应
第四章有机反应活性中间体介绍

轨道交盖
H 空的 p 轨道
CC H
H
Liaocheng University
Organic Advanced Chemistry
②共轭效应
CH2 CH CH2
CH
CH2
CH2
p-π共轭
共轭体系的数目越多,碳正离子越稳定
(CH2=CH)3C+ > (CH2=CH)2CH+ > CH2=CHCH2+
Ph3C+ > Ph2CH+ > PhCH2+
常见的活性中间体有:碳正离子、碳 离子、自由基、卡宾、乃春、苯炔等六种。
Liaocheng University
Organic Advanced Chemistry
一. 碳正离子 (Carbocations() 亲电反应中间体)
含义:带正电荷的三价碳原子的原子团。 最常见
特点:缺电子,∵带正电荷的碳有六个价电子。
Liaocheng University
C6H13CHCH 3 2BuLi I
C6H13CHCH 3 Li
1)CO2 2)H3O+
C6H13CHCH
3
COOH
-70℃时,60%构型保持;0 ℃时,外消旋化
2. 碳负离子稳定性
1)诱导效应
-I:分散负电荷,使碳负离子稳定;反之亦然
CH3- > MeCH2- > Me2CH- > Me3C-
Liaocheng University
Organic Advanced Chemistry
常见化合物的pKa值
化合物
CH4 CH2CH2
C6H6 PhCH3 Ph2CH2 CF3H CHCH CH3CN CH3COCH3 PhCOCH3
H 空的 p 轨道
CC H
H
Liaocheng University
Organic Advanced Chemistry
②共轭效应
CH2 CH CH2
CH
CH2
CH2
p-π共轭
共轭体系的数目越多,碳正离子越稳定
(CH2=CH)3C+ > (CH2=CH)2CH+ > CH2=CHCH2+
Ph3C+ > Ph2CH+ > PhCH2+
常见的活性中间体有:碳正离子、碳 离子、自由基、卡宾、乃春、苯炔等六种。
Liaocheng University
Organic Advanced Chemistry
一. 碳正离子 (Carbocations() 亲电反应中间体)
含义:带正电荷的三价碳原子的原子团。 最常见
特点:缺电子,∵带正电荷的碳有六个价电子。
Liaocheng University
C6H13CHCH 3 2BuLi I
C6H13CHCH 3 Li
1)CO2 2)H3O+
C6H13CHCH
3
COOH
-70℃时,60%构型保持;0 ℃时,外消旋化
2. 碳负离子稳定性
1)诱导效应
-I:分散负电荷,使碳负离子稳定;反之亦然
CH3- > MeCH2- > Me2CH- > Me3C-
Liaocheng University
Organic Advanced Chemistry
常见化合物的pKa值
化合物
CH4 CH2CH2
C6H6 PhCH3 Ph2CH2 CF3H CHCH CH3CN CH3COCH3 PhCOCH3
高等有机课件2活性中间体

度高
生物合成法的 缺点:反应速 度慢,需要较 长的反应时间
生物合成法的 应用:在药物 合成、天然产 物合成等领域
有广泛应用
物理法制备活性中间体
物理法制备活性中间体的原理
物理法制备活性中间体的注意事项
添加标题
添加标题
物理法制备活性中间体的步骤
添加标题
添加标题
物理法制备活性中间体的应用实例
06
高等有机化学课件2活 性中间体的检测与表征
结构特征
活性中间体: 有机化学反应 中的关键中间
产物
结构特点:具 有较高的反应 活性和选择性
稳定性:活性 中间体通常具 有较高的稳定 性,不易分解
反应性:活性 中间体具有较 高的反应活性, 易于发生化学
反应
应用:活性中 间体在合成化 学、药物化学 等领域具有广
泛的应用
04
高等有机化学课件2活 性中间体的应用
活性中间体的形成
反应条件:温 度、压力、催
化剂等
反应过程:反 应物转化为活
性中间体
活性中间体的 性质:不稳定、 易反应、易分
解
活性中间体的 应用:合成有 机化合物、研 究化学反应机
理
03
高等有机化学课件2活 性中间体的性质
稳定性
热稳定性:在高温下不易分解
化学稳定性:在酸碱条件下不 易发生化学反应
光稳定性:在光照条件下不易 发生化学反应
生物稳定性:在生物体内不易 发生化学反应
反应性
活性中间体具有较高的反应活性,能够参与化学反应 活性中间体在反应过程中容易发生化学反应,生成新的化合物 活性中间体在反应过程中容易发生化学反应,生成新的化合物 活性中间体在反应过程中容易发生化学反应,生成新的化合物
生物合成法的 缺点:反应速 度慢,需要较 长的反应时间
生物合成法的 应用:在药物 合成、天然产 物合成等领域
有广泛应用
物理法制备活性中间体
物理法制备活性中间体的原理
物理法制备活性中间体的注意事项
添加标题
添加标题
物理法制备活性中间体的步骤
添加标题
添加标题
物理法制备活性中间体的应用实例
06
高等有机化学课件2活 性中间体的检测与表征
结构特征
活性中间体: 有机化学反应 中的关键中间
产物
结构特点:具 有较高的反应 活性和选择性
稳定性:活性 中间体通常具 有较高的稳定 性,不易分解
反应性:活性 中间体具有较 高的反应活性, 易于发生化学
反应
应用:活性中 间体在合成化 学、药物化学 等领域具有广
泛的应用
04
高等有机化学课件2活 性中间体的应用
活性中间体的形成
反应条件:温 度、压力、催
化剂等
反应过程:反 应物转化为活
性中间体
活性中间体的 性质:不稳定、 易反应、易分
解
活性中间体的 应用:合成有 机化合物、研 究化学反应机
理
03
高等有机化学课件2活 性中间体的性质
稳定性
热稳定性:在高温下不易分解
化学稳定性:在酸碱条件下不 易发生化学反应
光稳定性:在光照条件下不易 发生化学反应
生物稳定性:在生物体内不易 发生化学反应
反应性
活性中间体具有较高的反应活性,能够参与化学反应 活性中间体在反应过程中容易发生化学反应,生成新的化合物 活性中间体在反应过程中容易发生化学反应,生成新的化合物 活性中间体在反应过程中容易发生化学反应,生成新的化合物
有机反应活性中间体 ppt课件

(1)碳负离子的结构
C
109°28′
..
90°
C
sp3杂化 棱锥型
烷基负碳离子为棱锥型
sp2杂化 平面三角型
孤对电子处于sp3杂化轨道上,轨道夹角为109°28′ 时,电子对间的排斥力小,利于负碳离子稳定。
(2)碳负离子的生成
C H+ B C+HB
共轭酸
共轭碱
负碳离子: 带有一对
孤对电子的三 价碳原子的原 子团。
(1)自由基的电子构型
●
●
●
sp3
sp2
sp3
是sp3杂化的棱锥构型或sp2杂化的平面构型。
自由基的稳定性顺序: 苄基,烯丙基自由基(sp2) > 叔碳自由基(sp3) > 仲碳自 由基(sp2) > 伯碳自由基(sp2) > 甲基自由基 > 芳基自 由基
(2)自由基的生成
主要有三种产生自由基的方法:热解、光解 和氧化还原。
三苯甲基正离子利用核磁共振谱证明为螺旋浆结构, 苯环之间的夹角为58°,这主要是因为苯环邻位氢原 子的相互排斥的结果。
+
C
- + -
(C6 H5)3 CAC l l4
+ (C6 H5)3 CCO l 4
(2)碳正离子的生成: a、直接离子化
通过化学键的异裂而产生。
RX
RX
Ph CHCl Ph
Ph2CH Cl
A、热解 在加热的情况下,共价键可以发生均裂而产生自
由基。
RR
2 R
均裂的难易主要取决于共价键的强度,即键的离解能。
自由基反应中常以过氧化物或偶氮化合物作为引发剂: 过氧化苯甲酰(BPO)、偶氮二异丁腈(AIBN),主要由 于其分子中含有较弱的键,容易均裂而产生自由基。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
正电荷分散程度大
共轭体系的数目越多,正碳离子越稳定:
CH2 CH 3C > CH2 CH 2CH > CH2 CHCH2
当共轭体系上连有取代基时,供电子基团使正碳离子 稳定性增加;吸电子基团使其稳定性减弱:
CH3
CH2 >
CH2 > O2N
CH2
环丙甲基正离子比苄基正离子还稳定:
3C >
2CH > CH2 >
含有带负电荷的三价碳原子的原子团。 是最早被确认的活性中间体
1、碳负离子的结构
两种构型: 未共用电子对占据p轨道
未共用电子对占据sp3杂化轨道
有利构型!
桥头碳负离子 角锥结构可以快速翻转,不具有手性
三元环碳负离子难于翻转 得到构型保持的氘代产物
当碳负离子与相邻的不饱和体系共轭时,平面结 构变为有利结构
CH2
环丙甲基正离子的结构:
C
其结果是使正电荷分散
CH2
空的 p 轨道与弯曲轨道的交盖
随着环丙基的数目增多,
CH2
CH2
正碳离子稳定性提高。
直接与杂原子相连的碳正离子结构:
氧上未共有电子对所 占 p 轨道 与中心碳原子上的空的 p轨道 侧面交盖,未共有电子对离域, 正电荷分散。
CH3 O CH2
CH3O CH2
HC CH
NaNH3 液 NH3
HC CNa
NH3
Ph3C H
NaNH3 液 NH3
Ph3CNa
NH3
CH3COCH2COOEt NaOEt CH3COCHCOOEt
常用的碱 ■ 有机锂试剂:n-BuLi, PhLi, MeLi ■ KOBut ■ LDA
不亲核碱
2)金属-卤素交换 3)碳负离子对双键的加成
H3C H3C
C CH3
sp2 - sp3 σbond
(CH3)3C + 的轨道结构
碳正离子的稳定性:
CH3 3C > CH3 2CH > CH3CH2 > CH3
σ- p超共轭效应:
轨道交盖在这里
H
空的 p 轨道
CC HH
共轭效应
烯丙型碳正离子:
CH2 CH CH2
CH
CH2
CH2
p-π共轭
电子离域
可发生邻基参与的几种情况: 1)σ-参与作用
外型和内型降冰片醇的对溴苯磺酸酯(2-OBs) 的醋酸解速度比为350倍
外型
HOAc OBs
HOAc OAc
内型 OBs
1,6- σ键的协助作用
2)n-参与作用
离去基团的邻位或更远位带有未共用电子 对的原子(团)时,所体现的参与作用,有 立体构型保留现象。
类似地,羰基正离子:
RCO R C O
乙烯型碳正离子:
HC C R
苯基正离子:
H
+ C原子进行sp2杂化, p轨道 用于形成π键,空着的是sp2 杂化轨道,使正电荷集中。
结构同乙烯型碳正离子,正电荷 集中在sp2杂化轨道上。
此两类碳正离子稳定性极差。(不离域)
溶剂效应:
1) 溶剂的诱导极化作用,利于底物的解离。
2) 溶剂使正碳离子稳定:
空的 p 轨道 易于溶剂化
3) 极性溶剂:溶剂化作用 强,利于底物的解离。
C CH3
溶剂
2、碳正离子的生成
1) 直接离子化
通过化学键的异裂而产生。
RX
RX
Ph CH Cl Ph
R OH H
Ph2CH Cl
ROH2
R H2O
2) 对不饱和键的加成
CZ H
C ZH Z: O,C,S,N
[-CH2CN]
3、碳负离子的稳定性 影响负碳离子稳定性的因素:
1) s-特性效应
HC C > CH2 CH > CH3CH2
2) 诱导效应和共轭效应 1o 2o 3o 超共轭效应不利于碳负离子的稳定
连有吸电子基的碳负离子一般比较稳定
常见的取代基对碳负离子的稳定性作用大小: NO2 > RCO > CN, CO2R, CONH2 > SO2R > SOR > X > Ph > SR >> H > R
3)-参与作用 C=C双键以及芳环的-键的邻基参与作用 反 7-降冰片烯的对甲基苯磺酸酯的醋酸解
通过烯丙基非经典碳正离子中间体,体现了C=C 双键的邻基参与作用。
双键上取代基的电子效应对邻基参与作用的影响
环丙烷代基对邻基参与作用的影响
芳环的-键的邻基参与作用
苯鎓离子中间体 AcO-
二、 碳负离子
CC
HCl
CC
Cl
C O H C OH
C OH
3) 由其它正离子转化而生成
NH2 NNaaNNOO32
N2
N2
HCl
4) 在超酸中制备碳正离子溶液 比100%的H2SO4的酸性更强的酸-超酸 (Super acid)
常见的超酸
与100%H2SO4的酸性比较
HSO3F (氟硫酸) HSO3F - SbF5 (魔酸) HF-SbF5
2、碳负离子的生成
1)碱夺氢
C H + B C + HB
共轭酸
共轭碱
碳氢酸的酸性一般很弱,被夺氢后可以生成其共 轭碱碳负离子。当碳氢酸的邻位没有可以稳定碳负 离子的基团时,较难从碳氢酸形成碳负离子。
碳氢酸的酸性较难确定,强碱是必要的。但是, 在质子溶剂中,强碱会更加容易夺取溶剂分子中的质 子。极性非质子溶剂通常作为测定碳氢酸酸性的介质。 如乙醚、THF、DMSO等 。
经典碳正离子指能用个别的lewis结构式表示的碳 正离子,其价电子层有6个电子,中心碳原子与3 个原子(团)相连。
非经典碳正离子指正电荷离域在3个碳原子之间 的桥型碳正离子。
实验表明: 反-7-原冰片烯基对甲苯磺酸酯 在乙酸中的溶剂解的速度比相应的饱 和化合物大 1011倍
TsO H
-TsO
AcO H
第四章 有机反应活性中间体
碳正离子 自由基
碳负离子
卡宾
乃春
苯炔
一、 碳正离子 含有带正电荷的三价碳原子的原子团。
1、碳正离子的结构和稳定性
构型: p轨道为空轨道
sp3杂化轨道为空轨道
sp3杂化轨道为空轨道的情况: 桥头碳正离子不可能为平面构型
H
C+
H
H
sp2 - s σbond
CH3+ 的轨道结构
1000倍 103倍 1016倍
叔丁醇在下列条件下完全转变成叔丁基正离子:
H3C
CH3 C OH CH3
HSO3F - SbF5 - SO2 -60℃
H3C
CH3 C + H3+O + SO3FCH3 + SbF5 + SO2
很多碳正离子的结构与稳定性的研究都是在超酸 介质中进行的。
3、非经典碳正离子
AcOH
Ts = CH3
SO2
+7
2电子3中心体系
5
1
3
4
2
4、邻基参与效应
邻基参与效应指在分子中邻近的亲核取代基参与 了在同一分子中另一部位上的取代反应
是除电子效应和空间立体效应之外的取代基效应 可从立体化学或反应速率上表现出来
有邻基参与的反应特点: 1)反应速率显著加快
2)反应中心的立体化学得以保留 3)可生成分子重排产物