奥思平(盐酸度洛西汀肠溶片)药品说明书
盐酸度洛西汀FDA说明书(英文)

1HIGHLIGHTS OF PRESCRIBING INFORMATIONdays of stopping an MAOI intended to treat psychiatric disorders.These highlights do not include all the information needed to use In addition, do not start Cymbalta in a patient who is being treated CYMBALTA safely and effectively. See full prescribing with linezolid or intravenous methylene blue (4.1) information for CYMBALTA. •Use in patients with uncontrolled narrow-angle glaucoma (4.2)CYMBALTA (duloxetine hydrochloride) Delayed-Release Capsules for Oral Use.Initial U.S. Approval: 2004WARNING: SUICIDAL THOUGHTS AND BEHAVIORS See full prescribing information for complete boxed warning. • Increased risk of suicidal thinking and behavior in children,adolescents, and young adults taking antidepressants (5.1) • Monitor for worsening and emergence of suicidal thoughtsand behaviors (5.1) • Cymbalta is not approved for use in pediatric patients (8.4) ---------------------------RECENT MAJOR CHANGES --------------------------Dosage and Administration:Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders (2.5) 10/2012 Use of Cymbalta with Other MAOIs such as Linezolid or Methylene Blue (2.6) 10/2012 Contraindications – Monoamine Oxidase Inhibitors (4.1) 10/2012 Warnings and Precautions: Serotonin Syndrome (5.4) 10/2012 Discontinuation of Treatment with Cymbalta (5.7) 08/2012 ----------------------------INDICATIONS AND USAGE ---------------------------Cymbalta ® is a serotonin and norepinephrine reuptake inhibitor (SNRI) indicated for: • Major Depressive Disorder (MDD) (1.1) • Generalized Anxiety Disorder (GAD) (1.2)• Diabetic Peripheral Neuropathic Pain (DPNP) (1.3)• Fibromyalgia (FM) (1.4)• Chronic Musculoskeletal Pain (1.5)------------------------DOSAGE AND ADMINISTRATION-----------------------• Cymbalta should generally be administered once daily withoutregard to meals. Cymbalta should be swallowed whole andshould not be chewed or crushed, nor should the capsule beopened and its contents be sprinkled on food or mixed with liquids (2) Indication Starting Dose Target DoseMaximumDose MDD (2.1, 2.2)40 mg/day to 60 mg/day Acute Treatment: 40 mg/day (20 mg twice daily) to 60 mg/day (once daily or as 30 mg twice daily); Maintenance Treatment: 60 mg/day 120 mg/day GAD (2.1) 60 mg/day 60 mg/day (once daily) 120mg/day DPNP (2.1) 60 mg/day 60 mg/day (once daily) 60 mg/day FM (2.1)30 mg/day 60 mg/day (once daily) 60 mg/day Chronic Musculoskeletal Pain (2.1)30 mg/day 60 mg/day (once daily) 60 mg/day • Some patients may benefit from starting at 30 mg once daily (2.1) • There is no evidence that doses greater than 60 mg/day confersadditional benefit, while some adverse reactions were observed to be dose-dependent (2.1) • D iscontinuing Cymbalta: A gradual dose reduction isrecommended to avoid discontinuation symptoms (2.4, 5.7) ----------------------DOSAGE FORMS AND STRENGTHS---------------------20 mg, 30 mg, and 60 mg capsules (3)-------------------------------CONTRAINDICATIONS ------------------------------• Serotonin Syndrome and MAOIs: Do not use MAOIs intended totreat psychiatric disorders with Cymbalta or within 5 days ofstopping treatment with Cymbalta. Do not use Cymbalta within 14------------------------WARNINGS AND PRECAUTIONS -----------------------• Suicidality: Monitor for clinical worsening and suicide risk (5.1) • Hepatotoxicity: Hepatic failure, sometimes fatal, has been reported in patients treated with Cymbalta. Cymbalta should be discontinued in patients who develop jaundice or other evidence of clinically significant liver dysfunction and should not be resumed unless another cause can be established. Cymbalta should not be prescribed to patients with substantial alcohol use or evidence of chronic liver disease (5.2)• Orthostatic Hypotension and Syncope: Cases have been reportedwith duloxetine therapy (5.3)• Serotonin Syndrome: Serotonin syndrome has been reported withSSRIs and SNRIs, including with Cymbalta, both when taken alone, but especially when co-administered with other serotonergic agents (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone and St. John’s Wort). If such symptoms occur, discontinue Cymbalta and initiate supportive treatment. If concomitant use of Cymbalta with other serotonergic drugs is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases (5.4) • Abnormal Bleeding: Cymbalta may increase the risk of bleedingevents. Patients should be cautioned about the risk of bleeding associated with the concomitant use of duloxetine and NSAIDs, aspirin, or other drugs that affect coagulation (5.5, 7.4)• Severe Skin Reactions: Severe skin reactions, including erythemamultiforme and Stevens-Johnson Syndrome (SJS), can occur with Cymbalta. Cymbalta should be discontinued at the firstappearance of blisters, peeling rash, mucosal erosions, or any other sign of hypersensitivity if no other etiology can be identified. (5.6)• Discontinuation: May result in symptoms, including dizziness,headache, nausea, diarrhea, paresthesia, irritability, vomiting, insomnia, anxiety, hyperhidrosis, and fatigue (5.7) • Activation of mania or hypomania has occurred (5.8)• Seizures: Prescribe with care in patients with a history of seizuredisorder (5.9)• Blood Pressure: Monitor blood pressure prior to initiatingtreatment and periodically throughout treatment (5.10)• Inhibitors of CYP1A2 or Thioridazine: Should not administer withCymbalta (5.11)• Hyponatremia: Cases of hyponatremia have been reported (5.12) • Hepatic Insufficiency and Severe Renal Impairment: Should ordinarily not be administered to these patients (5.13)• Controlled Narrow-Angle Glaucoma: Use cautiously in thesepatients (5.13)• Glucose Control in Diabetes: In diabetic peripheral neuropathicpain patients, small increases in fasting blood glucose, and HbA 1c have been observed (5.13)• Conditions that Slow Gastric Emptying: Use cautiously in these patients (5.13)• Urinary Hesitation and Retention (5.14)-------------------------------ADVERSE REACTIONS------------------------------• Most common adverse reactions (≥5% and at least twice theincidence of placebo patients): nausea, dry mouth, somnolence, constipation, decreased appetite, and hyperhidrosis (6.3). To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or /medwatch.-------------------------------DRUG INTERACTIONS ------------------------------• Potent inhibitors of CYP1A2 should be avoided (7.1). • Potent inhibitors of CYP2D6 may increase duloxetineconcentrations (7.2).• Duloxetine is a moderate inhibitor of CYP2D6 (7.9). ------------------------USE IN SPECIFIC POPULATIONS-----------------------• Pregnancy and Nursing Mothers: Use only if the potential benefitjustifies the potential risk to the fetus or child (2.3, 8.1, 8.3).See 17 for PATIENT COUNSELING INFORMATION and MedicationGuide Revised: 10/20122FULL PRESCRIBING INFORMATION: CONTENTS*WARNING: SUICIDAL THOUGHTS AND BEHAVIORS1 INDICATIONSANDUSAGE1.1 Major Depressive Disorder1.2 Generalized Anxiety Disorder1.3 Diabetic Peripheral Neuropathic Pain1.4 F ibromyalgia1.5 Chronic Musculoskeletal Pain2 DOSAGEANDADMINISTRATION2.1 I nitialTreatment2.2 M aintenance/Continuation/ExtendedTreatment2.3 Dosing in Special Populations2.4 D iscontinuingCymbalta2.5 Switching a Patient To or From a Monoamine OxidaseInhibitor (MAOI) Intended to Treat Psychiatric Disorders2.6 Use of Cymbalta with Other MAOIs such as Linezolid orMethylene Blue3 DOSAGE FORMS AND STRENGTHS4 CONTRAINDICATIONS4.1 Monoamine Oxidase Inhibitors (MAOIs)4.2 U ncontrolledNarrow-AngleGlaucoma5 WARNINGSANDPRECAUTIONS5.1 Suicidal Thoughts and Behaviors in Adolescents andYoung Adults5.2 H epatotoxicity5.3 Orthostatic Hypotension and Syncope5.4 S erotoninSyndrome5.5 A bnormalBleeding5.6 Severe Skin Reactions5.7 Discontinuation of Treatment with Cymbalta5.8 Activation of Mania/Hypomania5.9 S eizures5.10 Effect on Blood Pressure5.11 Clinically Important Drug Interactions5.12 H yponatremia5.13 Use in Patients with Concomitant Illness5.14 Urinary Hesitation and Retention5.15 L aboratoryTests6 ADVERSEREACTIONS6.1 Clinical Trial Data Sources6.2 Adverse Reactions Reported as Reasons forDiscontinuation of Treatment in Placebo-Controlled Trials6.3 Most Common Adverse Reactions6.4 Adverse Reactions Occurring at an Incidence of 5% orMore Among Duloxetine-Treated Patients in Placebo-Controlled Trials6.5 Adverse Reactions Occurring at an Incidence of 2% orMore Among Duloxetine-Treated Patients in Placebo-Controlled Trials6.6 Effects on Male and Female Sexual Function6.7 Vital Sign Changes6.8 W eightChanges6.9 L aboratoryChanges6.10 E lectrocardiogramChanges6.11 Other Adverse Reactions Observed During thePremarketing and Postmarketing Clinical Trial Evaluationof Duloxetine6.12 Postmarketing Spontaneous Reports7 DRUGINTERACTIONS7.1 Inhibitors of CYP1A27.2 Inhibitors of CYP2D67.3 Dual Inhibition of CYP1A2 and CYP2D67.4 Drugs that Interfere with Hemostasis (e.g., NSAIDs,Aspirin, and Warfarin)7.5 L orazepam7.6 T emazepam7.7 Drugs that Affect Gastric Acidity7.8 Drugs Metabolized by CYP1A27.9 Drugs Metabolized by CYP2D67.10 Drugs Metabolized by CYP2C97.11 Drugs Metabolized by CYP3A7.12 Drugs Metabolized by CYP2C197.13 Monoamine Oxidase Inhibitors (MAOIs)7.14 S erotonergicDrugs7.15 A lcohol7.16 C NSDrugs7.17 Drugs Highly Bound to Plasma Protein8 USE IN SPECIFIC POPULATIONS8.1 P regnancy8.2 Labor and Delivery8.3 N ursingMothers8.4 P ediatricUse8.5 G eriatricUse8.6 G ender8.7 S mokingStatus8.8 R ace8.9 H epaticInsufficiency8.10 Severe Renal Impairment9 DRUGABUSEANDDEPENDENCE9.2 A buse9.3 D ependence10 OVERDOSAGE10.1 Signs and Symptoms10.2 M anagementofOverdose11 DESCRIPTION12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action12.2 P harmacodynamics12.3 P harmacokinetics13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility14 CLINICAL STUDIES14.1 Major Depressive Disorder14.2 Generalized Anxiety Disorder14.3 Diabetic Peripheral Neuropathic Pain14.4 F ibromyalgia14.5 Chronic Musculoskeletal Pain16 HOW SUPPLIED/STORAGE AND HANDLING16.1 H owSupplied16.2 S torage17 PATIENT COUNSELING INFORMATION17.1 Information on Medication Guide17.2 Suicidal Thoughts and Behaviors17.3 M edicationAdministration17.4 Continuing the Therapy Prescribed17.5 H epatotoxicity17.6 A lcohol17.7 Orthostatic Hypotension and Syncope17.8 S erotoninSyndrome17.9 A bnormalBleeding17.10 Severe Skin Reaction17.11 Discontinuation of Treatment17.12 Activation of Mania or Hypomania17.13 Seizures17.14 Effects on Blood Pressure17.15 Concomitant Medications17.16 Hyponatremia17.17 Concomitant Illnesses17.18 Urinary Hesitancy and Retention17.19 Pregnancy and Breast Feeding17.20 Interference with Psychomotor Performance* Sections or subsections omitted from the full prescribing information are not listed.FULL PRESCRIBING INFORMATIONWARNING: SUICIDAL THOUGHTS AND BEHAVIORSAntidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term studies. These studies did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in patients over age 24; there was a reduction in risk with antidepressant use in patients aged 65 and older [see Warnings and Precautions (5.1)].In patients of all ages who are started on antidepressant therapy, monitor closely for worsening, and for emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber [see Warnings and Precautions (5.1)].Cymbalta is not approved for use in pediatric patients [see Use in Specific Populations (8.4)].USAGEAND1 INDICATIONS1.1 Major Depressive DisorderCymbalta is indicated for the treatment of major depressive disorder (MDD). The efficacy of Cymbalta was established in four short-term and one maintenance trial in adults [see Clinical Studies (14.1)].A major depressive episode (DSM-IV) implies a prominent and relatively persistent (nearly every day for at least 2 weeks) depressed or dysphoric mood that usually interferes with daily functioning, and includes at least 5 of the following 9 symptoms: depressed mood, loss of interest in usual activities, significant change in weight and/or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, increased fatigue, feelings of guilt or worthlessness, slowed thinking or impaired concentration, or a suicide attempt or suicidal ideation.AnxietyDisorder1.2 GeneralizedCymbalta is indicated for the treatment of generalized anxiety disorder (GAD). The efficacy of Cymbalta was established in three short-term trials and one maintenance trial in adults [see Clinical Studies (14.2)].Generalized anxiety disorder is defined by the DSM-IV as excessive anxiety and worry, present more days than not, for at least 6 months. The excessive anxiety and worry must be difficult to control and must cause significant distress or impairment in normal functioning. It must be associated with at least 3 of the following 6 symptoms: restlessness or feeling keyed up or on edge, being easily fatigued, difficulty concentrating or mind going blank, irritability, muscle tension, and/or sleep disturbance.1.3 Diabetic Peripheral Neuropathic PainCymbalta is indicated for the management of neuropathic pain (DPNP) associated with diabetic peripheral neuropathy [see Clinical Studies (14.3)].1.4 FibromyalgiaCymbalta is indicated for the management of fibromyalgia (FM) [see Clinical Studies (14.4)].PainMusculoskeletal1.5 ChronicCymbalta is indicated for the management of chronic musculoskeletal pain. This has been established in studies in patients with chronic low back pain (CLBP) and chronic pain due to osteoarthritis [see Clinical Studies (14.5)].2 DOSAGE AND ADMINISTRATIONCymbalta should be swallowed whole and should not be chewed or crushed, nor should the capsule be opened and its contents sprinkled on food or mixed with liquids. All of these might affect the enteric coating. Cymbalta can be given without regard to meals.Treatment2.1 InitialMajor Depressive Disorder — Cymbalta should be administered at a total dose of 40 mg/day (given as 20 mg twice daily) to 60 mg/day (given either once daily or as 30 mg twice daily). For some patients, it may be desirable to start at 30 mg once daily for 1 week, to allow patients to adjust to the medication before increasing to 60 mg once daily. While a 120 mg/day dose was shown to be effective, there is no evidence that doses greater than 60 mg/day confer any additional benefits. The safety of doses above 120 mg/day has not been adequately evaluated [see Clinical Studies (14.1)].Generalized Anxiety Disorder — For most patients, the recommended starting dose for Cymbalta is 60 mg administered once daily. For some patients, it may be desirable to start at 30 mg once daily for 1 week, to allow patients to adjust to the medication before increasing to 60 mg once daily. While a 120 mg once daily dose was shown to be effective, there is no evidence that doses greater than 60 mg/day confer additional benefit. Nevertheless, if a decision is made to increase the dose beyond 60 mg once daily, dose increases should be in increments of 30 mg once daily. The safety of doses above 120 mg once daily has not been adequately evaluated [see Clinical Studies (14.2)].Diabetic Peripheral Neuropathic Pain — The recommended dose for Cymbalta is 60 mg administered once daily. There is no evidence that doses higher than 60 mg confer additional significant benefit and the higher dose is clearly less well tolerated [see Clinical Studies (14.3)]. For patients for whom tolerability is a concern, a lower starting dose may be considered.Since diabetes is frequently complicated by renal disease, a lower starting dose and gradual increase in dose should be considered for patients with renal impairment [see Dosage and Administration (2.3), Use in Specific Populations (8.10), and Clinical Pharmacology (12.3)].Fibromyalgia — The recommended dose for Cymbalta is 60 mg administered once daily. Treatment should begin at 30 mg once daily for 1 week, to allow patients to adjust to the medication before increasing to 60 mg once daily. Some patients may respond to the starting dose. There is no evidence that doses greater than 60 mg/day confer additional benefit, even in patients who do not respond to a 60 mg dose, and higher doses are associated with a higher rate of adverse reactions [see Clinical Studies (14.4)].Chronic Musculoskeletal Pain — The recommended dose for Cymbalta is 60 mg once daily. Dosing may be started at 30 mg for one week, to allow patients to adjust to the medication before increasing to 60 mg once daily. There is no evidence that higher doses confer additional benefit, even in patients who do not respond to a 60 mg dose, and higher doses are associated with a higher rate of adverse reactions [see Clinical Studies (14.5)].Treatment2.2 M aintenance/Continuation/ExtendedMajor Depressive Disorder — It is generally agreed that acute episodes of major depression require several months or longer of sustained pharmacologic therapy. Maintenance of efficacy in MDD was demonstrated with Cymbalta as monotherapy. Cymbalta should be administered at a total dose of 60 mg once daily. Patients should be periodically reassessed to determine the need for maintenance treatment and the appropriate dose for such treatment [see Clinical Studies (14.1)].Generalized Anxiety Disorder — It is generally agreed that episodes of generalized anxiety disorder require several months or longer of sustained pharmacological therapy. Maintenance of efficacy in GAD was demonstrated with Cymbalta as monotherapy. Cymbalta should be administered in a dose range of 60-120 mg once daily. Patients should be periodically reassessed to determine the continued need for maintenance treatment and the appropriate dose for such treatment [see Clinical Studies (14.2)].Diabetic Peripheral Neuropathic Pain — As the progression of diabetic peripheral neuropathy is highly variable and management of pain is empirical, the effectiveness of Cymbalta must be assessed individually. Efficacy beyond 12 weeks has not been systematically studied in placebo-controlled trials.Fibromyalgia — Fibromyalgia is recognized as a chronic condition. The efficacy of Cymbalta in the management of fibromyalgia has been demonstrated in placebo-controlled studies up to 3 months. The efficacy of Cymbalta was not demonstrated in longer studies; however, continued treatment should be based on individual patient response.Chronic Musculoskeletal Pain — The efficacy of Cymbalta has not been established in placebo-controlled studies beyond 13 weeks.2.3 Dosing in Special PopulationsHepatic Insufficiency — It is recommended that Cymbalta should ordinarily not be administered to patients with any hepatic insufficiency [see Warnings and Precautions (5.13) and Use in Specific Populations (8.9)].Severe Renal Impairment — Cymbalta is not recommended for patients with end-stage renal disease or severe renal impairment (estimated creatinine clearance <30 mL/min) [see Warnings and Precautions (5.13) and Use in Specific Populations (8.10)].Elderly Patients — No dose adjustment is recommended for elderly patients on the basis of age. As with any drug, caution should be exercised in treating the elderly. When individualizing the dosage in elderly patients, extra care should be taken when increasing the dose [see Use in Specific Populations (8.5)].Pregnant Women — There are no adequate and well-controlled studies in pregnant women; therefore, Cymbalta should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus [see Use in Specific Populations (8.1)].Lilly maintains a pregnancy registry to monitor the pregnancy outcomes of women exposed to Cymbalta while pregnant. Healthcare providers are encouraged to register any patient who is exposed to Cymbalta during pregnancy by calling the Cymbalta Pregnancy Registry at 1-866-814-6975 or by visiting Nursing Mothers — Because the safety of duloxetine in infants is not known, nursing while on Cymbalta is not recommended [see Use in Specific Populations (8.3)].2.4 D iscontinuingCymbaltaSymptoms associated with discontinuation of Cymbalta and other SSRIs and SNRIs have been reported. A gradual reduction in the dose rather than abrupt cessation is recommended whenever possible [see Warnings and Precautions (5.7)].2.5 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat PsychiatricDisordersAt least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with Cymbalta. Conversely, at least 5 days should be allowed after stopping Cymbalta before starting an MAOI intended to treat psychiatric disorders [see Contraindications (4.1)].2.6 Use of Cymbalta with Other MAOIs such as Linezolid or Methylene BlueDo not start Cymbalta in a patient who is being treated with linezolid or intravenous methylene blue because there is an increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered [see Contraindications (4.1)].In some cases, a patient already receiving Cymbalta therapy may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue treatment are judged to outweigh the risks of serotonin syndrome in a particular patient, Cymbalta should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for 5 days or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with Cymbalta may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue [see Warnings and Precautions (5.4)].The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with Cymbalta is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use [see Warnings and Precautions (5.4)].3 DOSAGE FORMS AND STRENGTHSCymbalta is available as delayed release capsules:20 mg opaque green capsules imprinted with “Lilly 3235 20mg”30 mg opaque white and blue capsules imprinted with “Lilly 3240 30mg”60 mg opaque green and blue capsules imprinted with “Lilly 3237 60mg”60 mg opaque green and blue capsules imprinted with “Lilly 3270 60mg”4 CONTRAINDICATIONS4.1 Monoamine Oxidase Inhibitors (MAOIs)The use of MAOIs intended to treat psychiatric disorders with Cymbalta or within 5 days of stopping treatment with Cymbalta is contraindicated because of an increased risk of serotonin syndrome. The use of Cymbalta within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated [see Dosage and Administration (2.5) and Warnings and Precautions (5.4)].Starting Cymbalta in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration (2.6) and Warnings and Precautions (5.4)].4.2 Uncontrolled Narrow-Angle GlaucomaIn clinical trials, Cymbalta use was associated with an increased risk of mydriasis; therefore, its use should be avoided in patients with uncontrolled narrow-angle glaucoma [see Warnings and Precautions (5.13)].PRECAUTIONS5 WARNINGSAND5.1 Suicidal Thoughts and Behaviors in Adolescents and Young AdultsPatients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment.Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18-24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.The pooled analyses of placebo-controlled trials in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk of differences (drug vs placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1000 patients treated) are provided in Table 1.Table 1Age Range Drug-Placebo Difference in Number of Cases ofSuicidality per 1000 Patients TreatedIncreases Compared to Placebo<18 14 additional cases18-24 5 additional cases Decreases Compared to Placebo25-64 1 fewer case≥65 6 fewer casesNo suicides occurred in any of the pediatric trials. There were suicides in the adult trials, but the number was notsufficient to reach any conclusion about drug effect on suicide.It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, thereis substantial evidence from placebo-controlled maintenance trials in adults with depression that the use ofantidepressants can delay the recurrence of depression.All patients being treated with antidepressants for any indication should be monitored appropriately andobserved closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initialfew months of a course of drug therapy, or at times of dose changes, either increases or decreases.The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness,impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatricpatients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatricand nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening ofdepression and/or the emergence of suicidal impulses has not been established, there is concern that such symptomsmay represent precursors to emerging suicidality.Consideration should be given to changing the therapeutic regimen, including possibly discontinuing themedication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptomsthat might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset,or were not part of the patient’s presenting symptoms.If the decision has been made to discontinue treatment, medication should be tapered, as rapidly as is feasible,but with recognition that discontinuation can be associated with certain symptoms [see Dosage and Administration (2.4)and Warnings and Precautions (5.7) for descriptions of the risks of discontinuation of Cymbalta].Families and caregivers of patients being treated with antidepressants for major depressive disorder orother indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to health care providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for Cymbalta should bewritten for the smallest quantity of capsules consistent with good patient management, in order to reduce therisk of overdose.Screening Patients for Bipolar Disorder — A major depressive episode may be the initial presentation of bipolardisorder. It is generally believed (though not established in controlled trials) that treating such an episode with anantidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolardisorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior toinitiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened todetermine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including afamily history of suicide, bipolar disorder, and depression. It should be noted that Cymbalta (duloxetine) is not approvedfor use in treating bipolar depression.5.2 HepatotoxicityThere have been reports of hepatic failure, sometimes fatal, in patients treated with Cymbalta. These cases havepresented as hepatitis with abdominal pain, hepatomegaly, and elevation of transaminase levels to more than twenty times the upper limit of normal with or without jaundice, reflecting a mixed or hepatocellular pattern of liver injury. Cymbalta should be discontinued in patients who develop jaundice or other evidence of clinically significant liver dysfunction and should not be resumed unless another cause can be established.Cases of cholestatic jaundice with minimal elevation of transaminase levels have also been reported. Otherpostmarketing reports indicate that elevated transaminases, bilirubin, and alkaline phosphatase have occurred in patientswith chronic liver disease or cirrhosis.Cymbalta increased the risk of elevation of serum transaminase levels in development program clinical trials. Livertransaminase elevations resulted in the discontinuation of 0.3% (89/29,435) of Cymbalta-treated patients. In mostpatients, the median time to detection of the transaminase elevation was about two months. In placebo-controlled trials in any indication, for patients with normal and abnormal baseline ALT values, elevation of ALT >3 times the upper limit of normal occurred in 1.37% (132/9611) of Cymbalta-treated patients compared to 0.49% (35/7182) of placebo-treated patients. In placebo-controlled studies using a fixed dose design, there was evidence of a dose response relationship for ALT and AST elevation of >3 times the upper limit of normal and >5 times the upper limit of normal, respectively.。
盐酸度洛西汀治疗更年期抑郁症疗效评价

盐酸度洛西汀治疗更年期抑郁症疗效评价盐酸度洛西汀(商品名:奥思平)为5-羟色胺(5-HT)和去甲肾上腺素(NH)平衡和再摄取抑制剂(SNRI),与目前临床使用的5-HT再摄取抑制剂(SSRI)相比,具有更广谱、更快速的抗抑郁疗效,故可获得更高的临床治愈率。
盐酸度洛西汀也不同于现有SNRI类药物,具有平衡两种神经递质的作用机制,能使两种神经递质发挥更佳的协同作用,从而快速缓解抑郁情绪和躯体症状。
为客观评价盐酸度洛西汀的临床疗效及安全性,现将上海市第一妇婴保健院心理门诊和嘉定区妇婴保健院门诊中使用盐酸度洛西汀治疗更年期抑郁症妇女的情况报告、分析如下。
1 对象、方法和结果患者数:2009年1月-2010年3月间在上述两医院门诊治疗的16例更年期抑郁症妇女。
一般情况:平均年龄为(47.38±5.43)岁。
平均初潮年龄为(14.31±1.74)岁(9~19岁)。
未绝经6例(65.4%)、绝经10例(34.6%),其中自然绝经8例(占绝经者的80.0%)、手术绝经2例(占绝经者的20.0%),平均绝经年龄为(48.9±3.54)岁。
诊断依据:用《自评抑郁量表(SDS)》评定抑郁症状;用《焦虑自评量表(SAS)》进行更年期焦虑症状评分。
<50分为正常,50~59分为轻度、60~69分为中度、≥70分为重度抑郁和焦虑。
治疗方法:明确诊断后均给予盐酸度洛西汀20或40 mg口服,一般1周后复诊,了解患者症状的改善情况,然后给予下1周的药物治疗。
起效时间平均为2周,明显改善时间平均为4周。
治疗效果:16例患者经治疗,有4例痊愈,疗程平均为6个月;其他12例患者目前仍在治疗中,但抑郁情况都已明显改善。
2 讨论据统计,抑郁症的发病率高达6%~8%。
在综合性医院中,抑郁症占门诊就诊患者数的约10%,而住院患者中有25%~30%伴发抑郁症状。
统计表明,有近1/3的妇女曾出现抑郁症状。
妇女到更年期后,由于卵巢衰退、卵泡减少与老化、性激素合成减少、神经内分泌失调和免疫功能下降等,除月经紊乱外还会出现心烦易怒、头晕心悸、神疲乏力、失眠健忘、轰热汗多、关节酸痛、知觉过敏和小便增多等一系列以植物神经功能失调为主的症状,称为“更年期综合征”。
盐酸奥洛他定片说明书

核准日期:2010年10月13日修改日期:2012年09月14日 2013年10月15日 2015年02月09日 2015年12月24日 2016年07月20日2017年01月16日 2019年12月20日 2020年01月03日 2020年08月17日 2021年06月28日 2022年01月06日盐酸奥洛他定片说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称:盐酸奥洛他定片商品名称:阿洛刻英文名称:Olopatadine Hydrochloride Tablets汉语拼音:Yansuan Aoluotading Pian【成份】本品活性成份为盐酸奥洛他定。
化学名称:{11-[(1Z)-3-(二甲氨基) 亚丙基]-6, 11-二氢二苯并[b, e]恶庚英-2-基}乙酸盐酸盐化学结构式:分子式:C21H23NO3·HCl分子量:373.87辅料:乳糖水合物、结晶纤维素、交联羧甲基纤维素钠、聚乙烯醇(部分皂化物)、硬脂酸镁、羟丙基甲基纤维素、聚乙二醇6000、氧化钛、黄色氧化铁、氧化铁、巴西棕榈蜡【性状】本品为淡粉色的薄膜衣片,除去包衣后显白色。
【适应症】过敏性鼻炎、荨麻疹、瘙痒性皮肤病(湿疹、皮炎、痒疹、皮肤瘙痒症、寻常性银屑病、渗出性多形红斑)【规格】5mg【用法用量】本品为口服片剂。
成人用量通常为1日2次,1次5mg,早晨和晚上睡前各服1次。
根据年龄及症状适当增减。
【不良反应】在批准前临床试验、药物使用-结果调查及长期使用专项调查的9620例患者中,有1056例(发生率11.0%)发生不良反应共计1402件。
主要不良反应为嗜睡674件(7.0%),ALT (GPT)上升68件(0.7%),倦怠感53件(0.6%),AST (GOT)上升46件(0.5%),口渴36件(0.4%)等。
(在日本再审查结束时)1)有临床意义的不良反应暴发性肝炎、肝功能损害、黄疸(发生率不明):有可能发生暴发性肝炎、伴随AST (GOT)、ALT (GPT)、γ-GTP、LDH、Al-P上升等的肝功能损害、黄疸,故应注意观察。
度洛西汀片

盐酸度洛西汀肠溶胶囊产品简介一、产品介绍【适应症】治疗抑郁症【用法用量】起始治疗:推荐盐酸度洛西汀的起始剂量为40mg/日(20mg一日二次)至60mg/日(一日一次或30mg一日二次),不考虑进食情况。
现有的临床研究数据未证实剂量超过60mg/日将增加疗效。
维持/继续/长期治疗:一般认为抑郁症的急性发作需要数月或更长时间的药物治疗,但尚没有充足的试验资料来确定患者应该连续服用度洛西汀治疗达多长时间。
对此类患者,应对其接受维持治疗的必要性以及相应所需的剂量作定期评估。
特殊人群用药肾脏功能受损患者:用量一对于晚期肾脏疾病(需要透析的)患者,或有严重肾脏功能损害(估计肌酐清除率<30 mL/min的)患者,建议不用盐酸度洛西汀(见药理毒理)。
肝功能不全的患者:用量一建议有任何肝功能不全的患者避免服用盐酸度洛西汀(见药理毒理和注意事项)老年患者:用量一对于老年患者,建议不必根据年龄调整剂量。
与任何药物一样,治疗老年患者时应该慎重。
在老年患者中个体化调整剂量时,增加剂量时应该额外小心。
妊娠后三个月的女性患者:治疗一在妊娠后三个月内接触SSRIs或SNRls(五羟色胺和去甲肾上腺素再摄取抑制剂)的新生儿,产生的并发症会导致住院时间延长、需要呼吸支持和管道喂食(见注意事项)。
当孕期女性用度洛西汀治疗时,在妊娠后三个月,医生应对治疗的潜在风险和获益进行认真的评价。
医生应考虑在妊娠晚期逐渐减度洛西汀的用量。
度洛西汀停药已有报道盐酸度洛西汀及其他SSRI和SNRIs药物的停药反应(见注意事项)。
停药时应对这些症状进行监测。
建议尽可能的逐渐减药,而不是骤停药物。
由于减少药物剂量或停药而引起了无法耐受的症状时,可以考虑恢夏使用以往的的处方剂量。
随后再以更慢的的速度减药。
与单胺氧化酶抑制剂(MOA I)间的换药MOAT停药后至少14天才可开始盐酸度洛西汀的治疗。
盐酸度洛西汀停药后至少5天才可以开始MOAT的治疗(见禁忌和警告)。
度洛西汀(奥思平)说明书

百济新特药房提供度洛西汀(奥思平)说明书,让您了解度洛西汀(奥思平)副作用、度洛西汀(奥思平)效果、不良反应等信息。
百济新特药房—全国连锁专科药房,医保定点药房,消费者信得过商店,专家指导用药,度洛西汀(奥思平)说明书如下:【奥思平药品名称】商品名:奥思平通用名名:盐酸度洛西汀肠溶片英文名称:Duloxetine Hydrochloride EntericTablets【奥思平成份】奥思平主要成分为盐酸度洛西汀。
【奥思平适应症】用于治疗抑郁症。
【奥思平用法用量】吞服,不要咀嚼和压碎。
推荐起始剂量为40mg/日(40mg,一日一次或20mg,一日二次)至60mg/日(一日一次),不考虑进食影响。
【奥思平禁忌】1、禁用于已知对度洛西汀或产品中任何非活性成分过敏的患者。
2、禁止与单胺氧化酶抑制剂联用,也不可以在单胺氧化酶抑制剂停药14天内使用奥思平;根据度洛西汀的半衰期,停用度洛西汀后至少5天,才能开始使用MAOIs。
3、临床显示度洛西汀有增加瞳孔散大的风险,因此未经控制的闭角型青光眼患者应避免使用度洛西汀。
【奥思平药物相互作用】度洛西汀通过两种CYP2D6和CYP1A2代谢,中度抑制CYP2D6,但不抑制也不诱导CYP1A2和CYP3A4。
与其他主要通过CYP2D6代谢,且治疗窗狭窄的药物(如:TCAs、Ic类抗心律失常药物、吩噻嗪)时,应谨慎。
【奥思平规格】20mg(以度洛西汀计)【奥思平贮藏】密封,置阴凉干燥处。
【奥思平有效期】24个月【奥思平批准文号】国药准字H20061261【奥思平生产企业】上海中西制药有限公司。
抗抑郁药怡诺思、博乐欣、奥思平等说明书介绍2

口服常用量: 开始一次25mg(1 片),一日2~3 次,以后逐渐增加 至一日总量100~ 250mg(4~10片) 。 最高量: 一日不超过300mg (12片)。
盐酸阿米替林片
盐酸氯米帕明片
盐酸氯米帕明注射 盐酸多塞平片 液
治疗初期可能出现 抗胆碱能反应,如 多汗、口干、视物 模糊、排尿困难、 便秘等。中枢神经 不良反 系统不良反应可出 应 现嗜睡,震颤、眩 晕。可发生体位性 低血压。偶见癫痫 发作、骨髓抑制及 中毒性肝损害等。
盐酸度洛西汀肠 溶胶囊 肝脏毒性——度 洛西汀有增加血 清转氨酶水平的 风险。既往有躁 狂史的患者慎用 度洛西汀。既往 有癫痫发作史的 患者慎用度洛西 汀。度洛西汀慎 用于已稳定的窄 角型青光眼患者 。低钠血症--低钠血症可能表 现为非特异性的 体征和症状(如 头晕、虚弱、恶 心、呕吐、意识 模糊、嗑睡及昏 睡),较为严重 的体征和症状包 括晕厥发作、跌 倒和癫痫发作。 停用度洛西汀后 应注意观察患者 有无上述症状的 出现,建议尽可 能的逐渐减药, 而不是骤停药物 。
盐酸氯米帕明片 1. 本品与舒托必 利合用,有增加室 性心律失常的危 险,严重者可至尖 端扭转心律失常。 2. 本品与乙醇或 其他中枢神经系统 抑制药合用,中枢 神经抑制作用增强 。 3. 本品与肾上腺 素、去甲肾上腺素 合用,易致阵发性 高血压及心律失常 。 4. 本品与可乐定 合用,后者抗高血 压作用减弱。 5. 本品与抗惊厥 药合用,可降低抗 惊厥药的作用。 6. 本品与氟西汀 或氟伏沙明合用, 可增加两者的血浆 浓度,出现惊厥, 不良反应增加。 7. 本品与阿托品 类合用,不良反应 增加。
盐酸阿米替林片
盐酸氯米帕明片
盐酸氯米帕明注射 盐酸多塞平片 液
肝、肾功能严重不 全、前列腺肥大、 老年或心血管疾患 者慎用。使用期间 应监测心电图。本 品不得与单胺氧化 酶抑制剂合用,应 注意事 在停用单胺氧化酶 项 抑制剂后14天,才 能使用本品。患者 有转向躁狂倾向时 应立即停药。用药 期间不宜驾驶车辆 、操作机械或高空 作业。
Duloxetine HCl_Neuronal Signaling_CAS号136434-34-9说明书_AbMole中国
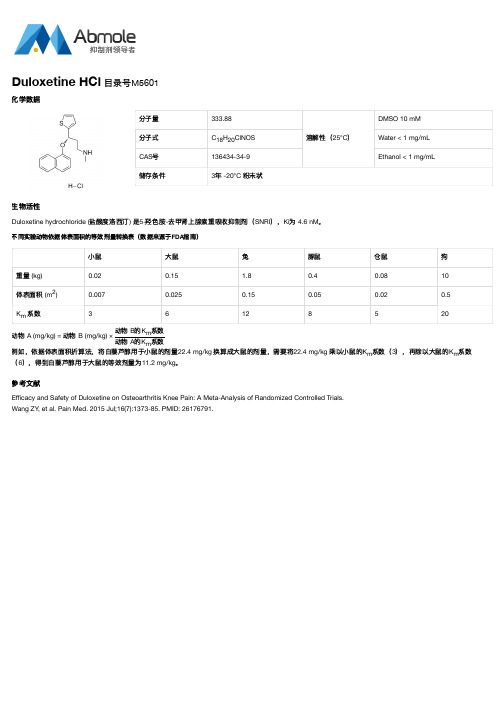
不同实验动物依据体表面积的等效剂量转换表(数据来源于FDA指南)
小鼠
大鼠
兔
豚鼠
仓鼠
狗
重量 (kg)
0.02
0.15
1.8
0.4
0.08
10
体表面积 (m2)
0.007
0.025
0.15
0.05
0.02
0.5
Km 系数
3
6
12
8
5
20
动例物如,A依(m据g/体kg表) =面动积物折算B (法m,g/k将g白) ×藜动动芦物物醇BA用的的于KK小mm系系鼠数数的剂量22.4 (6),得到白藜芦醇用于大鼠的等效剂量为11.2 。 mg/kg
mg/kg
换算成大鼠的剂量,需要将22.4
mg/kg
乘以小鼠的Km系数(3),再除以大鼠的Km系数
参考文献
Efficacy and Safety of Duloxetine on Osteoarthritis Knee Pain: A Meta-Analysis of Randomized Controlled Trials. Wang ZY, et al. Pain Med. 2015 Jul;16(7):1373-85. PMID: 26176791.
Duloxetine HCl 目录号M5601
化学数据
分子量 分子式 号 CAS136434-34-9
3年 -20°C 粉末状
溶解性(25°C)
DMSO 10 mM Water < 1 mg/mL Ethanol < 1 mg/mL
生物活性
Duloxetine hydrochloride (盐酸度洛西汀) 是5-羟色胺-去甲肾上腺素重吸收抑制剂(SNRI),Ki为 4.6 。 nM
盐酸度洛西汀肠溶胶囊20140728

盐酸度洛西汀肠溶胶囊项目立项论证意见稿1.简介盐酸度洛西汀(duloxetine hydrochloride/Cymbaha,以下简称为度洛西汀)是美国礼来Eli Lilly公司开发的一个5一羟色胺和去甲肾上腺素再摄取抑制剂。
5一羟色胺和去甲肾上腺素均属中枢神经递质,在调控情感和对疼痛的敏度方面起着重要作用。
度洛西汀能够抑制神经元对5一羟色胺和去甲肾上腺素的再摄取,由此提高这两种中枢神经递质在大脑和脊髓中的浓度,故可用于治疗某些心境疾病如抑郁症和焦虑症以及缓解中枢性疼痛如糖尿病外周神经病性疼痛和妇女纤维肌痛等。
度洛西汀也能作用于尿道中的5一羟色胺和去甲肾上腺素受体,从而增强尿道括约肌的神经性紧张程度和收缩能力,所以对妇女应激性尿失禁症治疗也有效。
度洛西汀为口服肠溶胶囊制剂,2004年8月首次在美国获得批准后,现已在70余个国家上市。
度洛西汀2006年的全球销售额即超过13亿美元,2007年和2008年的销售额又分别大幅增至2l亿和27亿美元,是近年世界范围内销售额增长最快的药物之一。
化学名称:盐酸度洛西汀;(S)-(+)-N-甲基-3-(1-萘氧基)-3-(2-噻吩)-丙胺盐酸盐Duloxetinehydrochloride;N-Methyl-gama-(1-naphthalenyloxy)-2-thiophenepropanamine分子式:C18H19NOS.HCl;C18H20ClNOS分子量:333.88结构式:CAS:136434-34-9【用途】对于抑郁症、糖尿病性周围神经病引起疼痛以及紧张性尿失禁有疗效。
2.原料药来源国家局已批2家企业生产,0家进口原料药。
如下:1.盐酸度洛西汀(国药准字H261262上海万代制药有限公司86977746)2.盐酸度洛西汀 (国药准字H20130055江苏恩华药业股份有限公司86901435000170)3.国内上市批准情况国内盐酸度洛西汀主要为肠溶片和肠溶胶囊。
度洛西汀改善老年躯体形式疼痛障碍的研究

度洛西汀改善老年躯体形式疼痛障碍的研究【摘要】目的:探讨度洛西汀改善老年躯体形式疼痛障碍的临床效果及安全性。
方法:将符合要求的75例老年躯体形式疼痛障碍患者随机分成2组,其中研究组36例,口服度洛西汀治疗,对照组39例,口服米氮平治疗,共8周;采用汉密尔顿抑郁量表、汉密尔顿抑郁量表、副反应量表评定临床疗效及不良反应。
结果:治疗8周末度洛西汀临床治愈率67.0%,有效率82.1%;米氮平临床治愈率56.1%,有效率69.2%,两组比较有显著性差异(P中国论文网/1/view-.htm【关键词】度洛西汀米氮平抑郁躯体形式疼痛障碍【中图分类号】R971.43 【文献标识码】A 【文章编号】2095-6851(2017)12-0-01目前,抑郁症在老年人中的发病率逐渐增高,老年抑郁症患者中有很多人以疼痛为主,主要表现为全身一处或多处疼痛不适[1],为此患者长期辗转于综合医院内外科各个科室,既浪费了医疗资源,又增加了患者的痛苦,严重影响老年人的生活质量。
研究发现新型抗抑郁药物对于改善伴有疼痛的抑郁症患者有较好的效果,本研究选取了度洛西汀及米氮平进行对照研究,以了解两种药物在抗抑郁、缓解疼痛方面的疗效以及安全性。
现报告如下。
1 对象和方法1.1 对象所有病例选自2015.9-2017.8月就诊于我院老年精神科有慢性躯体形式疼痛障碍的患者。
入组标准:(1)符合《国际疾病分类》第10版(ICD-10)躯体形式疼痛障碍诊断标准。
(2)年龄60岁以上。
(3)近2周内未使用过抗精神病或者抗抑郁药物。
(4)排除脑器质性疾病、严重躯体疾病等。
共有符合要求的被试75人,随机分为两组,其中实验组36人,男性17,女性19,年龄(67.1±4.1)岁,病程(3.2±1.4)年,口服度洛西汀治疗,对照组39例,男性19,女性20,年龄(68.3±5.8)岁,病程(3.5±1.6)年,口服米氮平治疗,共8周;两组患者在性别、年龄、病程、汉密尔顿抑郁量表(HAMD)评分及医学结局研究用疼痛量表(MOSPM)评分等方面均无显著性差异(P>0.05).所有病例均由本人签署知情同意书并且坚持至研究结束。
焦虑吃什么药

焦虑吃什么药文章目录*一、焦虑吃什么药*二、焦虑的典籍偏方*三、焦虑的护理知识焦虑吃什么药1、文拉法新1.1、商品名叫怡诺斯、博乐欣,有缓释制剂也有速释制剂。
该药为双受体作用的药物,作用于5-HT和NE两种神经递质,常用剂量是75-225mg/天。
抗抑郁抗焦虑效果好。
注意有可能在服药初期焦虑会加重,需要加用安定类药物,一般持续两周大多消失。
1.2、在服用本品过程中不宜饮酒。
1.3、起始剂量为75mg/d,分2~3次服用。
一般2周以内即可见效。
2、度洛西汀2.1、商品名叫欣百达、奥思平,有缓释制剂也有速释制剂,同样也是双受体作用原理的药物,作用于5-HT和NE两种神经递质, 常用剂量是60mg/天,这个药物最大的特点是在双受体的作用程度上有其优点,抗抑郁、抗焦虑效果比较好。
2.2、肾脏功能受损患者的用量——对于晚期肾脏疾病(需要透析的)患者,或有严重肾脏功能损害(估计肌酐清除率30mL/min 的)患者,建议不用本品。
2.3、推荐本品的起始剂量为40mg/日(20mg一日二次)至60mg/日(一日一次或30mg一日二次),不考虑进食情况。
现有的临床研究数据未证实剂量超过60mg/日将增加疗效。
3、艾司西酞普兰3.1、商品名叫来士普、百洛特,也属于SSRI类药物,作用于5-HT系统,常用剂量是10-20mg/天。
抗焦虑效果不错,副作用小是其优点,尤其适合老年人以及有心血管疾病的患者使用,价格偏贵。
3.2、不适用于儿童和18岁以下的青少年3.3、重症抑郁症:起始剂量 l0mg·d-1,qd,l周后可以增至20mg·d-1,早晨或晚上口服,持续几个月甚至更长。
广泛性焦虑:起始剂量l0mg·d-1,qd, 1周后可以增至20mg d-1,早晨或晚上口服。
至少停用单胺氧化酶抑制剂(MAOI)14d后才可以调换本药,同样,停用本药 14d后才可以用MAOI。
停药时应逐渐减量。
焦虑的典籍偏方1、银耳莲子汤水发银耳200g,莲子30g,冰糖适量。
奥卡西平片致皮疹合并重度血小板减少1例

奥卡西平片致皮疹合并重度血小板减少1例郭濠宁;吴思霖;张莉;何淼泉【期刊名称】《中国药物警戒》【年(卷),期】2018(015)006【摘要】病例:患者,男,80岁。
因“记忆力下降伴精神行为异常2年余,凭空闻声、视物2月余”于2017年3月22日入院。
查体:体温36.9℃,心率70次/min,呼吸19次/min,血压155/86 mmHg,生命体征平稳,心肺腹未见明显异常。
血小板(platelet count,PLT)305×109·L-1,电解质、肾功、肌钙蛋白、血脂、甲功三项、中枢神经特异蛋白等未见明显异常。
精神检查:神情,接触主动,注意力集中,反应尚可。
智能检查:理解力、判断力、一般常识、计算力、记忆力减退,可引出言语性幻听、幻视,思维正常,有阵性兴奋冲动,情感协调,自知力不全。
入院诊断为:①混合型痴呆;②痴呆伴发精神和行为异常。
给予富马酸喹硫平片0.15 g qn、(奥思平)盐酸度洛西汀肠溶片60 mg qd、丙戊酸镁缓释片0.25 g bid、盐酸多奈哌齐胶囊5 mg qn等药物治疗。
4月28日及29日,患者反复多次出现双眼凝视、四肢抽动,每次持续数秒到数分钟不等,查体无特殊补充。
复查脑电图示:轻度异常。
请神经内科会诊后,考虑补充诊断:癫痫,建议加用奥卡西平片治疗。
4月29日遵会诊意见,加用奥卡西平片(法国诺华制药有限公司,批号:T1883)0.15 g bid抗癫痫治疗。
5月6日将奥卡西平片加量至0.3 g bid。
5月8日患者全身出现散在红色皮疹,伴瘙痒,余未诉特殊不适,考虑为奥卡西平过敏,将其减量至0.15 g bid,并予以维生素C片0.1 g qd、氯雷他定片10 mg qd抗过敏治疗,于当日复查血常规。
5月9日血小板报危急值示:PLT 5×109·L-1,床旁查看患者,查体全身未见明显出血点、瘀斑,躯干部皮疹增多,颜面部有新增皮疹和水肿,请皮肤科急会诊后停用奥卡西平片,予以静脉滴注氢化可的松琥珀酸钠100 mg bid、维生素C注射液2 g qd,葡萄糖酸钙注射液1g qd,口服盐酸西替利嗪片10 mg qd、利可君片20 mg tid,炉甘石洗剂外涂对症处理。
临床度洛西汀适应证、用法用量、禁忌证、严重不良反应、相互作用及注意事项

临床度洛西汀适应证、用法用量、禁忌证、严重不良反应、相互作用及注意事项适应证与用法用量适应证①用于治疗抑郁症;②用于治疗广泛性焦虑障碍;③用于治疗慢性肌肉骨骼疼痛。
用法用量1)肠溶片:广泛性焦虑障碍:推荐的起始剂量为 60 mg/日,部分患者可能需要以 30 mg/日为起始剂量,一周后调整至 60 mg/日;对于 60 mg/日的剂量效果不佳的患者,可以考虑将剂量提高到 90 mg/日,最高可增至 120 mg/日。
维持治疗的剂量范围是 60~120 mg/日。
2)肠溶胶囊:①抑郁症:推荐的起始剂量为 40 mg/日(20 mg 一日二次)至60 mg/日(一日一次或 30 mg 一日二次),不考虑进食情况。
部分患者可能需要以 30 mg/日为起始剂量,一周后调整至 60 mg/日。
维持治疗的剂量范围是 60 mg/日。
②广泛性焦虑障碍:推荐的起始剂量为 60 mg/日,一些患者可能需要以 30 mg/日为起始剂量,一周后调整至 60 mg/日。
一些对于60 mg/日的剂量不能充分应答的患者,可以考虑将剂量提高到 90 mg/日,最高可增至 120 mg/日。
维持治疗的剂量范围是 60~120 mg/日。
禁忌证1)禁忌在已经用单胺氧化酶抑制剂(MAOI)治疗精神疾病的患者中使用度洛西汀,因为这会增加患者发生血清素综合征的风险。
停用 MAOI后14 日内禁用本药。
2)禁止对正在接受利奈唑胺或静脉注射亚甲蓝等单胺氧化酶抑制剂(MAOI)治疗的患者中使用度洛西汀治疗,因为这会增加患者发生血清素综合征的风险。
严重不良反应肝胆系统:肝毒性;血管系统:体位性低血压,跌倒和晕厥,血压升高;精神系统:躁狂症/低躁狂症的发作,癫痫发作,严重抑郁症,广泛性焦虑症;眼部:闭角型青光眼;泌尿系统:尿潴留;骨骼肌肉系统:纤维肌痛,骨关节炎引起的慢性疼痛,慢性腰背痛。
相互作用禁忌:度洛西汀-CYP1A2 抑制剂(强力)严重:度洛西汀-盐酸环丙沙星注意事项建议尽可能减少剂量而不是突然停止度洛西汀缓释胶囊停用后的不良反应包括:头晕、头痛、恶心、腹泻、感觉异常、烦躁不安、呕吐、失眠、焦虑、多汗症和疲劳等。
盐酸度洛西汀质量标准

盐酸度洛西汀YansuanDuluoxitingDuloxetine HydrochlorideC18H19NOS·HCl 333.88 本品为(S)-(+)-N-甲基-3-(1-萘氧基)-3-(2-噻吩基)丙胺盐酸盐。
按干燥品计算,含C18H19NOS·HCl应为97.0~102.0%。
【性状】本品为白色至近白色粉末。
本品在甲醇中易溶,在水中略溶,在己烷中几乎不溶。
【鉴别】(1)取本品约25 mg,加甲醇5 ml溶解,本品的红外光吸收图谱应与对照品的图谱一致(中国药典2010年版二部附录IV C)。
(2)供试品溶液主峰的保留时间应与度洛西汀杂质A测定项下的系统适用性试验的色谱图中S-异构体峰的保留时间一致。
(3)本品显氯化物的鉴别反应(中国药典2010年版二部附录III)。
【检查】异构体(杂质A)精密称取本品适量,用流动相溶解并稀释制成每1ml中含有0.1 mg的溶液,作为供试品溶液。
另取盐酸度洛西汀对照品适量,用流动相溶解并稀释制成每1 ml中含有0.1 µg的溶液,作为灵敏度测试溶液。
取盐酸度洛西汀对照品和杂质A 对照品各约5 mg,置于同一个50 ml容量瓶中,超声溶解,加流动相至刻度,作为系统适用性溶液。
照高效液相色谱法(中国药典2010年版二部附录V D)试验,用OD-H为填充剂(25 cm×4.6 mm,5 µm);移取2 ml二乙胺加入至正己烷:异丙醇=83:17(v/v)混合溶液1000ml中为流动相;检测波长为230 nm,流速为每分钟1.0 ml,柱温为40℃;取系统适用性溶液注入液相色谱仪,记录色谱图,度洛西汀峰和杂质A峰的分离度不小于3.5,度洛西汀峰和杂质A峰的拖尾因子应在0.8~1.5之间。
精密量取灵敏度测试溶液10 µl注入液相色谱仪,记录色谱图,信噪比不得小于3。
精密量取供试品溶液10 µl注入液相色谱仪,记录色谱图至主成分保留时间的2倍,供试品溶液色谱图中如有与杂质A峰保留时间一致的色谱峰,杂质A(相对保留时间约为1.3)的峰面积不得大于杂质A与度洛西汀峰面积之和的0.005倍(0.5%)。
奥思平说明书
奥思平(盐酸度洛西汀肠溶片)上海中西制药有限公司批准文号:国药准字H20061263已有1位医生分享药品名称:通用名称:盐酸度洛西汀肠溶片英文名称:Duloxetine Hydrochoride Enteric Capsules商品名称:奥思平成分:本品主要组成成分盐酸度洛西汀。
性状:片剂适应症:本品用于治疗抑郁症。
规格:20mg×20片/盒。
用法用量:1. 起始治疗。
推荐本品的起始剂量为40mg/日(20mg一日二次)至60mg/日(一日一次或30mg一日二次),不考虑进食情况。
现有的临床研究数据未证实剂量超过60mg/日将增加疗效。
一般认为抑郁症的急性发作需要数月或更长时间的药物治疗,但尚没有充足的试验资料来确定患者应该连续服用度洛西汀治疗达多长时间。
对此类患者,应对其接受维持治疗的必要性以及相应所需的剂量作定期评估。
2. 特殊人群。
肾脏功能受损患者的用量一对于晚期肾脏疾病(需要透析的)患者,或有严重肾脏功能损害(估计肌酐清除率〈30 mL/min的)患者,建议不用本品(见药理毒理)。
肝功能不全的患者的用量一建议有任何肝功能不全的患者避免服用本品(见药理毒理和注意事项)。
老年患者的用量一对于老年患者,建议不必根据年龄调整剂量。
与任何药物一样,治疗老年患者时应该慎重。
在老年患者中个体化调整剂量时,增加剂量时应该额外小心。
对妊娠后三个月的女性患者的治疗一在妊娠后三个月内接触SSRIs或SNRls(五羟色胺和去甲肾上腺素再摄取抑制剂)的新生儿,产生的并发症会导致住院时间延长、需要呼吸支持和管道喂食(见注意事项)。
当孕期女性用度洛西汀治疗时,在妊娠后三个月,医生应对治疗的潜在风险和获益进行认真的评价。
医生应考虑在妊娠晚期逐渐减度洛西汀的用量。
3. 度洛西汀停药。
已有报道本品及其他SSRI 和SNRIs药物的停药反应(见注意事项)。
停药时应对这些症状进行监测。
建议尽可能的逐渐减药,而不是骤停药物。
度洛西汀对躯体疼痛障碍的临床疗效研究

度洛西汀对躯体疼痛障碍的临床疗效研究发表时间:2016-11-01T16:37:29.760Z 来源:《医药前沿》2016年10月第30期作者:林卫苏冰王佳琳[导读] 躯体形式疼痛障碍一种心因性疼痛疾病,主要表现为无法用躯体障碍或生理过程来解释的一种严重疼痛。
(青岛市精神卫生中心山东青岛 266034)【摘要】目的:本文主要是对度洛西汀对躯体疼痛障碍的临床疗效进行研究。
方法:选择2014年4月到2016年4月我院收治的85例躯体疼痛障碍患者,将其随机分为研究组43例、参照组42例。
研究组患者进行口服度洛西汀片治疗,参照组患者进行口服安慰剂治疗,对两组患者在治疗前、治疗后的效果进行评定与比较。
结果:研究组患者治疗的总有效率为93.02%,参照组治疗的总有效率为80.95%,研究组患者治疗的有效率明显高于参照组,P<0.05。
结论:对躯体形式疼痛障碍患者通过度洛西汀进行治疗能够取得良好的治疗效果,值得临床进一步推广。
【关键词】度洛西汀;躯体疼痛障碍;临床治疗【中图分类号】R614 【文献标识码】A 【文章编号】2095-1752(2016)30-0200-02【Abstract】Objective This article is mainly for duloxetine to study the clinical effect of body pain disorders. Methods From April 2014 to April 2016 our hospital 85 cases of body pain disorder, the 43 cases were randomly divided into research group and control group, 42 cases. Research on patients with oral duloxetine treatment, control group patients with oral placebo treatment, before and after treatment in patients with two groups of effect evaluation and comparison. Results The total effective rate of treatment group patients was 93.02%, control group total effective rate of treatment was 80.95%, the effectiveness of the team treatment of patients with significantly higher than control group, P < 0.05). Conclusion In patients with somatoform pain disorder by duloxetine treatment can achieve good treatment effect, worth clinical further promotion.【Key words】Duloxetine; Body pain disorders; Clinical treatment躯体形式疼痛障碍一种心因性疼痛疾病,主要表现为无法用躯体障碍或生理过程来解释的一种严重疼痛。
度洛西汀

下列噻吩化合物经Marmieh反应,然后用Yarnaguchi-Masher-Pohland(YMP)复合试剂LiAl2H2在乙醚中,于 -78℃进行不对称还原,而后加入氢溴酸,使还原产物以氢溴酸盐形式得到。再和1-氟萘进行烷基化反应,最后 去甲基化,产物以草酸盐的形式得到。
相关药品说AOIs)均抑制5-HT代谢,两药合用易出现严重不良反应,如中枢神经毒性或5HT综合征(其临床表现为高血压、高热、肌阵挛、激惹及烦躁不安、反射亢进、出汗、寒战及震颤),甚至死亡。 禁止本药与MAOIs合用;停用MAOIs 14天后才能使用本药;停用本药5天后才能使用MAOIs。2.卷曲霉素、依诺沙 星、氟伏沙明及奎尼丁可抑制本药的代谢,增加本药血药浓度(或生物利用度)及毒性,两者合用须监测不良反应, 需要时减少本药剂量。3.本药与氟西汀、帕罗西汀合用,互相抑制代谢,两药的生物利用度、血药浓度均增加, 发生严重不良反应的危险性增加,合用时应调整两药剂量。4.本药可抑制三环类抗抑郁药(如阿米替林)的代谢, 两者合用,本药可增加后者的生物利用度、血药浓度及毒性。如必须合用,应密切监测三环类抗抑郁药的血药浓 度、中毒的症状及体征(抗胆碱能作用、过度镇静、意识混乱及心律失常)。5.本药可抑制吩噻嗪类药(奋乃静)的 代谢,增加后者的血药浓度及毒性(过度镇静、意识障碍、心律失常、直立性低血压、高热及锥体外系反应)。两 者合用应监测不良反应,必要时减少剂量。6.本药可抑制硫利达嗪的代谢,增加后者血药浓度及心脏毒性(QT间 期延长、尖端扭转性室性心动过速、心脏停搏),两者不应合用。7.本药可抑制Ic类抗心律失常药的代谢,增加 后者的血药浓度及心脏毒性。两者合用应密切监测Ic类抗心律失常药的血药浓度及心电图。8.本药与中枢神经系 统抑制药合用,可引起精神运动性障碍恶化,禁止两者合用。食物不影响本药的血药峰浓度,但可减慢吸收,并 降低吸收度10% 。
抗抑郁药怡诺思、博乐欣、奥思平等说明书介绍2资料

肝脏 肝脏 肝脏
主要代谢产物 去甲氯米帕明
去甲替林 去甲多塞平 去甲文拉法
排泄途径
肾脏、少量经粪便、乳 汁
肾脏、乳汁
肾脏
肾脏、少量 经粪便
肾脏
半衰期
19-37小时
9-25小时
8-25小时
8-25小时; 9-13小时
12小时
代谢酶
CYP3A4、2C19、2D6
CYP3A4、2C19 、2D6、1A2
CYP2D6、
类合用,不良反应 7. 本品与阿托品 合用,不良反应增 类合用,不良反应
增加。
类合用,不良反应 加。
增加。
8. 与单胺氧化酶 增加。 合用,可发生高血
8. 与单胺氧化酶 合用,可发生高血
氯米帕明 阿米替林 多塞平 文拉法辛 度洛西汀
血浆蛋白结合率 96%-97%
90%
代谢途径 肝脏,有首过效应 肝脏
端扭转心律失常。 2. 本品与乙醇或 其他中枢神经系统 抑制药合用,中枢 神经抑制作用增强 。 3. 本品与肾上腺 素、去甲肾上腺素 合用,易致阵发性 高血压及心律失常 。 4. 本品与可乐定 合用,后者抗高血 压作用减弱。 5. 本品与抗惊厥 药合用,可降低抗 惊厥药的作用。 6. 本品与氟西汀 或氟伏沙明合用, 可增加两者的血浆 浓度,出现惊厥, 不良反应增加。
清转氨酶水平的
风险。既往有躁
狂史的患者慎用
肝、肾功能严重不 度洛西汀。既往
全,前列腺肥大、 有癫痫发作史的
肝、肾功能严重不 肝、肾功能严重不 肝、肾功能严重不 老年或心血管疾病 患者慎用度洛西
全、前列腺肥大、 全、前列腺肥大、 全、前列腺肥大、 患者慎用,使用期 汀。度洛西汀慎 临床症状的恶化和自杀风
奥思平 说明书

奥思平说明书注意事项:一般注意事项肝脏毒性-度洛西汀有增加血清转氨酶水平的风险。
肝脏转氨酶升高导致0.4%(31/8454) 度洛西汀治疗的患者中断治疗。
这些患者出现转氨酶升高的时间中位数为2个月。
在抑郁症患者中进行的对照试验中,0.9%(8/930)用度洛西汀治疗的患者ALT升高超过正常上限3倍以上,而安慰剂组中为0.3%(2/652)。
所有安慰剂对照研究中,度洛西汀组中有1% (39/3732)的患者ALT升高超过正常上限3倍以上,而安慰剂组中为0.2%(6/2568)。
固定剂量的安慰剂对照研究中,有证据显示ALT升高超过正常上限3倍和AST升高超过正常上限5倍,与药物剂量有量效关系。
上市后监测还报道出现腹痛、肝肿大、伴有或无黄疸的转氨酶升高超过正常值上限20倍的肝炎病例,反映了混合性或肝细胞性损伤,也有出现转氨酶无明显升高的胆汁郁积型黄疸病例的报道。
在排除梗阻的情况下,通常认为转氨酶升高伴有胆红素升高,是严重肝脏损害的重要指标。
国外临床试验中,3名服用度洛西汀的患者,出现转氨酶、胆红素和碱性磷酸酶升高,提示存在梗阻情况。
上述患者有严重的过度饮酒的情况,这可能是出现上述异常指标的原因所在。
两名安慰剂治疗的患者也出现了转氨酶、胆红素升高的情况。
上市后报告显示转氨酶、胆红素和碱性磷酸酶升高也可以发生在患有慢性肝病或肝硬化患者中。
因为度洛西汀和酒精的相互作用可能引起肝损害或者加剧已有的肝病恶化,所以度洛西汀通常不用于有习惯性饮酒和慢性肝病患者的治疗。
在临床试验过程中观察到有些病人肝酶升高,这些现象通常是一过性的和自限性的,或者在停药恢复。
严重肝酶升高(>正常值上限的10倍)或肝损伤伴胆汁郁积,或混合型肝病罕有报道,有些病例与酒精滥用或既往肝病有关。
在酒精使用患者或既往有肝病史的患者中,度洛西汀应慎用。
对血压的影响-与安慰剂相比,度洛西汀治疗引起血压升高,平均升高:收缩压2 mm Hg,舒张压0.5 mm Hg ,偶尔有至少一次测量的收缩压大于140 mm Hg。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
奥思平(盐酸度洛西汀肠溶片)药品说明书药品名称通用名称:盐酸度洛西汀肠溶片商品名称:盐酸度洛西汀肠溶胶囊汉语拼音:YanSuanDuLuoXiDingChangRongJiaoNang剂型中国药典剂型胶囊剂性状本品内容物为白色或类白色小丸。
主要成份盐酸度洛西汀化学名:(+)-(S)-N-甲基-γ-(1-萘基氧)-2-噻吩丙醇胺盐酸盐分子式:C18H19NOS·HCL 分子量:333.88。
适应症用于治疗抑郁症。
规格20mg*20粒不良反应MDD急性期治疗的安慰剂对照研究中,接受度洛西汀治疗的患者中,发生率≧2%以及高于安慰剂组的不良反应。
其中最常见的不良反应(发生率≧5%,且至少是安慰剂组发生率的两倍)包括恶心、口干、便秘、食欲下降、疲乏、嗜睡、出汗增多(详情请咨询专科医生)尿急-度洛西汀属于已知的影响尿道阻力的药物。
如果应用度洛西汀治疗的过程中出现尿急,应当考虑药物导致的可能性。
实验室变化-9周MDD或13周DPNP安慰剂对照研究中,与基线比较,度洛西汀治疗结束后,ALT、AST、CPK和碱性磷酸(醋)酶均值出现轻度增高。
与安慰剂组比较,度洛西汀治疗的患者中出现这些异常值表现为偶发、中度、一过性的异常(见注意事项)。
生命体征改变-在为期9周的MDD安慰剂对照研究中,度洛西汀的剂量为40-120mg/日,导致血压升高,与安慰剂比较,收缩压平均升高2mrnHg,舒张压平均升高0.5mmHg,收缩压超过140mmHg的发生率增加。
9周的MDD和13周DPN尸安慰剂对照研究中,度洛西汀治疗导致心率轻度增加,与安慰剂比较约增加2次/分钟。
用法用量1. 起始治疗。
推荐本品的起始剂量为40mg/日(20mg一日二次)至60mg/日(一日一次或30mg一日二次),不考虑进食情况。
现有的临床研究数据未证实剂量超过60mg/日将增加疗效。
一般认为抑郁症的急性发作需要数月或更长时间的药物治疗,但尚没有充足的试验资料来确定患者应该连续服用度洛西汀治疗达多长时间。
对此类患者,应对其接受维持治疗的必要性以及相应所需的剂量作定期评估。
2. 特殊人群。
肾脏功能受损患者的用量一对于晚期肾脏疾病(需要透析的)患者,或有严重肾脏功能损害(估计肌酐清除率〈30 mL/min的)患者,建议不用本品(见药理毒理)。
肝功能不全的患者的用量一建议有任何肝功能不全的患者避免服用本品(见药理毒理和注意事项)。
老年患者的用量一对于老年患者,建议不必根据年龄调整剂量。
与任何药物一样,治疗老年患者时应该慎重。
在老年患者中个体化调整剂量时,增加剂量时应该额外小心。
对妊娠后三个月的女性患者的治疗一在妊娠后三个月内接触SSRIs 或SNRls(五羟色胺和去甲肾上腺素再摄取抑制剂)的新生儿,产生的并发症会导致住院时间延长、需要呼吸支持和管道喂食(见注意事项)。
当孕期女性用度洛西汀治疗时,在妊娠后三个月,医生应对治疗的潜在风险和获益进行认真的评价。
医生应考虑在妊娠晚期逐渐减度洛西汀的用量。
3. 度洛西汀停药。
已有报道本品及其他SSRI和SNRIs药物的停药反应(见注意事项)。
停药时应对这些症状进行监测。
建议尽可能的逐渐减药,而不是骤停药物。
由于减少药物剂量或停药而引起了无法耐受的症状时,可以考虑恢夏使用以往的的处方剂量。
随后再以更慢的的速度减药。
4. 与单胺氧化酶抑制剂(MOA I)间的换药。
MOAT停药后至少14天才可开始本品的治疗。
本品停药后至少5天才可以开始MOAT的治疗(见禁忌和警告)。
禁忌 1.度洛西汀肠溶胶囊禁用于已知对度洛西汀肠溶胶囊或产品中任何非活性成分过敏的患者。
2.禁止与单胺氧化酶抑制剂(MAOIs)联用。
(见警告)3.未经治疗的窄角型青光眼4.临床试验显示,度洛西汀有增加瞳孔散大的风险,未经治疗的窄角型青光眼患者应避免使用度洛西汀注意事项肝脏毒性-度洛西汀有增加血清转氨酶水平的风险。
肝脏转氨酶升高导致0.4%(31/8454)度洛西汀治疗的患者中断治疗。
这些患者出现转氨酶升高的时间中位数为2个月。
在抑郁症患者中进行的对照试验中,09%(8/930)用度洛西汀治疗的患者ALT升高超过正常上限3倍以上,而安慰剂组中为0.3%(2/652)。
所有安慰剂对照研究中,度洛西汀组中有1%(39/3732)的患者ALT升高超过正常上限3倍以上,而安慰剂组中为0.2%(6/2568)。
固定剂量的安慰剂对照研究中,有证据显示ALT升高超过正常上限3倍和AST升高超过正常上限5倍,与药物剂量有量效关系。
度洛西汀通常不用于有习惯性饮酒和慢性肝病患者的治疗。
治疗开始前应测量血压,治疗后应定期测量。
在酒精使用患者或既往有肝病史的患者中,度洛西汀应慎用。
既往有癫痫发作史的患者慎用度洛西汀。
既往有躁狂史的患者慎用度洛西汀停药-已对度洛西汀的停药症状做过系统研究。
在抑郁症患者中进行的为期9周的安慰剂对照试验中,骤停药物,观察到度洛西汀治疗的患者发生率2%或明显高于骤停安慰剂的症状包括:头晕,恶心,头痛,感孕妇及哺乳期妇女用药妊娠:妊娠分类C-在动物生殖研究中,发现度洛西汀对胚胎/胎儿和出生后的发育有不良影响。
大鼠和家兔在胚胎的器官发生期口服度洛西汀,剂量达45mg/kg/日时(大鼠剂量为7倍于[MRHD,60mg/kg/日],4倍于按照体重/体表面积指数计算的人体剂量120mg/日;家兔的剂量为15倍于[MRHD],7倍于按照体重/体表面积指数计算的人体剂量120mg/日),未发现有致畸作用。
这个剂量时,胎儿体重降低。
无效应剂量为10mg/kg/日(在大鼠为2倍于[MRHD],约1倍于按照体重/体表面积指数计算的人体剂量120mg/日;家兔的剂量为3倍于[MRHD],约2倍于按照体重/体表面积指数计算的人体剂量120mg/日)。
妊娠大鼠在整个孕期和哺乳期予以口服度洛西汀,在剂量30 mg/kg/日时(5倍于[MRHD],2倍于按照体重/体表面积指数mg/m2计算的人体剂量120mg/日),幼儿出生后存活1天、出生时和哺乳期的体重下降;无效应剂量为10 mg/kg/日。
而且,母体使用度洛西汀的剂量达到30 mg/kg/日时,幼儿的行为表现与反应性的增高一致,如对噪音的惊诧反应增强,自主活动的习惯性降低。
但母体使用度洛西汀对子代断奶后的生长和生殖行为没有负面影响。
由于缺乏足够的、设计良好的孕期女性对照研究,因此,只有在权衡对胎儿潜在的受益超过风险时,才考虑在母孕期使用度洛西汀。
近期服用度洛西汀的母亲,其新生儿可能发生如下停药症状,包括肌张力下降、震颤、紧张不安、喂养困难、呼吸窘迫和癫痫发作。
非致畸效应-新生儿在妊娠中期三个月暴露于SSRIs或SNRIs(五羟色胺和去甲肾上腺素再摄取抑制剂)后,产生的并发症会导致住院时间延长、需要呼吸支持和管道喂食。
这些并发症在出生后会立刻发生。
已有的临床发现包括呼吸窘迫、紫绀、呼吸暂停、癫痫发作和体温不稳、喂养困难、呕吐、低血糖症、肌张力不全、肌张力增高、反射亢进、震颤、反应过度、易怒、哭闹不止。
这些情况或者是SSRIs 和SNRIs的直接毒性作用所致或者可能是药物的戒断反应。
应当注意的是,在某些病例中,其临床表现与五羟色胺综合征一致(见警告,单胺氧化酶抑制剂)。
当妊娠女性在妊娠晚期三个月接受度洛西汀治疗时,医生应对治疗的潜在风险和获益进行认真地评价(见用法用量)。
分娩:度洛西汀对于人类分娩的影响尚不明确。
因此,只有证实度洛西汀对于胎儿的潜在获益超过风险时才考虑在分娩期应用。
哺乳:度洛西汀可通过哺乳期妇女乳汁分泌。
估计婴儿得到的日剂量大约为母亲服药剂量的0.14%(mg/kg)。
由于度洛西汀对婴儿的作用不明,因此服用度洛西汀的病人不推荐母乳喂养。
儿童用药对于儿童患者的疗效和安全性尚不清楚(见警告)。
如果考虑在儿童青少年中使用度洛西汀,必须权衡潜在的风险和临床需要。
老人用药在人体度洛西汀过量的研究资料很有限,临床资料中迅速吞下3000mg,单独或与其它药物一起,未报道有致死性事件发生。
然而,上市后有急性药物过量致死的报告,主要是混合性药物过量,也有单独服用度洛西汀约1000mg的。
过量的休征和症状(大多数是混合性药物过量)包括血清素综合症、嗜睡、呕吐和癫痫发作。
药物相互作用可能影响度洛西汀的其他药物度洛西汀的代谢与CYP1A2和CYP2D6有关CYP1A2抑制剂-度洛西汀与氟伏沙明(强CYP1A2抑制剂)联合应用于男性受试者(n=14)时,度洛西汀AUC增加超过5倍,Cmax增加约2.5倍,T1/2增加约3倍。
其他对CYP1A2代谢有抑制作用的药物包括:西米替丁,喹诺酮类抗生素例如环丙沙星, 依诺沙星。
CYP2D6抑制剂-由于CYP2D6参与度洛西汀的代谢,所以合并使用度洛西汀和强CYP2D6抑制剂时,盐酸度洛西汀的药物浓度将会增加,(见[注意事项]项下药物相互作用)。
度洛西汀可能影响的其他药物通过CYP1A2代谢的药物-体外药物相互作用研究显示:度洛西汀对CYP1A2活性无诱导作用。
因此,虽然未进行有关酶诱导作用的临床研究,预计不会因为酶诱导作用而使CYP1A2底物(例如茶碱、咖啡因)的代谢增加。
虽然体外研究显示度洛西汀是CYP1A2抑制剂,但是度洛西汀(60 mg 每日两次给药)与作为CYP1A2底物的茶碱联合使用时,茶碱的药代动力学没有明显变化。
因此,度洛西汀对CYP1A2底物的代谢不可能产生具有临床意义的明显影响。
通过CYP2D6代谢的药物-度洛西汀是CYP2D6中度抑制剂,能够增加经CYP2D6代谢药物的AUC和Cmax。
(见[注意事项])。
因而联合使用度洛西汀和其他主要经该酶代谢,并且治疗剂量范围狭窄的药物时,应该谨慎(见[注意事项]项下药物相互作用)。
通过CYP2C9代谢的药物-体外研究显示度洛西汀对CYP2C9酶的活性无抑制作用。
因而,虽然未进行相关的临床研究,预计度洛西汀对CYP2C9酶底物的代谢无抑制作用。
通过CYP3A代谢的药物-体外研究结果显示度洛西汀对CYP3A酶的活性无抑制或诱导作用。
因而,虽然未进行相关的临床研究,预计CYP3A酶底物(例如口服避孕药和其他甾体药物)不会因为酶诱导或抑制而产生代谢增强或抑制。
通过CYP2C19代谢的药物-体外研究结果显示治疗浓度的度洛西汀对CYP2C19酶活性无抑制作用。
因而,虽然未进行相关的临床研究,预计度洛西汀对CYP2C19酶底物的代谢无抑制作用。
联合应用苯二氮卓类药物的研究劳拉西泮-稳态浓度的度洛西汀(60mg Q 12 hrs)与劳拉西泮(2mg Q 12hrs)合用时,劳拉西泮的药代动力学不受联合治疗的影响。
替马西泮-稳态浓度的度洛西汀(20mg qhs)与替马西泮(30mg qhs)合用时,替马西泮的药代动力学不受联合治疗的影响。