Bioedit操作指南
bioedit中文使用手册

来自生物软件网谢谢jerry(xujz602@)和Huang(candycat719@)辛苦的翻译工作翻译说明1 原英文稿中附有许多示例如输出窗口序列等译文一般用参见英文稿表示在阅读此段时请参见英文中的图示对于一些较小的示例如等式的推导译文中保留2 原英文稿中也给出了许多算法和程序的原始文献和网址译者认为这是BioEdit的一个优点如果想深入的学习不能不读一读原始的文献译文中用REFERENCE表示请参考英文原稿3 译文中对专业的词汇采用以下办法处理即一般采用国内已有人使用的译法如果未见到则译者给出一种译法并在旁边列出英文译文中各节的标题都是这样处理的前者如最简单的例子Aligment一词有比对对比对排等多种翻译郝柏林院士建议译做联配见生物信息学手册p175方舟子译做排列对比见新语丝网页本译文采用联配的译法后者如mask一词文中专门有一节解释其含义此词的普通含义有面具遮饰译文中使用屏蔽并在旁边写mask总之此类词汇使用多了自然明了其内在的含义4 偶尔译者会对某处略做解释旁边用译者注表示表示译者的理解请注意5 翻译时在词汇的翻译和算法的理解上参考了以下资料A 生物信息学手册郝柏林等著上海科学技术出版社 2000年B 生物信息学基因和蛋白质分析的实用指南 Andreas D.Baxebanis等原著李衍达等译清华大学出版社 2000年6 由于译者占有的资料不多水平有限在译文中肯定有漏译译的不全面甚至理解完全错误的地方(尤其是算法上)敬请指正Email me at: xujz602@关于BioEdit介绍BioEdit版本5.0.6版权©1997-2001汤姆霍尔当前版本制作于2001.12.2BioEdit是一个生物序列编辑器可在Windows 95/98/NT/2000中运行它的基本功能是提供蛋白质核酸序列的编辑排列处理和分析 1.0α版本是最早的未完成的并有瑕疵的版本 1.0α版本也一直未完成并有很多问题但是比较前一个还是增加了一点东西修正了一些问题在2.0版本中在增加和配置附加分析应用程序上增加了一个界面使其能通过BioEdit得到一个图形界面而且还增加了位置排列的信息基础动态描影版本3中增加了疏水亲水面互交的2-D浮雕数据绘图和一些更多的序列操作法版本4为绘制和注解质粒载体增加了一个图形界面在4.7.1版本中修改了处理序列信息和存储方法而且增加了一个二进制文件格式允许快速保存和打开大的排列序列容量增加到20,000在版本5中增加了自动注解序列或手动使用所有的标准Genbank功能部件定义而且在Isis Pharmaceuticals公司的请求下增加了序列排序和分型组控制注解行以及残基和非残基字符的鉴别BioEdit并不打算成为一个强序列分析程序但是打算成为一个序列分析的友好用户界面并连接其他在局域网和万维网上的更多的序列分析程序它现在使用于大的排列>2000序列文件界面最初模仿于一个非常好的程序――Don Gilbert 编写的SeqApp and SeqPup印地安那州大学免费提供SeqApp (用于个人计算机) and SeqPup (用于交换平台)地址是ftp:///molbio/seqpup/GeneDoc是一个特别的排列程序能够自由的在Windows 9x 和NT上使用也是一个非常专业的程序有很好的蛋白质排列注解和分析描影和结构定义功能部件就象一个反映排列的内在的进化树而这些在BioEdit中是没有的GeneDoc的网址是/biomed/genedocGeneDoc有比BioEdit更好的描影和分类选项有助于手工排列序列还有更好的图形处理缠绕和伸展的排列视图选项动态共有序列和更平滑和更快速的排列卷曲和刷新BioEdit是用Borland's C++ Builder编写的C++程序我是北卡罗来纳州大学微生物系的研究生不是专业的程序员这是我学习C++语言的入门必然是个非专业的设计这不是我博士工作的一部分这个程序非常小而且很有效率BioEdit为序列排列输出和一些分析提供容易的工具BioEdit功能BioEdit的主要目的是为那些不愿意被迫详细了解一个程序的使用方法的生物学家提供一个有用的工具BioEdit是直观的菜单式的并有大量的图示提供用户一个外部分析程序的图形界面主要功能是提供明显的容易使用的菜单选项5.0.6版本提供以下功能用于序列处理和编辑的简单的图形界面使用编辑选项包括残基的select and drag选择和拖动和grab and drag抓取和拖动变量选择选项鼠标点击插入和删除缺口全框选择全屏编辑中剪切复制和粘贴编辑窗口的自动刷新固定序列框保护排列中的固定残基使用各种功能部件内含子外显子促进子CDS和所有标准GenBank功能部件类型自动的和手动的注解序列使用一个模板序列自动注解同一排列中的其他序列序列分组分为各个颜色编码家族为同步手动排列锁定组成员用户定义的适当功能部件能够设定考虑任何功能部件就像用于类似性描影序列同一性矩阵和保存图表视图的核酸或氨基酸序列中的相关碱基用户定义的基序搜索使用标准的Prosite命名法和IUPAC功能部件允许搜索核酸或氨基酸序列还有精确的文本搜索包括或忽略缺口程序行可以定义为DNA RNA核酸蛋白质未定义或注解注解可以用于保存普通的注释或东西就象二级结构模糊定义但是不能保存计算根本的多基因树图阅读器支持节点翻转和打印链接多基因树图到排列并保存到BioEdit格式排列文件在一个排列末端添加另一个排列配置附件应用程序界面进入一个有BioEdit产生的图形界面的外部分析程序在外部应用程序中自动提供信息和找回文件外部应用程序进入分开的调度单位允许同步应用BioEdit外部程序的输出文件可以自动被其他程序打开在ABI自动序列模型3773733700中显示打印和编辑ABI痕迹文件在版本2和3中有SCF文件就象用Licor序列输出文件RNA比较分析工具包括共变可能配对和互交信息分析使用鼠标指示的动态数据视图的互交信息输出2D矩阵图表关于互交信息矩阵行和框的互交式的1D图表用BioEdit或GanBank格式保存序列注解信息通过氨基酸翻译排列蛋白质编码核酸序列在排列中搜索保存的残基寻找好的PCR目标或帮助定义基序在核酸或蛋白质序列中搜索用户定义的基序或用通配符搜索精确的文本并选择包括或忽略缺口用支持最多20,000序列每个文档进行循环存储器分配最多可以成功测定四百六十万个碱基 E. coli基因组核糖体数据库中的原核细胞16SRNA排列29 Mb, 6205个序列将会被单独处理在配置为Pentium 233 Mhz80 Mb RAM的计算机中用BioEdit计划文件格式最多只需要10秒种可以写入一个16S RNA排列内部的读写GenBank Fasta Phylip和NBRF/PIR文件用Don Gilbert’s ReadSeq导入输出一些其他格式的文件使用BioEdit计划文件格式快速读写大排列文件使用自动更新的排列蛋白质全标题和GenBank区域信息进行ClustalW多序列排列Des Higgins et. al.编写的内部界面外部程序就象排列来自于核苷酸序列的蛋白质视图时的核苷酸编码序列将残基块状复制到剪贴板允许将全不排列或部分排列粘贴到文字处理器基本序列处理在文档之间复制粘贴序列翻译和还原编码RNAÆDNAÆRNA反转互补大写字母小写字母多文档界面最多同时打开20个文档但是在其他打开的窗口不能设置限制六框翻译核酸序列为Fasta格式ORF表用矢量图进行半自动质粒矢量绘图和注解自动酶切位点和位置标记自动多接头视图和用户控制绘图工具将质粒文件保存为可编辑的矢量图形文件如位图复制到其他图形程序并可以打印氨基酸和核苷酸成分摘要和图表Revert to Saved恢复保存和undo撤销功能编辑氨基酸和核酸序列简单的指定色彩表编辑蛋白质和核酸序列使用不同的色彩表排列易感的描影法以信息为根据其中包括排列位置BioEdit 能够读写GenBank, Fasta, NBRF/PIR, Phylip 3.2 和 Phylip 4格式能够读ClustalW 和 GCG格式.10个附加格式的导入输出过滤器使用Don Gilbert的ReadSeq导入/添加一个文件到最后的另一个文件上(不考虑文件格式)基本的多文本编辑器限制性内切酶图谱用于任何或所有形式的翻译复酶和输出选项包括酶的提供者和环状DNA选项游览限制性内切酶创造商自动连接到你喜欢的网页游览器如Netscape或Internet Explorer程序和程序组的概述BioEdit是用Borland C++ Builder 3.0编写的(开始时是用C++ Builder 1.0)这是曾经是Borland公司的最新C++产品它结合了Borland C++ 5和Delphi的可视要素库VCL允许用户界面的可视开发使用快速申请开发RAD环境的好处在于它能够容易的创造出大量的图形界面它的缺点是编码不轻便BioEdit只能在Windows 95, 98, NT and 2000中使用我原来计划可以使BioEdit在Win16使用但是自从Windows 3.x过时了以后我就不再计划这样做了组织BioEdit当前支持同时编辑最多50个文件主要的控制形式包括打开文件的菜单创建新文档调整球形选项如色彩表密码子表分析参数选择和一个窗口管理器最初每个文档有它自己的整套处理菜单可以限制文档然而这被一个更传统的多文档界面所替代BioEdit没有使用额外的物理存储器除非编辑大的排列但是它看起来像占用了很多资源BioEdit每个文档最多可以有20,000个序列但在序列大小上没有限制在80MbRAM的233MHz的个人计算机上可以很好的处理一个来自于核糖体数据库的完整的原核16S rRNA排列6205个序列每一个有3319个字符一旦用BioEdit格式保存这个文件可以在几秒钟打开用GenBank格式要几分钟才能打开程序文件(BioEdit.exe)可以在主安装目录中找到可能还有以下子目录apps附件程序网页和网页书签通常以下文件会出现在apps文件夹按名称排列accApp.ini (在首次安装时为accApp.def)Bblast.htmlBioEdit.htmlblast_adv.gifblast_form_0.gifblastall.exe (在没有BLAST的版本中不出现)blastcl3.exe (在没有BLAST的版本中不出现) blast.txtbookmark.txtcap.doccap.execlear_inp.gifclustalw.execlustalw.txtcutter.htmlDnadist.docDnadist.exeDnamlk.docDnamlk.exeDos4gw.exe (PHYLIP 程序需要)Expasy.giffastDNAml.docfastdnaml.exeFitch.docFitch.exeformatdb.exe (在没有BLAST的版本中不出现) IdPlot.exeisrecsmall.gifKitsch.docKitsch.exemod_ad.gifmod_submit.gifnnpredict.htmlNote.gifPFSCAN_form.htmlphi_blast.gifPHIBlast.htmlPhylip.mapProtdist.docProtdist.exeProtpars.docProtpars.exepsi_blast.gifPSIBlast.htmlReadseq.exeReadSeq.txtscnpsit1.htmlSiblogo.gifsmweb.gifdatabase (是局部的BLAST数据库安装的版本必须有BLAST工具).BioEdit (全版本) 有以下文件在database文件夹Ecoli.phrEcoli.pinEcoli.psqEcoli_ORFs.txt (E. coli 开放读码框架的文本文件).helptBioEdit.GID (不是安装来的出现在帮助文件第一次使用后)Bioedit.hlptablesBlosum62codon.tabcolor.tabdayhoffdefcolor.tabenzyme.tabGc.valgonnetIdentifymatchPam120Pam250Pam40Pam80Seqcode.val安装文件夹通常包括以下文件_deisreg.isr (安装相关文件)_isreg32.dll (安装相关文件)BioEdit.exe (BioEdit 执行文件)DeIsL1.isu (安装相关文件)RNaseP_prot.gb (蛋白质排列示例)RNaseP_prot_genes.gb (DNA排列示例)RNaseP_RNA.gb (RNA排列示例)PBSSK_plus.pmd (质粒绘图示例)bacterio.gb (附带GenBank 信息的蛋白质排列示例)bacterio.bio (附带GenBank信息图式注解记号标记和序列族的BioEdit文件示例) YopD.gb (附带GenBank信息的另一个示例文件)TreeView.zip (Roderic D.M. Page编写的极好的系统进化树阅读器完全安装才有) TreeView.txt (记录TreeView的安装信息和配置BioEdit与tree-generating附件的连接)license.txt (BioEdit 许可证协议)ReadMe.txt (总说明)重要的是文件夹和文件的名字不能更改如果更改了BioEdit将不能正确安装将会有一个BioEdit.ini文件出现在你的Windows主目录下它包含BioEdit的初始化默认值和参数选择虽然这个文件可以手动编辑但是我们推荐不要编辑和手动编辑这个文件当前被支持功能部件和已知问题的列表请看BioEdit的功能和已知问题局限性已知问题和局限性BioEdit想要成为一个处理个别简单序列的多用途界面带有适合于自动化多重排列选项的综合序列排列最佳成对排列并且着重于使手工排列更容易随着时间的推移增加了一些附件的功能质粒绘图限制性内切酶图谱ABI和SCF查阅RNA比较分析和其他功能中的图式注解然而常用的查找功能特殊化分析如蛋白质二级结构三级结构的预测RNA结构的热动力学预测排列性质的统计学分析序列模式的概率或神经网络模型排列和结构的预测不包括在这个程序之内虽然用户可以配置命令行附件应用软件有程序链接连到ClustalW局域BLAST和BLAST client 3但是在ClustalW程序或BLAST程序升级后不能保证这些链接正确工作虽然在BioEdit安装程序中提供的局域BLAST和Clustal程序将会继续工作但在下一次NCBI决定改变它的委托人时BLAST client 3将不能正常工作我也不再一直支持这个程序源代码将在稍后提供下载但是会有一些紊乱没有很好注释限制于Borland C++ Builder这是我毫无疑惑的发布源代码的原因同样自动网页链接为网页如BLAST PSI-BLAST PROSITE轮廓扫描网页提供一个选择序列它们的工作依赖于网页的局域HTML模板BioEdit编辑的资源包括查询文本区域的选择序列因为万维网的高度易变性这些也许不能长时间正常工作如果一些地址变化或者HTML界面充分改变这些将不再能正确工作它们可能可以在BioEdit/apps文件夹中局部的被新的同名更新网页所替代但是它们是否能正常工作将依赖于网页中必需的URL定位是否被指定为绝对路径或相对路径它们是否依赖于局域CGI或Java程序和其他潜在的问题想要配置命名行分析程序的界面很好的工作可能不需要复杂的scripting语言然而因为这个界面及其选项的静态特点可能有程序不能正确的通过BioEdit运行虽然绝大多数接受命令行的程序可以被设置总之许多人可能宁愿为了更好的控制选项而从命令行运行程序BioEidt可以很好显示合适大小的排列然而对于一次打开的排列文档数量有限制同样一个单一排列中的序列数量也有限制现在最多一次打开50个排列文档一个排列中的最多序列数是20,000序列数量的限制和序列长度是无关的排列的绝对大小是有效的系统内存决定的如果文档在系统中全部进入虚拟内存编辑将会变得很慢如果排列中有几千个rRNA基因或者全部基因组的序列列表在Win95/98或NT系统中至少需要64到128Mb的内存在Win2000系统中至少需要128Mb内存在排列矩阵N× M > 40,000,000 (N = 序列数M=最长序列长度)时Undo撤消选项自动失效BioEdit是由Borland C++ Builder编写的是100% Windows基础它是不可移植的因为这个程序的大部分是图形界面在UNIX或Mac中可能不好使用BioEdit使用手册序列编辑处理手工序列排列下面是基本的BioEdit排列文档窗口如果你不喜欢现在的样子不要当心字体大小背景颜色残基颜色和标题窗口宽度都可以改变鼠标箭头右下方的黄色条幅显示的是当前序列的绝对位置这同样显示在控制栏的Position标题选择关闭黄色条幅就进入View->show sequence position by mouse arrow总的手工排序功能是在编辑窗口有三个可应用的基本模式选项可在Sequence->Edit Mode中找到Select / Slide mode(选择/调整模式)用鼠标左键选择框住的残基用鼠标来回的拖动选择默认值是朝你滑动的方向忽略unlocked gaps并在所选择的另一边开启新的unlockedgaps为了移动所选择的全部序列的下游不管缺口在移动时按住shift键你也可以在按钮板上切换合适的按钮见后改变默认值为移动所选择的全部序列的下游选定选项后在滑动时用shift键忽略unlocked gaps用shift键选择所有在现在选定的和新选择的残基CTRL键可以在当前选择上增加一个新的选择例如你也许想在三个互不相连的序列中选择残基Edit mode编辑模式在编辑残基模式中你可以在文档的任何位置除了标题放置任何类型的光标用箭头你可以在序列中走来走去编辑有两种形式插入和改写当编辑器在编辑模式可以看见在编辑模式的下拉菜单中有一个选项在其它两个排列模式,这个选项不会出现.Grab & Drag mode(抓取/拖动模式)从mode目录中选择Grab & Drag或者切换G/D按钮见后你可以从屏幕上动态的抓取和拖动单个残基用shift键移动整个残基序列的下游或者在按钮板上切换成合适的按钮――见后Grouping of sequences序列分组Sequences may be grouped into groups (or"families").序列可以进行分组或分成家族一个组的序列排列可以相互锁定意味着手动调节用可调整的残基插入或和删除缺口将自动同步调节一个锁定的组This only applies to sliding resides (Select / slide mode or Grab & Drag mode), not to single insertions and deletions of gaps with right mouse clicks. For information on grouping sequences and locking the alignment of groups of sequences, see grouping sequences.这只适合于可调整的残基Select / slide mode或Grab & Drag mode不能用鼠标右键进行单个缺口的插入和删除想了解有关序列分组和其排列锁定的信息看grouping sequences工具条 / 加速按钮锁定和开启全部序列的所有缺口当打开一个排列这个按钮是在开启状态但是缺口是现在的虽然它们过去被保存在这个按钮被按下去后才能进行改变为了开启当前序列的所有缺口你必须按这个按钮两次进行切换到这个状态第一状态是锁定所有缺口上个按钮的锁定状态按下这个按钮可以用鼠标右键插入单个缺口用鼠标右键删除缺口在所有序列中插入缺口除了在用鼠标右键点击这个按钮的位置在所有序列中插入缺口除了在用鼠标右键点击这个按钮的位置在选择位置没有缺口的序列将不会改变但是有这个按钮在那儿缺口将始终被删除转换鼠标左键和右键的默认值功能切换Grab & Drag模式按下这个按钮可调整残基的默认值是忽略或扩展到下游缺口使用shift键可以调整转换这个功能按下这个按钮可调整残基的默认值是移动全部所选序列的下游胜过忽略或扩展到下游缺口使用shift键可以调整转换这个功能普通视图模式当序列颜色显示时残基根据当前的色彩表着色这个选项用于序列是单色视图时所有其他视图覆盖单色视图反转颜色视图模式背景栏根据每一个残基的色彩表描影残基的颜色是它们普通颜色的反转排列的强度――残基根据每一栏的信息内容灰度描影残基背景根据每一栏的信息内容描影把文档窗口中一致的和类似的残基描影按下这个按钮控制条上将会出现一个下拉菜单可以控制隐藏的百分比开端蛋白质排列的类似性隐藏的矩阵文件可以在Alignment->Similarity Matrix菜单中详细说明绘出功能部件其上有层次的序列只绘出功能部件没有序列根据当前的色彩表序列彩色视图根据当前选择的序列颜色序列单色视图只用于normal view按钮也被按下用一个字符默认值是.显示序列的同一性默认值是top.如果按下前一个按钮这个下拉菜单能够选择标记同一性的字符显示或隐藏交互信息检查器只用于RNA分析引出色彩表编辑对话窗切换ignore anchor points模式如果这个按钮没有按下固定栏限制排列的范围按下这个按钮固定栏被忽视卷屏速度控制器控制水平卷屏条卷屏是因残基增加增加或移去位置标记旗增加或移去一个栏的固定点在编辑盒中编辑在一个文本窗口中进行一个序列主要的编辑会十分方便为一个序列开启一个编辑窗口双击序列的标题或选中序列并从Sequence菜单中选择Edit Sequence为了使改变生效必须按下Apply或Apply and Close按钮取消将不会改变序列在一个序列第一次编辑时将会出现下面的窗口在Sequence Type下拉菜单中下列选项是可用的如果一个序列是未知的蛋白质色彩表通常是彩色的就像一个已经经过类似性底纹处理的蛋白质序列可以保留一个关于排列的每一行的屏幕信息的注解但是不能计算类似性和同一性不服从标准的处理如翻译互补自动排列等在单个序列编辑器中你可以用lock sequence选项选择锁定任何序列应用这个选项时selecting/dragging或抓取和拖动将不能使用但是用鼠标右键增加或删除缺口始终可以使用按下按钮可以展开窗口看相关的GenBank的信息窗口扩展如下按钮可以用于提出在大的编辑窗口中的相关领域**注意GenBank信息将只能用GenBank或BioEdit格式保存***注意GenBank信息包括功能部件领域是内部独立于用户定义的图示注解窗口隐藏一个文档可以进行窗口隐藏就是双击窗口的标题栏可以隐藏标题栏再次双击可以使其变回原来的大小它也可以最小化和最大化增加一个新序列通过以下方式增加新序列1.在Sequence菜单下选择New Sequence选项序列可以像原始文本一样被键入或复制进序列窗口按下Apply按钮可以在文档中增加序列2.通过Edit菜单的Copy Sequence(s)和Paste Sequence(s)命令复制或粘贴来自其他BioEdit文档的序列同样也可以使用当前菜单快捷键(默认值Ctrl+F8复制Ctrl+F9粘贴)全屏编辑序列可以在全屏编辑就像在一个文字处理器上一样必须首先设定Mode选项为Edit Residues(BioEdit在安装后默认模式为Slide Residue)在编辑模式下你可以使用箭头在屏幕上移动输入像在文本编辑器中一样编辑有两种选项插入模式和改写模式它们类似于在文字编辑器中的功能选择序列点击序列的标题可以选中序列拖划出一个方框可以选中多个序列或用shift键选择两个选择序列之间的所有序列用Ctrl键加鼠标可以分别选择标题或给选中的序列加上详细的标题双击标题将会打开一个单序列编辑器再次点击原先选中的标题使其进入全屏编辑模式你可以编辑标题后按下< return >或点击序列标题板的任何位置使对标题的改动生效移动序列想移动一个序列(或一些序列)选中它(用鼠标左键点击它的标题使其变亮)把它拖放到你想要的位置Cut Copy Paste剪切复制粘贴Copy复制编辑窗口的文本(序列残基)用鼠标选择文本并从Edit菜单选择Copy不像文字编辑器你可以复制你想选择的区域而不是复制文本的全部行这种方式复制的区域可以粘贴在任何能够进行文本编辑的程序中如果只是如果你没有选中在全部序列中任何残基序列的标题将会以BioEdit序列结构形式复制到BioEdit的剪贴板在选择Paste Sequence(s)时全部序列将会被粘贴到文档全部序列用鼠标选择序列标题并从Edit菜单选择Copy Sequence(s)标题被选中的序列将以Fasta格式被复制到Windows剪贴板多于一个被选中的序列将以Fasta序列目录的形式复制到剪贴板中并在BioEdit内部复制成一组全部BioEdit序列结构能够被粘贴在任何BioEdit文档中注意BioEdit剪贴板中包括所有序列相关数据Genbank信息图示注解是在BioEdit 同一步骤的内部它们不能在独立的步骤之间转移为了在BioEdit排列文档之间复制序列必须确定两个文档是在程序的同一步骤打开的只有Fasta格式的序列可以被复制到普通的Windows剪贴板Paste粘贴在编辑窗中的文本为了把一个序列粘贴入主编辑窗界面必须是Edit Residues模式见全屏编辑如果文本的一个区域被粘贴到一个序列只有第一行用回车键定义将会被粘贴这避免了在粘贴文本进入序列时可能出现的问题也避免了不注意的使错误的序列在其下为了把文本的片段粘贴到排列的一个区域片段必须一次一个的粘贴进序列如果文档在Slide Residues或Grab and Drag模式Paste粘贴的功能将会和Paste Sequence(s)粘贴序列的功能一样见后全部序列从文档菜单到粘贴序列从Edit菜单中选择Paste Sequence(s)序列将会增加到文档的最后它们可以移动到文档的任何位置Cut剪切和Cut Sequence(s)剪切序列就象Copy复制和CopySequences复制序列一样但是其功能是从文档中删除复制的信息然而只有在Edit Residues模式下残基才能从文档中删除同样当在没有选中任何残基的情况下使用剪切功能时标题被选中的序列将以Fasta格式被复制到Windows剪贴板并以序列结构的形式复制到BioEdit剪贴板中但是它们不能从文档中删除为了适当的从文档中剪切序列可以选择Cut Sequence(s)。
生物新版公式编辑器操作指南

生物新编辑器操作指南目录一、新编辑器特点 (2)二、文本编辑区 (2)1.文本编辑区界面按钮 (2)2.文本编辑区重要按钮详解 (2)三、新理科公式编辑器 (3)1.使用公式编辑器编辑公式 (3)2.修改公式 (3)3.公式编辑器界面简介 (5)4.符号分类详解 (5)1)常用符号 (5)2)运算符 (6)3)箭头 (8)4)希腊字母 (8)6)矩阵、行列式、方程不等式组大括号 (8)6)分式、根式及上下标预设 (9)7)括号及顶部符号预设 (10)8)连加连乘运算符 (10)9)微积分 (11)10)罗马数字 (11)四、公式编辑规范 (11)五、常见问题 (12)一、新编辑器特点1.可在公式编辑器内进行复制和粘贴;2.可将公式编辑器外的字母数字粘贴到公式编辑器内;3.操作更接近于MathType,对于习惯MathType编辑的老师更加友好;没听说过无需理会此条。
4.常用符号如上下标、分式的录入有快捷键可以使用;5.增加LaTex录入模式;二、文本编辑区1.文本编辑区界面按钮:2.文本编辑区重要按钮详解:1)【撤销及恢复撤销】:如果有误操作,比如误删等操作可通过撤销进行恢复;2)【插入图片】:点击可以从本地上传图片,但插入图片还可以使用QQ截图直接在文本编辑区内进行粘贴操作;3)【新理科公式编辑器】:点击可开启理科公式编辑器,所有公式、运算都需要在公式编辑器内进行编辑,后面有公式编辑器的详细介绍;4)【插入表格】:点击可插入表格,目前支持最大10×8的表格;5)其余按钮功能在解析中几乎没有机会使用,故不进行详细介绍。
三、新理科公式编辑器1.使用公式编辑器编辑公式2.修改公式3.公式编辑器界面简介4.符号分类详解1)常用符号①该区域是最常用的符号展示区,包括公式编辑器内的【撤销/取消撤销】、编辑器内的文本【加粗】和【正斜体】等按钮②【特殊符号集】包括【符号】、【数字】、【图形】三部分,点击之后可以选择,其中【数字】部分有带圈数字2)运算符此处数学运用较多,生物可忽略③【运算符】页中包含数学涉及到的所有运算符、集合运算符、逻辑运算符、几何关系及几何图形符号。
BioEdit和Clustalx进行序列比对步骤

1、将要进行序列比对的信息保存为.txt文本文档类型(如11.txt和21.txt)
2、打开BioEdit,选择“New Alignment”图1,在file下拉菜单中选择”import”→“sequence
alignment file”图2选择刚才新建的两个或多个文本,点击保存,保存类型为“Fasta”
生成文件名为.fas。
图1
图2
3、打开Clustalx软件,选择“文件”→“载入序列”,打开刚才保存的文件(注意,该文
件应该与ClustalX在同一文件夹中),选择“序列比对”→“完全序列比对”,这时会生成两个文件,.aln和.dnd文件,其中.aln为序列比对文件,而.dnd为树文件。
打开BioEdit软件,选择选择“New Alignment”,在file下拉菜单中选择”import”→“sequence alignment file”找到刚才生成的.aln文件打。
bioedit序列比对和mega制作系统发育树更新版

Bioedit程序操作1、将序列粘贴到记事本中。
每行以“>”为行头,后面可写上属种名称、模式菌株编号、序列登录号等等,回车,另起一行粘贴序列。
2、打开Bioedit程序,点击“File”,选择“Open”,在打开的对话框中选择文件类型为“All Files”,打开序列TXT文件,然后点击菜单栏的“Accessory Application”,在弹出菜单中选择“ClustalW Multiple alignment”,点击“Run ClustalW”,开始进行序列比对。
比对结束后,中间数据区的序列发生变化,头尾出现长短不一,这时在“mode”框里选择“edit”,然后点击序列上方的标尺,选择前端最短序列处,然后点击菜单栏“Edit”,弹出菜单中选择“Select to Begining”,这时序列前端不整齐的部分被选中,然后按“delete”删除;然后选择后端最短序列处,点击菜单栏“Edit”,弹出菜单中选择“Select to End”,删除,然后保存为Fasta格式文件。
Mega程序操作1、打开Mega程序,点击“File”,选择“Convert To MEGA Format”,选择上步保存的Fasta格式文件,点击“OK”,完成格式转换,然后点击保存,生成.meg格式文件。
2、然后点击“File”,选择“Open Data”,打开刚才保存的.meg格式文件,在弹出的对话框中选择“Nucleotide Sequences”,点击“OK”,然后选择“No”,这时比对的序列被打开。
然后点击菜单栏的“Phylogeny”,在弹出菜单中选择“Bootstrap Test of Phylogeny”,再选择“Neighbor-Joining”,在弹出的对话框里,点击“Test Phylogeny”标签栏,在“Replication”框里设置测试次数,默认是500,不要设置太高,然后点击左下角的红对勾,保存设置。
生物纯化系统操作规程(3篇)

第1篇一、前言生物纯化系统是一种用于分离、纯化和定量生物大分子的设备,广泛应用于生物化学、分子生物学、药物研发等领域。
为确保实验结果的准确性和安全性,以下为生物纯化系统的操作规程。
二、设备准备1. 检查设备状态:开机前,检查设备外观是否完好,各连接管道是否牢固,仪器各部分是否处于正常工作状态。
2. 开机:打开仪器主电源,启动电脑,进入操作界面。
3. 准备工作溶液和样品:所有工作溶液和样品需经过0.45μm的滤膜过滤,样品也可高速离心后取上清备用。
当缓冲液中含有有机溶剂时,需在使用前用低频超声脱气10分钟。
三、操作步骤1. 清洗及管道及装柱:- 将A1管道放入平衡液或banding buffering中,将B1管道放入高盐溶液中或elution buffering中。
- 在system control窗口点击manual pump pump wash basic,选中A1、B1管道为ON,execute。
- 泵清洗程序自动运行结束。
- 选择manual pump flow rate,输入流速1ml/ml,insert;选择manual Alarm & monitor alarm pressure,设置high alarm(输入填料的耐受压力,可在填料说明书中查到),insert,execute。
- 将进样阀的1号位管道接入柱子的柱头,稍微拧紧。
2. 加样:- 将样品溶液缓慢注入柱子,注意控制流速,避免冲击填料。
- 根据样品量和柱床体积,选择合适的上样体积。
3. 洗脱:- 根据实验目的和样品特性,选择合适的洗脱缓冲液和洗脱梯度。
- 调整流速,控制洗脱时间,收集洗脱液。
4. 检测与收集:- 使用合适的检测方法(如紫外检测、电导检测等)监测洗脱液。
- 收集目标蛋白峰对应的洗脱液,进行后续处理。
四、注意事项1. 操作过程中,严格遵守实验规程,确保实验安全。
2. 注意观察仪器运行状态,发现问题及时处理。
Bioedit操作指南PPT课件

1. 目的:纯菌种16S rRNA,利用Sanger方法双端测序,然后检查测序质量,将双端测序 的序列拼接成一条完整的序列,利用这个长序列去与数据库比对,判断这个序列最 可能是从哪个微生物来的。
2. 具体操作过程:以一个纯菌种的16S rRNA测序为例,练习序列拼接(assembly). 用Sanger方法,从两端测序,引物为27F和1492R。
正向引物27F测得的序列
Overlap
反向引物1492R测得的序列
拼接 (assembly)
Contig
.
1
Biedit软件的应用介绍:DNA序列基本操作
第一步:Sanger测序下机文件为.abi文件,可以用Bioedit打开查看测序质量。峰型好 的碱基质量较好,把质量好的碱基部分提取出来,存成fasta文件。
.
2
Biedit软件的应用介绍:DNA序列基本操作
第二步:将27F和1492R测得的序列都整接成 一个长的contig。
操作如下:file----new alignment-----file----import ------sequence alignment file---同时选择 Seq27F.fas和seq1492R.fas-----file-----save as 27F+1492R.fas-----file----open 27F+1492R.fas ---点击选择两个文件名-----accessory applications-----CAP assembly contig program-----run application-----enter----随即产生了contig序列,delete原始的序列(seq27F.fas和 Seq1492R.fas),给contig命名,另存为Seq27F+1492R_contig.fas。
经典的Bioedit使用说明书

选择的类型是“Normal Arrow ”、“Wide Arrow ”、“Normal Box” 和、“Wide Box ”。在上面例子中的所有特征是“常规”宽度的。如果特征是一个箭头,箭头的方向将是从起点位置到终点位置。
2
增加特征或酶时,他们各自的标记增加在外面,中心是可能的尺寸。标记可以被选择工具选择、移动、编辑和缩放。
综合序列分析软件BioEdit
PLEASE ENTER YOUR TITLE HERE
2003级 高芳銮
BioEdit是一个性能优良的免费的生物序列编辑器,可在Windows 95/98/NT/2000中运行,它的基本功能是提供蛋白质、核酸序列的编辑、排列、处理和分析。
与DNAMAN相比,其分析内容相对丰富一些,而且提供了很多网络程序的分析界面和接口,与DNAMAN等软件配合使用更好。
使用BioEdit质粒绘图功能,序列可以通过自动的位置标记,自动修改成环形质粒。特征、多连接位点和限制性位点可以通过使用对话框增加。当将一个序列进入质粒图时,在背景上出现一个限制性内切酶图谱,所以可以通过对话框选择可以增加限制性位点。它们自动增加到当前的位点。质粒功能提供简单的绘制和标记工具。标签和绘图可以通过鼠标移动和缩放。想要编辑目标性质,双击目标。
05
核酸分析:组成、互补、反转、翻译、质粒、限制性内切酶;
06
蛋白质分析:氨基酸成分、疏水性轮廓 、疏水力矩平均数
07
翻译或反翻译:把DNA或RNA翻译成蛋白质;
08
切换翻译:在核酸和编码蛋白质序列中切换核苷酸序列;
09
点图[成对比较]:相互比较两序列的矩阵,生成一个点图。
序列的常规操作:
BLAST
瞬间疏水性轮廓(hydrophobic moment profile)
BioEdit(一)多序列比对
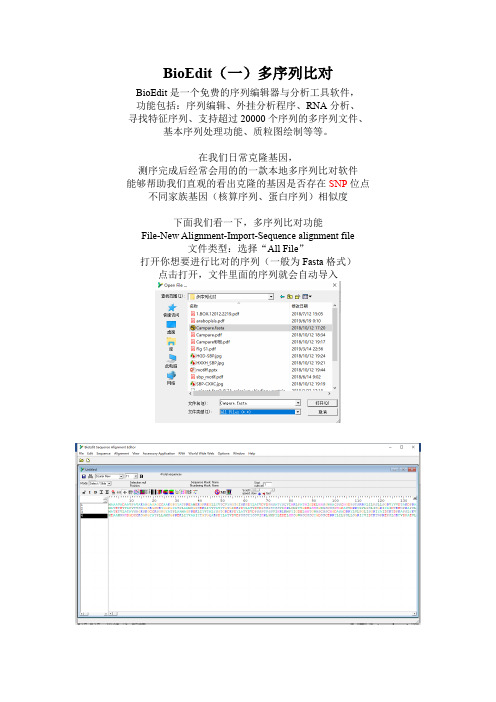
BioEdit(一)多序列比对BioEdit是一个免费的序列编辑器与分析工具软件,功能包括:序列编辑、外挂分析程序、RNA分析、寻找特征序列、支持超过20000个序列的多序列文件、基本序列处理功能、质粒图绘制等等。
在我们日常克隆基因,
测序完成后经常会用的的一款本地多序列比对软件
能够帮助我们直观的看出克隆的基因是否存在SNP位点不同家族基因(核算序列、蛋白序列)相似度
下面我们看一下,多序列比对功能
File-New Alignment-Import-Sequence alignment file
文件类型:选择“All File”
打开你想要进行比对的序列(一般为Fasta格式)点击打开,文件里面的序列就会自动导入
在最左边方框中双击序列名称
就可以直接命名
如果我们这个时候有新的序列想直接插入
点击Sequence-New sequence
将序列粘贴到下面大方框中
注意一定要选择“Sequence type”
点击Apply and Close,序列就会添加到比对界面
Ctrl+A可以将所有序列一键选中
Ctrl+左键,可以选择自己想比对的序列
最后依次点击
Accessory Application-Cluster W Multiple Alignment-Run Cluster W
为了我们方便我们快速寻找差异位点
点击方框所示,可以把相同的设置成“….”。
bioedit-phylip操作流程
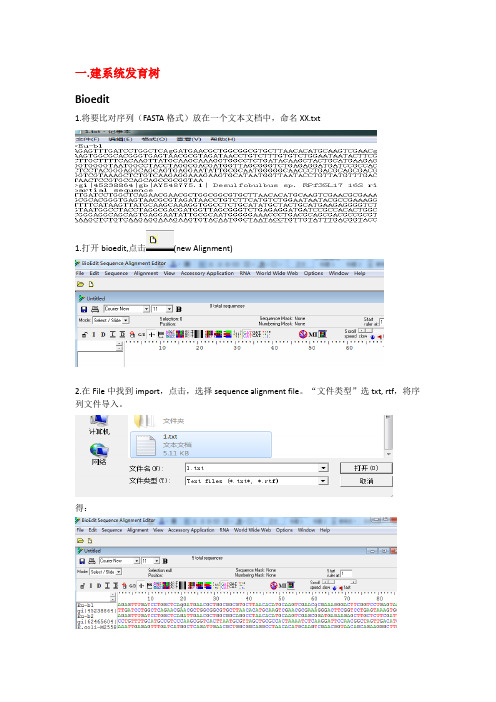
一.建系统发育树Bioedit1.将要比对序列(FASTA格式)放在一个文本文档中,命名XX.txt1.打开bioedit,点击(new Alignment)2.在File中找到import,点击,选择sequence alignment file。
“文件类型”选txt, rtf,将序列文件导入。
得:3.在中选择clustalw multiple alignment,点击点击Run Clustalw→OK 得:4.在mode中选择edit,对序列进行“掐头去尾”保存为xx.phy格式。
5.点击6.然后:可得:结果如下图所示:此即最终的系统进化树。
但还需对该树进行评价,这得用phylip 中的bootstrap(自展支持率:进化树可信度评估。
可信度一般超过66即认为可信,当然,如果是要判断二者是同一种,必须达到95以上,66-95只能说是一个亚种或者变种。
这个节点数不是同源性,注意了,是同源性可信度。
引导程序、辅助程序)进行。
来验证每个分支的支持率,可将支持率通过photoshop 软件添加到每个节点上。
二.系统树的评价Phylip[S(phy-outfile-1)→D(1-outfile-2)→N(2-outtree-3)→C(3-outtree)]1.将xx.phy复制到Phylip目录中exe文件夹中。
2.打开exe文件夹中seqboot,手动输入xx.phy→按回车键→再输入Y →回车→ 7(奇数均可)→回车→回车。
会在exe文件夹中生成outfile。
3.将outfile改为1,打开软件dnadist.exe,并将文件1 输入→回车→输入M→回车→D→回车→100→回车。
(将Analyze multiple data sets 项改为100,其余不变)。
输入Y →回车→ 7(奇数均可)→回车→回车。
会在exe文件夹中生成outfile。
4.生成一个outfile 文件,将其它改为2。
打开软件neighbor.exe,将刚才形成的文件2 输入:并将Analyze multiple data sets 项改为100,其余不变。
BioEdit

NBRF/PIR 格式
Phylip
Task1: 输入BCL2
登陆到GeneBank网站,搜索下载如下6个物种BCL2 protein,存成 *.gb文件。
[Bos taurus] [Mus musculus] [Rattus norvegicus] [Epinephelus coioides] [Felis catus] [Danio rerio]
Task1: 结果
序列编辑
操作主要在Edit菜单,但也有些快捷键
编辑模式
Sequence菜单里也有部分序列编辑选项,如
双序列比对
首先,选定待比对的两序列
其次,到Sequence->Similarity Matrix内选定打分矩阵
最后,到Sequence->Pairwise alignment里选择比对算 法。
结果
结果图形显示
以比对结果为活动框,打开File->Graph view,可进行更 精细人性化的显示。
Task 2: 序列编辑和比对
1. 对导入的BCL2序列尝试进行插入gap和删除gap,大小 写转换,序列彩色显示;
2. 任选两条BCL2蛋白序列,选择打分矩阵,进行双序列比 对;
3. 用graph view显示比对结果,并尝试对界面中的显示选 项做改变处理;
质粒图
质粒图:
编辑、修改和标识质粒序列; 编辑限定性定性内切酶标识的质粒图;
外挂程序
安装目录bioedit/apps/下
序列编辑比对
BCL2家族蛋白为例:
序列输入 序列编辑 序列比对 比对结果显示 其它
序列输入
手动输入 文件输入 剪切板输入 网络获取
bioedit 和Primer 使用软件

实验四使用Bioedit 和Primer 软件一、实验目的1、掌握Bioedit 一个核苷酸和蛋白质序列分析软件的使用,完成如序列比对、序列检索等内容。
2、掌握primer一个引物设计软件的使用,包括单双向引物、探针的设计与酶切位点的分析等。
二、实验器材计算机,Bioedit 和primer软件,核苷酸和蛋白质序列。
三、实验内容应用我们预先准备好的序列,然后在bioedit里先打开,然后进行序列的分析;将需要设计引物的序列在primer里打开,然后设置相关参数进行引物设计。
四、实验步骤1、在电脑里将以前下载好的FASTE格式的序列,全部复制到一个新的TXT格式文本中,保存到桌面方便使用。
2.在Bioedit里打开桌面上新建的TXT格式文本;进行序列分析Accessory application选择Clustalw Multiple alignment弹出的对话框点击Run clustalw等待分析结果;并根据需要保存实验结果。
3、打开NCBI主页在搜索框中,分别将相对的序列GI值输入,打开相关文章,将文章转换成genbank格式,在上面查找相关信息,填写相关表格对应信息。
4、在ncbi中选择5个氨基酸序列,下载成FASTE格式,在电脑上新建一个TXT格式文本,将下载的5个序列全部整理在一起,保存在我的桌面上,方便使用。
5、在bioedit中进行氨基酸序列分析,先打开保存的氨基酸序列文件,进行序列分析Accessory application选择Clustalw Multiple alignment弹出的对话框点击Run clustalw等待分析结果,保存结果图片,再根据相关表格要求填写相关的结果数据。
6、打开NCBI的主页,进入BIASTE选择BIASTE P 将下载在桌面上的氨基酸序列进行比对,在显示结果中找到显示的保守序列。
将相应名称填入表格中。
7、在bioedit进行序列分析的结果上,应用primer软件进行引物和探针的设计。
BioInsight版本0.3.1用户指南说明书

Package‘BioInsight’October12,2022Title Filter and Plot RNA BiotypesVersion0.3.1Description Analyze and plot the abundance of different RNA biotypes present in a count ma-trix,this evaluation can be useful if you want to test different strategies of normalization or ana-lyze a particular biotype in a differential gene expression analysis.License GPL(>=2)Encoding UTF-8RoxygenNote7.1.1Imports knitr,wordcloud,RColorBrewerDepends R(>=2.10),edgeR,limmaSuggests testthat(>=3.0.0),biomaRtNeedsCompilation noAuthor Luiz Antonio de Jesus Rocha[aut,cre,cph]Maintainer Luiz Antonio de Jesus Rocha<*********************>Repository CRANDate/Publication2021-06-2007:20:02UTCConfig/testthat/edition3R topics documented:describeRNA (2)Index41describeRNA describeRNADescriptionThis function providefilterByExpr with two customized options.See’Arguments’.Different strate-gies can be useful either if you are trying to compare different approaches of normalization or ifyou want to analyze a particular biotype where a different variation of expression is expected undercertain conditions.A BioInsight data frame will return with your new count matrix where you canproceed with your Differential Expression Analysis.UsagedescribeRNA(counts,biotypes,groups,report=FALSE,verbose=FALSE,filter=1)Argumentscounts data.frame where you have gene countsbiotypes data.frame where you have a gene_biotype columngroups factor with groups and samples(see Examples)report if TRUE will generate a.pdffile in/tmp/folder(’wordcloud’and’RColor-Brewer’are necessary)verbose if TRUE will print your result in your console(’knitr’is necessary)filter Your threshold(see edgeR::filterByExpr)DetailsForfilter you may want to use"1"if you want the default option fromfilterByExpr.If2"Slightly"above the default will be applied.If"3"A more restrictive option will be applied.NoteYou can use trace(describeRNA,edit=T)to set different values as threshold for"filter"option.ReferencesRobinson MD,McCarthy DJ,Smyth GK.edgeR:a Bioconductor package for differential expres-sion analysis of digital gene expression data.Bioinformatics.2010;26(1):139-140.doi:10.1093/bioinformatics/btp616Examplescounts=system.file("extdata","count_matrix.tsv",package="BioInsight")counts=read.table(counts,s=1,header=TRUE)biotypes=system.file("extdata","Rattus_Norvegicus_biomart.tsv",package="BioInsight") biotypes=read.table(biotypes,s=1,header=TRUE)groups=rep(as.factor(c("1","2")),each=5)describeRNA(counts=counts,biotypes=biotypes,groups=groups,filter=2)IndexdescribeRNA,24。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1. 目的:纯菌种16S rRNA,利用Sanger方法双端测序,然后检查测序质量,将双端测序 的序列拼接成一条完整的序列,利用这个长序列去与数据库比对,判断这个序列最 可能是从哪个微生物来的。 2. 具体操作过程:以一个纯菌种的16S rRNA测序为例,练习序列拼接(assembly). 用Sanger方法,从两端测序,引物为27F和1492R。
RDP 平台简单介绍:分类
1. 目的:利用RDP平台中的分类功能,对测定的高通量测序数据进行系统进化分类。 得到微生物群落的组成信息:门、纲、目、科、属、种。
Hale Waihona Puke 2. 具体操作过程: RDP pipeline Classifier test drive Select your file to upload
Submit for Classification
(Sample file name: Seq100)
Biedit软件的应用介绍:DNA序列基本操作
第二步:将27F和1492R测得的序列都整理成质量好的.fasta文件后,将这两个文件拼接成 一个长的contig。 操作如下:file----new alignment-----file----import ------sequence alignment file---同时选择 Seq27F.fas和seq1492R.fas-----file-----save as 27F+1492R.fas-----file----open 27F+1492R.fas ---点击选择两个文件名-----accessory applications-----CAP assembly contig program-----run application-----enter----随即产生了contig序列,delete原始的序列(seq27F.fas和 Seq1492R.fas),给contig命名,另存为Seq27F+1492R_contig.fas。 第三部:将contig序列拷贝到NCBI Blast中去比对, 看与数据库中哪些序列最相近。
正向引物27F测得的序列
Overlap
反向引物1492R测得的序列
拼接 (assembly) Contig
Biedit软件的应用介绍:DNA序列基本操作
第一步:Sanger测序下机文件为.abi文件,可以用Bioedit打开查看测序质量。峰型好 的碱基质量较好,把质量好的碱基部分提取出来,存成fasta文件。 具体操作流程: File -----Open----找到文件Sanger_Seq27f.abi。出现两个界面,根据测 序峰确定高质量测序区段;然后双击另一个界面的文件名字,即出现一个新的编辑 框。 将质量差的碱基区域去掉,然后另存为一个.fasta文件,存成seq27F.fas。另一个文 件存为seq1492R.fas.