实时荧光定量PCR——TaqMan探针法及设计原则
realtimePCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价

realtimePCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价real time PCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价一、实时荧光Taqman 探针设计总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。
即real-time PCR 的扩增片段是50bp----150bp。
当找不到150bp的保守片段时,必须确保探针的片段是保守的。
在设计探针和引物时,要同时考虑在两条链上设计引物与探针。
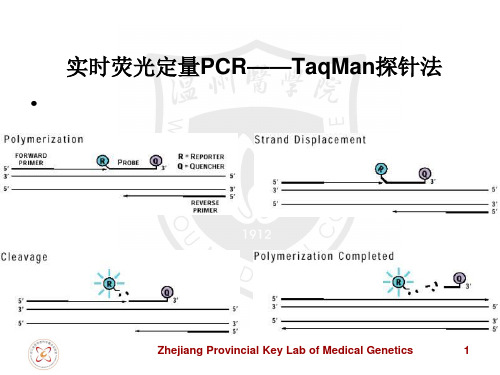
但要注意的是:在那条链上设计探针时,就应靠近在同一条链上设计的引物(即上游引物)。
这样,可保证在将来扩增时,即便没有完全扩增,也有荧光信号报告出来。
两者的距离最好是探针的5’端离上游引物的3’有一个碱基,但也可以重叠。
若在原序列中找不到合适的探针与引物(1主要是探针和上游引物的距离太远,而离下游引物的距离却较近时;2突变位点要求在探针的5’ 端也能检测到荧光信号,但却是在3’端),可在互补的序列中设计引物与探针。
另real-time PCR中的探针和引物的Tm值,均要高于平常PCR 的引物和杂交的探针的Tm值。
二、探针的设计探针设计的基本原则:1.保守:探针要绝对的保守,有时分型就单独依靠探针来决定。
理论上有一个碱基不配对,就可能检测不出来。
若找不到完全保守的片段,也只能选取有一个碱基不同的片段。
且这个不同的碱基最好在探针的中间,对探针与目的片段的杂交影响不大,不相同的碱基最好不要在两端,因为两端不利于探针的杂交。
且最好为A或T,而不能为G或A,因为A、T为双键,而G、A为三键。
2.探针长度Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,最好为70℃,确保探针的Tm 值要比引物的Tm值高出10℃,这样可保证探针在煺火时先于引物与目的片段结合。
因此探针最好是富含GC的保守片段,保证其的Tm值较高。
Taqman探针设计要领

如何设计荧光定量PCR实验方案 如何设计荧光定量PCR实验方案 PCR
根据仪器和个人的喜好,确定荧光定量PCR的方 法,选择荧光素的种类 合成引物和探针 收集样品、提取RNA或DNA(-80℃保存,避免反 复冻融 ) 制备标准品(质粒、化学合成的目的基因)
拷贝数=质量÷分子量×6.0×1023 标准曲线通常由4~5个点组成
定量—— 定量 绝对标准曲 线方法
标准品:纯化的质粒dsDNA,体外转录的RNA 准确配制标准系列并且注意其稳定性,最好 分装后保存于-80℃ 荧光材料:杂交探针、水解探针或SYBR Green I 不可以使用DNA作为标准品对RNA进行定量 应用于定量病毒荷载量等
定量—— 定量 相对 标准曲 线方法 指在测定目的基因的同时测定某一内对照基因,用
配制好的引物和探针,保存于-20℃,注意避光 保存探针 RT反应
如何设计荧光定量PCR实验Байду номын сангаас 如何设计荧光定量PCR实验方 PCR 案
不加探针的常规PCR反应,验证引物是否 合适、模板是否降解、模板纯度是否合适。 同时确定最佳反应体系和反应条件 每次实验都设置阴性对照和标准品,每个 样品设两个平行孔
• 引物和探针的设计原则的重要程度由上 往下越来越低 • 如果是设计SYBR Green I引物,也要选 择TaqMan Primer and Probe design并 遵守这些规则,但是只需要合成引物就 可以了。 • 为了确保引物和探针的特异性,最好将 设计好的序列在 www.ncbi.nlm.nih/blast 中核实一次, 如果发现由非特异性互补区,建议重新 设计引物探针。
一系列已知外参物做标准曲线,根据该标准曲线得 到目的基因和内对照基因的量,再将目的基因的同 内对照基因的比值作为定量的最后结果
实时荧光定量PCR技术

千里之行,始于足下。
Real-time PCR for mRNA quantitation一、原理实时荧光定量PCR技术是通过检测PCR产物中荧光讯号强度来达到定量PCR产物的目的,目前该技术已在动植物基因工程,微生物和医学领域中得到广泛应用。
实时定量PCR 包括探针法和染料法两种,探针法是利用与靶序列特异杂交的探针来指示扩增产物的增强,特异性高,如Taq Man TM技术;染料法则是利用染料来指示扩增的增强,特异性相对较低,但简便易行。
染料法的原理是在PCR 反应体系中,参加过量荧光染料,荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的染料分子不会发射任何荧光信号,从而保证荧光信号的增强与PCR产物的增强彻低同步。
荧光染料发射出的荧光讯号强度与DNA 产量成正比,检测PCR 过程中的荧光讯号便可得知靶序列初始浓度,从而达到定量目的。
目前染料法实时荧光定量PCR主要使用的是美国Molecular Probes 公司的SYBR Green 1 和SYBR Gold 染料。
二、实验步骤一)、单链cDNA 摸板的合成(参照相关资料)二)、Real-time PCR操作主意(TIANGEN 公司RealMasterMix(SYBR Green) PCR Kit)1、20×SYBR Green solution 在室温下平衡并彻底混匀。
2、将125μL 20×SYBR Green solution 参加至1.0 ml 2.5×ReaMasterMix 中并轻轻混匀。
3、照表1决定多个PCR反应混合物并分装到各个PCR管中。
4、将PCR管放入热循环仪并启动循环程序(表2)。
三、计算在定量PCR中,需要经过数个循环后荧光信号才干够被检测到。
荧光域值的缺省设置是3-15 个循环的荧光信号的标准偏差的10 倍。
在实际操作中普通以前15个循环的荧光信号作为荧光本底信号。
荧光定量PCR中一个关键的数据是“Ct(threshold cycle)值”,其中“t”是Threshold ,即PCR管内荧光超过本底(达到可检测水平)时的临界数值;第 1 页/共 3 页朽木易折,金石可镂。
实时荧光定量PCR方法简介

实时荧光定量PCR方法简介一.实时荧光定量PCR的基本原理理论上,PCR过程是按照2n(n代表PCR循环的次数)指数的方式进行模板的扩增。
但在实际的PCR反应过程中,随着反应的进行由于体系中各成分的消耗(主要是由于聚合酶活力的衰减)使得靶序列并非按指数方式扩增,而是按线性的方式增长进入平台期。
因此在起始模板量与终点的荧光信号强度间没有可靠的相关性.如采用常规的终点检测法(利用EB 染色来判断扩增产物的多少,从而间接的判断起始拷贝量),即使起始模板量相同经PCR扩增、EB染色后也完全有可能得到不同的终点荧光信号强度。
为了能准确判断样品中某基因转录产物(mRNA)的起始拷贝数,实时荧光定量PCR采用新的参数-—Ct值,定量的根本原理是Ct值与样品中起始模板的拷贝数的对数成线性反比关系。
Ct值是如何得到的在实时荧光定量PCR的过程中,靶序列的扩增与荧光信号的检测同时进行,定量PCR仪全程采集荧光信号,实验结束后分析软件自动按数学算法扣除荧光本底信号并设定阈值从而得到每个样品的Ct值。
Ct值的定义Ct值中的“C”代表Cycle(循环),“t”代表检测threshhold(阈值),其含义是PCR扩增过程中荧光信号强度达到阈值所需要的循环数;也可以理解为扩增曲线与阈值线交点所对应的横坐标。
Ct值与样品中模板的对应关系Ct值与样品中起始模板的拷贝数的对数成线性反比关系(y=ax+b,x代表起始模板拷贝数的对数,y代表Ct值)。
与终点法相比利用Ct值的优势由于Ct值是反映实际PCR反应过程中扩增即将进入指数期的参数,该参数几乎不受试剂消耗等因素的影响,因此利用Ct值判断的起始模板拷贝数更加精确,重复性也更好。
传统的终点检测法是在PCR扩增经历了指数扩增期进入平台期后利用EB等染料染色来判断扩增产物的多少,从而间接的判断起始拷贝量,这种方法的精确度不高、重复性也不好。
下图中是96个复孔的实时扩增曲线(完全相同的反应体系、相同的反应protocol、相同的样品起始浓度),可以看到Ct值具有很好的重复性,而终点的荧光信号强度差异达到300个单位。
定量PCR Taqman探针设计要领-2

定量PCR Taqman探针设计要领-2第三步:寻找一家信赖的公司合成引物和探针,一般引物合成大家比较熟悉,而且价格也比较便宜(特别是这两年便宜了许多),而探针则相对来说贵了许多,一般Taqman探针合成在1000到5000元不等(不同的合成要求价钱不同)——而这只是标记价钱,序列合成基本上和引物合成价钱相似。
第四、五、六步:一般的定量PCR反应体系与普通PCR其实也差不了多少,只是要加入Taqman探针,另外不同就是分步法的不同。
其中需要注意的是:* 扩增酶最好选用热启动酶* 引物和探针的浓度需要进行优化,有人建议从50nM开始,在50nM—900nM之间优化,一般为200nM(注意探针需要避光保存。
* 同样Mg+和酶量也需要进行优化,酶的推荐反应浓度是1.25-1.5U(50ul)* DNA模板的添加量通常在100 ng以下,因不同种类的DNA模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。
如果欲进行2 Ste p RT-PCR反应的第二步PCR扩增反应,第一步的RT反应液作为DNA模板时的添加量不要超过PCR反应液总体积的10%。
另外循环参数虽然在引物和探针设计完之后也就确定了,但是有时也需要进行优化。
第七步:在进行数据分析的时候,通常用不同浓度的标准样品的Ct值来产生标准曲线,然后计算相对方程式。
方程式的斜度可以用来检查PCR的效率,对于100%PCR效率来说,一个理想的斜率是3.32。
最佳的标准曲线是建立在PCR的扩增效率为90%-100 %(100%意味着在每个循环之后,模板的总数将增加为前一次的2倍)的基础上。
所有标准曲线的线性回归分析需要存在一个高相关系数(R2≥0.99) ,这样才能认为实验的过程和数据是可信的。
使用这个方程式我们可以计算出未知样本的初始模板量。
大多数定量PCR仪都有这样一个软件,它可以从标准曲线中自动地计算出未知样本的初始模板量。
taqman荧光定量pcr基本原理

taqman荧光定量pcr基本原理
TaqMan荧光定量PCR是指采用荧光标记的PCR技术,它可以实现对特定序列DNA的定量分析,即可以直接测量所检测序列在模板文库中的相对浓度。
TaqMan荧光定量PCR可以说是一种双通道的实时PCR技术,最初由PE Applied Biosystems(PE)推出。
它是通过特定的标记技术,开发了FRET和TaqMan技术,结合PCR技术,以达到定量分析和监测某一特定序列DNA的目的,这种技术比以往的技术有很大的优势,更易于完成。
TaqMan荧光定量PCR的基本原理比较简单,大致可以分为以下四步:
(1)样本处理:检测序列DNA及其作为模板的原始样品将在定量PCR试剂盒中经过一系列处理,得到模板文库。
(2)探针标记:样品处理完成后,将开发出一对对应该特殊序列的5'荧光探针—特殊碱基对相对应的序列和3'分子耦合(MolecularBeacon)。
探针内部具有双螺旋结构,当处于开放状态时,荧光物质才能折射发射自己特有的颜色荧光。
(3)反应物添加:在模板文库中添加进Taq DNA 聚合酶、核酸类型的合成引物,以及缓冲液,用水调节总体浓度。
(4)加热-冷却:完成上述步骤,将反应液放入定量PCR仪,按照一定的条件,经由一系列的加热、冷却过程,Taq DNA聚合酶将碱基对引物合成,产生复制模板文库。
同时在复制过程中,反应液中复制数量也不断增加,一旦达到一定温度,则表明文库复制到足够多,荧光探针能够在反应液中爆炸,产生光谱,得以直接测定检测序列DNA的定量。
此外,TaqMan荧光定量PCR技术能够检测DNA的扩增曲线,并由此提供准确的定量结果,同时也不受PCR技术的一些影响,更易于操作。
realtimePCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价

real-time-PCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价————————————————————————————————作者:————————————————————————————————日期:real time PCRTaqman探针设计、实时多重PCR探针的选择、引物的设计及评价一、实时荧光Taqman 探针设计总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。
即real-time PCR的扩增片段是50bp----150bp。
当找不到150bp的保守片段时,必须确保探针的片段是保守的。
在设计探针和引物时,要同时考虑在两条链上设计引物与探针。
但要注意的是:在那条链上设计探针时,就应靠近在同一条链上设计的引物(即上游引物)。
这样,可保证在将来扩增时,即便没有完全扩增,也有荧光信号报告出来。
两者的距离最好是探针的5’端离上游引物的3’有一个碱基,但也可以重叠。
若在原序列中找不到合适的探针与引物(1主要是探针和上游引物的距离太远,而离下游引物的距离却较近时;2突变位点要求在探针的5’ 端也能检测到荧光信号,但却是在3’端),可在互补的序列中设计引物与探针。
另real-time PCR中的探针和引物的Tm值,均要高于平常PCR的引物和杂交的探针的Tm值。
二、探针的设计探针设计的基本原则:1.保守:探针要绝对的保守,有时分型就单独依靠探针来决定。
理论上有一个碱基不配对,就可能检测不出来。
若找不到完全保守的片段,也只能选取有一个碱基不同的片段。
且这个不同的碱基最好在探针的中间,对探针与目的片段的杂交影响不大,不相同的碱基最好不要在两端,因为两端不利于探针的杂交。
且最好为A或T,而不能为G或A,因为A、T为双键,而G、A为三键。
2.探针长度Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,最好为70℃,确保探针的Tm 值要比引物的Tm值高出10℃,这样可保证探针在煺火时先于引物与目的片段结合。
实时荧光Taqman 探针设计

一、实时荧光Taqman 探针设计总原则:探针选择要保守,引物选择要保守,因此必须找一段100-200bp相对要保守的片段来设计引物与探针。
即real-time PCR的扩增片段是50bp----150bp。
当找不到150bp的保守片段时,必须确保探针的片段是保守的。
在设计探针和引物时,要同时考虑在两条链上设计引物与探针。
但要注意的是:在那条链上设计探针时,就应靠近在同一条链上设计的引物(即上游引物)。
这样,可保证在将来扩增时,即便没有完全扩增,也有荧光信号报告出来。
两者的距离最好是探针的5’端离上游引物的3’有一个碱基,但也可以重叠。
若在原序列中找不到合适的探针与引物(1主要是探针和上游引物的距离太远,而离下游引物的距离却较近时;2突变位点要求在探针的5’ 端也能检测到荧光信号,但却是在3’端),可在互补的序列中设计引物与探针。
另real-time PCR中的探针和引物的Tm值,均要高于平常PCR的引物和杂交的探针的Tm 值。
二、探针的设计探针设计的基本原则:1.保守:探针要绝对的保守,有时分型就单独依靠探针来决定。
理论上有一个碱基不配对,就可能检测不出来。
若找不到完全保守的片段,也只能选取有一个碱基不同的片段。
且这个不同的碱基最好在探针的中间,对探针与目的片段的杂交影响不大,不相同的碱基最好不要在两端,因为两端不利于探针的杂交。
且最好为A或T,而不能为G或A,因为A、T为双键,而G、A为三键。
2.探针长度Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,最好为70℃,确保探针的Tm值要比引物的Tm值高出10℃,这样可保证探针在煺火时先于引物与目的片段结合。
因此探针最好是富含GC的保守片段,保证其的Tm值较高。
现在有Taqman MGB探针,在TAMER之后再标记一个MGB,可使探针的Tm值较高,即使探针片段较短,也可达到Taqman探针的Tm值要求(68-70℃)。
定量PCR Taqman探针设计要领

定量PCR+Taqman探针设计要领自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR 荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:在第一步目的基因查找比对过程中可以利用NCBI genbank序列以及DNAstar等软件完成目的DNA 或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DN***段重叠的情况)内。
第二步:如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:总体原则* 先选择好探针,然后设计引物使其尽可能的靠近探针。
* 所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。
建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
* 扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。
较短的扩增片段也容易保证分析的一致性。
* 保持GC含量在20%和80%之间,GC富含区容易产生非特异反应,从而会导致扩增效率的降低,以及出现在荧光染料分析中非特异信号。
定量PCR引物探针设计原则完整版

定量P C R引物探针设计原则Document serial number【NL89WT-NY98YT-NC8CB-NNUUT-NUT108】定量PCR引物、探针设计原则自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR 要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:在第一步目的基因查找比对过程中可以利用NCBIgenbank序列以及DNAstar等软件完成目的DNA或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DNA片段重叠的情况)内。
第二步:如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:总体原则先选择好探针,然后设计引物使其尽可能的靠近探针。
所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。
建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。
实时荧光定量PCR——TaqMan探针法及设计原则
Taqman MGB 探针设计
• 探针的5’端避免出现G,即使探针水解为单个碱基,与报
告基团相相连的G碱基仍可淬灭基团的荧光信号。
• Tm值应为65-67℃。 • • 尽量缩短Taqman MGB探针,但探针长度不少于13bp。 尽量避免出现重复的碱基,尤其是G碱基,应避免出现4 个或4个以上的G重复出现。 • 原则上MGB探针只要有一个碱基突变,MGB探针就会检 测到(MGB探针将不会与目的片段杂交,不产生荧光信号)。
1
TaqMan技术引物设计原则
• 序列选取应在基因的保守区段; • 避免引物自身或与引物之间形成4个或4个以上连续配对, 避免引物自身形成环状发卡结构; • 典型的引物18到24个核苷长。引物需要足够长,保证序 列独特性,并降低序列存在于非目的序列位点的可能性。 • Tm值在55-65℃,GC含量在40%-60%; • 引物之间的TM相差避免超过2℃; • 引物的3’端避免使用碱基A,引物的3’端避免出现3个或3 个以上连续相同的碱基; • 为避免基因组的扩增,引物设计最好能跨两个外显子; • Taqman探针技术要求片段长度在50bp-150bp; • 引物末端(最后5个核苷酸)不能有超过2个的G和C。
Zhejiang Provincial Key Lab of Medical Genetics 4
尽量缩短t尽量缩短taqmanmgb探针但探针长度不少于13bpmgb探针但探针长度不少于13bzhejiangprovincialkeylabofmedicalgenetics4??尽量避免出现重复的碱基尤其是g碱基应避免出现4个或4个以上的g重复出现
实时荧光定量PCR——TaqMan探针法
两种定量分析方法的比较及Taqman探针引物设计原则

两种定量分析⽅法的⽐较及Taqman探针引物设计原则两种定量分析⽅法的⽐较及Taqman 探针、引物设计原则遗传物质DNA ⾸先要把所携带的遗传信息转录成为信使RNA (mRNA ),携带遗传信息的mRNA 从细胞核进⼊到细胞质中与核糖体结合,在核糖体中mRNA 携带的遗传信息被翻译成为多肽,多肽经过进⼀步加⼯后变成蛋⽩质,⾄此遗传物质DNA 完成了表达过程。
期间的转录过程是基因表达中⾮常重要的调节步骤,所转录的mRNA 的多少直接影响着相关最终蛋⽩质的多少,所以通过对细胞内某条基因mRNA 含量多少的分析,就能⼤致判断出该条基因的表达是否活跃。
定量PCR 仪是在普通PCR 仪的基础上加装了荧光激发装臵和荧光检测装臵,PCR 扩增和检测同时进⾏;在PCR 反应体系中加⼊荧光基团,利⽤荧光信号的积累实时监测整个PCR 进程,最后通过标准曲线对未知模板进⾏定量分析。
该技术于1996年由美国Applied Biosystems 公司推出,由于该技术不仅实现了PCR 从定性到定量的飞跃,⽽且与常规PCR 相⽐,它具有特异性更强、有效解决PCR 污染问题、⾃动化程度⾼等特点,⽬前已得到⼴泛应⽤。
定量PCR 常⽤的三个常⽤概念扩增曲线、荧光阈值、Ct 值扩增曲线:反映PCR 循环次数和荧光强度的曲线,定量PCR 仪每次轮PCR 扩增都会⾃动记录荧光强度的变化荧光阈值:样本的荧光背景值和阴性对照的荧光值,⼿动设臵的原则要⼤于样本的荧光背景值和阴性对照的荧光最⾼值,同时要尽量选择进⼊指数期的最初阶段,并且保证回归系数⼤于0.99。
CT 值: PCR 扩增过程中,扩增产物的荧光信号达到设定的阈值时所经过的扩增循环次数。
扩增曲线阈值及CT 值荧光定量PCR 的数学原理理想的PCR 反应:X=X0*2n⾮理想的PCR 反应:X=X0* (1+Ex)n(n :扩增反应的循环次数;X :第n 次循环后的产物量;X0:初始模板量;Ex :扩增效率)在扩增产物达到阈值线时: C(t) valueXCt=X0 (1+Ex)Ct =M (1)XCt:荧光扩增信号达到阈值强度时扩增产物的量,在阈值线设定以后,它是⼀个常数,我们设为M⽅程式(1)两边同时取对数得:log M=log X0 (1+Ex)Ct (2)整理⽅程式(2)得:log X0= - log(1+Ex) *Ct+ log M (3)由此可见,log X0浓度与循环数呈线性关系,根据样品扩增达到域值的循环数即Ct值就可计算出样品中所含的该基因的初始模板量。
实时荧光定量PCR原理及应用

实时荧光定量PCR原理及应用一、原理:1.荧光探针原理:a. TaqMan探针:TaqMan探针是由小分子荧光染料和一个捕获目标序列的DNA探针构成。
在PCR过程中,TaqMan探针会结合到特定的目标序列上,当DNA聚合酶在PCR反应中扩增特定序列时,探针被加性外切酶活性所降解,导致荧光信号逐渐降低,通过荧光信号的减弱来量化目标DNA的数量。
b. SYBR Green探针:SYBR Green探针是一种可以与双链DNA特异性结合的染料,当SYBR Green与PCR产物结合时,荧光信号增加。
通过测量荧光信号的增加来量化目标DNA的数量。
c. Molecular Beacons:Molecular Beacons是由在末端带有荧光分子和淬灭荧光的猝灭体构成的。
在PCR过程中,当Molecular Beacons与目标序列匹配时,荧光信号释放,通过测量荧光信号的释放来量化目标DNA的数量。
2.PCR反应原理:a.变性:将含有目标DNA序列的模板DNA样品与引物和荧光探针混合,加热至高温,使DNA双链解除成两股单链DNA。
b.引物结合:将反应体温度降低,引物结合到目标DNA序列的特定区域,并与模板DNA进行互补组装。
c.扩增:在DNA聚合酶的作用下,引物在模板上逐渐沿着DNA链延伸,产生新的DNA片段。
每一轮PCR循环结束后,荧光信号会相应地增加。
二、应用:1.目标基因表达分析:可以用实时荧光定量PCR测定特定目标基因的表达水平,从而研究基因的功能、调控机制或者生理功能的变化。
2.病原体检测:实时荧光定量PCR可以检测和定量各种病原体,例如病毒、细菌、真菌等。
常见的应用包括检测呼吸道病原体、性传播疾病病原体、食物中污染的细菌等。
3.肿瘤检测:实时荧光定量PCR可以用于肿瘤相关标志物的检测,帮助早期筛查和诊断肿瘤。
4.遗传突变检测:可以通过实时荧光定量PCR检测人类基因中的突变位点,提供遗传病检测和个体基因组分析的支持。
taqman探针原理

taqman探针原理TaqMan探针原理。
TaqMan探针是一种广泛应用于分子生物学研究中的探针技术,它能够准确、快速地检测目标DNA序列的存在和数量。
TaqMan探针原理的核心是通过荧光信号实时监测PCR反应的过程,从而实现对目标DNA的定量检测。
本文将详细介绍TaqMan探针的工作原理及其在实验室研究中的应用。
TaqMan探针是一种双标记的寡核苷酸探针,通常由三部分组成,5'端的荧光素(reporter dye)、3'端的类似荧光素的化合物(quencher dye)以及连接两者的寡核苷酸序列。
在PCR反应中,TaqMan探针与目标DNA序列特异性结合,当Taq DNA聚合酶在扩增过程中到达TaqMan探针的结合位点时,它会切割探针,导致荧光素和类似荧光素的化合物分离,从而释放出荧光信号。
PCR仪器会实时检测并记录荧光信号的强度,通过信号强度的变化可以确定目标DNA序列的存在和数量。
TaqMan探针的工作原理非常简单直观,但其在实验室研究中的应用却十分广泛。
首先,TaqMan探针可以用于定量PCR(qPCR),通过实时监测PCR反应过程中的荧光信号变化,可以准确地确定起始DNA模板的数量。
这使得TaqMan探针在基因表达分析、病原体检测、基因定量分析等领域得到了广泛的应用。
其次,TaqMan探针还可以用于检测单核苷酸多态性(SNP),通过设计特异性的TaqMan探针,可以快速、准确地鉴定DNA序列中的SNP位点,为基因组学研究提供了重要的工具。
除了在实验室研究中的应用,TaqMan探针在临床诊断、食品安全监测、环境污染检测等领域也发挥着重要作用。
例如,在临床诊断中,TaqMan探针可以用于检测病原体的存在和数量,为临床诊断提供了重要的分子学依据。
在食品安全监测中,TaqMan探针可以用于检测食品中的致病微生物,保障食品安全。
在环境污染检测中,TaqMan探针可以用于监测环境中的致病微生物和污染物,为环境保护提供了重要的技术支持。
实时荧光定量PCR-TaqMan探针法及设计原则

03
Taqman探针的合成与制备
探针的合成方法
化学合成法
通过化学反应将荧光基团和淬灭基团分别连接到DNA或RNA的5'和3'末端,形成 Taqman探针。
酶促合成法
利用DNA聚合酶将荧光基团和淬灭基团分别添加到DNA或RNA的特定位置,形成 Taqman探针。
探针的质量检测与纯化
质量检测
通过电泳、质谱和光谱分析等方法检测探针 的长度、荧光基团和淬灭基团的数量和位置 ,以及探针的纯度。
定义与原理
定义
实时荧光定量PCR-Taqman探针法是 一种基于荧光信号的实时监测技术, 用于定量分析DNA或RNA的拷贝数。
原理
通过在Taq酶催化下,利用荧光染料 标记的特异性探针与待测核酸进行特 异性结合,在PCR循环过程中实时监 测荧光信号的增强,从而实现对核酸 的定量分析。
发展历程与现状
02
Taqman探针的设计原则
探针的长度与结构
长度
通常为20-30bp,确保特异性并减少非特异性扩增。
结构
由报告基团、淬灭基团和连接臂组成,连接臂长度一般为5-6个脱氧核糖核苷酸。
探针的特异性
针对目标序列
确保探针与目标序列完全匹配,避免 交叉反应。
序列选择
选择基因特异性区域,避免基因组中 的高变区。
04
Taqman探针在实时荧光定量 PCR中的应用
样本处理与PCR反应体系建立
样本处理
确保样本质量,去除杂质和抑制剂,提 取高质量的DNA或RNA。
Taqman探针设计
根据目标基因序列设计Taqman探针, 确保探针的特异性和荧光信号的稳定
性。
引物设计
根据目标基因序列设计特异性引物, 确保引物与模板的结合效率和特异性。
Taqman设计总结

一、单重Taqman引物探针设计:(为了确保引物和探针的特异性,最好将设计好的序列在www.ncbi.nlm.nih/blast中核实一次)1、探针:探针的设计应该在引物的设计之前①长度:18~35(18~30之间最好、最常见),最长37;太长淬灭效果不好。
② TM值:Primer Express软件计算出来的Tm值在66~72℃之间(最常见68~70℃),最好为70℃,确保探针的Tm值要比引物的Tm值高出5~10℃,GC含量在30~80%(最好40%~70%),因此探针最好是富含GC的保守片段;(Primer Express:Do not expand the Tm range by more than 2 °C from the default range)。
③探针位置:尽可能地靠近上游引物,上游引物的3' 端离探针的5' 端为1-20bp(一般10个以内),最好是探针的5'端离上游引物的3' 有1个碱基。
④5'端应避免使用碱基G,5' G会有淬灭作用,即使被切割下来这种淬灭作用也还会存在;如果选择FAM-dye在5'端第二个序列也不能为G(G in the second position on the 5' end in FAM dye-labeled probes can reduce fluorescent normalized reporter signal)。
⑤可选:整条探针中,碱基C的含量要明显高于G的含量,G的含量多于C会降低反应效率,这时就应选择配对的另一条链作为探针。
要同时考虑在正反两条链上设计引物与探针。
若找不到完全保守的片段,也只能选取有一个碱基不同的片段,且这个不同的碱基最好在探针的中间,且最好为A或T。
⑥Repeating oligonucleotides:a. 避免探针中同一碱基重复过多,尤其是要避免4个或超过4个的G碱基出现,即≦3(Avoid runs of identical nucleotides. If repeats are present, there must be fewer than four consecutive G residues.);b. 避免连续的6个A出现(Consecutive A residues:Avoid six consecutive A residues anywhere in the probe.Consecutive A residues can cause a high No Template Control (NTC) signal);c. 避免探针的中间区域含有2个或以上的CC dinucleotides(Avoid two or more CC dinucleotides in themiddle of the probe, which can sometimes reduce signal)。
taqman探针实时荧光定量rt-pcr法检测siv2fshiv病毒rna拷贝数方法的建立及..

第三节结果1.TaqMan探针real.timeRT-PCR方法及SYBRGreenIreal.timeRT-PCR方法测定定量曲线各个稀释度RS标准品在第一个循环结束后测到一个基础吸光值,图1-a中一直到第15个循环结束时都没有明显变化,从第15个循环开始lX107copies/pLRS标准品的吸光值增大,定量曲线开始抬头,18个循环(Ct值)后,曲线迅速上扬,至第28个循环前后达到峰值,随后进入平台期,一直到反应结束,此后的标准品的Ct值依次延迟约3个循环,但除10个拷贝Rs标准品外,各扩增曲线的斜率相同,间距相等,相互平行,阴性对照PCR级水的扩增曲线稍有起伏。
而图l-b中从第11个循环开始lX107copies/pLRS标准品的吸光值增大,15个循环后,曲线迅速上扬,至第30个循环前后达到峰值,随后同样进入平台期,至反应结束,102和10个拷贝RS标准品几乎同时在33个循环左右出现曲线,但斜率不同,阴性对照PCR级水也出现了扩增曲线,但吸光值不高(见图1)。
图1-aTaqMan探针real—timeRT-PCR测得10倍系列稀释RNA标准品的荧光定量曲线。
由左至右分别是1×107copies/I_tL、l×106copies/pL、1×l05copies//aL、l×104eopies/I.tL、1×103copies/I.tL、l×102copies/pL、1×101copies/BL,最下面一条线是PCR级水做的阴性对照。
横坐标显示的是循环数,纵坐标是荧光值。
Fig.1-aAmplificationcurveobtainedwith10一foldserialdilutionsoftheRNAstandards(RS)byreal-timeRT-PCRmethodwithTaqManprobe.Fromlefttorightarestandardsfroml×107copies/lJLto1×101copies/lxL.Asanegativecontr01.thetemplateRNAwasreplacedwithPCR-gradewater.Thex-axisshowsthenumberofPCRcyclesandthey-axisshowsthenormalizedfluorescenceintensity.b图1.bSYBRGreenIreal—timeRT-PCR测得10倍系列稀释RNA标准品的荧光定量曲线。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。