先天性无痛无汗综合症七例-2019年精选文档
Evan综合征9例分析

Evan综合征9例分析
张启明;朱学习;王叶亮;孔雪琴;乔强;绳俊海
【期刊名称】《中国优生与遗传杂志》
【年(卷),期】1991(0)2
【摘要】Evan综合征主要表现为免疫溶血性贫血和血小板减少,临床少见,现将我院1973年以来收住的9例报告如下。
【总页数】2页(P88-87)
【关键词】免疫溶血性贫血;Evan综合征9;血红蛋白尿;出血点;红细胞脆性;冷抗体;红细胞膜;磺胺药;温抗体;免疫作用
【作者】张启明;朱学习;王叶亮;孔雪琴;乔强;绳俊海
【作者单位】河南省宁陵县人民医院
【正文语种】中文
【中图分类】R394
【相关文献】
1.儿童 Evans 综合征19例临床分析 [J], 王欣;乔丽津
2.儿童Evans综合征8例临床分析 [J], 刘婵娟;刘玉峰;李白;魏林林;苏淑芳
3.疑似Evans综合征并发脑梗:"血栓性血小板减少性紫癜"文献重新分析之结果——亦与何志义、武剑等作者商榷 [J], 王志湖;张海鹏;李瑞霄;刘建平;张芳;杨子军
4.Evans综合征6例临床分析 [J], 张伟;蒋丽君;王小婕;马燕萍;包慎
5.系统性红斑狼疮合并Evans综合征的临床特征分析 [J], 王丛丛;王卫敏;田文亮;王菲;孙慧;孙玲
因版权原因,仅展示原文概要,查看原文内容请购买。
中国近年来的自然灾害事件-2019年精选文档

中国近年来的自然灾害事件1950年7月,淮河大水。
1954年7月,长江、淮河大水。
1959到1961三年自然灾害1966年河北邢台地震1970年通海7.8级大地震1975年8月,河南大水。
1976年河北唐山大地震1978—1983年北方大旱1991年安徽江淮水灾长江2019年特大洪水2019年非典疫情2019年度“十大自然灾害事件”2019年度中国十大自然灾害事件1、5.12汶川特大地震导致重大人员伤亡和财产损失2、南方雪灾:年初特大低温雨雪冰冻灾害影响21省(区、市、兵团)3、台风“黑格比”严重影响两广地区4、6月上中旬华南、中南地区发生严重洪涝灾害5、新疆出现历史上第二个严重干旱年6、长江沿线及江南地区发生严重秋涝7、四川攀枝花-会理地震导致川滇两省损失严重8、9月下旬四川发生严重暴雨洪涝和泥石流灾害9、宁夏严重干旱致夏秋粮减产10、10月末西藏发生强降雪,10万余人受灾9、2009年自然灾害(五个“历史罕见”)10、2019自然灾害4月14日青海玉树地震2019年全国十大自然灾害事件1950年7月,淮河大水。
由于泥沙淤积,河床高涨,加上国民党军队在淮海战役时对沿淮堤坝的大肆破坏,这年汛期,淮河流域全面告急,河南、皖北许多地方一片汪洋,水灾淹没土地3400余万亩,灾民1300万。
淮北地区受灾惨重,为百年所罕见。
1954年7月,长江、淮河大水。
长江中下游、淮河流域降水量普遍比常年同期偏多一倍以上,致使江河水位猛涨,汉口长江水位高达29.73米,较历史最高水位的1931 年高出14.5米。
虽然沿江人民做出了极大努力保卫荆江大堤,从而保证了武汉市和南京市的安全,但却淹没农田4755万亩,1888万人受灾,财产损失在100亿元以上。
由于农产品减少,也影响了人民的生活和1955年的工业生产。
1、1959到1961三年自然灾害在1959年7月,华东地区长江发洪水。
据灾害中心数据, 因为淹水和接下来歉收所带来的饥荒,洪水直接带来的死亡人数估计达两百万,而且别的地区也多少受到影响。
【参考文档】儿童阿奇霉素说明书(共7篇-精选word文档 (25页)

本文部分内容来自网络整理,本司不为其真实性负责,如有异议或侵权请及时联系,本司将立即删除!== 本文为word格式,下载后可方便编辑和修改! ==儿童阿奇霉素说明书(共7篇篇一:儿童阿奇霉素说明书 (共7篇)篇一:阿奇霉素干混悬剂说明书核准日期:201X年10月22日阿奇霉素干混悬剂说明书请仔细阅读说明书并在医师指导下使用【药品名称】通用名称:阿奇霉素干混悬剂英文名称:azithromycin for suspension 汉语拼音:aqimeisu ganhunxuanji 【成份】活性成份:阿奇霉素化学名称:(2r,3s,4r,5r,8r,10r,11r,12s,13s,14r)-13-[(2,6-二脱氧-3-c-甲基-3-o-甲基-?-l-核-已吡喃糖基)氧]-2-乙基-3,4,10-三羟基-3,5,6,8,10,12,14-七甲基-11-[[3,4,6-三脱氧-3-(二甲氨基)-β-d-木-已吡喃糖基]氧]-1-氧杂-6-氮杂环十五烷-15酮化学结构式:分子式:c38h72n2o12 分子量:749.00 【性状】本品为白色或类白色混悬颗粒:气芳香,味甜。
【适应症】本品适用于敏感细菌所引起的下列感染:支气管炎、肺炎等下呼吸道感染;皮肤和软组织感染;急性中耳炎;鼻窦炎、咽炎、扁桃体炎等上呼吸道感染(青霉素是治疗化脓性链球菌咽炎的常用药,也是预防风湿热的常用药物。
阿奇霉素可有效清除口咽部链球菌,但目前尚无阿奇霉素治疗和预防风湿热疗效的资料)。
阿奇霉素可用于男女性传播疾病中由沙眼衣原体所致的单纯性生殖器感染。
阿奇霉素亦可用于由非多重耐药淋球菌所致的单纯性生殖器感染及由杜克嗜血杆菌引起的软下疳(需排除梅毒螺旋体的合并感染)。
【规格】0.1g(10万单位) 【用法用量】每日口服药一次。
溶于水中,服用前搅拌均匀。
可与食物同时服用。
以阿奇霉素干混悬剂治疗各种感染性疾病,其疗程及使用方法如下:成人:对沙眼衣原体、杜克嗜血杆菌或敏感淋球菌所致的性传播疾病,仅需单次口服本品1.0g。
【最新】入职体检报告单模板(共8篇-精选word文档 (26页)

本文部分内容来自网络整理,本司不为其真实性负责,如有异议或侵权请及时联系,本司将立即删除!== 本文为word格式,下载后可方便编辑和修改! ==入职体检报告单模板(共8篇篇一:入职体检报告单图片入职体检报告合格标准第一条风湿性心脏病、心肌病、冠心病、先天性心脏病、克山病等器质性心脏病,不合格。
先天性心脏病不需手术者或经手术治愈者,合格。
遇有下列情况之一的,排除心脏病理性改变,合格:(一)心脏听诊有生理性杂音;(二)每分钟少于6次的偶发期前收缩(有心肌炎史者从严掌握);(三)心率每分钟50-60次或100-110次;(四)心电图有异常的其他情况。
第二条血压在下列范围内,合格:收缩压90mmhg-140mmhg(12.00-18.66kpa);舒张压60mmhg-90mmhg(8.00-12.00kpa)。
第三条血液病,不合格。
单纯性缺铁性贫血,血红蛋白男性高于90g/l、女性高于80g/l,合格。
第四条结核病不合格。
但下列情况合格:(一)原发性肺结核、继发性肺结核、结核性胸膜炎,临床治愈后稳定1年无变化者;(二)肺外结核病:肾结核、骨结核、腹结核、淋巴结核等,临床治愈后2年无复发,经专科医院检查无变化者。
第五条慢性支气管炎伴阻塞性肺气肿、支气管扩张、支气管哮喘,不合格。
第六条严重慢性胃、肠疾病,不合格。
胃溃疡或十二指肠溃疡已愈合,1年内无出血史,1年以上无症状者,合格;胃次全切除术后无严重并发症者,合格。
第七条各种急慢性肝炎,不合格。
乙肝病原携带者,经检查排除肝炎的,表面说合格,过后或不加工资歧视或找莫须有的原因辞退。
第八条各种恶性肿瘤和肝硬化,不合格。
第九条急慢性肾炎、慢性肾盂肾炎、多囊肾、肾功能不全,不合格。
第十条糖尿病、尿崩症、肢端肥大症等内分泌系统疾病,不合格。
甲状腺机能亢进治愈后1年无症状和体征者,合格。
第十一条有癫痫病史、精神病史、癔病史、夜游症、严重的神经官能症(经常头痛头晕、失眠、记忆力明显下降等),精神活性物质滥用和依赖者,不合格。
无脾综合征二例报告

无脾综合征二例报告
曹洪晓;孙萍;吴星恒
【期刊名称】《南昌大学学报(医学版)》
【年(卷),期】2009(049)006
【摘要】@@ 1 临床资料rn例1,男,5个半月,因全身皮肤青紫5个半月,加重伴气急、呻吟1 d于2005年12月9日入院.患儿系第2胎第2产,足月顺产,生后即有口唇及甲床青紫、哭闹时青紫加剧伴气急;2个月时曾患肺炎,查心脏彩超示右位心、复杂性先天性心脏病.
【总页数】2页(P122,125)
【作者】曹洪晓;孙萍;吴星恒
【作者单位】南昌大学第一附属医院儿科,南昌,330006;南昌大学第一附属医院儿科,南昌,330006;南昌大学第一附属医院儿科,南昌,330006
【正文语种】中文
【中图分类】R726.2
【相关文献】
1.成年无脾综合征伴阵发性室上速射频消融术后1例报告并文献复习 [J], 刘靖;陈曦;李学斌;董强
2.新生儿无脾综合征1例报告 [J], 刘莺;周卫萍;王玲;周媚
3.新生儿无脾综合征1例报告 [J], 刘莺;周卫萍;王玲;周媚
4.儿童无脾综合征一例报告并文献复习 [J], 赵宇东;黄蕊;李晓峰
5.无脾综合征一例报告 [J], 杨金龙
因版权原因,仅展示原文概要,查看原文内容请购买。
MEF2C单倍剂量不足综合征4例病例系列报告
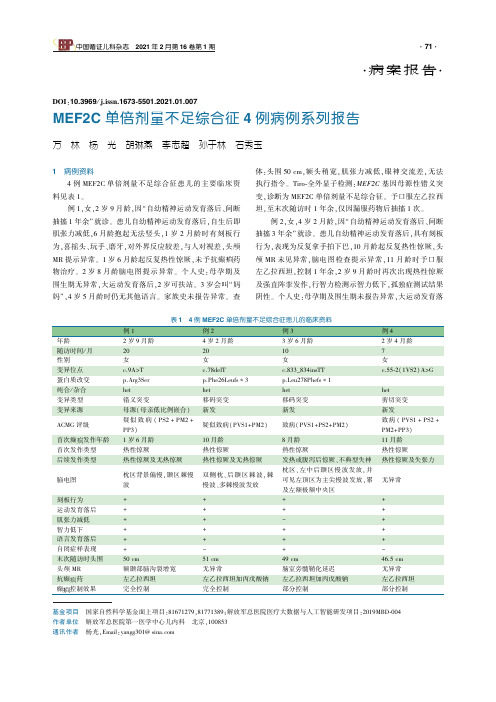
基金项目㊀国家自然科学基金面上项目:81671279,81771389;解放军总医院医疗大数据与人工智能研发项目:2019MBD⁃004作者单位㊀解放军总医院第一医学中心儿内科㊀北京,100853通讯作者㊀杨光,Email:yangg301@sina.com㊃病案报告㊃DOI:10.3969/j.issn.1673⁃5501.2021.01.007MEF2C单倍剂量不足综合征4例病例系列报告万㊀林㊀杨㊀光㊀胡琳燕㊀李志超㊀孙于林㊀石秀玉1㊀病例资料㊀㊀4例MEF2C单倍剂量不足综合征患儿的主要临床资料见表1㊂㊀㊀例1,女,2岁9月龄,因 自幼精神运动发育落后㊁间断抽搐1年余 就诊㊂患儿自幼精神运动发育落后,自生后即肌张力减低,6月龄抱起无法竖头,1岁2月龄时有刻板行为,喜摇头㊁玩手㊁磨牙,对外界反应较差,与人对视差,头颅MR提示异常㊂1岁6月龄起反复热性惊厥,未予抗癫药物治疗㊂2岁8月龄脑电图提示异常㊂个人史:母孕期及围生期无异常,大运动发育落后,2岁可扶站㊂3岁会叫 妈妈 ,4岁5月龄时仍无其他语言㊂家族史未报告异常㊂查体:头围50cm,额头稍宽,肌张力减低,眼神交流差,无法执行指令㊂Tiro⁃全外显子检测:MEF2C基因母源性错义突变,诊断为MEF2C单倍剂量不足综合征㊂予口服左乙拉西坦,至末次随访时1年余,仅因漏服药物后抽搐1次㊂㊀㊀例2,女,4岁2月龄,因 自幼精神运动发育落后㊁间断抽搐3年余 就诊㊂患儿自幼精神运动发育落后,具有刻板行为,表现为反复拿手拍下巴,10月龄起反复热性惊厥,头颅MR未见异常,脑电图检查提示异常,11月龄时予口服左乙拉西坦,控制1年余,2岁9月龄时再次出现热性惊厥及强直阵挛发作,行智力检测示智力低下,孤独症测试结果阴性㊂个人史:母孕期及围生期未报告异常,大运动发育落随访时间/月2020107性别女女女女变异位点c.9A>Tc.78delTc.833_834insTTc.55⁃2(1VS2)A>G蛋白质改变p.Arg3Serp.Phe26Leufs∗3p.Leu278Phefs∗1纯合/杂合hethethethet变异类型错义突变移码突变移码突变剪切突变变异来源母源(母亲低比例嵌合)新发新发新发ACMG评级疑似致病(PS2+PM2+PP3)疑似致病(PVS1+PM2)致病(PVS1+PS2+PM2)致病(PVS1+PS2+PM2+PP3)首次癫发作年龄1岁6月龄10月龄8月龄11月龄首次发作类型热性惊厥热性惊厥热性惊厥热性惊厥后续发作类型热性惊厥及无热惊厥热性惊厥及无热惊厥发热或腹泻后惊厥㊁不典型失神热性惊厥及失张力脑电图枕区背景偏慢,颞区棘慢波双侧枕㊁后颞区棘波,棘慢波㊁多棘慢波发放枕区㊁左中后颞区慢波发放,并可见左顶区为主尖慢波发放,累及左额极额中央区无异常刻板行为++++运动发育落后++++肌张力减低++-+智力低下++++语言发育落后++++自闭症样表现+-+-末次随访时头围50cm51cm49cm46.5cm头颅MR额颞部脑沟裂增宽无异常脑室旁髓鞘化延迟无异常抗癫药左乙拉西坦左乙拉西坦加丙戊酸钠左乙拉西坦加丙戊酸钠左乙拉西坦癫控制效果完全控制完全控制部分控制部分控制后,2岁可独走㊂1岁余会叫 爸爸 ㊁ 妈妈 ,后退步,5岁9月龄时仍仅无意义发声㊂家族史未报告异常㊂查体:头围51cm,额头宽,耳廓增大,与人可眼神交流,能听懂及执行简单指令㊂Tiro⁃全外显子检测:MEF2C基因新发移码突变,诊断为MEF2C单倍剂量不足综合征㊂加用丙戊酸钠,至末次随访时,无抽搐发作1年余㊂㊀㊀例3,女,3岁6月龄,因 自幼精神运动发育落后㊁间断抽搐2年余 就诊㊂患儿自幼精神运动发育落后伴刻板行为,反复翻书㊁转动书本,对外界反应差,不与周边人玩耍㊂8月龄起出现热性惊厥及腹泻后惊厥,脑电图检查提示异常,未予抗癫治疗㊂3岁2月龄时无明显诱因出现癫持续状态,同期脑电图提示异常,头颅MR示髓鞘化延迟,血㊁尿代谢检查未见异常㊂3岁2月龄时予口服丙戊酸钠及左乙拉西坦,发作较前明显减少㊂个人史:母孕期及围生期未报告异常㊂大运动发育落后,2岁时可独走,至4岁4月龄无语言发育,仅喉中无意识发声㊂家族史无异常㊂查体:头围49cm,额头宽,与人无眼神交流,无法执行指令㊂Tiro⁃全外显子检测:MEF2C基因新发移码突变,诊断为MEF2C单倍剂量不足综合征㊂继续原有抗癫治疗,至末次随访时,无抽搐发作半年㊂㊀㊀例4,女,2岁4月龄,因 自幼精神运动发育落后㊁间断抽搐1年余 就诊㊂患儿自幼精神运动发育落后,4月龄时仍无法抬头,心脏超声检查示室间隔缺损,头颅MR及血㊁尿代谢未见异常㊂四肢肌张力低下,发育商低下,11月龄起出现反复热性惊厥,15月龄时出现重复刻板行为,表现为频繁摇头及双手不停抓放,并对算珠表现出特殊偏好,喜独自大笑,不喜与人玩耍,脑电图检查未见异常,1岁4月龄时口服左乙拉西坦及康复治疗,仍发热伴抽搐发作,并出现频繁头部后仰表现㊂个人史:母孕期及围生期未报告异常,大运动发育落后,2岁时可扶走㊂2岁时曾叫过 爸爸 ,1周后退步,2岁11月龄时仅喉中无意识发声㊂家族史未报告异常㊂查体:头围46.5cm(<-1SD),额头宽,耳廓增大,与人有眼神交流,无法执行指令㊂Tiro⁃全外显子检测:MEF2C基因新发剪切突变,诊断为MEF2C单倍剂量不足综合征㊂予左乙拉西坦加量继续抗癫治疗,至末次随访时仍有抽搐发作,较前明显减少㊂2㊀文献复习㊀㊀以 MEF2Chaploinsufficiencysyndrome ㊁ 5q14.3 ㊁ MEF2C 为关键词,检索至2020年3月5日在PubMed数据库中收录的文献,以 MEF2C单倍剂量不足综合征 ㊁5q14.3 ㊁ MEF2C 为关键词在万方医学数据网及中国知网数据库中检索截至2020年10月12日的文献㊂因既往染色体5q14.3微缺失综合征报道中,除MEF2C基因异常外,多包含其他基因片段异常,故人工筛选仅有致病和可能致病的MEF2C基因变异或微缺失,而不涉及其他邻近或远距离基因的文献,共获得10篇文献[1⁃10],其中英文9篇,俄文1篇㊂㊀㊀共报告MEF2C单倍剂量不足综合征30例(包括本文4例),其中男性11例(37.9%),女性19例,7例为MEF2C基因部分缺失,23例为MEF2C基因点突变,其中剪切突变2例㊁无义突变7例㊁错义突变7例㊁移码突变7例㊂在已报道的18份脑电图中13份结果异常,已报道的29份头颅MR结果中20份可见结构异常,以髓鞘化延迟(12例,60%)多见㊂㊀㊀MEF2C单倍剂量不足综合征最常见的表现:均有不同程度的精神运动发育的落后㊁96.7%(29例)语言功能障碍㊁86.7%(26例)婴儿期肌张力障碍㊁83.3%(25例)癫发作㊁80%(24例)刻板行为及56.7%(17例)行走受限㊂86.7%(26例)语言功能完全缺失,10%(3例)仅会几个有限的词语㊂首次癫发作常见于婴儿期(18例,60%),各种发作形式中,以热性惊厥最为多见(16例,53.3%)㊂进行抗癫药物治疗并长期随访的14例患儿中,11例至末次随访完全控制发作㊂在已评估自闭倾向相关表现的25例中,76%(19例)存在无眼神交流或对外界无反应,社交行为受限,男性患儿均出现孤独症样表现㊂3㊀讨论㊀㊀MEF2C单倍剂量不足综合征既往也称染色体5q14.3微缺失综合征,在识别了5q14.3微缺失患者相似的表型特征后,有研究提出了最小的共同缺失区域仅包含MEF2C基因[9,11]㊂无论是MEF2C基因的点突变还是包含有该基因的染色体5q14.3微缺失,都可以造成类似的临床表现,即智力障碍㊁刻板动作㊁癫和/或脑畸形㊂其发病机制是基因突变或者缺失后,只有正常水平50%的蛋白质产生,而不足以维持细胞正常的生理功能,即单倍体剂量不足效应[9]㊂因此,2013年Paciorkowski在此基础上提出,将MEF2C基因点突变或染色体5q14.3微缺失导致MEF2C基因异常引起的一组临床表现命名为MEF2C单倍剂量不足综合征[9]㊂㊀㊀MEF2C基因位于染色体5q14.3区域,是肌细胞增强因子⁃2(MEF2)转录因子家族的成员,调节兴奋性突触数量㊁树突形态的发生和树突的突触后分化[12⁃15]㊂在小鼠胚胎中将皮质和海马兴奋性神经元中的Mef2c基因条件敲除后,引起抑制性神经突触的急剧增加和兴奋性突触传递的减少,从而导致体内皮质网络活动的急剧减少[16]㊂另一项实验中显示,Mef2c基因杂合缺失的小鼠出现突触前抑制性标志物泡状γ⁃氨基丁酸转运体转录水平明显减低,兴奋性突触标志物的囊泡谷氨酸转运体2的水平明显增高[17]㊂同时MEF2C基因参与了CDKL5及MECP2基因的转录调控,有研究表明,在人体中MEF2C功能的缺失,可以导致CDKL5及MECP2基因表达水平的显著减低[18,19],表现为早期婴儿癫性脑病,包括婴儿期起病的癫发作㊁严重的肌张力低下㊁Rett综合征样特征(头围增长缓慢㊁手部失用及刻板动作㊁运动功能异常及睡眠障碍)[20]㊂迄今为止,在神经系统疾病中报道的MEF2C突变位点共有20个(包括本文)[1⁃4,6⁃10],突变类型似乎与症状表型以及轻重无关,有研究[2]认为,突变位点越靠近N端,临床表型越重,越靠近C端,症状表现越轻,本研究中例3的突变位点更靠近C端,而症状相对更重㊂本文例2 4所发现的3个新的突变位点c.78delT(p.Phe26LeufsTer3)㊁c.833_834insTT(p.Leu278Phefs∗1)和c.55⁃2(1VS2)A>G此前均未见报道,例1遗传自其母亲,其母为低比例嵌合体(10/119),故患儿母亲可能单倍体剂量未受严重影响,因此未发病㊂㊀㊀根据既往文献[1⁃10]报道,MEF2C单倍剂量不足综合征患儿大部分语言功能完全缺失,少数仅会几个有限的词语,多数患儿表现为自闭症样症状㊂首次癫发作常见于婴儿期,各种发作形式中,以热性惊厥最为多见,亦可见局灶性发作㊁全面强直阵挛发作及多种发作形式,大部分抗癫药物治疗效果较好㊂本文4例中,3例在婴儿期㊁1例在幼儿期出现癫发作,首次发作均以热性惊厥起病,除例4无脑电图异常外,例1 3脑电图均存在癫样放电,使用抗癫药物治疗后,例1 3均无临床发作,例4仍有发作但较前明显减少,持续时间较前明显缩短,频次减少,总体癫控制效果较好㊂例1㊁2和4存在肌张力减低,例2和3存在脑结构的异常,4例患儿均有语言功能的丧失并伴有刻板动作㊂头颅MR常提示异常以髓鞘化延迟为主要表现,本文2例MR异常,其中1例为髓鞘化延迟,相对文献报道比例较低㊂值得注意的是,既往文献中面容的描述中均存在宽额头的表现[12⁃15],本文4例患儿均可见额头较宽㊂㊀㊀因本病与Rett综合征在表型中多有重叠,需要予以鉴别㊂有研究指出,Rett综合征患者合并癫的患病率随着年龄的增长逐渐增加,2岁以前出现癫的可能性较小[21],而MEF2C单倍剂量不足综合征正好相反,癫多发生在婴儿期㊂Rett综合征的诊断标准中必须具备发育的倒退[22],本文例2及4曾有意识发音而后退步,即语言发育的倒退,例1及3则为语言发育迟滞并无倒退现象㊂认为癫发病时间以及语言功能的倒退可能是两者间的不同之处㊂㊀㊀目前除了控制癫症状,临床对于MEF2C单倍剂量不足综合征的治疗尚无有效手段㊂动物实验发现[17],Mef2c基因杂合突变小鼠出现海马内神经元数量减低及兴奋性/抑制性神经元比例失调,表现出空间记忆障碍㊁异常焦虑和异常重复运动,以及与人类MEF2C单倍剂量不足综合征一致的其他体征㊂研究人员用NitroSynapsin(一种氨基金刚烷硝酸盐)试验性治疗了MEF2C单倍剂量不足模型小鼠3个月,发现可以显著减少小鼠的神经元凋亡,改善兴奋性/抑制性神经元比例,并表现出对认知缺陷㊁社交障碍和刻板行为的治疗效果[17]㊂在国内一项孤独症大鼠模型的研究中发现,雷帕霉素可通过抑制mTOR通路刺激细胞内自噬,导致MEF2C表达上调,激活p38/MEF2C通路[23]㊂㊀㊀综上所述,本文4例MEF2C单倍剂量不足综合征患儿有3个新发现的MEF2C基因突变位点,扩大了MEF2C单倍剂量不足综合征的基因诊断图谱㊂在儿童早期出现以热性惊厥起病的癫发作,并伴有刻板动作㊁语言功能丧失及大运动发育迟缓的情况下,应考虑MEF2C单倍剂量不足综合征,及时进行相应的遗传学诊断以避免误诊及漏诊㊂参考文献1 BorlotF WhitneyR CohnRD etal.MEF2C⁃relatedepilepsy Delineatingthephenotypicspectrumfromanovelmutationandliteraturereview.Seizure 2019 67 86⁃90. 2 WangJ ZhangQ ChenY etal.NovelMEF2CpointmutationsinChinesepatientswithRett ⁃like syndromeornon⁃syndromicintellectualdisability insightsintogenotype⁃phenotypecorrelation.BMCMedGenet 2018 19 1 191. 3 Vrec㊅arI InnesJ JonesEA etal.FurtherclinicaldelineationoftheMEF2Chaploinsufficiencysyndrome Reportonnewcasesandliteraturereviewofsevereneurodevelopmentaldisorderspresentingwithseizures absentspeech andinvoluntarymovements.JPediatrGenet 2017 6 3 129⁃141.4 RochaH SampaioM RochaR etal.MEF2Chaploinsufficiencysyndrome ReportofanewMEF2Cmutationandreview.EurJMedGenet 2016 59 9 478⁃482. 5 TantelesGA AlexandrouA EvangelidouP etal.PartialMEF2CdeletioninaCypriotpatientwithsevereintellectualdisabilityandajugularfossamalformation reviewoftheliterature.AmJMedGenetA 2015 167A 3 664⁃669. 6 PaciorkowskiAR TraylorRN RosenfeldJA etal.MEF2CHaploinsufficiencyfeaturesconsistenthyperkinesis variableepilepsy andhasaroleindorsalandventralneuronaldevelopmentalpathways.Neurogenetics 2013 14 2 99⁃111.7 BienvenuT DieboldB ChellyJ etal.RefiningthephenotypeassociatedwithMEF2Cpointmutations.Neurogenetics 2013 14 1 71⁃75.8 ZweierM GregorA ZweierC etal.MutationsinMEF2Cfromthe5q14.3q15microdeletionsyndromeregionareafrequentcauseofseverementalretardationanddiminishMECP2andCDKL5expression.HumMutat 2010 31 6 722⁃733.9 LeMeurN Holder⁃EspinasseM JaillardS etal.MEF2Chaploinsufficiencycausedbyeithermicrodeletionofthe5q14.3regionormutationisresponsibleforseverementalretardationwithstereotypicmovements epilepsyand/orcerebralmalformations.JMedGenet 2010 47 1 22⁃29.10 AnisimovaIV DadaliEL KonovalovFA etal. Newallelicvariantsofnon⁃syndromicmentalretardationoftype20causedbymutationsintheMEF2Cgene .ZhNevrolPsikhiatrImSSKorsakova 2018 118 4 70⁃75.11 NovaraF BeriS GiordaR etal.RefiningthephenotypeassociatedwithMEF2Chaploinsufficiency.ClinGenet 2010 78 5 471⁃477.12 LiZ McKercherSR CuiJ etal.Myocyteenhancerfactor2Casaneurogenicandantiapoptotictranscriptionfactorinmurineembryonicstemcells.JNeurosci 2008 28 26 6557⁃6568.13 BarbosaAC KimMS ErtuncM etal.MEF2C atranscrip⁃tionfactorthatfacilitateslearningandmemorybynegativeregulationofsynapsenumbersandfunction.ProcNatlAcadSciUSA 2008 105 27 9391⁃9396.14 ShaliziA GaudillièreB YuanZ etal.Acalcium⁃regulatedMEF2sumoylationswitchcontrolspostsynapticdifferentiation.Science 2006 311 5763 1012⁃1017.15 FlavellSW CowanCW KimTK etal.Activity⁃dependentregulationofMEF2transcriptionfactorssuppressesexcitatorysynapsenumber.Science 2006 311 5763 1008⁃1012.16 HarringtonAJ RaissiA RajkovichK etal.MEF2Cregulatescorticalinhibitoryandexcitatorysynapsesandbehaviorsrelevanttoneurodevelopmentaldisorders.Elife2016 5 e20059.17 TuS AkhtarMW EscorihuelaRM etal.NitroSynapsintherapyforamouseMEF2Chaploinsufficiencymodelofhumanautism.NatCommun 2017 8 1 1488.18 ZweierM RauchA.TheMEF2C⁃relatedand5q14.3q15microdeletionsyndrome.MolSyndromol 2012 2 3⁃5 164⁃170.19 KhaliliAlashtiS FallahiJ MohammadiS etal.TwonovelmutationsintheMECP2geneinpatientswithRettsyndrome.Gene 2020 732 144337.20 KatoT MorisadaN NagaseH etal.SomaticmosaicismofaCDKL5mutationidentifiedbynext⁃generationsequencing.BrainDev 2015 37 9 911⁃915.21 GlazeDG PercyAK SkinnerS etal.EpilepsyandthenaturalhistoryofRettsyndrome.Neurology 2010 74 11 909⁃912.22 GoldWA KrishnarajyR EllawayC etal.Rettsyndrome Ageneticupdateandclinicalreviewfocusingoncomorbidities.ACSChemNeurosci 2018 9 2 167⁃176.23 罗瑜平周波刘芬等.增强自噬激活p38/MEF2C通路调节突触相关蛋白的表达改善孤独症大鼠的症状.细胞与分子免疫学杂志2019 35 3 236⁃242.(收稿日期:2020⁃06⁃18㊀修回日期:2021⁃01⁃28)(本文编辑:张崇凡)。
新生儿22q13缺失综合征1例报告

现及判断心律失常类型并处理,可使胎儿病死率下降至10%以内。
胎儿心动过速的治疗途径目前首选经胎盘转运药物。
本例患儿采用母亲口服地高辛治疗,控制心率后心脏有所缩小,符合国外报道。
经治疗后胎儿仍为持续性房性心动过速,亦可考虑经胎盘转运或经脐静脉注射索他洛尔、胺碘酮等二线治疗药物。
但本例胎儿胎龄已达37周,经评估胎儿成熟度满意,决定对孕妇终止妊娠,继续对新生儿进行治疗。
右心耳起源的局灶性房速表现为持续性无休止发作,通常药物治疗无效,故决定行射频消融术。
右心耳房速的射频消融难度大、复发率较高,发生心脏穿孔的风险高。
本例患儿的房速起源于右心耳分叶顶部,消融大头到达真正的靶点困难,消融能量亦难以有效地到达局灶兴奋灶,术后复发与心耳解剖结构有关。
术中仅使用单根消融大头标测及放电,有一定的局限性,但在血管穿刺困难、导管入路有限的情况下,该标测方法可行。
心耳切除后,随访3个月患儿再无心动过速发作,心脏缩小,达到根治目的。
综合上述结果,提示对于这类顽固性心律失常,产前生后一体化治疗有利于患儿的短期恢复,降低病死率,但要求在经验丰富的心脏中心进行。
[广东省心血管病研究所广东省华南结构性心脏病重点实验室广东省人民医院广东省医学科学院心儿科 (广东广州 510800) 刘 甜 李渝芬 许 刚 潘 微 张 旭 张智伟 梁东坡](本文编辑:邹 强)doi:10.3969/j.issn.1000-3606.2019.01.018新生儿22q13缺失综合征1例报告22q13缺失综合征,也称Phelan- McDermid综合征,是指22号染色体长臂末端完全或者不完全缺失导致的一种微小染色体缺失综合征。
在22q13区域的典型终端的任何异常的基因变异可能被诊断为22q13缺失综合症。
22q13缺失综合征最常见的表现为:中重度智力缺陷、发育迟缓、语言发育落后、新生儿期肌张力低下、特殊外观特征、自闭症样表现等。
1985年Watt 首次报道,目前为止全世界仅有1200多例;国内也曾报道几例,而新生儿22q13缺失综合征尚未见报道。
22q11.2缺失综合征一例

22q11.2缺失综合征一例摘要22q11.2缺失综合征是人类常见的染色体缺失综合征,不同患者之间的临床症状表现出高度的异质性,极易造成误诊、漏诊。
本文报道1例经全外显子组测序确诊的22q11.2缺失综合征患儿的临床特点。
女,3岁,以“不会说话,行走姿势异常”为代主诉就诊河南省儿童医院康复科。
患儿偶会发“奶奶”音,主动言语少,叫名有反应,可完成简单指令,会指认五官及日常用品,不会示意大小便。
弓背坐,不能从仰卧位直接坐起,坐位、站立、行走时头偏向左侧,行走姿势异常。
不会上下楼梯,不会独跑,不会双脚蹦离地面。
咀嚼能力欠佳,易流涎。
查体无特殊面容,头颅外观无畸形,头围47.8cm。
神经系统检查双下肢肌张力略高,病理征无异常。
辅助检查:头颅核磁示双侧额颞叶巨脑回,局部多小脑回畸形;双侧侧脑室后角旁斑点状异常,考虑脱髓鞘或髓鞘化不良;双侧侧脑室轻度扩张,双侧侧脑室周围血管间隙稍扩张。
婴幼儿感觉发育检测:视觉、听觉反应未见明显异常,前庭觉、本体觉失常。
OT:1.双上肢力量差,肌耐力不足,肩关节稳定性差,关节囊松弛;2.双侧拇示指对捏小物体不灵活,指尖捏无发完成,手指分离动作欠佳,3.双手协调性差,手指控制能力欠佳,手肌协调欠佳。
精神科B类量表:运动相当于21个月,社会适应相当于15个月,智力相当于21个月。
核型分析:46,XX。
心脏彩超、甲状腺功能、血尿遗传代谢筛查、肝肾功能、乳酸、血氨、同型半胱氨酸等均无异常。
既往体质较差,易患呼吸道感染,在外院间断康复治疗1年余,运动逐渐进步,但进步缓慢。
全外显子组测序结果显示22号染色体q11.21区域(chr22:18893887-21823635)存在2.93Mb的杂合缺失。
该区域包含蛋白编码基因55个,包括明确单倍剂量不足基因TBX1,22q11.2缺失综合征诊断明确。
入院以来,给予经颅磁刺激治疗、平衡功能训练、作业疗法、小儿捏脊治疗、低频、言语训练等综合康复治疗,患儿运动机能进步,但仍存在语言、运动和智力全面发育落后。
Mohr综合征一例报告

Mohr综合征一例报告
陈京奕;陈振东;张学强;靳书滨;田云霄
【期刊名称】《华西口腔医学杂志》
【年(卷),期】2002(020)006
【摘要】@@ Mohr综合征属口-面-指综合征Ⅱ型(orofacial-digital syndrome typeⅡ,OFDⅡ)非常罕见.邯郸市中心医院收治1例,报告如下.
【总页数】1页(P457)
【作者】陈京奕;陈振东;张学强;靳书滨;田云霄
【作者单位】056001,河北省邯郸市中心医院;056001,河北省邯郸市中心医
院;056001,河北省邯郸市中心医院;056001,河北省邯郸市中心医院;056001,河北省邯郸市中心医院
【正文语种】中文
【中图分类】R78
【相关文献】
1.早期复极综合征猝死一例报告(Brugada综合征变异) [J], 陈玉珍;马维勇;郑更生
2.噬血细胞综合征误诊为肾病综合征一例报告 [J], 熊昕;赵琳
3.肾病综合征合并假Bartter`s综合征一例报告及文献复习 [J], 戴勇;陈世件
4.产后大出血致席汉氏综合征及肝肾综合征一例报告 [J], 吴桂英;黎明秀
5.RS3PE综合征伴发骨髓增生异常综合征一例报告 [J], 赵金荣;袁伟壮;黄璨;赵丽丹
因版权原因,仅展示原文概要,查看原文内容请购买。
“怪症”困惑医学界八:“无痛症”:这个女童永远不怕痛

来说 , 一 怪 病 就 这
像 是 一 种 噩 梦般 的 诅 咒— — 因 为 它 让 患 者 任 何 时 候 都 处 于 性 冲 动 性 亢 奋 状 态 中。 一 怪 病 是 如 此 罕 见 , 这 直到 2 0 年 才 01 正 式 获 得 了 自 己的 医学 名 称 。 现年 3 5岁的 美 国 南卡 罗 莱 纳 州女 性 海 瑟 ・ 尔蒙 就 是 迪
一
个“ 续性 冲 动 综 合 征 ” 者 , 总是 处 于性 亢 奋 状 态 中 , 持 患 她
任何 声音 , 包括 汽车喇叭 声、 火车轰鸣 声都能让她 引发性 亢 奋。 海瑟痛苦地说 :我 看过 各种 医生, “ 包括妇科学家、 心理学
家 、 神 病 学 家 , 果 没人 知 道 我 患 了什 么疾 病 。 ” 精 结 美 国加 州 大 学 外科 教 授 欧 文 ・ 戈德 斯 坦 博 士 是 研 究 “ 持
班 的 歌 声也 会 令她 癫 痫 发 作 。
茜 ・ 尔来说 却 是 一 盖
个恐怖的噩 梦 ,因为 她 惠 有 一 种 罕 见 的
“ 乐癫痫症” 音 ,她 只
不过去年 。美 国医生通 过一种先锋性 手术移 除
了斯 塔 茜 大 脑 右 前 颞 叶 中可 能 引 发 癫 痫 的 脑 组 织 ,
要 一 听到 音 乐 就 会 癫
从此 以后 ,斯塔 茜再也没有发作过癫痫 , 年 来她 也 s
可 以首 次 享 受 音 乐 、 不 必 害怕 昏倒 了 。 而
痫发作 。 陷入 昏厥 。斯 塔 茜的 怪 病 让 美 国 医 学 专 家们
深感 困惑。
“ 症” 怪 困惑 医学 界 八 :
“ 症” 怪 阑惑 医学 界 八 :
2019年最新-PD性综合征课件-精选文档

左旋多巴撤药MS
散发性 撤药(左旋多巴)
存在 明显增高 可能障碍
明显 明显增高 可能存在
精神药物MS
散发性 精神药物
存在 明显增高 可能障碍
明显 明显增高 可能存在
恶性高热
常显和散发性 吸入性麻醉
不存在 明显增高 可能障碍
明显 明显增高 可能存在
DEPARTMENT OF GERIATRIC NEUROLOGY
难或胃吸收差时可胃管给药; • (2)多巴胺受体(DR)激动剂; • 4.DIC的治疗:肝素静脉输液泵输入 • 5. 急性肾功能衰竭:血液透析
DEPARTMENT OF GERIATRIC NEUROLOGY
• 八 、帕 金 森病撤药MS的预后
• MS是一种致死性高的疾病,除非早期诊断 后给予相应的治疗。帕金森病患者发生MS 时为老年、病程长、Hoehn和Yahr分期高、 UPDRS评分高等会影响预后。
• 左旋多巴撤药MS的临床表现如下:
1. 体温升高(至40℃)
2. 明显肌强直。
3. 意识障碍
4. 自主神经障碍:心动过速;呼吸急促;多汗或无汗; 非阻塞性肠梗阻;血压波动;声带麻痹。
5. 血清CK增高。
6. 横纹肌溶解
7. 弥散性血管内凝血(DIC)
8. 急性肾功能衰竭(肌红蛋白阻塞)。
DEPARTMENT OF GERIATRIC NEUROLOGY
• 据文献统计,2/3患者在MS前期进行相应的 治疗有效,其病死率为4%。所有发展成 DIC和急性肾功能衰竭患者的病情十分严重。 这两种并发症的病死率都很高。
• 六 、帕金森病撤药MS的预防
• 帕金森病患者中体温升高至38℃时虽未进行抗帕
金森病药物的减量,也应考虑是MS的早期症状。
威廉姆斯综合征疾病PPT演示课件

在诊断过程中,需要与类似症状的疾病进行鉴别,如唐氏综合征、天使综合征 等。这些疾病虽然有一些相似的症状,但通过详细的临床检查和遗传学分析可 以进行区分。
02
遗传学与发病机制
Chapter
遗传学特点
01
02
03
染色体微缺失
威廉姆斯综合征是由7号 染色体长臂上特定区域微 缺失引起的遗传性疾病。
误诊原因分析
症状不典型
威廉姆斯综合征患者的症状可能因个体差异而不典型,导致医生在 初步诊断时难以准确识别。
遗传学检查误差
遗传学检查在威廉姆斯综合征的诊断中具有重要作用,但检查结果 可能受到多种因素的影响,如样本污染、实验操作失误等,导致误 诊。
医生经验不足
威廉姆斯综合征是一种相对罕见的疾病,部分医生可能对其认识不足 ,缺乏诊断经验,从而导致误诊。
精神药物
对于伴有精神症状的患者,如焦虑 、抑郁等,可酌情使用抗焦虑、抗 抑郁等药物。
其他药物
根据患者具体情况,可选用改善认 知功能、促进生长发育等药物。
非药物治疗措施
心理治疗
通过心理咨询、行为疗法等方 式,帮助患者调整心态,增强
自我认知和自我管理能力。
教育干预
针对患者的学习困难和社交障 碍等问题,制定个性化的教育 计划,提供特殊教育支持。
神经心理评估
采用智力测验、行为评估等方法,了解患者的认 知、情感和行为状况。
生活质量评估
运用生活质量量表等工具,综合评估患者在生理 、心理、社会功能等方面的生活质量。
生活质量改善措施建议
个性化教育计划
根据患者的智力水平和兴趣爱好,制定个性化的教育计划,提高 患者的认知能力和社会适应能力。
心理干预
复旦大学历年哲学考研试题(2019-2019年)-5页精选文档

2019 年硕士研究生入学考试西方哲学史试题1.概述亚里士多德的“实体” (sustance)理论,并简要评述。
2 .概述斯多亚派(亦译斯多葛派)的伦理学。
3 .简述巴门尼德的存在论。
4.托马斯.阿奎那的经院哲学的基本观点述评。
5.你认为笛卡尔对后世哲学影响最大的观点是什么?6 .评休谟的因果观。
7 .从本体论、认识论、伦理学的统一谈斯宾诺莎哲学的基本特征。
8 .概述康德的批判哲学中关于感性、知性和理性的基本观点。
9 .简述萨特的自在自为理论和自由观。
10.胡塞尔从事哲学追求的根本目的或基本纲领是什么?他创立什么方法和理论来实现其目的?11.维也纳学派为何要拒斥形而上学?如何拒斥形而上学?2019 年西方哲学史考研试题1 芝诺关于运动的难题。
2.简述苏格拉底的“美德就是知识”3.中世纪唯名论与唯实论之争。
4.培根的归纳法。
5.笛卡尔的“我思故我在”。
6.洛克第一性质和第二性质的学说。
7.莱布尼茨的预定和谐说。
8.康德说休谟把他从独断论的迷梦中惊醒,是何意,你如何理解?9 简述斯宾诺莎的实体观。
10 以某一实用主义哲学家为例说明实用主义真理观。
11 新康德主义复活和发挥了康德哲学中哪些因素?2019 年西方哲学史考研试题一,名词解释 (15 分*5 个)兼爱大乘三位一体此在人类历史的史前时期二,论述 (25 分*3 个)1 ,试评述孔子关于仁与礼关系的思想。
2 ,简单介绍康德范畴的先验演绎,并评述在其思想中的地位和作用。
3,简单评述《费尔巴哈提纲》中“社会生活在本质上是实践的” 。
2019 年西方哲学史考研试题一,名词解释 (6 分*8 个)逻各斯教父哲学四假相反思(黑格尔) 奥卡姆剃刀绝对命令证实原则解释学循环二,简答 (15 分*4 个)1 ,简述亚里士多德的实体论。
2 ,康德的先天综合判断。
3 ,唯名论与实在论的异同。
4 ,简述黑格尔的真理观。
三,论述 (42 分)1 ,从笛卡尔到斯宾诺莎、莱布尼兹实体论的演进发展。
婴儿神经母细胞瘤术后合并霍纳综合征一例

* )7B*
婴儿神经母细胞瘤术后合并 霍纳综合征一例
宋元华!杨!震
作者单位! 7&###!云南$昆明市儿童医院肿瘤科 作者简介! 宋元华" $()$ * # $女$医学硕士$主治医师$研究方向!儿科肿瘤学% + , . / 0 !$'&BB"(()8$7'9 : ; -
A6讨论 A@ ?6根据患儿病史&手术情况及病理等资料$诊断! " $ # 右后纵膈神经母细胞瘤术后! 期低危'" " # 术后
患儿$ 男$ 7 个 月$ 因 呛 咳 & P 入 院% 患 儿 于
"#$%, $", #& 吃米线后出现咳嗽$ 阵发性$ 无喘息& 气
促$无面色青紫$无犬吠样咳及鸡鸣样尾声$ 无呼吸 困难% 在当地县医院 k检查考虑支气管异物可能$来
A@ A6经验及教训6基层医师对于此病认识不够$误
四胸椎旁$宽约 %M # : $ 切开后纵膈胸膜沿着肿瘤 锐钝交替分离$最后完整切除肿瘤$周围未见肿大淋 巴结瘤$未见胸壁肋骨破坏征象$剖开肿物见鱼肉样 组织% 病检结果!神经母细胞瘤伴坏死&钙化% 骨穿 检查未提示骨髓转移% 术后第 ' 天患儿出现霍纳综 合征$表现为右侧眼裂变小$ 眼球内陷$ 右侧颜面部 无汗% 患儿年龄 L $ 岁$ 肿瘤局限在一侧肉眼完整 切除$但因同侧非粘连性淋巴结未活检$根据国际神
神经母细胞瘤!期低危$行 D A fq + G方案化疗 " 个 疗程后患儿右侧眼裂与正常侧基本相同$ 右眼球无 明显内陷$右侧颜面仍无汗$现患儿已完成 % 个疗程 化疗$霍纳综合征较术后明显改善%
无汗症有哪些症状?

无汗症有哪些症状?
*导读:本文向您详细介绍无汗症症状,尤其是无汗症的早期症状,无汗症有什么表现?得了无汗症会怎样?以及无汗症有哪些并发病症,无汗症还会引起哪些疾病等方面内容。
……
*无汗症常见症状:
面色潮红、出汗异常、虚脱
*一、症状
1.患者全身皮肤或某一部位终年无明显汗液。
全身性无汗的患者常感全身不适,极度疲劳,在运动时最明显。
在天热季节中,体温往往升高,心率加快,全身皮肤潮红,甚至出现虚脱、中暑等症状。
2.局限性皮肤干燥、粗糙,或在某些皮肤病皮损上出现,一般症状轻微。
3.发汗实验可确定。
*二、诊断
根据临床表现的特征性即可诊断。
*以上是对于无汗症的症状方面内容的相关叙述,下面再看下无汗症并发症,无汗症还会引起哪些疾病呢?
*无汗症常见并发症:
淋巴瘤、尿毒症
*一、并发病症
后天性无汗症见于硬皮病、肿瘤、烧伤、皮肤移植、放射性皮炎、淋巴瘤、Sjogren综合征和慢性萎缩性肢端皮炎。
麻风、酒精中毒性神经炎、淀粉样变、糖尿病、痛风可发生无汗症。
甲状腺功能低下患者、肿瘤、尿毒症、肝硬化、内分泌疾病(如Addison病、糖尿病、尿崩症)以及罕见的遗传病(如Fabry病、Franceschetti-Jatassohn综合征和Helweg-Larssen综合征)均可出现无汗症
*温馨提示:以上就是对于无汗症症状,无汗症并发症方面内容的介绍,更多疾病相关资料请关注疾病库,或者在站内搜索“无汗症”可以了解更多,希望可以帮助到您!。
SATB2相关综合征13例病例系列报告

基金项目㊀国家自然科学基金面上项目:81471483;上海市出生缺陷防治重点实验室项目:13DZ2260600作者单位㊀复旦大学附属儿科医院㊀上海,201102,1儿科研究所分子医学中心,2儿童保健科;3共同第一作者通讯作者㊀吴冰冰,email:bingbingwu2010@163.com;徐秀,email:xuxiu@shmu.edu.cn;王慧君,email:huijunwang@fudan.edu.cn㊃论著㊃DOI:10.3969/j.issn.1673⁃5501.2020.06.011SATB2相关综合征13例病例系列报告王㊀晴1,3㊀徐㊀琼2,3㊀肖非凡1㊀钱琰琰1㊀刘仁超1㊀李㊀刚1㊀周文浩1㊀吴冰冰1㊀徐㊀秀2王慧君1㊀㊀摘要㊀目的㊀总结SATB2相关综合征(SAS)的临床及遗传学特征,为早期干预及产前咨询提供依据㊂方法㊀回顾性分析2016年1月至2020年6月于复旦大学附属儿科医院就诊且基因诊断为SAS患儿的临床资料和基因检测结果㊂结合人类基因突变数据库(HGMD)和PubMed,对SAS的临床表型及遗传特征进行文献复习㊂结果㊀13例SAS患儿进入本文分析,男7例,女6例,样本检测年龄为生后3d至9岁7月(M为14月龄)㊂13例均存在发育迟缓,>1岁的患儿均出现语言发育障碍㊂8例行头颅MR检查,7例提示脑成像异常㊂4例癫发作㊂2例骨发育不良,4例四肢肌张力低下,2例心血管畸形㊂13例均有小颌和牙齿畸形,4例腭裂,3例体重低于同龄儿2SD,2例流涎,1例角膜白斑㊂4例并发肺部反复感染,1例合并先天性喉软化及声带麻痹㊂13例患儿检测到13种SATB2基因杂合变异,包括6种错义变异(E436A㊁L261P㊁L626P㊁R399C㊁A590T㊁E566K),2种无义变异(Q666Ter㊁R239Ter),5种染色体2q32⁃2q37区缺失/重复导致的拷贝数变异,其中R239Ter和E566K为HGMD已收录的致病位点,其余11种均为新变异㊂结论㊀SAS可累及多系统,需通过基因检测协助明确诊断㊂建议将SATB2基因作为原因不明的神经发育障碍性疾病的重要候选基因进行筛查和诊断㊂关键词㊀SATB2相关综合征;㊀SATB2基因;㊀临床特征;㊀基因型13childrenwithSATB2⁃associatedsyndrome:AcaseseriesreportWANGQing1,3,XUQiong2,3,XIAOFeifan1,QIANYanyan1,LIURenchao1,LIGang1,ZHOUWenhao1,WUBingbing1,XUXiu2,WANGHuijun1(Children'sHospitalofFudanUniversity,Shanghai201102,China,1CenterforMolecularMedicine,2DepartmentofChildHealthcare;3Co⁃firstauthors)CorrespondingAuthor:WUBingbing,email:bingbingwu2010@163.com;XUXiu,email:xuxiu@shmu.edu.cn;WANGHuijun,email:huijunwang@fudan.edu.cnAbstractObjectiveTosummarizetheclinicalandgeneticcharacteristicsofSATB2⁃associatedsyndrome(SAS),andtoprovideevidenceforearlyinterventionandprenatalcounseling.MethodsChildrendiagnosedasSASbygenetictestingwererecruitedfromJanuary2016toJune2020inChildren'sHospitalofFudanUniversity.Weanalyzedtheclinicalandgeneticfeaturesofthesepatientsretrospectively.AccordingtoHumanGeneMutationDatabase(HGMD)andPubMed,literatureonclinicalphenotypeandgeneticcharacteristicsofSASwerereviewed.ResultsThirteenchildrenwithSASwereincludedinthisstudy,consistingof7malesand6females.Theageofgenetictestrangedfrom3daysto9yearsand7months(medianageof14months).Afterpsychomotordevelopmentwasevaluatedin13patientsduringthefollow⁃up,allofthempresentedwithdevelopmentaldelayandallchildrenover1yearoldhadlanguageretardation.Ofthe8casesundergoingbrainMRexaminations,7casesshowedabnormalbrainimaging.Fourcasespresentedepilepsy.Twopatientspresentedbonedysplasiaand4individualswerehypotonia.Congenitalheartdefectswereidentifiedin2children.Physicalexaminationshowedthatallof13childrenhadmalformationofsmalljawandteeth,4caseswerediagnosedwithcleftpalate,3patients'weightwas2standarddeviationslowerthanthoseoftheirpeers,2childrenpresentedsalivation,and1caseoccurredcornealleukoplakia.Therewere4casesofrecurrentpulmonaryinfectionand1patientaccompaniedwithcongenitallaryngomalaciaandvocalcordparalysis.ThirteenmutationsofSATB2weredetectedin13patients,including6missensesmutations(p.E436A,p.L261P,p.L626P,p.R399C,p.A590T,p.E566K),2nonsensemutation(Q666Ter,R239Ter),and5copynumbervariationsresultingfromdeletion/duplicationinregion3ofthelongarmofchromosome2.ThemutationsofR239TerandE566KwererecordedinHGMD,andothervariationswerenovel.ConclusionSATB2⁃associatedsyndromeinvolvesmultiplesystemabnormalities,whichneedstobeconfirmedbygenetictesting.SATB2genecanberegardedasanimportantcandidategeneforthescreeninganddiagnosisofneurodevelopmentaldisorderswithunknowncauses.Keywords㊀SATB2⁃associatedsyndrome;㊀SATB2gene;㊀Clinicalcharacteristics;㊀Genotype㊀㊀SATB2相关综合征(SAS)是SATB2基因突变导致的可累及多系统异常的一类疾病,又称Glass综合征㊂1989年Glass等[1]首次报告1例老年患者,以严重智力障碍为主要表现㊂SAS的临床表现以神经发育障碍为主,表现为生长发育迟缓㊁智力障碍㊁语言发育迟缓等㊂近5年来,陆续有SAS患者的病例报告,但国内仅可检索到3篇文献共报道4例[2⁃4]㊂本文回顾性分析复旦大学附属儿科医院(我院)近年来诊断的SAS患儿的临床资料和基因检测结果,以提高临床对该病的认识㊂1㊀方法1.1㊀病例纳入标准㊀2016年1月至2020年6月于我院分子诊断中心行基因检测明确为SAS的连续病例㊂1.2㊀基因检测方法㊀我院对患有原因不明的神经发育障碍性疾病的患儿,经家长知情同意后行全外显子组测序(WES)或临床外显子组测序(Panel)㊂本文患儿的基因测序㊁变异解读和报告分析基于我院分子诊断中心建立的第2版高通量测序数据分析和临床诊断流程[5]㊂1.3㊀资料截取㊀从我院病历系统中截取母孕产史,患儿围生期情况㊁出生喂养史㊁生长发育史㊁家族史㊁起病年龄㊁性别㊁基因检测年龄㊁住院病历信息等,患儿就诊时身高㊁体重㊁头围㊁面容㊁骨骼㊁关节㊁神经系统查体㊁智力发育测试㊁口腔颌面检查㊁听力筛查㊁视力检测及头颅影像学检查㊁心脏彩超㊁脑电图等㊂2㊀结果2.1㊀一般情况㊀13例SAS的患儿进入本文分析,表1显示,男7例,女6例,样本送检年龄为生后3d至9岁7个月(M为14月龄)㊂就诊原因:早产伴ARDS3例,发育迟缓伴语言发育障碍9例,发育迟缓伴癫1例㊂7例有母亲孕产情况及出生体重,妊娠年龄为孕34 39(M为36)周,患儿出生体重为1940 3000(M为2000)g㊂1女2岁+++--未查指/趾甲凹凸不平,甲上白斑,双侧髋臼角偏大2女2岁++---未查早产儿,窒息史,孤独症3男3月+----部分脑外间隙增宽㊁右额硬膜下积液癫,肺炎4男9岁++---未见异常消瘦5女14月++-+-侧脑室饱满,侧脑室旁白质内少许稍高信号影下颌偏短6男10月+--+-顶枕叶侧脑室旁白质髓鞘化不良出牙延迟7男7月+---+脑外间隙稍宽,脑髓鞘化不良鼻梁平,招风耳,癫8女8岁++---未查孤独症㊁反应慢㊁理解力差9男3d+-+-+早产儿脑改变;双侧侧脑室周围异常信号,髓鞘化异常可能早产儿,喂养困难伴呕吐,肺炎10男3d+--++早产儿脑改变;右侧脑室前角旁及左侧脑室体旁小软化灶早产儿,喂养困难,小下颌,吐泡沫,癫,贫血,肺炎11男17月++-+-胼胝体薄,白质髓鞘化略延迟癫,流涎,小下颌12女17月++---未查早产儿,房间隔缺损,先天性喉软骨软化,声带麻痹13女6d+---+未查房间隔缺损,室间隔缺损,角膜白班,双手通贯掌,呼吸衰竭,肺炎2.2㊀神经发育情况㊀13例患儿在随访时进行了精神运动发育评估,均提示发育迟缓,且>1岁患儿均出现语言发育障碍㊂例9和10为孕34+4周行剖宫产娩出的双胞胎,生后于暖箱中鼻饲喂养困难,原始反射未引出㊂例2和8有孤独症行为,易怒和苦笑㊂例3㊁7㊁10和11癫发作㊂2.3㊀头颅MR影像检查㊀8例患儿行头颅MR,7例提示脑成像异常,例5㊁6㊁9和10侧脑室扩大及同侧脑室旁白质高信号,例6㊁9和11髓鞘发育异常,例3脑血管周围间隙变宽,例11薄胼胝体㊂2.4㊀其他系统表现㊀例1㊁9骨发育不良,例1双侧髋臼角偏大;例7㊁9㊁10㊁13四肢肌张力低下;例12房间隔缺损,例13房间隔和室间隔缺损㊂2.5㊀体格检查㊀例9㊁10㊁12体重低于同龄儿2SD,例10㊁11流涎,例13角膜白斑㊂㊀㊀随访观察发现,13例均有小颌和牙齿畸形表现,例5㊁6㊁10㊁11腭裂㊂2.6㊀并发症㊀例3㊁9㊁10㊁13肺部反复感染病史,例10有哮喘表现,例12有先天性喉软化及声带麻痹病史㊂2.7㊀基因检测㊀在13例患儿中,检测到13种SATB2基因杂合变异,分别为6种错义变异㊁2种无义变异㊁5种缺失/重复导致的拷贝数变异(表2)㊂㊀㊀经HMGD㊁Clinvar数据库及文献检索,例6和8携带的1c.1996C>Tp.Q666Ter杂合novelNA/NA/D0/0/0致病2c.1307A>Cp.E436A杂合novelT/D/D0/0/0可疑致病3c.782T>Cp.L261P杂合novelT/D/D0/0/0可疑致病4c.1877T>Cp.L626P杂合novelD/D/D0/0/0可疑致病5c.1195C>Tp.R399C杂合novelD/D/D0/0/0致病6c.715C>Tp.R239Ter杂合PMID17377962NA/NA/D0/0/0致病7c.1768G>Ap.A590T杂合novelT/B/N0/0/0可疑致病8c.1696G>Ap.E566K杂合PMID28151491T/P/D0/0/0致病9NA2q32.2⁃2q33.1del杂合novelNANA致病10NA2q32.2⁃2q33.1del杂合novelNANA致病11NA2q33.1del杂合novelNANA致病12NA2q32.2⁃2q33.1del杂合novelNANA致病13NA2q33.1⁃2q37.1dup杂合novelNANA致病SATB2基因变异位点为已报道的致病变异,例1Q666Ter是无义突变,致病性明确㊂错义突变中,除A590T预测为良性变异(例7),例2 5经PolyPhen⁃2㊁MutationTaster软件预测均为有害突变㊂SIFT软件显示E436A和L261P的预测分数>0.05,结论为可容忍变异T(tolerant);L626P㊁R399C突变体的预测分数<0.05,结论为有害变异D(deleterious)㊂5种染色体拷贝数变异包括2q32.2⁃2q33.1区域的4种缺失突变及1种重复突变(2q33.1⁃2q37.1dup)㊂上述变异评级结果中,例2㊁3㊁4和7为可疑致病变异,其余为致病变异㊂3㊀讨论㊀㊀截至2020年4月,国内外文献共报道了165例SATB2基因变异/缺失的患者㊂所有患儿均出现发育迟缓,且>2岁患儿均存在语言发育障碍㊂本文13例SAS患儿,在随访中发现均出现发育迟缓,且>1岁患儿均有语言落后表现㊂提示发育迟缓和语言发育障碍是SATB2基因突变的主要表型[6]㊂㊀㊀另外,上颌㊁牙齿畸形也是SAS患者的常见临床表现㊂SATB2基因可通过促进颌面部成骨细胞特异性基因(RUNX2㊁ATF4㊁SOX9)的表达,调控颌骨发育[6]㊂SATB2基因变异可影响牙囊细胞破骨能力,导致恒牙迟萌及牙列紊乱㊂牙齿畸形在SATB2基因缺失表型中有着较高的发生率㊂Zarate等[6]报告98.5%的SAS患者出现牙齿畸形,本文13例在随访检查中发现均存在牙列拥挤㊂SATB2基因单倍剂量不足可能是腭裂形成的重要原因[7,8]㊂Zhao等[9]研究表明,SATB2蛋白结合HOXA2基质连接区,负调节HOXA2基因在造骨细胞中的表达㊂在SATB2基因敲除小鼠中,Hoxa2基因过度表达,抑制骨质生成,改变腭突上抬过程的信号传递,形成腭裂[10]㊂Zarate等[11]报告约40%的病例有腭裂,本文4例患儿(30.7%)存在腭裂㊂㊀㊀SAS其他较罕见的表型包括:先天性心脏病㊁癫等㊂目前报道的SAS患者中,30例既往存在癫发作,癫发作的中位年龄为8.5岁㊂Leoyklang等[12]报告了1例36岁男性携带SATB2基因的无义突变(c.715C>T,p.R239Ter),患者童年期有多次热性惊厥,26岁发展成全身强直性痉挛发作㊂Usui等[13]报告了1例5岁9月龄的男孩,染色体2q32.1⁃2q33.3区域发生重复突变,25月龄出现惊厥发作,57月龄后抽搐频繁,几乎每周发作1次,卡马西平治疗效果不佳㊂本文4例有癫的患儿均发病较早,目前服用丙戊酸钠控制㊂值得注意的是,Li等[14]的体内外实验发现,小鼠海马中SATB2表达水平在神经元过度活跃后会降低,而SATB2突变小鼠海马CA1区锥体神经元兴奋性和冲动发放均减少,提示SATB2可能成为治疗癫的新靶点㊂SAS患者的先天性心脏病表型描述较少㊂在已报道的2例存在心脏间隔部位缺损的患者中,1例是Cormack等[15]报告的新西兰毛利人,在染色体2q32⁃2q35区域约24.7Mb片段缺失,超声心动图显示室间隔膜部缺损㊁房间隔缺损㊁动脉导管未闭㊁严重的右心室容量超负荷和持续性肺动脉高压;另1例为VanBuggenhout等[16]报告的3岁8月龄的男孩,染色体2q32⁃2q33区域约21.6Mb片段缺失,存在室间隔缺损㊂Zarate等[17]总结已报道的病例,发现有先天性心脏缺陷的SAS患者均存在拷贝数异常㊂本文2例先天性心脏病的患儿,经基因检测证实存在含SATB2基因的片段缺失或重复㊂位于染色体2q31⁃2q33区域的CREB2㊁INPP1㊁NAB1等是SATB2基因的邻近基因,这些基因与心肌细胞凋亡㊁增殖相关[18]㊂推测SATB2及邻近基因共同缺失/重复导致上述先天性心脏病表型㊂㊀㊀目前,已报道的165例患者共有124种变异类型,包括错义变异32种㊁无义变异20种㊁移码变异32种㊁剪接位点变异6种㊁染色体易位6种㊁框内插入1种㊁拷贝数变异27种(缺失24种㊁重复3种)㊂131例有患者父母测序数据的病例中,来自5个家庭的9例先证者父亲/母亲验证为嵌合体[6,19⁃22],其余父母均未检测到SATB2基因突变,推测为患者的新发变异㊂这些变异主要分布在外显子8和外显子9区域(氨基酸起止位置:232⁃459),本文4例点突变位于该区域,提示此区域为SATB2基因突变的 热点 区域㊂㊀㊀近5年来,随着越来越多的病例出现,研究者开始关注SAS基因型与临床表型之间的关系㊂Zarate等[6]总结158例SAS患者临床表型与基因型,发现>4岁完全丧失语言表达能力的SAS患者,无义突变组的比例为27.6%(8/29),但在所有变异组中占比最低;错义变异组的比例为51.3%(20/39),在所有变异组中占比最高(P=0.0496)㊂SATB2基因发生错义变异的SAS患者发生腭裂的比例为22.5%(11/49),较其他变异组56.2%(59/105)低(P<0.0001);但错义突变组患者有癫发作的比例为30.4%(14/46),较其他组15.5%(15/97)高(P=0.0375)㊂移码突变组患者存在喂养困难的比例为96.2%(25/26),明显高于其他组61.5%(64/104),P=0.0007㊂Zarate等[11]发现,在72例存在大片段缺失的SAS患儿中,16例患儿的平均诊断年龄为2.5岁,较其他变异组更早获得诊断(P<0.0006)㊂本文分析,片段缺失患儿的早期诊断可能得益于自2010年起国际上推荐将基因芯片作为智力发育迟缓和畸形患儿的首选检测手段[23]㊂㊀㊀SAS是一种累及多系统的遗传病,目前尚无有效的治疗方法,以对症和支持治疗为主[11]㊂临床治疗主要强调对患儿的支持治疗,修复腭裂等颌面部缺陷;在神经系统发育方面,建议早期干预,进行康复训练,注意语言能力训练;需要长期监测患儿牙齿㊁眼睛㊁骨骼等方面的发育情况㊂参考文献1 GlassIA SwindlehurstCA AitkenDA etal.Interstitialdeletionofthelongarmofchromosome2withnormallevelsofisocitratedehydrogenase.JMedGenet 1989 26 2 127⁃130.2 靳春雷 雷永良 刘姣 等.2q33.1微缺失致SATB2基因部分缺失兄妹的表型及遗传学分析.中华医学遗传学杂志 2019 36 6 628⁃631.3 林美丽 姚如恩 陆静 等.一例Glass综合征患儿的SATB2基因突变分析.中华医学遗传学杂志 2019 36 7 712⁃715.4 梅道启 梅世月 陈国洪 等.SATB2相关综合征一例报道并文献复习.中华神经科杂志 2019 52 12 1059⁃1060. 5 杨琳 董欣然 彭小敏 等.复旦大学附属儿科医院高通量测序数据分析流程 第二版 对遗传疾病候选变异基因筛选用时和准确性分析.中国循证儿科杂志 2018 13 2 118⁃123.6 ZarateYA BosankoKA CaffreyAR etal.MutationupdatefortheSATB2gene.HumMutat 2019 40 8 1013⁃1029. 7 BritanovaO DepewMJ SchwarkM etal.Satb2haploinsufficiencyphenocopies2q32⁃q33deletions whereaslosssuggestsafundamentalroleinthecoordinationofjawdevelopment.AmJHumGenet 2006 79 4 668⁃678.8 TalkowskiME RosenfeldJA BlumenthalI etal.Sequencingchromosomalabnormalitiesrevealsneurodevelopmentallocithatconferriskacrossdiagnosticboundaries.Cell 2012 149 3 525⁃537.9 ZhaoY GuoYJ TomacAC etal.IsolatedcleftpalateinmicewithatargetedmutationoftheLIMhomeoboxgenelhx8.ProcNatlAcadSciUSA 1999 96 26 15002⁃15006. 10 NazaraliA PuthucodeR LeungV etal.TemporalandspatialexpressionofHoxa⁃2duringmurinepalatogenesis.CellMolNeurobiol 2000 20 3 269⁃290.11 ZarateYA Smith⁃HicksCL GreeneC etal.Naturalhistoryandgenotype⁃phenotypecorrelationsin72individualswithSATB2⁃associatedsyndrome.AmJMedGenetA 2018 176 4 925⁃935.12 LeoyklangP SuphapeetipornK SiriwanP etal.HeterozygousnonsensemutationSATB2associatedwithcleftpalate osteoporosis andcognitivedefects.HumMutat 2007 28 7 732⁃738.13 UsuiD ShimadaS ShimojimaK etal.Interstitialduplicationof2q32.1⁃q33.3inapatientwithepilepsy developmentaldelay andautisticbehavior.AmJMedGenetA 2013 161A 5 1078⁃1084.14 LiY HuangWY LvCY etal.Satb2ablationdecreasesPTZ-inducedseizuresusceptibilityandpyramidalneuronalexcitability.BrainRes 2018 1695 102-107.15 AdrianMC JulietT NerineG etal.Delineationof2q32q35deletionphenotypes twoapparent"proximal"and"distal"syndromes.CaseRepGenet 2013 2013 823451.16 VanBuggenhoutG VanRavenswaaij⁃ArtsC McMN etal.Thedel 2 q32.2q33 deletionsyndromedefinedbyclinicalandmolecularcharacterizationoffourpatients.EurJMedGenet 2005 48 3 276⁃289.17 ZarateYA FishJL.SATB2⁃associatedsyndromeMechanisms phenotype andpracticalrecommendations.AmJMedGenetA 2017 173 2 327⁃337.18 RampazzoA NavaA MiorinM etal.ARVD4 anewlocusforarrhythmogenicrightventricularcardiomyopathy mapstochromosome2longarm.Genomics 1997 45 2 259⁃263. 19 BenganiH HandleyM AlviM etal.Clinicalandmolecularconsequencesofdisease⁃associateddenovomutationsinSATB2.GenetMed 2017 19 8 900⁃908.20 GreletM MortreuxJ AlazardE etal.SATB2⁃associatedsyndrome firstreportofagonadalandsomaticmosaicismforanintrageniccopynumbervariation.ClinDysmorphol 2019 28 4 205⁃210.21 QianY LiuJ YangY etal.PaternalLow⁃LevelMosaicism⁃CausedSATB2⁃AssociatedSyndrome.FrontGenet 2019 10 630.22 ScottJ AdamsC SimmonsK etal.Dentalradiographicfindingsin18individualswithSATB2⁃associatedsyndrome.ClinOralInvestig 2018 22 8 2947⁃2951.23 MillerDT AdamMP AradhyaS etal.Consensusstatementchromosomalmicroarrayisafirst⁃tierclinicaldiagnostictestforindividualswithdevelopmentaldisabilitiesorcongenitalanomalies.AmJHumGenet 2010 86 5 749⁃764.(收稿日期:2020⁃06⁃30㊀修回日期:2020⁃08⁃05)(本文编辑:张崇凡)。
婴儿Evans综合征的临床观察与护理

婴儿Evans综合征的临床观察与护理发表时间:2014-01-03T11:13:55.090Z 来源:《医药前沿》2013年11月第32期供稿作者:金向群[导读] 本人就最近我科一例婴儿Evans综合征,经过及时的诊断,治疗及护理。
病情得以控制。
现将护理报告如下。
金向群(湖北省咸宁市中心医院儿内湖北咸宁 437100)【中图分类号】R473.72 【文献标识码】A 【文章编号】2095-1752(2013)32-0090-02 Evans综合征(Evans syndrome,ES)是一种自身免疫疾病,系体内出现自身抗体,引起红细胞和血小板破坏增加,从而导致相继或同时发生自身免疫性溶血性贫血(AIHA)和免疫性血小板减少症(ITP)[1]。
儿童ES多数以ITP起病,随后发展为AIHA,其临床表现为贫血、出血、黄疸、肝脾肿大等。
患儿病情凶险,病死率及复发率极高。
目前在国内外儿童报道极少,婴儿罕见。
因此对本病存在一定的认识不足。
本人就最近我科一例婴儿Evans综合征,经过及时的诊断,治疗及护理。
病情得以控制。
现将护理报告如下。
1.资料简介患儿,男,3月,因“反复面色苍黄、皮肤瘀点40余天,再发1天”于2012年4月10日入院,40余天前无明显诱因下出现面色苍黄、皮肤瘀点在某医院住院治疗,诊断为“溶血性贫血”、“婴儿肝炎综合征”。
给予输注红细胞,血小板,丙种球蛋白,糖皮质激素等治疗,病情好转症状消失出院,出院时患儿皮肤无黄染、无紫癜,带强的松口服,服用一周后自行停药。
今患儿再次出现面色苍黄,皮肤瘀点。
急来我院门诊查血常规异常,以“贫血、黄疸原因待查?”收住我科。
患儿系G1P1孕足月剖宫产,出生体重3600克,纯母乳喂养。
否认过敏病史。
入院查体:T:36.5℃,HR150bpm,R48bpm,体重5Kg,神清,精神反应一般,轻度贫血貌,颜面及双下肢可见散在出血点,浅表淋巴结无肿大,咽无充血,双肺、心、腹未见异常,神经系统无阳性体征。
噬血细胞综合征-liaoyang

添加标题
肾上腺糖皮质激素可以减轻炎 性细胞因子的释放和抑制噬血 细胞对血细胞的吞噬。糖皮质 激素选择地塞米松,该药透过 血脑屏障的效果好,对中枢神 经系统受累有预防和治疗作用。
预后
上海复旦大学18例就诊HLH患儿有14例接受HLH-2004治疗方案。 治疗结果:持续活动4例,4例临床缓解,失访2例,复发4例(仅 有1例按复发方案治疗2周后重新缓解,其余均死亡),总共有7例
第9~40周: 10mg/m2 ×3天,隔周1次
第40周后视病情而定
依托泊苷(VP-16) 150mg/m2/次 静滴
第1、2周
每周2次
第3~8周
每周1次
第9~40周 隔周1次
第40周后停药
环孢素A (CsA) 6mg/kg.d 口服 一直服用。
如果有神经系统症状,在第3~6周每周各加用1次鞘注
病案
单击此处可添加副标题
给予积极抗感染,大剂量丙种球蛋白,保肝及对症支持治疗。 患儿住院第3天昏迷,作腰椎穿刺术测脑脊液压力正常,细胞 数及蛋白正常。患儿住院3 d,仍发热,体温波动在39-42℃, 常规降温措施无效,肝脾进行性增大,肝右肋下5 cm,剑突 下7cm,脾左肋下6cm。复查血常规:白细胞0.4 x109/L, 血红蛋白52 g/L,血小板12×109/L。肝功能:ALT404 U/L,AST 1024 U/L,TBIL 110.4 mmol/l,DBIL 97.0 mmol/l,ALB19.8 g/L。电解质:K+3.5mmol/L。 Na+125 mmol/L。后经积极治疗抢救无效于住院第6天死亡。 死亡当天查凝血酶原时间 (PT)25.6 8,PA 67%;血常规: 白细胞0.3×109/L。血红蛋白49 g/L,血小板7×109/L。肝 功能:ALT 360 U/L,AST1383 U/L,TBIL 49.5 mmol/l ,DBIL 39.2 mmol/l ,ALB 14.8 g/l 电解质: K+ 3.4 mmol/L,Na+ 118 mmol/L。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
先天性无痛无汗综合症七例
先天性无痛无汗综合症(fam ilial non-sweatingand nonpainful syndrome)又称遗传性感觉自律神经病,是一种少见的染色体隐形遗传性疾病,临床少见。
目前该病的发生机制还不清楚。
其主要缺乏传导痛觉的感受器,传递特异性痛觉通道损害。
电镜下观察缺少周围神经,神经纤维无髓鞘,临床表现主要为自幼全身痛觉减退或缺失,伴无汗症或智力稍差,但温觉、触觉、深感觉及位置觉皆完好。
本病无特殊治疗方法,主要加强监护和自我保护。
现将我院收治的七例报告如下。
1.一般情况
七例均为男孩,发病年龄为生后2-15天,发热为首发症状。
七例患儿的父母身体均为健康,且非近亲婚配。
其中一例有明显的家族史,此患儿为第三胎第三产,第一和第二胎生后发热、无汗,分别于生后一个月和四个月死亡。
七例中随访一例于生后一周岁死亡。
2.临床表现
2.1发热:七例均反复发热。
于出生后2-15天开始发热,体温最高达40-41度,表现为弛张热。
体温明显受环境温度的影响,室温升高,体温升高,物理降温有效,其中较大的患儿表现为天热时喜平卧于水泥地板上降温。
2.2无汗:七例均全身无汗,皮肤干燥皲裂、脱屑。
全身皮
肤做碘发汗定性实验和热发汗定性实验均证明无汗。
2.3感觉障碍:七例中六例全身性痛觉消失,一例痛觉反应迟钝。
七例中有五例是在发热输液时患儿不哭发现其无痛觉,两例是在幼儿期有自残行为,表现为反复咬伤手指、嘴唇及舌头,被发现其无痛觉,其中有一患儿现已7岁,表现为经常从高处往下跳,脚踝骨折3次,双脚踝已肿胀畸形,牙齿碰掉3颗,下颌骨脱位,七例中五例温度觉消失,两例减低,温度觉中对冷的感觉障碍较对热的感觉障碍更明显。
2.4智力障碍:七例中六例均有明显的精神运动发育迟缓。
其中一例7个月不会翻身,一例1岁不会坐、不会说话,一例新生儿双目不能视物,眼底检查显示视神经萎缩,三例于幼儿期自控能力差,20项行为精神测定,评分均低于正常同龄儿。
2.5创伤及感染:两例舌尖咬伤后舌下溃疡、增生。
两例头皮、甲沟致伤后继发感染,指甲脱落。
一例脚踝陈旧性骨折肿胀畸形,下颌骨脱位,牙齿脱落(外伤),一例食指端残缺,一例烫伤感染。
2.6其他:(1)七例毛发正常,内脏未见畸形;(2)三例出现惊厥,脑电图及CT未见异常。
3.实验室检查:
七例患儿的血、尿、便常规正常,血、尿、便培养、C-反应蛋白均阴性。
血IgA、IgG、IgM正常。
两例到北京儿童医院做皮肤活检:真皮可见皮脂腺、汗腺及部分小血管,未见神经组织及
环房小体。
4.随访:
一例生后一周死亡,五例失访,一例已7岁。
5.讨论:
先天性无痛无汗综合症为常染色体隐性遗传性疾病。
兄弟姐妹中可数名发病,男女比例为8:1,病因尚不确切,可能为胎儿期神经系统发育异常所致。
皮肤和末梢神经活检:皮肤组织结构正常,汗腺形态无异常。
周围神经髓鞘及细小髓鞘纤维消失。
先天性无痛无汗综合症的主要表现为:(1)无痛觉:为全身性,婴幼儿萌牙后有自残行为。
温度觉减低或消失,易发生烫伤,触觉正常。
(2)无汗:全身无汗,皮肤干燥皲裂,全身皮肤做碘发汗定性实验证明无汗。
(3)发热:大部分患儿以发热为主诉而就诊,热型为弛张热或不规则热,体温受环境温度的影响。
(4)智力迟缓:精神运动发育落后,部分患儿视神经萎缩,双目不能视物。
(5)多发性骨折。
痛觉、温度觉试验及碘发汗定性试验是本病的主要诊断依据。
本病需与感觉障碍性疾病相鉴别,如先天性感觉性神经病、遗传性感觉根性神经病及先天性无痛症,后三者临床表现痛觉障碍、出汗均正常,而且不伴智力迟缓。
本病无特殊疗法,高热时物理降温,加强保育,应以防止自残及外伤为重点,20%左右患儿3岁前因高热死亡。