注射用雷替曲塞说明书
肿瘤科常用化疗药物输注注意事项
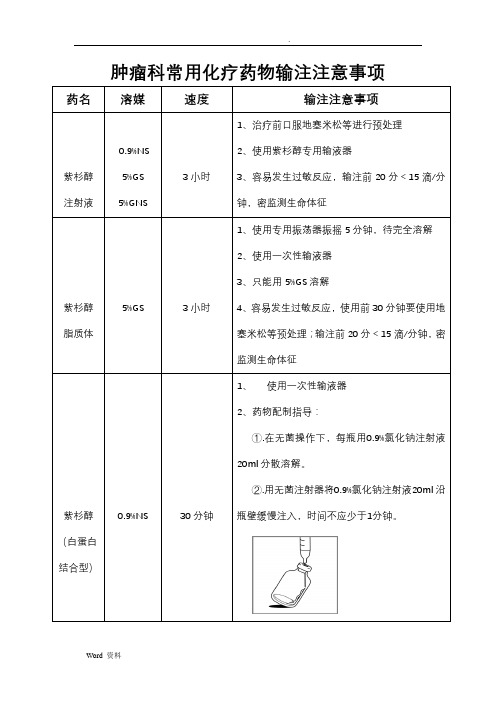
肿瘤科常用化疗药物输注注意事项药名溶媒速度输注注意事项
紫杉醇注射液0.9%NS
5%GS
5%GNS
3小时
1、治疗前口服地塞米松等进行预处理
2、使用紫杉醇专用输液器
3、容易发生过敏反应,输注前20分<15滴/分
钟,密监测生命体征
紫杉醇脂质体5%GS 3小时
1、使用专用振荡器振摇5分钟,待完全溶解
2、使用一次性输液器
3、只能用5%GS溶解
4、容易发生过敏反应,使用前30分钟要使用地
塞米松等预处理;输注前20分<15滴/分钟,密
监测生命体征
紫杉醇(白蛋白结合型)0.9%NS 30分钟
1、使用一次性输液器
2、药物配制指导:
①.在无菌操作下,每瓶用0.9%氯化钠注射液
20ml分散溶解。
②.用无菌注射器将0.9%氯化钠注射液20ml沿
瓶壁缓慢注入,时间不应少于1分钟。
药物说明书drins-雷替曲塞-正大

目前尚无确切有效解毒剂。一旦超量使用可考虑使用亚叶酸治疗。根据经验,每6小时静脉注射25mg/m2亚叶酸,越晚使用亚叶酸效果越差。超量用药预期不良反应容易扩大。应仔细监测有关胃肠道和血液学的毒性征兆并有针对性采取措施治疗。
【药理作用】
雷替曲塞为抗代谢类叶酸类似物,特异性地抑制胸苷酸合酶(TS)。与5-FU或氨甲喋呤相比,霍替曲塞是直接的和特异性的TS抑制剂。TS是胸腺嘧啶脱氧核苷三磷酸盐(TTP)合成过程的关键酶,而TTP又是DNA合成的必须核苷酸。抑制TS可导致DNA断裂和细胞凋亡。雷替曲塞经还原叶酸载体摄入细胞被叶酰聚谷氨酸合成酶转化成聚谷氨酸盐形式贮存细胞中,发挥更强TS抑制作用。雷替曲塞聚谷氨酸盐通过增强TS抑制能力、延长抑制时间而提高其抗肿瘤活性。但其在正常组织中的贮留可能会使毒性增加。
【适应症】
在患者无法接受联合化疗时,本品可单药用于治疗不适合5-Fu/亚叶酸钙的晚期结直肠癌患者。
【规格】
2mg。
【用法用量】
成人:推荐剂量为3mg/m2,用50-250ml 0.9%氯化钠注射液或5%葡萄糖注射液溶解稀释后静脉输注,给药时间15分钟,如果未出现毒性,可考虑按上述治疗每3周重复给药1次。本药应避免与其它药物混合输注。增加剂量会致使危及生命或致死性毒性反应的发生率升高,所以不推荐剂量大于3mg/m2。每次用药治疗前需检查全血细胞计数(包括白细胞分类计数和血小板计数)和肝、肾功能。治疗前应该白细胞计数>4.0×108/L、中性粒细胞计数>2.0×109/L和血小板计数>1.0×1011/L。出现毒性反应时,下一周期用药需延迟至不良反应消退;尤其是胃肠道毒性(腹泻或粘膜炎)及血液学毒性(中性粒细胞减少或血小板减少)需完全恢复才可进行后续治疗,出现胃肠道毒性者应至少每周检查一次全血细胞计数以监测血液学毒性。根据前一治疗周期观察到的最严重的胃肠道及血液学毒性等级,如果此类毒性已完全缓解,推荐按前一周期最严重的胃肠道、血液学毒性(以下毒性均按WHO标准分级)进行剂量调整:剂量减少25%:血液学毒性(中性粒细胞减少或者血小板减少)3级或胃肠道毒性2级(腹泻或粘膜炎);剂量减少50%:血液学毒性(中性粒细胞减少或者血小板减少)4级或胃肠道毒性3级(腹泻或粘膜炎)。一旦减量,后续治疗的剂量也须按减量后给药。出现4级胃肠道毒性(腹泻或粘膜炎),或3级胃肠道毒性伴4级血液学毒性时必须中止治疗;同时迅速给予标准支持治疗,包括静脉补水和造血功能支持。临床前研究提示可以使用亚叶酸治疗,按照临床经验需每6小时静脉注射25mg/m2亚叶酸直至症状缓解。对于此类患者建议停用本药。肾功能不全:血清肌酐异常者,每次用药治疗前应监测肌酐清除率。对于因年龄或体重下降等因素使血清肌酐可能与肌酐清除率相关性不好而血清肌酐正常的患者,应按相同程序操作。如果肌酐清除率<65ml/min,作如下剂量调整:肝功能不全:对于轻到中度的肝功能损害患者不需调整剂量,但是因为部分药物经粪便排出(见药代动力学),且这些患者预后较差,故应慎用本药。本药未在重度肝功能损害患者中进行研究,因此对于显性黄疸或肝功能失代偿的患者不推荐使用。
注射剂药物输注时间(滴速)要求汇总(2023)

注射剂药物输注时间(滴速)要求汇总(2023)注射剂药物说明书提到有滴速要求的,临床实际操作中建议根据输液器说明中的点滴系数进行换算。
静脉输液中ImI有多少滴?输液皮条厂家不同滴速不同,约在15-20滴/毫升左右,精密输液器为15滴/毫升,普通输液器20滴/毫升,压力套装为30滴/毫升。
加入一些药物,如头袍类、红霉素等药物后,药液变浓稠,液滴张力随之变大,液珠变小,这样的液体滴注时可为22滴/毫升。
一般输液器包装上标明:规格0.7mm的针头,滴蒸储水20滴/毫升。
药品说明书对输注时间(滴速)有要求的药物:一、抗菌药物1左氧氟沙星(扬子江药业)每250m1不得少于2小时,500m1不得少于3小时2莫西沙星(拜耳先灵医药)静脉给药0.4g的滴注时间应为90分钟3万古霉素(V IANEXSA)以至少IOOm1溶媒进行稀释溶解后,静脉滴注时间应60分钟以上4亚胺培南西司他丁(杭州默沙东)剂量W500mg,给药时,静脉滴注时间应不少于20〜30分钟;剂量>500mg,给药时,应不少于40〜60分钟。
如病人滴注时出现恶心状态,可减慢滴注速度5阿奇霉素(辉瑞制药)配置成浓度为2mg∕m1,25On11的溶液滴注时间应不少于1小时;配置成浓度为1mg∕m1,50Om1的溶液时滴注时间应不少于3小时6两性霉素B(华北制药)浓度不超过IOmg∕100m1,避光缓慢静滴,每次滴注时间6小时以上7伏立康哇(PharmaciaUpjohn)浓度不应高于5mg∕m1,滴注速度最快不超过每小时3mg∕kg,每瓶滴注时间须1至2小时8注射用更昔洛韦(科益药业)不少于1小时9注射用醋酸卡泊芬净(MerckSharpDohme)静脉缓慢输注约1小时以上10注射用替加环素(惠氏制药)成人:静脉滴注时间约30〜60分钟;儿童:静脉滴注时间至少60分钟二、抗肿瘤药物1吉西他滨(江苏豪森)30分钟2注射用奥沙利钻(赛诺菲)2〜6小时3白消安注射液(浙江大冢制药)2小时4盐酸多柔比星脂质体注射液(石药集团)起始给药速率应不大于1mg∕min o如果无滴注反应,以后的滴注可在60分钟完成。
注射用雷替曲塞产品介绍

。
雷替曲塞药理
做为新一代水溶性胸苷酸合酶(TS)抑制剂, 该药通过细胞膜外还原型叶酸盐载体系统 将本品主动摄入细胞内,而后迅速代谢为 多谷氨酸类化合物抑制胸苷酸合酶活性, 并能在细胞内潴留,长时间发挥作用。它 对结肠直肠癌细胞系抑制作用强于5-氟尿嘧 啶.
雷替曲塞药理
雷替曲塞IC50(50%抑制浓度)长期给药为 1.3~3.9nmol/L,短期给药为80nmol/L,而 5-氟尿嘧啶与甲酰四氢叶酸合用长期给药 IC50为330~5800nmol/L,短期给药为 150000nmol/L。
注射用雷替曲塞产品介绍
南京正大天晴 广州办事处
通用名:注射用雷替曲塞 英文名:Raltitrexed Injection(Tumudex)
O
HN H N N CH3 H3 C N
O OH
S
O
O
OH
被列入《国家级化学医药新产品开发指南》
〖功用主治〗:
用于晚期结直肠肠癌患者。
雷替曲塞药理
高选择性的胸腺嘧啶合成酶(TS)抑制剂 其代谢物为多聚谷氨酸类化合物,比母药发挥更 强的酶抑制作用
结论
综上所述:雷替曲赛作为近几十年来第一 个新的一线细胞毒治疗药,可成为5-FU为 主的化疗方案的较好替代药。他的问世可 为医生和患者提供更多选择。为患者带来 方便,治疗效益显著。
注意事项
注意事项:本能只做单独给药,避免与其 它药物混合使用。本品用0.9%生理盐水或 5%葡萄糖水溶液稀释后应避光保存,在24 小时内使用。轻度和中毒肝损伤患者使用 时无需调整本品剂量,但本品部分经粪便 排泄,严重肝损伤患者使用时应注意。孕 妇及哺辱期妇女禁用本品。
雷替曲塞

雷替曲塞雷替曲塞,Raltitrexed英文化学名:S)-2-[(1-{5-[Methyl-(2-methyl-4-oxo-3,4-dihydro-quinazolin-6-ylm ethyl)-amino]-thiophen-2-yl}-methanoyl)-amino]-pentanedioic acid 中文化学名:N-[5-[N-甲基-N-(2-甲基-4-氧代-3,4-二氢喹唑啉-6-基甲基)氨基]-2-噻吩甲酰基]-L-谷氨酸分子式: C21H22N4O6S分子量:458.49质量标准:企标成分:雷替曲塞、甘露醇、氢氧化钠和磷酸氢二钠。
性状:白色或类白色疏松块状物或粉末规格2mg/支【药物名称】雷替曲塞粉针 Raltitrexed Injection【分子式成分】N-[5-[N-甲基-N-(2-甲基-4-氧代-3,4-二氢喹唑啉-6-基甲基)氨基]-2-噻吩甲酰基]-L-谷氨酸【制剂规格】本品为白色冻干粉末,每瓶含雷替曲塞2 mg。
【药理毒理】药理学研究表明,雷替曲塞为新一代水溶性胸苷酸合酶抑制剂,该药通过细胞膜外还原型叶酸盐载体系统将本品主动摄入细胞内,而后迅速代谢为多谷氨酸类化合物抑制胸苷酸合酶的活性,并能在细胞内潴留,长时间发挥作用。
它对结肠直肠癌细胞系的抑制作用强于5-氟尿嘧啶,雷替曲塞的IC50长期给药为1.3~3.9 nmol/L,短期给药为80 nmol/L,而5-氟尿嘧啶与甲酰四氢叶酸合用长期给药IC50为330~5800 nmol/L,短期给药为150000 nmol/L。
体外研究观察到雷替曲塞与5-氟尿嘧啶联合用药有协同作用,这种作用依赖于给药方案和剂量。
对176例晚期结肠直肠癌患者进行的II期临床试验,给予雷替曲塞3 mg/㎡,每3周1次,有25.6%产生综合疗效,从治疗到病情进展平均时间为4.2周,存活期平均为11.2月。
1300多例晚期结肠直肠癌患者的3项III期临床研究结果表明,雷替曲塞治疗组(每3周1次,每次3 mg/㎡)与5-氟尿嘧啶加甲酰四氢叶酸治疗组(5-氟尿嘧啶425mg/㎡加甲酰四氢叶酸20 mg/㎡或5-氟尿嘧啶400 mg/㎡加甲酰四氢叶酸200 mg/㎡,每天1次,连续5天,每4~5周重复1次),所产生的客观有效率相似,分别为14.3%~19.3%和15.2%~18.1%,中位缓解时间分别为3.1~4.8和3.6~5.3个月,中位生存期分别为9.7~10.9和10.2~12.7个月。
雷替曲塞临床研究

雷替曲塞临床研究进展雷替曲塞(ZD1694,'Tomudex')是一种新型的,直接的和特异的TS抑制剂,由Zeneca 医药和肿瘤研究中心(UK)共同研发,1991年进行临床试验。
1992年开始进行II期临床试验,推荐雷替曲塞 3.0 mg/m2,每3周一次,15分钟静脉滴注。
一、雷替曲塞作用机制雷替曲塞为抗代谢类叶酸类似物,是胸苷酸合成酶(TS)特异性选择性抑制剂。
TS是胸腺嘧啶脱氧核苷三磷酸盐(TTP)合成过程的关键酶,而TTP又是DNA合成的必需核苷酸。
抑制TS可导致DNA断裂和细胞凋亡。
肿瘤细胞DNA从头合成途径的关键酶是TS,因此抑制TS的活性常常作为抗肿瘤治疗的一个靶点。
雷替曲塞经还原叶酸载体摄入细胞被叶酰聚谷氨酸合成酶转化成聚谷氨酸盐形式贮存细胞中,聚谷氨酸盐通过较强地抑制TS活性、延长抑制时间而提高其抗肿瘤活性。
5-FU也是一种TS抑制剂,它在体内代谢成很多活性物质,包括氟尿嘧啶脱氧核苷单磷酸盐,结合在酶的嘧啶结合区域,抑制TS的活性。
其他的5-FU代谢产物有很多的效应,比如非特异性地干扰RNA和DNA的合成。
5-FU是进展期结直肠癌标准治疗方案的基本药物之一,但是治疗肿瘤的有效率往往不尽人意。
此外,用药的选择也不明确,例如联合生物调节剂特别是亚叶酸钙类(LV)等。
5-FU/LV联合用药可应用在很多肿瘤领域,但是目前联合奥沙利铂或伊立替康是治疗进展期结直肠癌的标准治疗方案。
但是一些5-FU为基础的治疗方案由于毒性持续存在等问题,而迫切需要5-FU的替代物单药或联合化疗。
标准5-FU/LV方案是一线治疗进展期结直肠癌的基础方案,而雷替曲塞是替代药物。
4个大型的III期临床研究结果显示,雷替曲塞的中位总生存期在9.7-10.7个月,而5-FU/LV在10.0-12.7个月,没有统计学差异;总有效率两种方案相似。
四个临床试验中有两个雷替曲塞治疗的PFS明显短于5-FU/LV,但是总生存期没有差异。
雷替曲塞用于腹腔镜结直肠癌根治术中腹腔灌注化疗的安全性分析

雷替曲塞用于腹腔镜结直肠癌根治术中腹腔灌注化疗的安全性分析1. 引言1.1 背景介绍雷替曲塞是一种新型的抑制肿瘤血管生成的药物,在肿瘤治疗中展现出了较好的疗效。
在腹腔镜结直肠癌根治术中,结合雷替曲塞进行腹腔灌注化疗可以有效杀灭残留的肿瘤细胞,进而降低复发率,提高患者的生存率。
腹腔灌注化疗的安全性一直是一个备受关注的问题。
本研究旨在探讨雷替曲塞在腹腔镜结直肠癌根治术中的应用,分析腹腔灌注化疗的作用机制,评估雷替曲塞在腹腔灌注化疗中的安全性,为临床治疗提供更为科学的依据。
愿通过本研究的开展,为结直肠癌的治疗和预后带来新的突破。
1.2 研究目的参考文献的引用格式等。
本研究的目的是探讨雷替曲塞用于腹腔镜结直肠癌根治术中腹腔灌注化疗的安全性。
具体包括评估雷替曲塞在结直肠癌手术中腹腔灌注化疗的安全性和有效性,分析雷替曲塞对手术患者的不良反应及并发症,为临床医生提供更安全有效的治疗方案,降低手术并发症的发生率,提高手术成功率。
本研究旨在为进一步明确雷替曲塞在结直肠癌根治术中的作用机制提供实验依据,为临床实践提供科学依据,为将来的研究提供参考。
通过本研究的开展,希望为临床医生提供更加全面的治疗方案,提高患者的生存率和生活质量,对结直肠癌的治疗提供新的思路和方法。
1.3 研究意义本研究的意义在于深入探讨雷替曲塞在腹腔镜结直肠癌根治术中腹腔灌注化疗的安全性,为临床医生提供更多治疗选择和决策依据。
通过对雷替曲塞在腹腔镜手术中的应用、腹腔灌注化疗的作用机制以及安全性分析等方面的研究,可以更好地评估该治疗方法的疗效和安全性,为患者的治疗提供更加科学、有效的方案。
本研究也有助于推动相关领域的学术研究和临床实践,为进一步完善结直肠癌治疗方案和提高患者生存率贡献力量。
通过本研究的开展,可以为临床医生提供更多选择和决策依据,促进结直肠癌治疗的进步,为患者提供更好的治疗效果和生存质量。
2. 正文2.1 雷替曲塞在腹腔镜结直肠癌根治术中的应用雷替曲塞是一种新型的抗肿瘤药物,已在腹腔镜结直肠癌根治术中得到广泛应用。
雷替曲塞二线治疗晚期结直肠癌临床护理体会

当使 用 思 密达 及 漱 口液 等 对 症 药 物 , 密 切 观 察 腹 泻 的 次 数 及性质 , 有无发热及感染 , 必 要 时 使 用 抗 生 素 。 适 当 使 用 降 温食品 , 减低 口腔 温 度 , 可 减 少 黏 膜 炎 的发 作 或 症 状 。注 意
~
3 0 mi n , 与 氟尿 嘧 啶 持 续 静 点 相 比 , 用 药时 间短 , 刺 激 血
管轻 , 静脉炎发生率低 、 程度轻 , 患者的耐受性 好 , 可 减 少 药
评价 1 次, 直 到疾 病进展 或 毒性不 能耐 受 , 最多 4 ~ 6个 周 期 。按 实 体 瘤 的 疗 效 评 价 标 准 : 完全 缓解 ( C R) 、 部 分 缓 解
止 吐药 物 ( 包 括 静 脉 及 口服 药 物 ) 可 明 显 减 轻 相 关 症 状 。 对
1 . 1 临床资料 : 本组 3 O例 , 男性 1 8例 , 女性 1 2 例, 年龄 4 7
~
8 6 岁, 平均 7 3 . 6 岁, 结肠癌 1 1 例, 直肠癌 1 9例 。 初 治 患
者 9 例, 复治患者 2 1 例 。均 有 明 确 的 病 理 学 诊 断 及 影 像 学
表 现 和 可 观 察 及 测 量 病 灶 。3 O例 患 者 均 接 受 过 F o l { o x或
呕 吐剧 烈 、 难 以进 食 者 应 注 意 补 液 及 电解 质 , 注 意 生 命 体 征
的变 化 , 防 止 脱 水 情 况 出现 。② 腹 泻 及 口腔 黏 膜 炎 : 本 组 发 病率分别 为 2 3 和 4 7 , 绝大多数 均为轻度 , 可 以耐 受 , 适
肿瘤血液科化疗药物使用方案

小细胞肺癌,有效率达40%-85%。恶性淋巴瘤,前列腺癌,胃癌,绒毛膜上皮癌,卵巢癌,恶性葡萄胎。
规格:0.1g。实体瘤:60-100mg/㎡。连续使用3-5天。
骨髓抑制,食欲减退,恶心,口腔炎,脱发,滴速过快<30min可会有低血压、喉痉挛等过敏反应。
骨髓抑制,血小板、细胞减少者。心、肝肾功能严重障碍者,不宜静推。不能过快,滴注时间>30min。不做胸、腹腔和鞘注射。
氟尿嘧啶化疗失败者,转移性大肠癌。
规格:100mg。350mg/㎡。滴注<30>90分钟,避光应用。
迟发性腹泻,在用药后第五天出现。胃肠道反应,厌食,腹痛。少见发生肠梗阻,中性粒细胞减少,低血压,视力障碍,瞳孔缩小,早期有呼吸困难痉挛等。
肾功能不全者不宜用,慢性肠炎或肠梗阻禁用,对其药物过敏者禁用,胆红素>正常1.5倍,严重骨髓功能衰竭的。不能静推。
规格:2mg。3 mg/㎡。不推荐>mg/㎡。用无菌注射用水溶解。避光使用。
骨髓抑制,窦性心动过速,皮疹,发热,伴有流感症状,乏力,败血症,胀气,口干,失眠,味觉异常。
孕妇,肾功能损伤重度,造血功能底下,腹泻,粘膜炎。
西妥昔单抗(爱必妥)
与伊立替康联合用药的转移性直肠癌。
规格;100mg
初始剂量;40 mg/㎡。而后250 mg/㎡。初始滴注时间120min,而后60min,最大速度不超过5ml/min
不可静推或者滴速过快。对本品过敏者禁用。左心室功能不全,心律失常,心悸怔,支气管痉挛,过敏反应,血管性水肿。
利妥昔单抗(美罗华)
复发或耐药型的中央性淋巴瘤,非霍奇金淋巴瘤。
规格:100mg/10ml。500mg/50ml。必须稀释,不能静推。流泡型非霍奇金淋巴瘤,每次用药前应用扑热息痛,苯海拉明。375 mg/㎡。,每周一次,22天给4次。复发后:375mg/㎡。用4周,每周一次,避光使用。
雷替曲塞治疗晚期恶性肿瘤

雷替曲塞治疗晚期恶性肿瘤-安全有效(基础知识与进展)发表者:刘连科(访问人次:406)由于既往国外研究认为雷替曲塞较5-FU存在较高死亡患者,而且雷替曲塞的疗效并不优于5—FU,在国外未得到足够的重视.国内2010年由南京正大天晴上市(赛维健),研究基于一项设计严格的多中心、随机盲法、阳性药物平行对照,雷替曲塞的适应症为:在患者无法接受联合化疗时,本品可单药用于治疗不适合5—Fu/亚叶酸钙的晚期结直肠癌患者。
目前已在国内广泛应用,逐渐得到大家的认可。
事实上,雷替曲塞不但可单药化疗,而且更多的学者采用雷替曲塞联合其他药物。
越来越多的文献支持,对于中国肿瘤患者,雷替曲塞可能较西方人更安全.1. 雷替曲塞为氟尿嘧啶类药物发生心脏毒性者的最佳替代药物临床上,氟尿嘧啶类药物(5-FU、卡培他滨、替吉奥胶囊等)可引起严重的心脏毒性,既往只能放弃氟尿嘧啶类药物。
2012年欧洲肿瘤内科学会(ESMO)发布ARCTIC试验,对于接受氟尿嘧啶类药物出现心脏毒性的患者,可用雷替曲塞(一种胸甘酸合成酶(TS)的特异性抑制剂)来替代,并且是一个安全的替代药品.文献报道5—FU/卡培他滨引起的心脏毒性发生率为0。
55%~ 19%(平均值: 5。
0%,中位数: 3.85%),而与雷替曲塞相关的心脏毒性未见报道。
但最近有学者报道有心脏病史或者发生5—FU/卡培他滨引起心脏毒性的患者,再用雷替曲塞,可有4.5%的患者再发生心脏毒性。
2. 不良反应较多,但易处理国内大型III临床研究(临床肿瘤学杂志2012年):“雷替曲塞或氟尿嘧啶/亚叶酸钙联合奥沙利铂治疗局部晚期或复发转移性结直肠癌的随机对照多中心Ⅲ期临床试验",试验组1~2 级中性粒细胞减少( 48.2% vs.29.4%, P= 0.005) 和转氨酶升高( 49.1% vs。
35.3%, P = 0.041) 的发生率明显高于对照组。
对照组呕吐的发生率明显高于试验组( 61.8% vs. 40.2%, P = 0。
雷替曲塞临床研究者手册

化学药品注册分类3.1 资料编号30注射用雷替曲塞临床研究者手册研究机构名称(盖章):研究机构地址:研究机构电话:资料整理者:原始资料保存地点:联系人:联系电话:申请机构(盖章):XXXXXXXXXX制药有限公司一、注射用雷替曲塞(Raltitrexed)化学名:N-〔5-〔N-甲基-N-(2-甲基-4-氧代-3,4-二氢喹唑啉-6-基甲基)氨基〕-2-噻吩基〕-L-谷氨酸二、命名依据雷替曲塞的英文名称为Raltitrexed,中文译文为雷替曲塞。
我们研制的为其注射用无菌冷冻干燥产品,依据我国药典委员会和《新药审批办法》的命名原则,将本品的正式名称定为注射用雷替曲塞。
三、分类依据根据国家新药审批办法规定,本品应为化学药品3.1类。
四、选题目的与依据结肠直肠癌是世界上第四大常见肿瘤,并且新增病例数从1975年开始快速增长。
在1990年估计的新增病例数为783000,而在1975年,世界范围内只有500000例新病例。
研究者指出,“它对那性和女性的影响几乎是一致的”。
结肠直肠癌发病在世界各个地方是不同的。
在西方国家(北美、欧洲的大部分地区、澳大利亚和新西兰),结肠直肠癌占全部男性癌症的12.6%,女性的14.1%,而在别的地方,分别表现为7.7%和7.9%。
生存率有很大差异,这主要依赖于疾病的分期。
世界上估计每年有394000人因此丧生,在欧盟国家,它占男性癌症死因的第二位。
国内大肠癌的发病率仅次于胃癌和食管癌,而且随着人民生活水平的提高,本病的发病率有逐年上升的趋势,所以值得重视。
结肠直肠癌在人类癌症常见死因中居第三位。
结肠直肠癌的治疗是以手术为主的综合治疗,根治术后的化疗即辅助化疗是结肠直肠癌综合治疗的一个重要组成部分。
辅助化疗的机理在于化疗控制减灭根治术后的残留病灶。
术后机体荷瘤减轻,远处微小转移灶的增值导致其对化疗的敏感性增高,同时化疗也有利于减少抗药克隆产生。
5-FU应用于结肠直肠癌临床治疗已有40余年,是治疗结肠直肠癌的一线用药。
注射用雷替曲塞说明书

注射用雷替曲塞说明书药品名称通用名称:注射用雷替曲塞商品名称:_____英文名称:Raltitrexed for Injection成份本品主要成份为雷替曲塞。
性状本品为白色或类白色疏松块状物或粉末。
适应症在患者无法接受联合化疗时,本品可单药用于治疗晚期结直肠癌。
规格_____mg/瓶用法用量推荐成人每次用量为 3mg/m²,用 50-250ml 09%氯化钠注射液或 5%葡萄糖注射液溶解稀释后静脉输注,输注时间约 15 分钟。
每 3 周重复给药一次。
具体剂量应根据患者的身高、体重、体表面积等因素进行计算,并在医生的指导下使用。
不良反应在临床试验中,常见的不良反应包括恶心、呕吐、腹泻、食欲不振、乏力、骨髓抑制(如白细胞减少、血小板减少、贫血)等。
较严重的不良反应可能包括肝功能异常、肾功能损害等。
如果出现严重不良反应,应立即停药并采取相应的治疗措施。
禁忌对雷替曲塞或其任何辅料过敏者禁用。
严重肾功能损害(肌酐清除率<30ml/min)者禁用。
孕妇及哺乳期妇女禁用。
注意事项1、用药前应进行血常规、肝肾功能等检查,治疗期间应定期监测。
2、本品可能导致骨髓抑制,治疗后应密切观察血象变化。
如果出现严重的骨髓抑制,如白细胞减少、血小板减少等,应暂停用药并给予相应的治疗。
3、对于有肝肾功能损害的患者,应谨慎使用,并根据肝肾功能调整剂量。
4、本品应在有经验的医生指导下使用,用药期间如出现任何不适,应及时告知医生。
孕妇及哺乳期妇女用药孕妇禁用。
雷替曲塞可能对胎儿造成损害。
哺乳期妇女禁用。
尚不清楚雷替曲塞是否会经乳汁分泌,为避免对婴儿造成潜在危害,哺乳期妇女应停止哺乳。
儿童用药儿童使用本品的安全性和有效性尚未确立,不建议儿童使用。
老年用药老年患者使用本品时,无需调整剂量,但应密切观察不良反应。
药物相互作用1、与其他骨髓抑制药物合用时,可能会增加骨髓抑制的风险。
2、与具有肝毒性的药物合用时,应注意监测肝功能。
HPLC法测定注射用雷替曲塞的含量

HPLC法测定注射用雷替曲塞的含量作者:乙永林来源:《中国保健营养·下旬刊》2013年第10期【摘要】目的建立反相高效液相色谱法测定雷替曲塞含量的方法。
方法采用KromasilC18柱,流动相为甲醇:0.15molL乙酸水溶液(42:58,流速为0.9mLmin,检测波长为348nm。
雷替曲塞的线性范围为0.0125-0.0638mgmL(r=0.9993。
雷替曲塞平均回收率为99.12%(RS=0.56%。
结论该方法简便、准确、专属,可用于注射用雷替曲塞的含量测定。
【关键词】雷替曲塞;HPLC;含量测定雷替曲塞是一种用于治疗晚期结直肠癌和恶性胸膜间皮瘤的抗肿瘤药物。
本品为高选择性抗代谢药物,是新一代水溶性胸苷酸合酶抑制剂,在体内被主动摄入细胞内,而后代谢为一系列多聚谷氨酸类化合物,这些代谢物抑制胸苷酸合酶的活性更强,从而抑制细胞NA的合成,且能潴留在细胞内长时间发挥细胞毒作用。
雷替曲塞具有疗效确切,副反应发生率低,方法简便,患者易于接受等优点。
1仪器与试剂日本岛津ShimadzuLC-10A高效液相色谱仪,SP-10Avp UV-VIS etector,LC-10Avp两元泵,7725i手动进样阀;UV2450紫外分光光度计(日本岛津制作所;自动双重纯水蒸馏器(上海亚荣生化仪器厂,型号SZ一93。
甲醇(色谱纯,美国Fisher Scientific公司,乙醇(分析纯,济南元通化工有限公司,水为双重蒸馏水。
雷替曲塞对照品(河南天方药业股份有限公司药物研究所,批号为000752,含量100.0%和注射用雷替曲塞(河南天方药业股份有限公司药物研究所,批号为010804。
2方法与结果2.1色谱条件色谱柱:KromasilC18(250mm×4.6mm,5μm,流动相:A(甲醇:B(0.15molL乙酸水溶液=42:58,检测波长:226nm,柱温:40℃,进样量:20μL,流速:0.9mLmin。
注射用雷替曲塞说明书

注射用雷替曲塞说明书说明书来源:南京正大天晴制药有限公司【药品名称】通用名称:注射用雷替曲塞英文名称:RaltitrexedforInjection 商品名称:赛维健【成份】本品活性成份为雷替曲塞辅料:甘露醇、磷酸氢二钠及氢氧化钠。
【性状】本品为白色或类白色疏松块状物或粉末。
【适应症】在患者无法接受联合化疗时,本品可单药用于治疗不适合5-Fu/亚叶酸钙的晚期结直肠癌患者。
【规格】2mg。
【用法用量】成人:推荐剂量为3mg/m2,用50-250ml 0.9%氯化钠注射液或5%葡萄糖注射液溶解稀释后静脉输注,给药时间15分钟,如果未出现毒性,可考虑按上述治疗每3周重复给药1次。
本药应防止与其它药物混合输注。
增加剂量会致使危及生命或致死性毒性反应的发生率升高,所以不推荐剂量大于3mg/m2。
每次用药治疗前需检查全血细胞计数(包含白细胞分类计数和血小板计数)和肝、肾功能。
治疗前应该白细胞计数>4.0×108/L、中性粒细胞计数>2.0×109/L和血小板计数>1.0×1011/L。
出现毒性反应时,下一周期用药需延迟至不良反应消退;尤其是胃肠道毒性(腹泻或粘膜炎)及血液学毒性(中性粒细胞减少或血小板减少)需完全恢复才可进行后续治疗,出现胃肠道毒性者应至少每周检查一次全血细胞计数以监测血液学毒性。
根据前一治疗周期观察到的最严重的胃肠道及血液学毒性等级,如果此类毒性已完全缓解,推荐按前一周期最严重的胃肠道、血液学毒性(以下毒性均按WHO尺度分级)进行剂量调整:剂量减少25%:血液学毒性(中性粒细胞减少或者血小板减少)3级或胃肠道毒性2级(腹泻或粘膜炎);剂量减少50%:血液学毒性(中性粒细胞减少或者血小板减少)4级或胃肠道毒性3级(腹泻或粘膜炎)。
一旦减量,后续治疗的剂量也须按减量后给药。
出现4级胃肠道毒性(腹泻或粘膜炎),或3级胃肠道毒性伴4级血液学毒性时必须中止治疗;同时迅速给予尺度支持治疗,包含静脉补水和造血功能支持。
注射用雷替曲塞说明书

注射用雷替曲塞说明书说明书来源:南京正大天晴制药有限公司【药品名称】通用名称:注射用雷替曲塞英文名称:RaltitrexedforInjection 商品名称:赛维健【成份】本品活性成份为雷替曲塞辅料:甘露醇、磷酸氢二钠及氢氧化钠。
【性状】本品为白色或类白色疏松块状物或粉末。
【适应症】在患者无法接受联合化疗时,本品可单药用于治疗不适合5-Fu/亚叶酸钙的晚期结直肠癌患者。
【规格】2mg。
【用法用量】成人:推荐剂量为3mg/m2,用50-250ml 0.9%氯化钠注射液或5%葡萄糖注射液溶解稀释后静脉输注,给药时间15分钟,如果未出现毒性,可考虑按上述治疗每3周重复给药1次。
本药应避免与其它药物混合输注。
增加剂量会致使危及生命或致死性毒性反应的发生率升高,所以不推荐剂量大于3mg/m2。
每次用药治疗前需检查全血细胞计数(包括白细胞分类计数和血小板计数)和肝、肾功能。
治疗前应该白细胞计数>4.0×108/L、中性粒细胞计数>2.0×109/L和血小板计数>1.0×1011/L。
出现毒性反应时,下一周期用药需延迟至不良反应消退;尤其是胃肠道毒性(腹泻或粘膜炎)及血液学毒性(中性粒细胞减少或血小板减少)需完全恢复才可进行后续治疗,出现胃肠道毒性者应至少每周检查一次全血细胞计数以监测血液学毒性。
根据前一治疗周期观察到的最严重的胃肠道及血液学毒性等级,如果此类毒性已完全缓解,推荐按前一周期最严重的胃肠道、血液学毒性(以下毒性均按WHO标准分级)进行剂量调整:剂量减少25%:血液学毒性(中性粒细胞减少或者血小板减少)3级或胃肠道毒性2级(腹泻或粘膜炎);剂量减少50%:血液学毒性(中性粒细胞减少或者血小板减少)4级或胃肠道毒性3级(腹泻或粘膜炎)。
一旦减量,后续治疗的剂量也须按减量后给药。
出现4级胃肠道毒性(腹泻或粘膜炎),或3级胃肠道毒性伴4级血液学毒性时必须中止治疗;同时迅速给予标准支持治疗,包括静脉补水和造血功能支持。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
TomudexSummary of Product Characteristics Updated 04-Dec-2017 | Hospira UK Ltd1. Name of the medicinal product'Tomudex'2. Qualitative and quantitative composition'Tomudex' contains 2 mg raltitrexed in each vial.3. Pharmaceutical formPowder for solution for infusion.4. Clinical particulars4.1 Therapeutic indicationsThe palliative treatment of advanced colorectal cancer where 5-Fluorouracil and folinic acid based regimens are either not tolerated or inappropriate.4.2 Posology and method of administrationFor instructions on reconstitution and dilution of the product before administration, see section 6.6 Instructions for use/handling.Adults: - The dose of 'Tomudex' is calculated on the basis of the body surface area. The recommended dose is 3 mg/m2 given intravenously, as a single short, intravenous infusion in 50 to 250 ml of either 0.9% sodium chloride solution or 5% dextrose (glucose) solution. It is recommended that the infusion is given over a 15 minute period. Other drugs should not be mixed with 'Tomudex' in the same infusion container. In the absence of toxicity, treatment may be repeated every 3 weeks.Dose escalation above 3 mg/m2 is not recommended, since higher doses have been associated with an increased incidence of life-threatening or fatal toxicity.Prior to the initiation of treatment and before each subsequent treatment a full blood count (including a differential count and platelets), liver transaminases, serum bilirubin and serum creatinine measurements should be performed.The total white cell count should be greater than 4,000/mm3, the neutrophil count greater than 2,000/mm3 and the platelet count greater than 100,000/mm3 prior to treatment. In the event of toxicity the next scheduled dose should be withheld until signs of toxic effects regress. In particular, signs of gastrointestinal toxicity (diarrhoea or mucositis) and haematological toxicity (neutropenia or thrombocytopenia) should have completely resolved before subsequent treatment is allowed. Patients who develop signs of gastrointestinal toxicity should have their full blood counts monitored at least weekly for signs of haematological toxicity.Based on the worst grade of gastrointestinal and haematological toxicity observed on the previous treatment and provided that such toxicity has completely resolved, the following dose reductions are recommended for subsequent treatment:● 25% dose reduction: in patients with WHO grade 3 haematological toxicity (neutropenia or thrombocytopenia) or WHOgrade 2 gastrointestinal toxicity (diarrhoea or mucositis).● 50% dose reduction: in patients with WHO grade 4 haematological toxicity (neutropenia or thrombocytopenia) or WHOgrade 3 gastrointestinal toxicity (diarrhoea or mucositis).Once a dose reduction has been made, all subsequent doses should be given at the reduced dose.Treatment should be discontinued in the event of any WHO grade 4 gastrointestinal toxicity (diarrhoea or mucositis) or in the event of a WHO grade 3 gastrointestinal toxicity associated with WHO grade 4 haematological toxicity. Patients with such toxicity should be managed promptly with standard supportive care measures including i.v. hydration and bone marrow support. In addition, preclinical data suggest that consideration should be given to the administration ofleucovorin (folinic acid). From clinical experience with other antifolates, leucovorin may be given at a dose of 25 mg/m2i.v. every 6 hours until the resolution of symptoms. Further use of 'Tomudex' in such patients is not recommended.It is essential that the dose reduction scheme should be adhered to since the potential for life threatening and fatal toxicity increases if the dose is not reduced or treatment not stopped as appropriate.Elderly: - Dosage and administration as for adults. However, 'Tomudex' should be used with caution in elderly patients (see section 4.4 Special warnings and precautions for use).Children: - 'Tomudex' is not recommended for use in children as safety and efficacy have not been established in this group of patients.Renal impairment:- For patients with abnormal serum creatinine, before the first or any subsequent treatment, a creatinine clearance should be performed or calculated.For patients with a normal serum creatinine when the serum creatinine may not correlate well with the creatinine clearance due to factors such as age or weight loss, the same procedure should be followed. If creatinine clearance is≤65 ml/min, the following dose modifications are recommended:Dose modification in the presence of renal impairmentCreatinine Clearance Dose as % of 3.0 mg/m2Dosing Interval> 65 ml/min Full dose3-weekly55 to 65 ml/min75%4-weekly25 to 54 ml/min50%4-weekly< 25 ml/min No therapy Not applicableSee Contraindications for use in patients with severe renal impairmentHepatic Impairment:- No dosage adjustment is recommended for patients with mild to moderate hepatic impairment. However, given that a proportion of the drug is excreted via the faecal route, (see section 5.2 Pharmacokinetic Properties) and that these patients usually form a poor prognosis group, patients with mild to moderate hepatic impairment need to be treated with caution (see section 4.4 Special warnings and special precautions for use).'Tomudex' has not been studied in patients with severe hepatic impairment, clinical jaundice or decompensated liver disease and its use in such patients is not recommended.4.3 Contraindications'Tomudex' should not be used in pregnant women, in women who may become pregnant during treatment or women who are breast feeding. Pregnancy should be excluded before treatment with 'Tomudex' is commenced. (see section 4.6 Pregnancy and lactation).'Tomudex' is contraindicated in patients with severe renal impairment (creatinine clearance < 25ml/min).Administration of leucovorin (folinic acid), folic acid or vitamin preparations containing these agents with 'Tomudex' is contraindicated (see section 4.5 Interaction with other medicinal products and other forms of interaction).4.4 Special warnings and precautions for use'Tomudex' must only given by or under the supervision of a physician who is experienced in cancer chemotherapy, and in the management of chemotherapy-related toxicity. Patients undergoing therapy should be subject to appropriate supervision so that signs of possible toxic effects or adverse reactions (particularly diarrhoea) may be detected and treated promptly (see section 4.2 Posology and method of administration).In common with other cytotoxic agents of this type, caution is necessary in patients with depressed bone marrow function, poor general condition, or prior radiotherapy.Patients whose disease progressed on previous treatment for advanced disease with 5-Fluorouracil based regimens may also be resistant to the effects of 'Tomudex'.Elderly patients are more vulnerable to the toxic effects of 'Tomudex'. Since renal function tends to decline with age and the plasma clearance of raltitrexed is reduced with renal function impairment, there is a potential for accumulation of raltitrexed in elderly patients. Extreme care should be taken to ensure adequate monitoring of adverse reactions especially signs of gastrointestinal toxicity (diarrhoea or mucositis) and myelosuppression (neutropenia, thrombocytopenia, infection) and dose should be reduced and /or delayed as appropriate. A proportion of the 'Tomudex' is excreted via the faecal route, (see section 5.2 Pharmacokinetic properties) therefore, patients with mild to moderate hepatic impairment should be treated with caution.Treatment with 'Tomudex' in patients with severe hepatic impairment is not recommended.It is recommended that pregnancy should be avoided during treatment and for at least 6 months after cessation of treatment if either partner is receiving 'Tomudex' (see also section 4.6 Pregnancy and lactation).There is no clinical experience with extravasation. However, perivascular tolerance studies in animals did not reveal any significant irritant reaction.'Tomudex' is a cytotoxic agent and should be handled according to normal procedures adopted for such agents (see section 6.6 Instructions for use/handling).4.5 Interaction with other medicinal products and other forms of interactionNo specific clinical drug - drug interaction studies have been conducted in man.Leucovorin (folinic acid), folic acid or vitamin preparations containing these agents must not be given immediately prior to or during administration of 'Tomudex', since they may interfere with its action.Clinical trials evaluating the use of Tomudex in combination with other antitumour therapies are currently ongoing.'Tomudex' is 93% protein bound and while it has the potential to interact with similarly highly protein bound drugs, no displacement interaction with warfarin has been observed in vitro. Data suggest that active tubular secretion may contribute to the renal excretion of raltitrexed, indicating a potential interaction with other actively secreted drugs such as non-steroidal antiinflammatory drugs (NSAIDS). However, a review of the clinical trial safety database did not reveal evidence of clinically significant interaction in patients treated with 'Tomudex' who also received concomitant NSAIDS, warfarin and other commonly prescribed drugs.4.6 Pregnancy and lactationPregnancyPregnancy should be avoided if either partner is receiving 'Tomudex'. It is also recommended that conception should be avoided for at least 6 months after cessation of treatment.'Tomudex' should not be used during pregnancy or in women who may become pregnant during treatment (see section 5.3 Preclinical safety data). Pregnancy should be excluded before treatment with 'Tomudex' is started.Breastfeeding'Tomudex' should not be given to women who are breast feeding.FertilityFertility studies in the rat indicate that 'Tomudex' can cause impairment of male fertility. Fertility returned to normal three months after dosing ceased. 'Tomudex' caused embryolethality and foetal abnormalities in pregnant rats.4.7 Effects on ability to drive and use machines'Tomudex' may cause malaise or asthenia following infusion and the ability to drive/use machinery could be impaired whilst such symptoms continue.4.8 Undesirable effectsAs with other cytotoxic drugs, 'Tomudex' may be associated with certain adverse drug reactions. These mainly include reversible effects on the haemopoietic system, liver enzymes and gastrointestinal tract. Table 1 presents the possible adverse drug reactions occurring with 'Tomudex' treatment.In this section undesirable effects are defined as follows: Very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to ≤1/100); rare (≥1/10,000 to ≤1/1,000); very rare (≤1/10,000), not known (cannot be estimated from the available data).Table 1: Adverse drug reactions in patients treated with Tomudex for advanced colorectal carcinoma divided by System Organ Class and frequencySystem Organ Class Frequency Adverse drug reactionInfections & infestations Common CellulitisSepsisFlu-like syndrome Blood and lymphatic disorders Very Common Leucopenia (neutropenia in particular) a, bAnaemia aCommon Thrombocytopenia a, b Metabolism and Nutrition Disorders Very Common AnorexiaCommon DehydrationNervous system disorders Common HeadacheHypertonia (usually muscular cramps)Taste perversion Eye disorders Common ConjunctivitisGastrointestinal disorders Very Common Nausea cDiarrhoea d,eVomiting c,eConstipationAbdominal PainCommon StomatitisDyspepsiaMouth ulcerationFrequency unknown Gastrointestinal Bleeding f,gHepato-biliary disorder Common HyperbilirubinemiaSkin & subcutaneous tissue disorders Very Common RashCommon AlopeciaPruritusSweatingUncommon DesquamationCommon ArthralgiaMusculoskeletal, Connective tissue & bonedisordersVery Common Asthenia hGeneral disorders and administration siteconditionsFever hMucositisCommon Peripheral oedemaPainMalaiseInvestigations Very Common AST increased iALT increased iCommon Weight lossAlkaline phosphatase increaseda Leucopenia (neutropenia in particular), anaemia and thrombocytopenia, alone or in combination, are usually mild to moderate and occur in the first or second week after treatment and recover by the third week.b Severe (WHO grade 3 and 4) leucopenia (neutropenia in particular) and thrombocytopenia of WHO grade 4 can occur and may be life-threatening or fatal especially if associated with signs of gastrointestinal toxicity.c Nausea and Vomiting are usually mild (WHO grade 1 and 2), occur usually in the first week following the administration of 'Tomudex', and are responsive to antiemetics.d Diarrhoea is usually mild or moderate (WHO grade 1 and 2) and can occur at any time following the administration of 'Tomudex'. However, severe diarrhoea (WHO grade 3 and 4) can occur, and may be associated with concurrent haematological suppression especially leucopenia (neutropenia in particular). Subsequent treatment may need to be discontinued or dose reduced according to the grade of toxicity (see Section 4.2 Posology and method of administration).e Diarrhoea and vomiting may be severe and if untreated may proceed to dehydration, hypovolaemia and renalimpairmentf from spontaneous reportingg Gastrointestinal bleeding may be associated with mucositis and/or thrombocytopenia.h Asthenia and fever were usually mild to moderate following the first week of administration of 'Tomudex' and reversible.Severe asthenia can occur and may be associated with malaise and a flu-like syndrome.i Increases in AST and ALT have usually been asymptomatic and self-limiting when not associated with progression ofthe underlying malignancy.Reporting of suspected adverse reactionsReporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions viaYellow Card SchemeWebsite: /yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store4.9 OverdoseThere is no clinically proven antidote available. In the case of inadvertent or accidental administration of an overdose, preclinical data suggest that consideration should be given to the administration of leucovorin. From clinical experience with other antifolates leucovorin may be given at a dose of 25mg/m2 i.v. every 6 hours. As the time interval between 'Tomudex' administration and leucovorin rescue increases, its effectiveness in counteracting toxicity may diminish.The expected manifestations of overdose are likely to be an exaggerated form of the adverse drug reactions anticipated with the administration of the drug. Patients should, therefore, be carefully monitored for signs of gastrointestinal and haematological toxicity. Symptomatic treatment and standard supportive care measures for the management of this toxicity should be applied.5. Pharmacological properties5.1 Pharmacodynamic propertiesRaltitrexed is a folate analogue belonging to the family of anti-metabolites and has potent inhibitory activity against the enzyme thymidylate synthase (TS). Compared to other antimetabolites such as 5-Fluorouracil or methotrexate, raltitrexed acts as a direct and specific TS inhibitor. TS is a key enzyme in the de novo synthesis of thymidine triphosphate (TTP), a nucleotide required exclusively for deoxyribonucleic acid (DNA) synthesis. Inhibition of TS leads to DNA fragmentation and cell death. Raltitrexed is transported into cells via a reduced folate carrier (RFC) and is then extensively polyglutamated by the enzyme folyl polyglutamate synthetase (FPGS) to polyglutamate forms that are retained in cells and are even more potent inhibitors of TS. Raltitrexed polyglutamation enhances TS inhibitory potency and increases the duration of TS inhibition in cells which may improve antitumour activity. Polyglutamation could also contribute to increased toxicity due to drug retention in normal tissues.In clinical trials, 'Tomudex' at the dose of 3mg/m2 i.v. every 3 weeks has demonstrated clinical antitumour activity with an acceptable toxicity profile in patients with advanced colorectal cancer.Four large clinical trials have been conducted with 'Tomudex' in advanced colorectal cancer. Of the three comparative trials, two showed no statistical difference between 'Tomudex' and the combination of 5-fluorouracil plus folinic acid for survival while one trial showed a statistically significant difference in favour of the combination of 5-fluorouracil plus folinic acid. 'Tomudex' as a single agent was as effective as the combination of 5-fluorouracil and folinic acid in terms of objective response rate in all trials.5.2 Pharmacokinetic propertiesFollowing intravenous administration at 3.0 mg/m2, the concentration-time profile in patients was triphasic: Peak concentrations, found at the end of the infusion, were followed by a rapid initial decline in concentration. This was followed by a slow elimination phase. The key pharmacokinetic parameters are presented below:Summary of mean pharmacokinetic parameters in patients administered 3.0 mg/m2 Raltitrexed byintravenous infusionC m ax (ng/ml)AUC o-∞(ng.h/ml)CL(ml/min)CL r(ml/min)V ss(l)t1/2β(h)t1/2γ(h)656185651.625.1548 1.79198 Key:C m ax: Peak plasma concentration.AUC: Area under plasma concentration-time curve.CL: Clearance.V ss: Volume of distribution at steady state. t ½γ: Terminal half life.CL r: Renal clearancet½β: Half life of the second (β) phase.The maximum concentrations of raltitrexed increased linearly with dose over the clinical dose range tested.During repeated administration at three week intervals, there was no clinically significant plasma accumulation of raltitrexed in patients with normal renal function.Apart from the expected intracellular polyglutamation, raltitrexed was not metabolised and was excreted unchanged, mainly in the urine, 40 - 50%. Raltitrexed was also excreted in the faeces with approximately 15% of the radioactive dose being eliminated over a 10 day period. In the [14C] - raltitrexed trial approximately half of the radiolabel was not recovered during the study period. This suggests that a proportion of the raltitrexed dose is retained within tissues, perhaps as raltitrexed polyglutamates, beyond the end of the measurement period (29 days). Trace levels of radiolabel were detected in red blood cells on Day 29.Raltitrexed pharmacokinetics are independent of age and gender. Pharmacokinetics have not been evaluated in children.Mild to moderate hepatic impairment led to a small reduction in plasma clearance of less than 25%.Mild to moderate renal impairment (creatinine clearance of 25 to 65 ml/min) led to a significant reduction (approximately 50%) in raltitrexed plasma clearance.5.3 Preclinical safety dataPerivascular tolerance in studies in animals did not reveal any significant irritant reaction.Acute toxicityThe approximate LD50 values for the mouse and rat are 875-1249 mg/kg and >500 mg/kg respectively. In the mouse, levels of 750 mg/kg and above caused death by general intoxication.Chronic toxicityIn one month continuous and six month intermittent dosing studies in the rat, toxicity was related entirely to the cytotoxic nature of the drug. Principal target organs were the gastrointestinal tract, bone marrow and the testes. In similar studies in the dog, cumulative dose levels similar to that used clinically, elicited only pharmacologically-related changes to proliferating tissue. Target organs in the dog were therefore similar to the rat.Mutagenicity'Tomudex' was not mutagenic in the Ames test or in supplementary tests using E. coli or Chinese hamster ovary cells.'Tomudex' caused increased levels of chromosome damage in an in vitro assay of human lymphocytes. This effect was ameliorated by the addition of thymidine, thus confirming it to be due to the anti-metabolic nature of the drug. An in vivo micronucleus study in the rat indicated that at cytotoxic dose levels, 'Tomudex' is capable of causing chromosome damage in the bone marrow.Reproductive toxicologyFertility studies in the rat indicate that 'Tomudex' can cause impairment of male fertility. Fertility returned to normal three months after dosing ceased. 'Tomudex' caused embryolethality and foetal abnormalities in pregnant rats.CarcinogenicityThe carcinogenic potential of 'Tomudex' has not been evaluated.6. Pharmaceutical particulars6.1 List of excipientsMannitol Ph Eur, USPDibasic sodium phosphate (heptahydrate USP or dodecahydrate Ph Eur)Sodium hydroxide Ph Eur, USNF6.2 IncompatibilitiesThere is no information on incompatibilities at present and therefore 'Tomudex' should not be mixed with any other drug.6.3 Shelf lifeThe expiry life of 'Tomudex' is 36 months when stored below 25°C, protected from light.Once reconstituted, 'Tomudex' is chemically stable for 24 hours at 25°C exposed to ambient light. For storagerecommendation, see Instructions for Use/Handling.6.4 Special precautions for storageAddressHorizon, Honey Lane, Hurley, Maidenhead, SL6 6RJ, UK WWWMedical Information Direct LineUnopened vial - Do not store above 25°C. Keep container in the outer carton.Reconstituted vial - Refrigerate at 2-8°C.6.5 Nature and contents of container'Tomudex' is packed in 5ml clear neutral type I glass vials, with a bromobutyl rubber closure and an aluminium crimp seal with a plastic flip-off cover.The vials are packed in individual cartons to protect the product from light.6.6 Special precautions for disposal and other handlingEach vial, containing 2mg of raltitrexed, should be reconstituted with 4ml of sterile water for injections to produce a 0.5 mg/ml solution.The appropriate dose of solution is diluted in 50 - 250 ml of either 0.9% sodium chloride or 5% glucose (dextrose) injection and administered by a short intravenous infusion over a period of 15 minutes.There is no preservative or bacteriostatic agent present in 'Tomudex' or the materials specified for reconstitution or dilution. 'Tomudex' must therefore be reconstituted and diluted under aseptic conditions and it is recommended that solutions of 'Tomudex' should be used as soon as possible. Reconstituted 'Tomudex' solution may be stored refrigerated(2 - 8°C) for up to 24 hours.In accordance with established guidelines, when diluted in 0.9% sodium chloride or 5% glucose (dextrose) solution, it is recommended that administration of the admixed solution should commence as soon as possible after admixing. The admixed solution must be completely used or discarded within 24 hours of reconstitution of 'Tomudex' intravenous injection.Reconstituted and diluted solutions do not need to be protected from light.Do not store partially used vials or admixed solutions for future patient use.Any unused injection or reconstituted solution should be discarded in a suitable manner for cytotoxics.'Tomudex' should be reconstituted for injection by trained personnel in a designated area for the reconstitution of cytotoxic agents. Cytotoxic preparations such as 'Tomudex' should not be handled by pregnant women.Reconstitution should normally be carried out in a partial containment facility with extraction e.g. a laminar air flow cabinet, and work surfaces should be covered with disposable plastic-backed absorbent paper.Appropriate protective clothing, including normal surgical disposable gloves and goggles, should be worn. In case of contact with skin, immediately wash thoroughly with water. For splashes in the eyes irrigate with clean water, holding the eyelids apart, for at least 10 minutes. Seek medical attention.Any spillages should be cleared up using standard procedures.Waste material should be disposed of by incineration in a manner consistent with the handling of cytotoxic agents.7. Marketing authorisation holderHospira UK LimitedHorizon, Honey Lane, HurleyMaidenhead,SL6 6RJUnited Kingdom8. Marketing authorisation number(s)PL 04515/02259. Date of first authorisation/renewal of the authorisation25th June 200010. Date of revision of the text11/2017Ref: gxTM 3_1Company Contact DetailsHospira UK Ltd+44 (0)1304 616161 Fax+44 (0)800 098 8653Medical Information Fax+44 (0)1304 656221。