三代单分子实时测序与细菌全基因组修饰分析

三代单分子实时测序与细菌全基因组修饰分析三代测序,除了具有读长更长之外,还有一个很重要的优势,就是能够同时测得基因组甲基化修饰的信息。这带来了一个新的视角,让我们在获得细菌完成图的同时,还可以了解基因组在表观层面的变化,从更多角度来解析细菌基因组的基因密码。
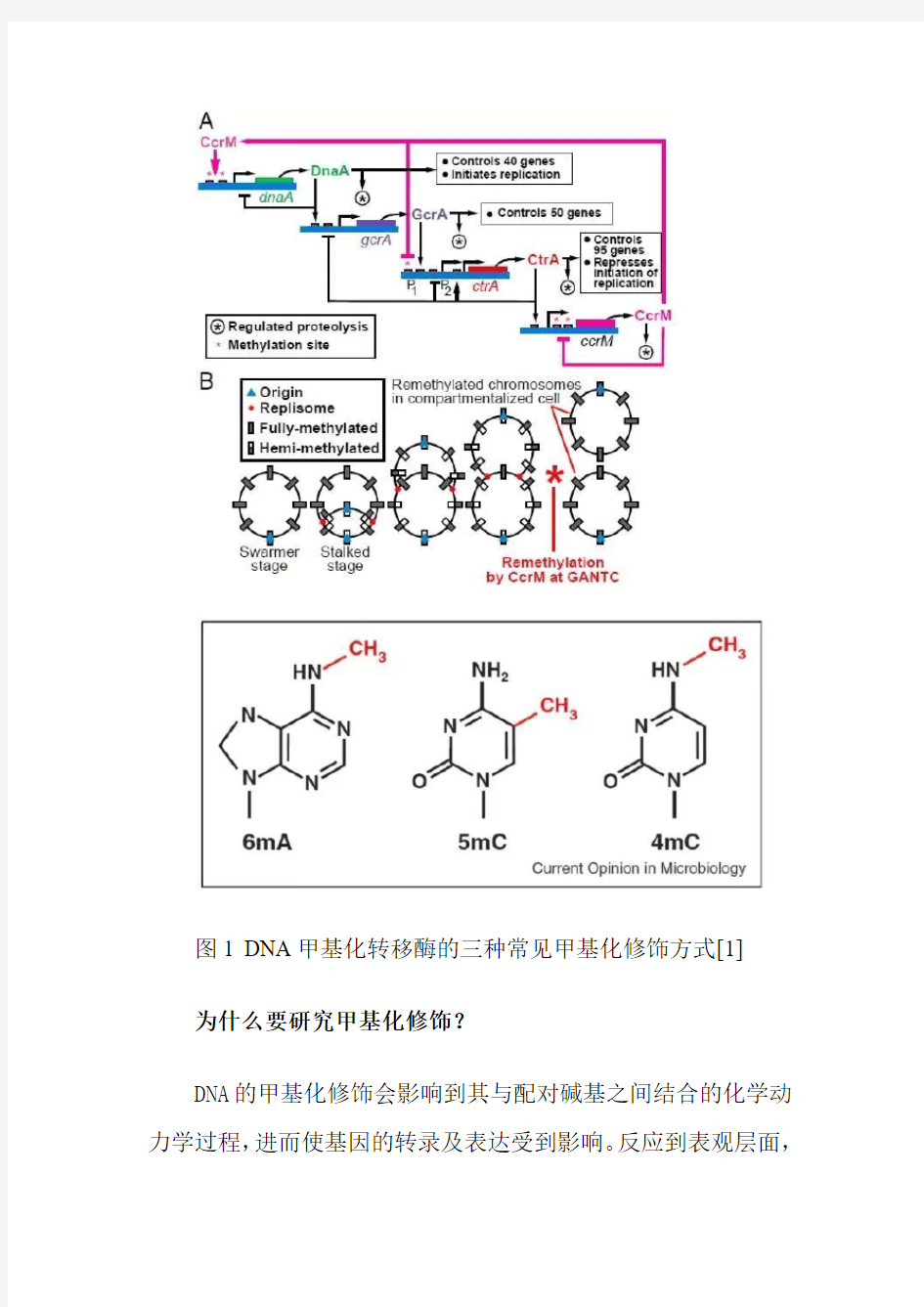
甲基化修饰是什么?
我们都知道,常见的生命遗传编码是由DNA承载的,DNA 的双链结构,编码核心就是带有ATGC四种不同碱基的脱氧核糖核酸。而实际上在生物体内很多时候DNA的碱基形式不是单纯的ATGC碱基,其中A和C两种碱基经常会存在一些甲基化修饰的现象,如下图所示:
图1 DNA甲基化转移酶的三种常见甲基化修饰方式[1]
为什么要研究甲基化修饰?
DNA的甲基化修饰会影响到其与配对碱基之间结合的化学动力学过程,进而使基因的转录及表达受到影响。反应到表观层面,
细胞不同生长周期下,不同位置的甲基化修饰起到了重要的辅助功能[2,3]。
图2 细菌细胞不同周期的甲基化改变[3]
同时DNA的甲基化修饰对于部分细菌的毒力表现也起着相当重要的作用。某些基因的改变,甚至会影响到全基因组的甲基化水平,进而影响到整个菌株的毒力性状表现[4]。
图 3 6个不同菌株的基因差异(A)和不同菌株毒力基因甲基化水平的改变(B)[4]
三代测序如何测得甲基化信息?
三代PacBio平台的测序方法为单分子实时测序(Single Molecule Real Time, SMRT),是基于单分子底物边合成边测序的方法。由于甲基化修饰过的碱基,其同配对碱基结合的化学动力学会有所差异,在底物碱基被甲基化修饰时,检测到的测序信号就会发生改变,如下图所示:
从图中可以看到,底物甲基化位点的检测信号会有一个明显的延时。PacBio的测序仪就是通过这个特征,辨认出底物中的甲基化位点的。同时,由于甲基化的信号取决于底物的甲基化特征,所以实际上我们最终获得的不仅有甲基化位点信息,还可以同时得知甲基化具体是发生在基因组的哪条链上。这也为后续的分析提供了更准确的数据基础。
细菌完成图的甲基化修饰分析
有了三代测序这个方便的平台,我们能够在拼接得到完整的细菌基因组同时,还能同时获得两条链上的甲基化修饰信息,具体展示示例如下图:
图5 完成图圈图展示,由内至外各圈依次为:GC偏离,GC 含量,ncRNA,表观修饰情况位点分布情况,GO功能分类,COG 功能分类,基因分布情况.
天津生物芯片细菌基因组完成图测序最新解决方案:
解决方案优点:
测序周期短;
测序成本低;
测序准确性高:PacBio长读长减少大尺度连接错误,Illumina 数据减少单碱基错误;
实验范围广泛:该策略不受GC含量影响,特别适合于极端环境来源细菌,如放线菌、粘细菌等
天津生物芯片真菌基因组测序最新解决方案:
该解决方案优点:
测序结果准确性高:结合不同测序平台,减少测序和拼接错误; 拼接结果完整性高:该测序方案可获得NGS测序无法获得的基因组区域,基因组完整性高,利用光学图谱平台,甚至可拼接获得真菌着丝粒和端粒序列信息,获得染色体级别拼接结果;
适用范围广泛:该方案很好低适用于GC异常或基因组中存在大量重复区的真菌,能够很好GC异常区域序列信息及跨越重复区;
参考文献
1Davis BM, Chao MC, Waldor MK. Entering the era of bacterial epigenomics with single molecu le real time DNA sequencing. Current opinion in microbiology. 2013, 16(2), 192-198.
2Gonzalez D, Kozdon JB, McAdams HH, et al. The functions of DNA methylation by CcrM in Cau lobacter crescentus: a global approach. Nucleic a cids research. 2014, 42(6), 3720-3735.
3Kozdon JB, Melfi MD, Luong K, et al. Globa l methylation state at base-pair resolution of t he Caulobacter genome throughout the cell cycle. Proceedings of the National Academy ofSciences. 2013, 110(48), E4658-E4667.
4Manso AS, Chai MH, Atack JM, et al. A ran dom six-phase switch regulates pneumococcal virule nce via global epigenetic changes. Nature communi cations. 2014, 5.
5Pacific Biosciences official website: http://w https://www.360docs.net/doc/f117244991.html,/applications/base_modification/
第三代测序技术的三种技术平台介绍
第三代测序技术的三种技术平台介绍 随着生物学的发展,人们对基因的功能研究更加透彻,为了进一步研究和改造基因的目的需要详细了解生物的基因组全序列,因为DNA序列是改造基因的基础,这就要求具有高效的DNA测序技术。DNA测序技术到目前为止已经发展到了第三代测序技术。 最早的Sanger测序在人类基因组计划中立下赫赫战功,但也给基因组测序贴上了数亿美元的价格标签,让人生畏。这两年发展迅猛的第二代测序仪——Illumina的Genome Analyzer、Roche 454的GS系列以及ABI的SOLiD系统——让人类基因组重测序的费用蹭地降低到10万美元以下。现在,能对单个DNA分子进行测序的第三代测序仪也加入到这场比赛中,让竞争更加激烈。 目前,第三代测序主要有三种技术平台。两种通过掺入并检测荧光标记的核苷酸,来实现单分子测序。Helicos的遗传分析系统已上市,而Pacific Biosciences准备在明年推出单分子实时(SMRT)技术。第三种Oxford Nanopore的纳米孔(nanopore)测序还尚未有推出的时间表,但有可能是这三种当中最便宜的。纳米孔测序的优势在于它不需要对DNA进行标记,也就省去了昂贵的荧光试剂和CCD照相机。 最近,Oxford Nanopore T echnologies的Hagan Bayley及他的研究小组正致力于改善纳米孔。根据他们之前的工作,他们以a-溶血素来设计纳米孔,并将环式糊精共价结合在孔的内侧(下图)。当核酸外切酶消化单链DNA后,单个碱基落入孔中,它们瞬间与环式糊精相互作用,并阻碍了穿过孔中的电流。每个碱基ATGC以及甲基胞嘧啶都有自己特有的电流振幅,因此很容易转化成DNA序列。每个碱基也有特有的平均停留时间,它的解离速率常数是电压依赖的,+180 mV的电位能确保碱基从孔的另一侧离开。
全基因组关联分析的原理和方法
全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中 数以百万计的单核苷酸多态性(single nucleotide ploymorphism ,SNP)为分子 遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。 随着基因组学研究以及基因芯片技术的发展,人们已通过GWAS方法发现并鉴定了大量与复杂性状相关联的遗传变异。近年来,这种方法在农业动物重要经济性状主效基因的筛查和鉴定中得到了应用。 全基因组关联方法首先在人类医学领域的研究中得到了极大的重视和应用,尤其是其在复杂疾病研究领域中的应用,使许多重要的复杂疾病的研究取得了突破性进展,因而,全基因组关联分析研究方法的设计原理得到重视。 人类的疾病分为单基因疾病和复杂性疾病。单基因疾病是指由于单个基因的突变导致的疾病,通过家系连锁分析的定位克隆方法,人们已发现了囊性纤维化、亨廷顿病等大量单基因疾病的致病基因,这些单基因的突变改变了相应的编码蛋白氨基酸序列或者产量,从而产生了符合孟德尔遗传方式的疾病表型。复杂性疾病是指由于遗传和环境因素的共同作用引起的疾病。目前已经鉴定出的与人类复杂性疾病相关联的SNP位点有439 个。全基因组关联分析技术的重大革新及其应用,极大地推动了基因组医学的发展。(2005年, Science 杂志首次报道了年龄相关性视网膜黄斑变性GWAS结果,在医学界和遗传学界引起了极大的轰动, 此后一系列GWAS陆续展开。2006 年, 波士顿大学医学院联合哈佛大学等多个研究机构报道了基于佛明翰心脏研究样本关于肥胖的GWAS结果(Herbert 等. 2006);2007 年, Saxena 等多个研究组联合报道了与2 型糖尿病( T2D ) 关联的多个位点, Samani 等则发表了冠心病GWAS结果( Samani 等. 2007); 2008 年, Barrett 等通过GWAS发现了30 个与克罗恩病( Crohns ' disrease) 相关的易感位点; 2009 年, W e is s 等通过GWAS发现了与具有高度遗传性的神经发育疾病——自闭症关联的染色体区域。我国学者则通过对12 000 多名汉族系统性红斑狼疮患者以及健康对照者的GWAS发现了5 个红斑狼疮易感基因, 并确定了4 个新的易感位点( Han 等. 2009) 。截至2009 年10 月, 已经陆续报道了关于人类身高、体重、 血压等主要性状, 以及视网膜黄斑、乳腺癌、前列腺癌、白血病、冠心病、肥胖症、糖尿病、精神分 裂症、风湿性关节炎等几十种威胁人类健康的常见疾病的GWAS结果, 累计发表了近万篇 论文, 确定了一系列疾病发病的致病基因、相关基因、易感区域和SNP变异。) 标记基因的选择: 1)Hap Map是展示人类常见遗传变异的一个图谱, 第1 阶段完成后提供了 4 个人类种族[ Yoruban ,Northern and Western European , and Asian ( Chinese and Japanese) ] 共269 个个体基因组, 超过100 万个SNP( 约1
全基因组重测序数据分析
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
Ion torrent微生物(细菌)全基因组重测序文库构建实验方案
微生物(细菌)全基因组重测序文库构建实验方案 一、重测序原理 全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。 二、技术路线 ↓基因组DNA提取 细菌DNA(纯化) ↓超声波打断 DNA片段化 ↓ 文库构建 ↓Ion OneTouch 乳液PCR、ES ↓Ion PGM、Ion Proton 上机测序 ↓ 生物信息学分析 三、实验方案 1.细菌总DNA的提取 液氮速冻、干冰保存的细菌菌液:若本实验室可以提供该细菌生长的条件,则对菌液进行活化,培养至对数期时,对该细菌进行DNA提取;若本实验室不能提供该细菌的生长条件,则应要求客户提供尽可能多的样本,以保证需要的DNA量。 细菌DNA采用试剂盒提取法(如TianGen细菌基因组提取试剂盒)。 取对数生长期的菌液,按照细菌DNA提取试剂盒操作步骤进行操作。提取完成后,对基因组DNA进行纯度和浓度的检测。通过测定OD260/280,范围在1.8-2.0之间则DNA较纯,使用Qubit对提取的DNA进行定量,确定提取的DNA 浓度达到文库构建的量。
2.DNA片段化 采用Covaris System超声波打断仪(Covaris M220),将待测DNA打断 步骤: 1)对待打断的DNA进行定量,将含量控制在100ng或者1μg 2)打开Covaris M220安全盖,将Covaris AFA-grade Water充入水浴容器内,至液面到最高刻度线(约15mL),软件界面显示为绿色 3)将待打断DNA装入Ep LoBind管中,其中DNA为100ng或1μg,加入Low TE 至总体积为50mL 4)将稀释的DNA转移至旋钮盖的Covaris管中(200bp规格),转移过程中不能将气泡带入,完成后旋紧盖子 5)选择Ion_Torrent_200bp_50μL_ScrewCap_microTube,将对应的小管放入卡口,关上安全盖,点击软件界面“RUN” 6)打断结束后,将混合液转移至一支新的1.5mL离心管中 3.末端修复及接头连接 3.1 末端修复 使用Ion Plus Fragment Kit进行,以100ng DNA量为例,各组分使用前瞬时离心2s 步骤: 1)加入核酸酶free水至装有DNA片段的1.5mL离心管中,至总体积为79μL 2)向体系中加入20μL 5×末端修复buffer,1μL末端修复酶,总体积为100μL 3)室温放置20min 3.2 片段纯化 片段纯化使用Agencourt AMpure XP Kit进行 步骤: 1)加入180μL Agencourt AMpure XP Reagent beads于经过末端修复的1.5mL离心管中,充分混匀,室温放置5min
一代、二代、三代测序技术
一代、二代、三代测序技术 (2014-01-22 10:42:13) 转载 第一代测序技术-Sanger链终止法 一代测序技术是20世纪70年代中期由Fred Sanger及其同事首先发明。其基本原理是,聚丙烯酰胺凝胶电泳能够把长度只差一个核苷酸的单链DNA分子区分开来。一代测序实验的起始材料是均一的单链DNA分子。第一步是短寡聚核苷酸在每个分子的相同位置上退火,然后该寡聚核苷酸就充当引物来合成与模板互补的新的DNA链。用双脱氧核苷酸作为链终止试剂(双脱氧核苷酸在脱氧核糖上没有聚合酶延伸链所需要的3-OH基团,所以可被用作链终止试剂)通过聚合酶的引物延伸产生一系列大小不同的分子后再进行分离的方法。测序引物与单链DNA模板分子结合后,DNA聚合酶用dNTP延伸引物。延伸反应分四组进行,每一组分别用四种ddNTP(双脱氧核苷酸)中的一种来进行终止,再用PAGE分析四组样品。从得到的PAGE胶上可以读出我们需要的序列。 第二代测序技术-大规模平行测序 大规模平行测序平台(massively parallel DNA sequencing platform)的出现不仅令DNA测序费用降到了以前的百分之一,还让基因组测序这项以前专属于大型测序中心的“特权”能够被众多研究人员分享。新一代DNA测序技术有助于人们以更低廉的价格,更全面、更深入地分析基因组、转录组及蛋白质之间交互作用组的各项数据。市面上出现了很多新一代测序仪产品,例如美国Roche Applied Science公司的454基因组测序仪、美国Illumina公司和英国Solexa technology公司合作开发的Illumina测序仪、美国Applied Biosystems公司的SOLiD测序仪。Illumina/Solexa Genome Analyzer测序的基本原理是边合成边测序。在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。以Illumina测序仪说明二代测序的一般流程,(1)文库制备,将DNA用雾化或超声波随机片段化成几百碱基或更短的小片段。用聚合酶和外切核酸酶把DNA片段切成平末端,紧接着磷酸化并增加一个核苷酸黏性末端。然后将Illumina测序接头与片段连接。(2)簇的创建,将模板分子加入芯片用于产生克隆簇和测序循环。芯片有8个纵向泳道的硅基片。每个泳道内芯片表面有无数的被固定的单链接头。上述步骤得到的带接头的DNA 片段变性成单链后与测序通道上的接头引物结合形成桥状结构,以供后续的预扩增使用。通过不断循环获得上百万条成簇分布的双链待测片段。(3)测序,分三步:DNA聚合酶结合荧光可逆终止子,荧光标记簇成像,在下一个循环开
三代测序原理技术比较
导读从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到长。 摘要:从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测序 技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到长。虽然就当前形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势位置,但第三和第四代测序技术也已在这一两年的时间中快速发展着。测序技术的每一次变革,也都对基因组研究,疾病医疗研究,药物研发,育种等领域产生巨大的推动作用。在这里我主要对当前的测序技术以及它们的测序原理做一个简单的小结。 图1:测序技术的发展历程 生命体遗传信息的快速获得对于生命科学的研究有着十分重要的意义。以上(图1)所描述的是自沃森和克里克在1953年建立DNA双螺旋结构以来,整个测序技术的发展历程。 第一代测序技术 第一代DNA测序技术用的是1975年由桑格(Sanger)和考尔森(Coulson)开创的链终止法或者是1976-1977年由马克西姆(Maxam)和吉尔伯特(Gilbert)发明的化学法(链降解). 并在1977年,桑格测定了第一个基因组序列,是噬菌体X174的,全长5375个碱基1。自此,人类获得了窥探生命遗传差异本质的能力,并以此为开端步入基因组学时代。研究人员在Sanger法的多年实践之中不断对其进行改进。在2001年,完成的首个人类基因组图谱就是以改进了的Sanger法为其测序基础,Sanger法核心原理是:由于ddNTP的2’和3’都不含羟基,其在DNA的合成过程中不能形成磷酸二酯键,因此可以用来中断DNA 合成反应,在4个DNA合成反应体系中分别加入一定比例带有放射性同位素标记的ddNTP(分为:ddATP,ddCTP,ddGTP和ddTTP),通过凝胶电泳和放射自显影后可以根据电泳带的位置确定待测分子的DNA序列(图2)。这个网址为 sanger测序法制作了一个小短片,形象而生动。 值得注意的是,就在测序技术起步发展的这一时期中,除了Sanger法之外还出现了一些其他的测序技术,如焦磷酸测序法、链接酶法等。其中,焦磷酸测序法是后来Roche公司454技术所使用的测序方法2–4,而连接酶测序法是后来ABI公司SOLID技术使用的测序方法2,4,但他们的共同核心手段都是利用了Sanger1中的可中断DNA合成反应的dNTP。
全基因组从头测序(de novo测序)
全基因组从头测序(de novo测序) https://www.360docs.net/doc/f117244991.html,/view/351686f19e3143323968936a.html 从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动物、植物、细菌、真菌的全基因组序列,从而推进该物种的研究。一个物种基因组序列图谱的完成,意味着这个物种学科和产业的新开端!这也将带动这个物种下游一系列研究的开展。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。华大科技利用新一代高通量测序技术,可以高效、低成本地完成所有物种的基因组序列图谱。包括研究内容、案例、技术流程、技术参数等,摘自深圳华大科技网站 https://www.360docs.net/doc/f117244991.html,/service-solutions/ngs/genomics/de-novo-sequencing/ 技术优势: 高通量测序:效率高,成本低;高深度测序:准确率高;全球领先的基因组组装软件:采用华大基因研究院自主研发的SOAPdenovo软件;经验丰富:华大科技已经成功完成上百个物种的全基因组从头测序。 研究内容: 基因组组装■K-mer分析以及基因组大小估计;■基因组杂合模拟(出现杂合时使用); ■初步组装;■GC-Depth分布分析;■测序深 度分析。基因组注释■Repeat注释; ■基因预测;■基因功能注释;■ ncRNA 注释。动植物进化分析■基因家族鉴定(动物TreeFam;植物OrthoMCL);■物种系统发育树构建; ■物种分歧时间估算(需要标定时间信息);■基因组共线性分析; ■全基因组复制分析(动物WGAC;植物WGD)。微生物高级分析 ■基因组圈图;■共线性分析;■基因家族分析; ■CRISPR预测;■基因岛预测(毒力岛); ■前噬菌体预测;■分泌蛋白预测。 熊猫基因组图谱Nature. 2010.463:311-317. 案例描述 大熊猫有21对染色体,基因组大小2.4 Gb,重复序列含量36%,基因2万多个。熊猫基因组图谱是世界上第一个完全采用新一代测序技术完成的基因组图谱,样品取自北京奥运会吉祥物大熊猫“晶晶”。部分研究成果测序分析结果表明,大熊猫不喜欢吃肉主要是因为T1R1基因失活,无法感觉到肉的鲜味。大熊猫基因组仍然具备很高的杂合率,从而推断具有较高的遗传多态性,不会濒于灭绝。研究人员全面掌握了大熊猫的基因资源,对其在分子水平上的保护具有重要意义。 黄瓜基因组图谱黄三文, 李瑞强, 王俊等. Nature Genetics. 2009. 案例描述国际黄瓜基因组计划是由中国农业科学院蔬菜花卉研究所于2007年初发起并组织,并由深圳华大基因研究院承担基因组测序和组装等技术工作。部分研究成果黄瓜基因组是世界上第一个蔬菜作物的基因组图谱。该项目首次将传
微生物基因组研究进展及意义
微生物基因组研究进展及其意义 近年来,病原微生物的基因组研究取得了飞速的进展。所谓基因组研究是指对微生物的全基因进行核苷酸测序,在了解全基因的结构基础上,研究各个基因单独或数个基因间相互作用的功能。由于过去人们大多从表型分析入手,寻找已知功能的编码基因,实际只了解微生物中极少数的基因,如链球菌的链激酶基因、结核杆菌编码的热休克蛋白基因等。还有大量未知基因未被发现。通过基因组研究,则从根本上揭示了微生物的全部基因,不仅可发现新的基因,还可发现新的基因间相互作用、新的调控因子等。这一研究将使人类从更高层次上掌握病原微生物的致病机制及其规律,从而得以发展新的诊断、预防及治疗微生物感染的制剂、疫苗及药品。此外,新发现的微生物酶及蛋白还可能有在工农业生产上的应用价值。因此,全球除已完成了70余株覆盖重要病毒科的病毒代表株全基因组研究外,据美国基因组研究所(The Institute for Genomic Research, TIGR)报道,目前已完成了19种微生物基因组测序,其中11种与人类及疾病相关(嗜血流感杆菌,生殖道支原体,肺炎支原体,幽门螺杆菌,枯草杆菌,伯氏疏螺旋体,结核杆菌,梅毒螺旋体,沙眼衣原体,普氏立克次体)。另外,还有40余种微生物已被登记正在进行测序,预计在1999~2000年完成〔1〕。 病毒基因组研究进展 病毒因其基因组小,是进行基因组研究最早的生物体。早在1977 年已完成了噬菌体DNA的全基因测序。存在于脊髓灰质炎疫苗中的SV40,是最早完成全基因测序的与疾病相关的病毒;此后,许多病毒均已完成了全基因测序,并根据序列的开放阅读框架(ORF)对编码蛋白进行了推导。已对相当一些病毒蛋白进行了重组表达,还对一些病毒基因编码的调控序列进行了研究。除一般大小的病毒已完成了基因组测序,对大基因组病毒,疱疹病毒科,如水痘病毒基因组为0.125Mb(Mega-basepair,兆碱基对)〔2〕。巨细胞病毒,基因组为0.229Mb〔3〕。我国已对痘苗病毒天坛株(约0.2Mb)进行了全基因测序,发现与国外的痘苗毒株序列有明显的差异〔4〕。我国还对甲、乙、丙、丁、戊、庚型肝炎病毒进行了国内毒株的全基因测序。近来还对国内2株发现的虫媒病毒毒株完成了全基因测序。我国从不同来源的标本中发现了不少乙肝病毒变异株,有的具有特殊的生物学特性〔5〕。对病毒基因中调控因子的分析,发现了与乙肝病毒增强子作用的新细胞核因子〔6〕。 因此,目前对病毒的基因组研究已进入了后基因组阶段,即从全基因水平研究病毒的生物学功能,同时发现新的基因功能。对于医学病毒学当前主要方向是研究病毒基因组中与致病及诱生免疫应答相关的基因,从而揭示和解决迄今尚未解决的问题,以达到控制或消灭一些重要病毒感染的目的。 建议目前可进行后基因组研究的领域为: 1.病毒持续性感染:基因组中与持续性感染相关的基因,基因变异或调控因子研究。已报道的乙肝病毒的前核心基因出现终止密码突变,
一、二、三代测序技术
一代、二代、三代测序技术 第一代测序技术-Sanger链终止法 一代测序技术是20世纪70年代中期由Fred Sanger及其同事首先发明。其基本原理是,聚丙烯酰胺凝胶电泳能够把长度只差一个核苷酸的单链DNA分子区分开来。一代测序实验的起始材料是均一的单链DNA分子。第一步是短寡聚核苷酸在每个分子的相同位置上退火,然后该寡聚核苷酸就充当引物来合成与模板互补的新的DNA链。用双脱氧核苷酸作为链终止试剂(双脱氧核苷酸在脱氧核糖上没有聚合酶延伸链所需要的3-OH基团,所以可被用作链终止试剂)通过聚合酶的引物延伸产生一系列大小不同的分子后再进行分离的方法。测序引物与单链DNA模板分子结合后,DNA聚合酶用dNTP延伸引物。延伸反应分四组进行,每一组分别用四种ddNTP(双脱氧核苷酸)中的一种来进行终止,再用PAGE分析四组样品。从得到的PAGE胶上可以读出我们需要的序列。 第二代测序技术-大规模平行测序 大规模平行测序平台(massively parallel DNA sequencing platform)的出现不仅令DNA测序费用降到了以前的百分之一,还让基因组测序这项以前专属于大型测序中心的“特权”能够被众多研究人员分享。新一代DNA测序技术有助于人们以更低廉的价格,更全面、更深入地分析基因组、转录组及蛋白质之间交互作用组的各项数据。市面上出现了很多新一代测序仪产品,例如美国Roche Applied Science公司的454基因组测序仪、美国Illumina公司和英国Solexa
technology公司合作开发的Illumina测序仪、美国Applied Biosystems公司的SOLiD测序仪。Illumina/Solexa Genome Analyzer测序的基本原理是边合成边测序。在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。以Illumina测序仪说明二代测序的一般流程,(1)文库制备,将DNA用雾化或超声波随机片段化成几百碱基或更短的小片段。用聚合酶和外切核酸酶把DNA片段切成平末端,紧接着磷酸化并增加一个核苷酸黏性末端。然后将Illumina测序接头与片段连接。(2)簇的创建,将模板分子加入芯片用于产生克隆簇和测序循环。芯片有8个纵向泳道的硅基片。每个泳道内芯片表面有无数的被固定的单链接头。上述步骤得到的带接头的DNA 片段变性成单链后与测序通道上的接头引物结合形成桥状结构,以供后续的预扩增使用。通过不断循环获得上百万条成簇分布的双链待测片段。(3)测序,分三步:DNA 聚合酶结合荧光可逆终止子,荧光标记簇成像,在下一个循环开始前将结合的核苷酸剪切并分解。(4)数据分析 第三代测序技术-高通量、单分子测序 被称为第三代的测序的He-licos单分子测序仪,PacificBioscience的SMRT技术和 Oxford Nanopore Technologies 公司正在研究的纳米孔单分子测序技术正向着高通量低成本长读取长度的方向发展。不同于第二代测序依赖于DNA模板
人类全基因组测序
1 技术优势 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对人类不同个体或群体进行全基因组测序,并在个体或群体水平上进行生物信息分析。可全面挖掘DNA 水平的遗传变异,为筛选疾病的致病及易感基因,研究发病及遗传机制提供重要信息。 全基因组测序 平台优势 HiSeq X 测序平台 读长:PE150 通量:1.8T/run 测序周期:3 天 专为人全基因组测序准备、测序周期短、通量高
生物信息分析 技术路线 技术参数 样品要求 样本类型:DNA 样品 样本总量:≥1.0 μg DNA (提取自新鲜及冻存样本) ≥1.5 μg DNA (提取自FFPE 样本)样品浓度:≥ 20 ng/μl 测序平台及策略HiSeq X PE150 测序深度 肿瘤:癌组织(50X),癌旁组织/血液样本(30X)遗传病:30~50 X 项目周期37天
3 案例解析 该研究选取3个家系中6个患者和1个正常个体,首先使用基因芯片寻找纯合突变位点,然后对其中无亲缘关系的2例患者采用全基因组测序研究,在2例患者非编码区域均发现相同的变异,10号染色体PTF1A 末端发生一个点突变(chr10:23508437 A>G),且变异在患病人群和细胞试验中均得到了验证。研究解释了生长发育启动子隐性变异是罕见孟德尔遗传病的常见致病原因,同时说明许多疾病的致病突变也可能位于非编码区。 图1 检出的变异信息 智力障碍是影响新生儿心智发育的一类疾病。这项研究选取50个经过基因芯片和全外显子测序未确诊致病因子的trio 家系,全基因组测序检出84个de novo SNVs 和8个de novo CNVs,及一些结构变异(如VPS13B、STAG1、IQSEC2-TENM3),检出率为42%。揭示编码区的de novo SNVs 和de novo CNVs 是导致智力障碍的主要因素,全基因组测序可以作为可靠的遗传性检测应用工具。 案例一 单基因病研究——全基因组测序鉴定PTF1A末端增强子常染色体隐性突变导致胰腺 发育不全[1] 案例二 复杂疾病研究——全基因组测序解析智力障碍的主要致病因素[2] 图2 PTF1A 的家系图谱
三代测序原理技术比较
导从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测导序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从读长到短,再从短到长。 摘要:从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到 长。虽然就当前形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势 位置,但第三和第四代测序技术也已在这一两年的时间中快速发展着。测序技术的每一次变 革,也都对基因组研究,疾病医疗研究,药物研发,育种等领域产生巨大的推动作用。在 这里我主要对当前的测序技术以及它们的测序原理做一个简单的小结。 图1 :测序技术的发展历程 生命体遗传信息的快速获得对于生命科学的研究有着十分重要的意义。以上(图1)所描述的是自沃森和克里克在1953年建立DNA双螺旋结构以来,整个测序技术的发展历程。 第一代测序技术 第一代DNA测序技术用的是1975年由桑格(Sanger)和考尔森(Coulson )开创的链终止法或者是1976-1977年由马克西姆(Maxam和吉尔伯特(Gilbert )发明的化学法(链降解)?并在1977年,桑格测定了第一个基因组序列,是噬菌体X174的,全长5375个碱 基1。自此,人类获得了窥探生命遗传差异本质的能力,并以此为开端步入基因组学时代。 研究人员在Sanger法的多年实践之中不断对其进行改进。在2001年,完成的首个人类基 因组图谱就是以改进了的Sanger法为其测序基础,Sanger法核心原理是:由于ddNTP的2' 和3'都不含羟基,其在DNA的合成过程中不能形成磷酸二酯键,因此可以用来中断DNA 合成反应,在4个DNA合成反应体系中分别加入一定比例带有放射性同位素标记的ddNTP分为:ddATP,ddCTP,ddGTP和ddTTP),通过凝胶电泳和放射自显影后可以根据电泳带的位置确定待测分子的DNA序列(图2)。这个网址为san ger测序法制作了一个小短片,形象而生动。 值得注意的是,就在测序技术起步发展的这一时期中,除了San ger法之外还出现了一 些其他的测序技术,如焦磷酸测序法、链接酶法等。其中,焦磷酸测序法是后来Roche公司454技术所使用的测序方法2 - 4,而连接酶测序法是后来ABI公司SOLID技术使用的测序方 法2,4,但他们的共同核心手段都是利用了Sanger1中的可中断DNA合成反应的dNTP 图2: Sanger法测序原理
三代测序技术的比较
一代、二代、三代测序技术 张祥瑞 2013/04/22 11:43 第一代测序技术-Sanger链终止法 一代测序技术是20世纪70年代中期由Fred Sanger及其同事首先发明。其基本原理是,聚丙烯酰胺凝胶电泳能够把长度只差一个核苷酸的单链DNA分子区分开来。一代测序实验的起始材料是均一的单链DNA分子。第一步是短寡聚核苷酸在每个分子的相同位置上退火,然后该寡聚核苷酸就充当引物来合成与模板互补的新的DNA链。用双脱氧核苷酸作为链终止试剂(双脱氧核苷酸在脱氧核糖上没有聚合酶延伸链所需要的3-OH基团,所以可被用作链终止试剂)通过聚合酶的引物延伸产生一系列大小不同的分子后再进行分离的方法。测序引物与单链DNA模板分子结合后,DNA聚合酶用dNTP延伸引物。延伸反应分四组进行,每一组分别用四种ddNTP(双脱氧核苷酸)中的一种来进行终止,再用PAGE分析四组样品。从得到的PAGE胶上可以读出我们需要的序列。 第二代测序技术-大规模平行测序 大规模平行测序平台(massively parallel DNA sequencing platform)的出现不仅令DNA测序费用降到了以前的百分之一,还让基因组测序这项以前专属于大型测序中心的“特权”能够被众多研究人员分享。新一代DNA测序技术有助于人们以更低廉的价格,更全面、更深入地分析基因组、转录组及蛋白质之间交互作用组的各项数据。市面上出现了很多新一代测序仪产品,例如美国Roche Applied Science公司的454基因组测序仪、美国Illumina公司和英国Solexa technology公司合作开发的Illumina测序仪、美国Applied Biosystems公司的SOLiD测序仪。Illumina/Solexa Genome Analyzer测序的基本原理是边合成边测序。在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。以Illumina测序仪说明二代测序的一般流程,(1)文库制备,将DNA用雾化或超声波随机片段化成几百碱基或更短的小片段。用聚合酶和外切核酸酶把 DNA片段切成平末端,紧接着磷酸化并增加一个核苷酸黏性末端。然后将Illumina测序接头与片段连接。(2)簇的创建,将模板分子加入芯片用于产生克隆簇和测序循环。芯片有8个纵向泳道的硅基片。每个泳道内芯片表面有无数的被固定的单链接头。上述步骤得到的带接头的DNA 片段变性成单链后与测序通道上的接头引物结合形成桥状结构,以供后续的预扩增使用。通过不断循环获得上百万条成簇分布的双链待测片段。(3)测序,分三步:DNA 聚合酶结合荧光可逆终止子,荧光标记簇成像,在下一个循环开始前将结合的核苷酸剪切并分解。(4)数据分析 第三代测序技术-高通量、单分子测序
三代基因组测序技术原理(简介)
三代基因组测序技术原理简介 【写在前面的话】:首先,这一篇博文中的内容并非原创,而是对多篇文献中内容的直接摘录,有些图片和资料还来自身边的同事(在此深表谢意!),再夹杂自己的零星想法,写在这里分享与大家,同时也是为了方便自己日后若有需要能够方便获得,文章比较长。 摘要:从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到长。虽然就当前形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势位置,但第三和第四代测序技术也已在这一两年的时间中快速发展着。测序技术的每一次变革,也都对基因组研究,疾病医疗研究,药物研发,育种等领域产生巨大的推动作用。在这里我主要对当前的测序技术以及它们的测序原理做一个简单的小结。 图1: 测序技 术的发 展历程 生命体 遗传信 息的快 速获得 对于生 命科学 的研究 有着十分重要的意义。以上(图1)所描述的是自沃森和克里克在1953年建立DNA双螺旋结构以来,整个测序技术的发展历程。 第一代测序技术 第一代DNA测序技术用的是1975年由桑格(Sanger)和考尔森(Coulson)开创的链终止法或者是1976-1977年由马克西姆(Maxam)和吉尔伯特(Gilbert)发明的化学法(链降解). 并在1977年,桑格测定了第一个基因组序列,是噬菌体X174的,全长5375个碱基1。自此,人类获得了窥探生命遗传差异本质的能力,并以此为开端步入基因组学时代。研究人员在Sanger法的多年实践之中不断对其进行改进。在2001年,完成的首个人类基因组图谱就是以改进了的Sanger法为其测序基础,Sanger法核心原理是:由于ddNTP的2’和3’都不含羟基,其在DNA的合成过程中不能形成磷酸二酯键,因此可以用来中断DNA合成反应,在4个DNA合成反应体系中分别加入一定比例带有放射性同位素标记的ddNTP(分为:ddATP,ddCTP,ddGTP和ddTTP),通过凝胶电泳和放射自显影后可以根据电泳带的位置确定待测分子的DNA序列(图2)。这个网址为sanger测序法制作了一个小短片,形象而生动。
一代、二代、三代测序技术
三代基因组测序技术原理简介 摘要:从1977年第一代DNA测序技术(Sanger法)1,发展至今三十多年时间,测序技术已取得了相当大的发展,从第一代到第三代乃至第四代,测序读长从长到短,再从短到长。虽然就当前形势看来第二代短读长测序技术在全球测序市场上仍然占有着绝对的优势位置,但第三和第四代测序技术也已在这一两年的时间中快速发展着。测序技术的每一次变革,也都对基因组研究,疾病医疗研究,药物研发,育种等领域产生巨大的推动作用。在这里我主要对当前的测序技术以及它们的测序原理做一个简单的小结。 图1:测序技术的发展历程 生命体遗传信息的快速获得对于生命科学的研究有着十分重要的意义。以上(图1)所描述的是自沃森和克里克在1953年建立DNA双螺旋结构以来,整个测序技术的发展历程。 第一代测序技术 第一代DNA测序技术用的是1975年由桑格(Sanger)和考尔森(Coulson)开创的链终止法或者是1976-1977年由马克西姆(Maxam)和吉尔伯特(Gilbert)发明的化学法(链降解). 并在1977年,桑格测定了第一个基因组序列,是噬菌体X174的,全长5375个碱基1。自此,人类获得了窥探生命遗传差异本质的能力,并以此为开端步入基因组学时代。研究人员在Sanger法的多年实践之中不断对其进行改进。在2001年,完成的首个人类基因组图谱就是以改进了的Sanger法为其测序基础,Sanger法核心原理是:由于ddNTP的2’和3’都不含羟基,其在DNA的合成过程中不能形成磷酸二酯键,因此可以用来中断DNA合成反应,在4个DNA合成反应体系中分别加入一定比例带有放射性同位素标记的ddNTP(分为:ddATP,ddCTP,ddGTP和 ddTTP),通过凝胶电泳和放射自显影后可以根据电泳带的位置确定待测分子的DNA序列(图2)。这个网址为sanger测序法制作了一个小短片,形象而生动。 值得注意的是,就在测序技术起步发展的这一时期中,除了Sanger法之外还出现了一些其他的测序技术,如焦磷酸测序法、链接酶法等。其中,焦磷酸测序法是后来Roche公司454技术所使用的测序方法2–4,而连接酶测序法是后来ABI公司SOLID技术使用的测序方法2,4,但他们的共同核心手段都是利用了Sanger1中的可中断DNA合成反应的dNTP。
第三代基因测序技术比较与总结
第三代基因测序技术比较与总结 [摘要]在第二代测序技术的协助下,个人基因组图谱正在如火如荼地绘制中。 在第二代测序技术的协助下,个人基因组图谱正在如火如荼地绘制中。但第二代测序技术很快就遇上了强劲的对手——第三代测序技术,也被称为“下、下一代的测序(next-next-generation sequencing)”。第三代测序技术是基于纳米孔(nanopore)的单分子读取技术,有着更快的数据读取速度,应用潜能也势必超越测序。 2012年2月5日,基因组科学家们齐聚美国佛罗里达州的基因组生物和技术进展会议,来了解哪家公司的第三代测序技术能实现人类基因组的3分钟测序或以5000美元的价格出售。尽管科学家们对公布的数据表示谨慎乐观,但他们对于此类测序仪的优越之处仍心存疑虑。 Complete Genomics 在2008年10月,美国加利福尼亚州的Complete Genomics公司曾宣称他们将在2009年以5000美元的价格售卖人类基因组,但当时没有公布支持数据。在这次会议上,该公司公布了一个人类基因组,据称是用9台仪器在8天内完成的。 该公司的CEO,Clifford Reid表示,他们将254GB的数据拼接成草图,覆盖某个匿名男性基因组的92%,每个碱基平均读取了91次。与目前应用中的高速测序,即第二代测序类似,Complete Genomics也产生短的DNA读长。通过对每个碱基的多次测序,它的目标是排除悄悄混入的可能错误。Reid认为这项技术非常准确,碱基错误的概率低于0.33%。这与目前的测序仪相当。 Complete Genomics并不出售测序仪,但用自己的测序仪来完成所有的内部工作。这让某些科学家质疑,但另一些却深受鼓舞。 速度和费用成为Complete Genomics的最大卖点。该公司并没有透露基因组测序的确切费用,但据称每个基因组的原材料费用低至1000美元。它的目标是在上个月推出市场,今年对1000个基因组进行测序,明年测序数量达到20000个。 Pacific Biosciences 在Complete Genomics做报告前的一小时,Pacific Biosciences的首席技术官Stephen Turner展示了大肠杆菌的完整基因组,并称每个碱基的平均读取了38次,准确率大于99.9999%。 Pacific Biosciences利用了单分子技术和DNA聚合酶,在反应的同时读取测序产物。尽管目前仪器的读取速度仅为3 碱基/秒,但它的目标是在2013年前实现三分钟读完人类基因组。它还有望实现更长的读长。Tuner表示大肠杆菌
基因组测序术语解释
DNA关键词: WG-BSA (全基因组重测序BSA) 对已有参考基因组序列的物种的所有作图群体(F1、F2、RIL、DH 和BC1等),对亲本进行个体重测序,对某个极端性状材料混池测序,检测SNP,获得与性状紧密关联的分子标记和精细定位区域,是目前最高效的基因定位方法。通过选取某个极端性状,利用高效率低成本的混池测序技术,勿需开发分子标记进行遗传图的构建,快速定位与性状相关的候选QTL。 MP-Reseq (多混池全基因组重测序) 针对特有的优良地方品种中的不同品种/品系,通过群体内pooling 建库的方法,进行全基因组重测序,采用生物信息学方法全基因组范围内扫描变异位点,能快速的定位不同混池样品基因组中明显经过人工或自然选择的区域,检测与性状相关的基因区域及其功能基因。 全基因组个体重测序 基于全基因组重测序的变异图谱通过测序手段结合生物信息分析研究同一物种不同个体之间的变异情况,获得大量的变异信息,如SNP、Indel、SV 等。主要可以快速地获得大量的分子标记以及不同个体在基因组水平上的差异。 全基因组关联分析-GWAS 通过重测序对动植物重要种质资源进行全基因组基因型鉴定,与关注的表型数据进行全基因组关联分析,找出与关注表型相关的SNP位点,定位数量性状基因,与数量性状相关的基因紧密连锁的SNP标记,后续可用于分子标记辅助育种,助力育种进程。 全基因组重测序-遗传进化 通过对来自全国各地、具有代表性的XX 份XX 材料进行全基因组重测序,检测SNP、Indel、SV,并利用获得的SNP 与SV 数据进行群体多样性分析,包括连锁不平衡分析、群体进化分析、群体结构分析、群体主成分分析等。 全基因组重测序-遗传图谱 基于全基因组重测序技术对已有参考基因组序列的物种进行个体或群体的全基因组测序,利用高性能计算平台和生物信息学方法,检测单核苷酸多态性位点(SNP),并计算多态性标记间的遗传连锁距离,绘制高密度的遗传图谱。通过与表型性状进行关联分析,利用获得的强关联性标记进行下游基因的精细定位。遗传图可用于分子标记辅助育种,重要性状候选基因克隆,辅助基因组组装,比较基因组学等研究。 细菌基因组de novo 测序 细菌是生物的主要类群之一,是所有生物中数量最多的一类。细菌广泛分布于土壤和水中,或者与其他生物共生,也有部分种类分布在极端环境中,例如温泉,甚至是放射性废弃物中。由于细菌自身的营
07年完成基因组测序的生物
07年完成基因组测序的生物 生物通报道:在即将过去的2007年,动物、植物、微生物的基因组测序工作进行的如火如荼,多项基因组测序结果被公布,包括第一个个人基因组图谱、马基因组图谱、肺癌基因组图谱和多种致病性细菌的基因组测序结果。 人类基因组测序的进一步深入 世界首份个人DNA图谱出炉 57年前,美国生物学家詹姆斯·沃森与弗朗西斯·克里克共同发现了脱氧核糖核酸(DNA)分子结构的双螺旋模型,并因这项基因研究领域的重大突破获得诺贝尔奖。今天,沃森成为自己研究的受益者--他将成为世界第一份完全破译的“个人版”基因组图谱的拥有者。 第一个个体基因组序列公布 来自美国克莱格凡特研究所(J. Craig Venter Institute,由TIGR所建立),加拿大多伦多大学,加州大学圣地亚哥分校,西班牙巴塞罗那大学(Universitat de Barcelona)的研究人员近期公布了单个个体二倍体基因组序列,为未来的基因组比较打开了一道门,也开创了个体基因组信息的新纪元。 杜克大学公布第一张人类基因组印记基因图谱
来自杜克大学的研究人员创造了第一张人类基因组印记基因(imprinted genes)图谱,并且他们表示其成功的关键在于一个称为机器学习(machine learning)的人工智能形式:modern-day Rosetta stone。这项研究新发现了四倍于之前识别的印记基因,并即将公布在12月3日《Genome Research》封面上。 完成测序的动物 第一张马基因组图谱草图公布 国际马类基因组序列计划(the international Horse Genome Sequencing Project)宣布,科学家们首次完成家马((Equus caballus))的基因图谱草图,得到了270万个DNA碱基对的数据,全部数据已经进入公共数据库,可免费供全世界的生物学家和兽医学家使用。 《自然》封面:首个有袋动物基因组序列公布 一种灰色短尾负鼠(Monodelphis domestica)的基因组测序的完成则为这一推测给出了切实的证据。负鼠是第一个完成基因组测序的有袋动物,测序结果公布在4月10日的《自然》杂志上,而且这种小动物还登上了该期杂志的封面。 家猫基因组测序完成