Bio Rad电泳仪操作规程
BIO-RAD ChemiDocXRS+化学发光凝胶成像仪中文简易说明书
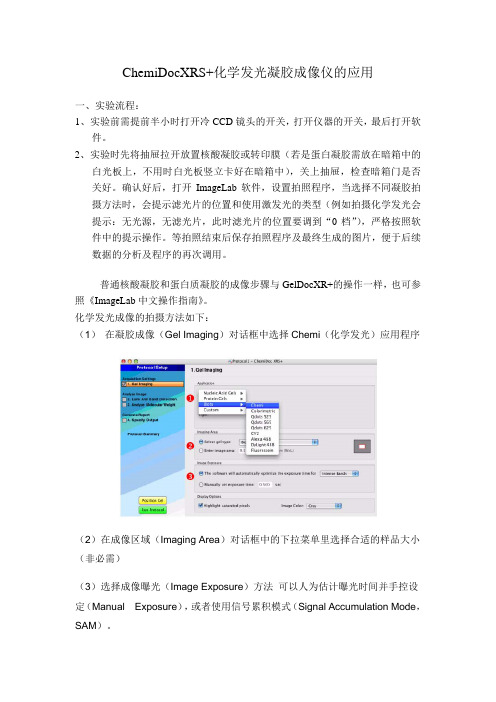
ChemiDocXRS+化学发光凝胶成像仪的应用一、实验流程:1、实验前需提前半小时打开冷CCD镜头的开关,打开仪器的开关,最后打开软件。
2、实验时先将抽屉拉开放置核酸凝胶或转印膜(若是蛋白凝胶需放在暗箱中的白光板上,不用时白光板竖立卡好在暗箱中),关上抽屉,检查暗箱门是否关好。
确认好后,打开ImageLab软件,设置拍照程序,当选择不同凝胶拍摄方法时,会提示滤光片的位置和使用激发光的类型(例如拍摄化学发光会提示:无光源,无滤光片,此时滤光片的位置要调到“0档”),严格按照软件中的提示操作。
等拍照结束后保存拍照程序及最终生成的图片,便于后续数据的分析及程序的再次调用。
普通核酸凝胶和蛋白质凝胶的成像步骤与GelDocXR+的操作一样,也可参照《ImageLab中文操作指南》。
化学发光成像的拍摄方法如下:(1) 在凝胶成像(Gel Imaging)对话框中选择Chemi(化学发光)应用程序(2)在成像区域(Imaging Area)对话框中的下拉菜单里选择合适的样品大小(非必需)(3)选择成像曝光(Image Exposure)方法可以人为估计曝光时间并手控设定(Manual Exposure),或者使用信号累积模式(Signal Accumulation Mode,SAM)。
在建立一个化学发光的程序时最难的任务就是图像曝光的确定,因为您想获得一个充分利用了相机大的动态范围的图像。
过短的图像曝光时间可能使高于膜背景的微弱信号不能被识别;过长的图像曝光时间能够使得某些条带饱和(也就是说,超出了相机准确报告信号的能力)。
如果这是您第一次用ChemiDoc XRS+分析化学发光样品,您需要在使用手控或信号累积模式(SAM)之前决定一个合适的成像时间框架。
获得这个信息的最好的方式是取得一个相对短的手工曝光并利用Image Lab里的工具估计最适的曝光时间。
手控曝光1. 选择手控设定曝光时间(Manually setexposure time)并在读秒框中输入10秒。
电泳仪操作及维护保养作业指导书

广州东盛生物科技有限公司
实验仪器
编号:
操作及维护保养作业指导书
电泳仪
1.0目的:保证实验仪器的有效使用,确保产品实现过程的质量。
2.0适用范围:本公司电泳仪(百晶BG-Power600i)。
3.0工作环境:保持电泳仪环境温度在15℃~30℃;相对湿度不大于70%;周围空气中无腐
蚀性的气体存在。
4.0操作流程:
4.1打开电源,按电泳仪面板上的“SET”按钮,使仪器进入电压及电泳时间设置状态,根
据上下箭头设置所要的电压及电泳时间。
4.2将需要点样的DNA样品以移液器点进琼脂糖胶孔中。
4.3按下电泳仪面板的“start”按钮,运行指示亮起红色,电泳进行。
4.4按照预定时间进行电泳后,电泳仪鸣叫,提示电泳结束,按压电泳仪面板上的“stop”
按钮,电泳停止,运行指示灯灭。
4.5从电泳仪中取出琼脂糖胶,进行下一步的凝胶成像系统分析。
5.0维护保养方法:
5.1维护保养周期:两周。
5.2维护保养条件:依照3.0工作环境实施。
5.3外观检查:
5.3.1 仪器需保持清洁,表面外周干净清洁。
5.3.2 保证运行指示灯正常亮与灭。
5.3.3 检查电泳插头是否完后无损。
5.3.4 观察液晶屏字幕显示是否清晰,外观是否完整无损。
5.4 注意事项:
5.4.1 电泳时,防止无DNA样品空跑。
5.4.2 防止电极正反反插。
6.0 表单记录:电泳仪维护保养记录表格。
编制/时间:审批/时间:。
(完整版)Bio_Rad电泳仪操作规程

Bio—rad小电泳槽及电泳仪操作规程一.目的为规范Bio—rad小电泳槽及电泳仪的基本操作、维护保养、异常处理程序,防止人为操作失误,确保Bio-rad小电泳槽及电泳仪正常运转,特制定本程序。
二.适用范围本程序适用于Bio—rad小电泳槽及电泳仪.三.责任1. 本程序的实施者为Bio-rad小电泳槽及电泳仪操作者,各实验室负责人对本程序的实施情况进行监督。
2. 日常运行及维护、定期维护、定期点检及保养由Bio-rad小电泳槽及电泳仪操作者负责。
四.内容1.灌胶a、取出灌胶模具,打开封底盘至完全开放的状态,松开两侧螺丝,向外侧打开夹子。
b、取长方形和带凹槽的长玻板各一块,从内到外依次按带凹槽玻板及长方形玻板的顺序排列,形成gel sandwich.注意正确排列以防漏胶。
c、短玻板朝外将sandwich放入模具中,检测sandwich的底是否紧靠底面.旋紧螺丝。
将封底盘锁至密闭状态。
d、将模具放在桌面上准备灌胶.e、根据需要配制一定浓度的胶.1、分离胶的灌制:用加样枪慢慢将胶加入sandwich中,注意不要有气泡。
其上加0.1%SDS 封顶。
2、浓缩胶的灌制:待分离胶凝后弃去SDS,开始灌浓缩胶,最后轻轻插入梳子。
f、待胶聚合后,拔出梳子,用电泳缓冲液清洗加样槽,去除未聚合的胶。
g、模具放入电泳槽中。
2.电泳a、加适量的电泳缓冲液b、准备样品,加样。
c、盖上电泳槽的盖子,注意红色对正极,黑色对负极。
打开电源,开始电泳.初始电压100V, 15分钟后改为120v。
d、直至指示剂距前沿1cm处结束电泳,拔掉电源,小心取出胶,根据需要进行染色或转膜,回收电泳缓冲液(可重复利用3次)。
五.注意事项:a、每次用完后,立即清洗电泳槽,灌胶模具,玻璃板(先用自来水洗干净,再用蒸馏水冲一遍后放在架子上晾干)。
b、扣紧或打开灌胶模具时要用力均匀,以免压坏玻璃板。
c、盖上电泳槽的盖子时,务必注意正负极不要接反。
Bio-Rad电泳仪操作规程

Bio-Rad电泳仪操作规程一、实验室准备及前置操作1.确认所需试剂齐全,按照试剂说明书正确配制试剂。
2.准备电泳池:将电泳池放在水平台上,并在池底加入适量的电泳缓冲液,上层电泳缓冲液加入清水至电泳池上部。
3.安装电泳模板:将电泳模板安装于电泳池内,使用扳手或洛氏钳拧紧螺丝未水密。
4.准备样品:将样品于电泳缓冲液中混合均匀,加入电泳模板中的样品槽中,尽量避免气泡。
5.准备DNA标尺:选择合适的DNA标尺,并按照说明书添加至样品中。
二、电泳仪的操作流程1.打开电源:按照电泳仪说明书将电泳仪电源插座插入电源接口,按下电源开关按钮打开电泳仪电源。
2.设置温度:根据实验需要,在电泳仪面板上按照说明书设置电泳仪的工作温度。
3.设置电压和电流:根据实验需要,在电泳仪面板上按照说明书设置电泳仪的电压和电流,通常情况下,实验初期需要较低电流和电压,随后增加直到试样达到最佳分离效果。
4.加载样品:将样品及DNA标尺加入电泳模板,并注意样品槽的填充水平是否均匀,注意避免样品和标尺之间的干扰。
5.开始电泳:确认样品准备和电泳仪设置无误后,按下“开始电泳”按钮启动电泳过程。
6.结束电泳:确定电泳时间到达后,按下“停止电泳”按钮停止电泳过程。
7.处理电泳后试样:将电泳模板取出,使用染色剂染色并传递至显微镜下观察或进行下一步实验步骤。
8.关闭电源:关闭电泳仪电源,拔下电源接口。
三、维护和清洁电泳仪1.频繁检查电泳仪电路板的情况,并用除尘器清洁电泳仪的内部环境,保持电泳仪清洁干净。
2.经常检查电泳仪的液位是否充足、密封是否完好,电泳模板螺钉是否松动等问题,及时维护。
3.定期对电泳仪进行全面的清洁、保养和维修,确保电泳仪的准确性和稳定性。
四、实验注意事项1.电泳缓冲液的pH值要控制在所选用DNA电泳缓冲液的限度内。
2.电泳的时间、电压、电流、电极介质、电泳芯板质量等参数的选择应根据实验目的、样品性质和操作经验等综合考虑。
3.操作时注意安全,不要将电泳仪中的试样触碰到电极、线路和电泳芯板,以免引起放电等事故。
BIO-RAD凝胶电泳成像系统的操作方法

BIO-RAD凝胶电泳成像系统的操作方法BIO-RAD凝胶电泳成像系统的操作方法
1.接通电源和照相机电源
2.打开开关,预热30分钟
3.打开软件,文件菜单中gel-doc.xr
4.按live/focus,30s后按autoexpose or manualexpose
5.调iris,zoom ,focus调节图像清晰度,之后按freeze拍照,保存。
BIO-RAD 凝胶成像系统Universal Hood Ⅱ
仪器性能:
拍摄对象:核酸琼脂糖凝胶电泳
X光胶片
软件功能:获取并处理图像
测定图像光密度
操作步骤:
1. 开启成像系统,电脑电源
2. 打开Quality One软件,调整光源所示图像
3. 观察并调整曝光时间,获得图像并保存
4. 调整图像,并对图像进行光密度分析
5. 关闭所有电源。
电泳纯度(非还原型SDS-PAGE)测定标准操作规程

细胞因子电泳纯度(非还原型SDS-PAGE)测定标准操作规程依据:《中华人民共和国药典》2005年版第三部。
范围:适用于细胞因子纯度的检测。
目的:检测细胞因子蛋白质纯度。
原理:蛋白质具有不同的电荷和分子量,在经过阴离子去污剂SDS处理后,蛋白质分子上的电荷被中和,在聚丙稀酰胺凝胶电泳时,不同的蛋白质按照其分子量大小进行分布,电泳迁移率仅取决于蛋白质的分子量。
由于不连续的PH梯度作用,样品被压缩成一条狭窄区带,采用染色液染色,经扫描仪扫描胶片可及时观察结果。
内容:1 材料1.1样品:经二人复核批号无误后检测1.2试剂甲醇 CH3OH 分析纯无水乙醇 CH3CH2OH 分析纯浓盐酸 HCl 分析纯甘油 C3H8O3分析纯硝酸银 AgNO3分析纯重铬酸钾 K2Cr2O7分析纯正丁醇 C4H9OH 分析纯甲醛 HCHO 分析纯丙烯酰胺 CH2CHCONH2分析纯甲叉双丙烯酰胺(CH2CHCONH2)2CH2分析纯过硫酸铵(NH4)2S2O8分析纯TEMED(四甲基乙二胺)(CH3)2N(CH2)2N(CH3)2分析纯甘氨酸 C2H5NO2分析纯SDS(十二烷基硫酸钠)C12H25O4SNa 分析纯乙酸 CH3COOH 分析纯碳酸钠 Na2CO3分析纯溴酚蓝 C19H10Br4O5S 分析纯1.3注射用水:符合《中华人民共和国药典》2005年版要求。
1.4设备1.4.1扭力天平上海第二天平仪器厂1.4.2垂直板状电泳槽北京东方仪器厂1.4.3恒温恒流电泳仪法玛西亚公司1.4.4快速电泳槽BIORAD USA1.4.5全自动扫描仪CS-930 日本岛津1.4.6TS-1型摇床1.5器皿:500ml瓶、100ul移液器、1ml吸管、10ml吸管3000ml三角瓶、平皿、1000ml烧杯、80ml烧杯,以上器皿经本室洗刷组处理。
2 方法2.1准备工作2.1.1工作环境:控制区,确认场地清场合格,摘下“清场合格”标志牌,挂上“使用中”标志牌。
BIORAD电泳仪及SDS-PAGE凝胶电泳操作规范

使用BIO-RAD电泳仪进行SDS-PAGE凝胶电泳的操作规范实验原理根据蛋白分子量亚基的不同而分离蛋白,在样品介质和丙烯酰胺凝胶中加入离子去污剂和强还原剂后,蛋白质亚基的电泳迁移速率主要取决于亚基分子量的大小。
实验所用仪器FR-200A全自动紫外与可见分析装置上海复日科技有限公司电泳仪 BIO-RAD公司TS-1型脱色摇床江苏海门市其林贝尔仪器制造有限公司实验用试剂低分子量蛋白Maker TAKARA4*上样缓冲 TAKARAPagn Blue protein staining solution Fermentas试剂的配制1.贮液的配制(1)凝胶储液取30g丙烯酰胺+0.8g甲叉双丙烯酰胺0.8g ,先用35ml双蒸水溶解,搅拌,直到溶液变成透明,再用双蒸水稀释至100ml,过滤。
棕色瓶4℃保存一个月。
(2)1 mol/l Tris-HCL (PH 8.8)12.1g Tris(三羟甲基氨基甲烷)溶解在80ml双蒸水中,用4mol/l盐酸调PH至8.8。
再用双蒸水稀释至100ml,保存在4℃冰箱。
(3)1.0 mol/l Tris-HCL (PH 6.8)6.06g Tris溶解在40ml双蒸水中,用用4mol/l盐酸调PH至6.8。
再用双蒸水稀释至50ml,保存在4℃冰箱。
(4)10%过硫酸铵(APS)0.1g过硫酸铵+1ml双蒸水。
使用前新鲜配制(5)Tris–甘氨酸电泳缓冲液的配制(25mmol/L Tris;250mmol/L甘氨酸(pH8.3))30.3gTris+ 144.2g甘氨酸+ 10gSDS,双蒸水定容至1L。
每次使用时10倍稀释。
(6)样品缓冲液使用4*SDS-PAGE loading buffer(Takara公司),上样缓冲与样品比例1:3混匀,之后煮沸5min。
2.凝胶的配制注:上表所标体积为配制两块胶的用量。
若配制一块或多块,可按比例减半或加倍。
具体步骤如下:1、样品制备:40 µL蛋白+5*上样缓冲液10 µL,煮沸5 min,冷却后放冰箱下层保存,备用2、制胶1)用专用的医用棉口罩将胶板擦拭干净,电泳槽清洗干净,组装模具。
BIORAD-2DE中文操作说明

目录第一章样品制备 21.1 一般性原则 21.2 样品制备程序 3 1.2.1 培养细胞样品处理方法 31.2.2 组织样品处理方法 31.2.3文献报道较多的裂解液配方 4第二章第一向等电聚焦(IEF) 72.1 IPG胶条的水化和电泳 72.1.1 仪器 72.1.2 试剂 72.1.3 实验步骤 72.2 IPG胶条的平衡 92.2.1 仪器 102.2.2 试剂 102.2.3 实验步骤 10第三章第二向SDS电泳 123.1 垂直SDS-PAGE 123.1.1 溶液 123.1.2 灌胶步骤 123.1.3 电泳步骤 14第四章 2-DE胶蛋白质点的检测 154.1 考马斯亮兰染色 154.1.1 经典的考马斯亮兰染色程序 154.1.2 Neuhoff胶体考染法 154.1.3 热考马斯亮兰染色及二次染色法 164.2 硝酸银染色 16Appendix I Troubleshooting 18 Appendix II Solutions 232DE分析流程:样品制备(Sample preparation)固相pH梯度胶条的水化(IPG strip rehydration)第一向等电聚焦(IEF)第一向胶条的平衡( IPG strip equilibration)第二向SDS电泳(SDS-PAGE)检测染色(Detection/Staining)第一章样品制备(Sample Preparation)1.1一般性原则:样品制备是双向电泳中最为关键的一步,这一步处理的好坏将直接影响2-DE结果。
目前并没有一个通用的制备方法,尽管处理方法是多种多样,但都遵循几个基本的原则:1)尽可能的提高样品蛋白的溶解度,抽提最大量的总蛋白,减少蛋白质的损失;2)减少对蛋白质的人为修饰;3)破坏蛋白质与其他生物大分子的相互作用,并使蛋白质处于完全变性状态。
根据这一原则,样品制备需要四种主要的试剂:离液剂(chaotropes),主要包括尿素(Urea)和硫脲(thiourea);表面活性剂(sufactants),也称去垢剂,早期常使用NP-40、TritonX-100等非离子去垢剂,近几年较多的改用如CHAPS与Zwittergent系列等双性离子去垢剂;还原剂(reducing agents),最常用的是二硫苏糖醇(DTT),也有用二硫赤藓糖醇(DTE)以及磷酸三丁酯(TBP)等。
BIO-RAD MYCYCLER PCR 操作步骤

BIO-RAD MyCycler PCR仪操作步骤(一)接通电源,打开开关,如果仪器处于备用状态,可直接按显示屏上的standby键,在通过一个快速自动诊断程序后,初始界面将会出现。
(二)按F2键,初始界面上将会出现选择菜单。
(三)选择“Custom”项,并按Enter键以建立新的运行程序。
如果菜单已有的程序与你需要的程序相近,那么你也可以在选择菜单对已有的模板程序进行编辑。
(四)编辑运行程序(1)仪器默设的运行程序有3个简单的步骤组成。
95℃30秒,55℃30秒,72℃30秒。
你将根据需要按如下步骤更改。
(2)按F4键增加或删除部分步骤,注意竖线把不同的循环周期搁开,正在运行的循环次数在第一次循环的左下脚标出。
(3)采用方向键在不同的步骤之间转换,调定点温度将显示在温度曲线上方。
相反运行时间将显示在温度曲线下方。
循环次数将在每一个循环最后一步的右下角,后跟随“x”符号。
(4)在用方向键选择要更改的温度调定点以及运行时间后,按F3,选择要更改的操作,此时会弹出一个新的界面,在此界面下,可以输入所需的时间或温度。
(5)程序编辑完成后按F5。
(五)在选择菜单中点“Sae Protocol As…”保存编辑程序,以字母与数字组合命名。
然后按Enter键完成操作。
(六)此时初始界面会出现,按F1键进入程序储存库,用箭头键选择你新编辑的程序,随后按Enter键,将会出现一个选择菜单。
选择“Run Protocol”(七)随之出现运行方案界面。
提示是否想热启动(如果你选择“YES”将会进一步提示你所需要的热启动温度),如果你选择“Algorithmic Measurement”,界面上还会让你选择温度标准以及取样容积标准。
(八)按F5开始运行程序。
(九)运行完毕后按Stop键完成操作。
电泳仪操作规程

一、目的:建立电泳仪的操作规程,保证实验结果的有效、准确性。
二、适用范围:适用于电泳仪的使用。
三、职责:设备管理人员、操作人员。
四、内容:
1.电泳前,禁止将电泳仪附带的电泳导线连接到电泳仪电源上。
2.把U型凝胶托盘放入制胶器中,然后把配好的琼脂糖凝胶倒入制胶器中(注意凝胶液体温度控制在60℃左右),再把试样格插入凝胶托盘的两边槽内。
3.待凝胶聚合后,轻轻拔掉试样格,注意先拔一侧。
垂直拔出会使胶孔产生真空,导致部分凝胶被带出。
把凝胶托盘移入电泳仪,加入缓冲液,使缓冲液没过凝胶1-2mm。
(注意:加样孔靠近负极一端)
4.用移液器将样品加入加样孔中。
5.盖好上盖,连接电泳仪电源与电泳仪之间的电泳导线。
6.注意不要接错正负极,检查无误后接通电泳仪电源,根据实际情况选择适宜的电压(一般情况200V-250V,电压过高会产生液体过热影响电泳结果)即可电泳。
7.电泳实验结束后,先关闭电泳仪电源,随之拔出电泳导线,然后打开电泳仪上盖,取出凝胶托盘。
把凝胶轻轻推入到用于照相的凝胶托板上准备照相。
8.仪器使用登记。
SDS聚丙烯酰胺凝胶电泳步骤

【跑电泳的步骤】1、取出电泳仪器和四个烧杯;2、将超纯水倒入烧杯中,保鲜膜封口;3、配制过硫酸铵溶液(AP):+0.9g超纯水,保鲜膜封口;4、取出所有药品。
Tris-HCL(88,,TEMED,SDS,Acry-bis,按次序排好;5、拿卷纸平铺,移液枪枪头:三个蓝,两个黄,一个白,移液枪3只;6、先配分离胶7、组装凝胶模具,插好板(1)海绵用自来水润湿,插在下面槽里;(2)板:Bio-Rad前后玻璃板,注意前后顺序,夹子夹好,塞枪头于夹子上。
8、配胶见配方,用2号蓝混匀所有试剂;9、加液至支架上方,用超纯水液封(不能用气泡);10、待界面清晰后,用滤纸将水吸干;11、配浓缩胶;12、插梳子:一个个斜着插,有字面朝向我;13、拔下梳子,转移(有字朝里,白色架子垫黄纸,将玻璃板卡在绿卡下面,不要有空隙,装去离子水,不能漏(1h),等待。
),样品煮沸3分钟;14、用10uL移液枪移液,移液,每个依次加入梳槽内;15、电压(浓缩胶)90V,电流20mV;16、盖上盖子:红对红,黑对黑;插上电源插头:红对红,黑对黑。
【附配方】分离胶separating gel (5mL) ( 1个板1板2板Prescription12%10%%mL mLDel H2O mL mL3mL1.5M Tris-HCL mL mL mL mL mL30%Acry/bis 2 mL mL mL mL单体胶(29.2g+0.8g/100mL)10%SDS50uL50uL50uL35uL70uL10%AP50uL50uL50uL35uL70uLTEMED2uL2uL(5 uL)2uL 7uL(夏)~8uL浓缩胶Stacking gel ( 2板3mL)Prescription Volume(2 mL)(2 mL)Del H2O0.5M Tris-HCL 250uL375uL30%单体胶330uL495uL10%SDS20uL30uL10%AP20uL30uLTEMED2uL(5 uL)3uL(8 uL冬,5 uL夏)【附所需试剂配制】1、30%单体胶(30%单体胶即30%T,%C)配制50mL:丙烯酰胺Acry:14.6g,甲叉双丙烯酰胺bis:0.4g;(100mL:Acry 29.2g,bis 0.8g) 超纯水定容至50mL,uM膜过滤,4℃保存(一个月);说明:单体浓度%T是指单体总质量(丙烯酰胺+双丙烯酰胺)在溶液中的百分比(m/V),可选择的范围为3%~30%;%C是指双丙烯酰胺与单体总质量的比值(m/m),也指交联度。
BIORAD电泳仪及SDS-PAGE凝胶电泳操作规范
使用BIO-RAD电泳仪进行SDS-PAGE凝胶电泳的操作规范实验原理根据蛋白分子量亚基的不同而分离蛋白,在样品介质和丙烯酰胺凝胶中加入离子去污剂和强还原剂后,蛋白质亚基的电泳迁移速率主要取决于亚基分子量的大小。
实验所用仪器FR-200A全自动紫外与可见分析装置上海复日科技有限公司电泳仪 BIO-RAD公司TS-1型脱色摇床江苏海门市其林贝尔仪器制造有限公司实验用试剂低分子量蛋白Maker TAKARA4*上样缓冲 TAKARAPagn Blue protein staining solution Fermentas试剂的配制1.贮液的配制(1)凝胶储液取30g丙烯酰胺+0.8g甲叉双丙烯酰胺0.8g ,先用35ml双蒸水溶解,搅拌,直到溶液变成透明,再用双蒸水稀释至100ml,过滤。
棕色瓶4℃保存一个月。
(2)1 mol/l Tris-HCL (PH 8.8)12.1g Tris(三羟甲基氨基甲烷)溶解在80ml双蒸水中,用4mol/l盐酸调PH至8.8。
再用双蒸水稀释至100ml,保存在4℃冰箱。
(3)1.0 mol/l Tris-HCL (PH 6.8)6.06g Tris溶解在40ml双蒸水中,用用4mol/l盐酸调PH至6.8。
再用双蒸水稀释至50ml,保存在4℃冰箱。
(4)10%过硫酸铵(APS)0.1g过硫酸铵+1ml双蒸水。
使用前新鲜配制(5)Tris–甘氨酸电泳缓冲液的配制(25mmol/L Tris;250mmol/L甘氨酸(pH8.3))30.3gTris+ 144.2g甘氨酸+ 10gSDS,双蒸水定容至1L。
每次使用时10倍稀释。
(6)样品缓冲液使用4*SDS-PAGE loading buffer(Takara公司),上样缓冲与样品比例1:3混匀,之后煮沸5min。
2.凝胶的配制注:上表所标体积为配制两块胶的用量。
若配制一块或多块,可按比例减半或加倍。
具体步骤如下:1、样品制备:40 µL蛋白+5*上样缓冲液10 µL,煮沸5 min,冷却后放冰箱下层保存,备用2、制胶1)用专用的医用棉口罩将胶板擦拭干净,电泳槽清洗干净,组装模具。
Bio-Rad 核酸蛋白测定仪中文操作说明

SmartSpec™ Plus 核酸蛋白测定仪中文操作指南(本指南仅供参考,以英文说明书为准)第一章仪器介绍SmartSpec Plus 核酸蛋白测定仪比其它许多台式分光光度仪拥有更完善的特点和功能,其性能优越,运行稳定,功能强大。
特别适用于生命科学研究SmartSpec Plus工作波长在200-800nm,是核酸和蛋白样品常规定量的完美工具。
SmartSpec Plus可用于●DNA,RNA 和寡核苷酸的定量●用Bradford,Lowry和BCA检测法定量蛋白●监控细胞的生长状况●简易的动力学分析●波长扫描和峰检测更简单的样品分析SmartSpec Plus 的设计充分考虑了用户的需求。
简易的菜单式界面简化了测试过程,只需触摸一下按键就可以提供常用样品的计算结果。
转换因子可以储存和修改。
SmartSpec Plus能提供以下计算结果,如:●显示核酸纯度的A260/A280比率●定量分析(考虑稀释因子)●μg/ml样品浓度(寡核苷酸pmol/μl)●寡核苷酸的摩尔消光系数和分子量在测试结束时,打印显示使用者,日期和结果的报告核酸定量SmartSpec Plus能满足定量PCR产物、核酸制备或细胞转染样品的定量检测要求。
选择定量dsDNA、ssDNA或RNA,并从预设的转换因子中选择或输入一个最适合代测样品的数值。
SmartSpec Plus能提供吸收值、浓度和纯度值,确保下游工作的顺利进展。
SmartSpec Plus简化了DNA、RNA寡核苷酸的定量过程。
当你输入序列、长度或组成时,SmartSpec Plus会以μg/ml或pmol/μl为单位显示出样品浓度,并计算摩尔消光系数和分子量。
蛋白定量SmartSpec Plus安装了Bradford,Lowry和BCA蛋白定量检测方法的预编程序,每个检测方法都具有其独特的特性,方便数据收集及对测试结果进行全面分析。
简单明了的菜单引导你完成整个测试流程,确保检测到所有标准品和重复样品。
毛细管电泳仪的使用(原创)电泳仪的使用方法

毛细管电泳仪的使用(原创)*电泳仪的使用方法毛细管, 电泳仪, 原创概要:准备工作1.毛细管的制备:①剪略长于需要长度的毛细管(使用专用的裁剪纱布),断端利用镊子小心移除。
②烧窗:从毛细管进液端开始到实验者需要长度的位置烧窗,窗口尽量小,过程中可用防护工具对窗口两端.准备工作1.毛细管的制备:①剪略长于需要长度的毛细管(使用专用的裁剪纱布)。
断端利用镊子小心移除。
②烧窗:从毛细管进液端开始到实验者需要长度的位置烧窗,窗口尽量小,过程中可用防护工具对窗口两端的毛细管进行防护。
烧好的窗需用粘有酒精的棉花擦拭。
注:检测的有效长度为从毛细管进液端到检测窗之间的距离,这个距离一般根据实验者的需要自己掌握。
③取出卡槽:打开安放卡槽区域的门。
旋转固定卡槽的杠杆90度,按卡槽中央的释放按钮,握住卡槽的两端向上抽出卡槽。
④安装毛细管:将进液端的毛细管小心的从卡槽的出口插入。
进入时可轻轻旋转。
当窗口到达检测点时停止插入,将游离的进液端毛细管从卡槽的入口引出。
并固定好毛细管外端的橡胶管。
注:插入毛细管时小心操作,窗口位置的毛细管非常容易破裂,需格外谨慎。
过程中切忌使用暴力。
⑤安放卡槽:用与取出的步骤相反顺序的操作安放卡槽。
2.添加样品,缓冲液及冲洗液:①为了减少样品及缓冲液的挥发。
在插入加液管前可在加样管固定器的小孔内加入少量的ddH2O。
②已添加好的加液管,在插入转盘上的固定槽前先离心2min加液时注意加样枪头紧贴管壁,防止气泡的产生。
③为了减少虹吸作用,需要保持入口处和出口处的running buffer量一致,对于500ul体积的加液管,推荐加入500ul的剂量;对1.5ml体积的加液管,推荐1.3ml的剂量。
④推荐加入样品的最小剂量为50ul检测需要的最小剂量为15ul。
⑤在出口处的waste管中加入50ul的ddH2O,可以防止检测端的毛细管被冲洗液的残留污染。
电泳仪的操作1.插上电源。
检查电泳仪的连接情况。
Bio-rad 双向电泳系统标准操作规程

Bio-rad 双向电泳系统标准操作规程1、目的:正确使用Bio-rad 双向电泳系统,确保Bio-rad 双向电泳系统正常运行。
2、适用范围:Bio-rad 7cm/11cm/17cm双向电泳系统。
3、责任人:双向电泳系统操作人员。
4、程序:4.1、第一向等电聚焦4.1.1、准备工作及注意事项用标配的刷子小心将聚焦盘清洗干净,注意聚焦盘两端的两根电极丝,晾干后备用;根据样品使用合适的水化上样液,对于不同的聚焦盘的上样体积可参考下表:4.1.2、上样4.1.2.1、从冰箱中取-20℃冷冻保存的水化上样缓冲液(不含DTT,不含Bio-Lyte)一小管,置室温溶解,加入合适的DTT与Bio-Lyte,充分混匀。
4.1.2.2、从小管中取出适量水化上样缓冲液与样品充分混匀。
4.1.2.3、取出-20℃冷冻保存的IPG预制胶条,室温中放置10分钟。
4.1.2.4、沿着聚焦盘或水化盘中槽的边缘至左而右线性加入样品。
在槽两端各1cm左右不要加样,中间的样品液一定要连贯。
注意:不要产生气泡。
否则影响到胶条中蛋白质的分布。
4.1.2.5、当所有的蛋白质样品都已经加入到聚焦盘或水化盘中后,用镊子轻轻的去除预制IPG胶条上的保护层。
4.1.2.6、分清胶条的正负极,轻轻地将IPG胶条胶面朝下置于聚焦盘或水化盘中样品溶液上,使得胶条的正极(标有+)对应于聚焦盘的正极。
确保胶条与电极紧密接触。
不要使样品溶液弄到胶条背面的塑料支撑膜上,因为这些溶液不会被胶条吸收。
同样还要注意不使胶条下面的溶液产生气泡。
如果已经产生气泡,用镊子轻轻地提起胶条的一端,上下移动胶条,直到气泡被赶到胶条以外。
4.1.2.7、在每根胶条上覆盖1-3ml矿物油,防止胶条水化过程中液体的蒸发。
需缓慢的加入矿物油,沿着胶条,使矿物油一滴一滴慢慢加在塑料支撑膜上。
4.1.2.8、对好正、负极,盖上盖子。
设置等电聚焦程序。
4.1.3、设置程序4.1.3.1、打开电源;4.1.3.2、根据情况选择水化(REHYDRATION),预设的程序(PRESET METHOD),储存的程序(STORED METHOD),新的程序(NEW METHOD);4.1.3.3、如果只需要水化,选择水化(REHYDRATION)选项,在接下来的界面选择主动水化或者被动水化、水化温度、水化时间;4.1.3.4、如果需要跑完整的程序,选择新的程序(NEW METHOD), 在接下来的界面选择是否水化,并设置相应的等电聚焦程序,设置完成后,在最后的界面选择总的胶条数、限电流和聚焦温度,然后开始运行程序。
BIO-RAD双向电泳中文手册

ProteomicsBio-Rad蛋白质组双向电泳实验操作手册 ProteomeWorks TM System双向电泳实验流程z样品制备(Sample preparation)z固相预制胶条的水化(IPG strip rehydration)z第一向等电聚焦(IEF)z胶条的平衡(IPG strip equilibration)z第二向SDS-PAGE电泳(SDS-PAGE electrophresis) z凝胶的染色及检测(Detection/Staining)z PDQuest软件分析(Software analysis)z质谱鉴定(Protein identification)目 录第一章 实验材料1.1 IPG预制胶条及载体两性电解质1.2 蛋白质定量试剂盒及其试剂1.3 试剂盒及其试剂1.4 化学试剂1.5 蛋白质Marker1.6 染色试剂1.7 注意事项第二章 SDS-PAGE聚丙烯酰胺凝胶电泳 2. 1 溶液的配制2. 2 SDS-PAGE凝胶的配制2. 3 操作方法2. 4 注意事项第三章 双向电泳3. 1 溶液配制3. 2 操作步骤3. 3 注意事项附录1 双向电泳完整的操作步骤附录2 聚丙烯酰胺凝胶电泳凝胶的配置 附录3 细胞样品的一般处理步骤附录4 组织样品的一般处理步骤第一章 实验材料1.1 IPG预制胶条及载体两性电解质(一)IPG预制胶条(美国Bio-Rad公司),-20℃冰箱保存cm 163-2000 IPG预制胶条 pH 3-10,7,nonlinear(NL) 163-2002cmIPG预制胶条 pH 3-10, 7cm 163-2001 IPG预制胶条 pH 4-7,7cm 163-2003 IPG预制胶条 pH 3-6,7cm 163-2004 IPG预制胶条 pH 5-8,7cm 163-2005 IPG预制胶条 pH 7-10,7IPG预制胶条 pH 3-10,17cm 163-2007cm,nonlinear(NL) 163-2009 IPG预制胶条 pH 3-10, 17IPG预制胶条 pH 4-7,17cm 163-2008 IPG预制胶条 pH 3-6,17cm 163-2010 IPG预制胶条 pH 5-8,17cm 163-2011 IPG预制胶条 pH 7-10,17cm 163-2012 IPG预制胶条 pH 3-10,18cm 163-2032 IPG预制胶条 pH 3-10, 18cm,nonlinear(NL) 163-2033 IPG预制胶条 pH 4-7,18cm 163-2034 IPG预制胶条 pH 3-6,18cm 163-2035 IPG预制胶条 pH 5-8,18cm 163-2036 IPG预制胶条 pH 7-10,18cm 163-2037 IPG预制胶条 pH 3-10,11cm 163-2014 IPG预制胶条 pH 3-10, 11cm,nonlinear(NL) 163-2016 IPG预制胶条 pH 4-7,11cm 163-2015 IPG预制胶条 pH 3-6,11cm 163-2017 IPG预制胶条 pH 5-8,11cm 163-2018 IPG预制胶条 pH 7-10,11cm 163-2019 IPG预制胶条 pH 3.9-5.1,17cm163-2020163-2021 IPG预制胶条 pH 4.7-5.9,17cmIPG预制胶条 pH 5.5-6.7,17cm163-2022163-2023 IPG预制胶条 pH 6.3-8.3,17cm163-2038 IPG预制胶条 pH 3.9-5.1,18cm163-2039 IPG预制胶条 pH 4.7-5.9,18cm163-2040 IPG预制胶条 pH 5.5-6.7,18cm163-2041 IPG预制胶条 pH 6.3-8.3,18cm163-2024 IPG预制胶条 pH 3.9-5.1,11cm163-2025 IPG预制胶条 pH 4.7-5.9,11cm163-2026 IPG预制胶条 pH 5.5-6.7,11cm163-2027 IPG预制胶条 pH 6.3-8.3,11cmIPG预制胶条 pH 3.9-5.1,7cm 163-2028 IPG预制胶条 pH 4.7-5.9,7cm 163-2029 IPG预制胶条 pH 5.5-6.7,7cm 163-2030 IPG预制胶条 pH 6.3-8.3,7cm 163-2031 (二)载体两性电解质(美国Bio-Rad公司),4℃冰箱保存Bio-Lyte 3/10 Ampholyte,40%,10ml 163-1112 Bio-Lyte 3/10 Ampholyte,40%,25ml 163-1113 Bio-Lyte 3/5 Ampholyte,20%,10ml 163-1132 Bio-Lyte 4/6 Ampholyte,40%,10ml 163-1142 Bio-Lyte 4/6 Ampholyte,40%,25ml 163-1143 Bio-Lyte 5/7 Ampholyte,40%,10ml 163-1152 Bio-Lyte 5/7 Ampholyte,40%,25ml 163-1153 Bio-Lyte 6/8 Ampholyte,40%,10ml 163-1162 Bio-Lyte 6/8 Ampholyte,40%,25ml 163-1163 Bio-Lyte 7/9 Ampholyte,40%,10ml 163-1172 Bio-Lyte 8/10 Ampholyte,20%,10ml 163-1182 Bio-Lyte 5/8 Ampholyte,40%,10ml 163-1192 Bio-Lyte 5/8 Ampholyte,40%,25ml 163-1193 (三)等电聚焦上样缓冲液(美国Bio-Rad公司),4℃冰箱保存100×ReadyStrip Buffer,for pH 7-10 IPG Strips,1ml 163-2093 Bio-Lyte 3/10 Ampholyte,100×,1ml 163-2094163-2095 100×ReadyStrip Buffer,for pH 6.8-8.3 IPG Strips,1ml100×ReadyStrip Buffer,for pH 5.5-6.7 IPG Strips,1ml163-2096163-2097 100×ReadyStrip Buffer,for pH 4.7-5.9 IPG Strips,1ml163-2098 100×ReadyStrip Buffer,for pH 3.5-5.1 IPG Strips,1ml1.2 蛋白质定量试剂盒及其试剂Protein Assay Kit I,bovine γ-globulinstandard 500-0001standard 500-0002 Protein Assay Kit II,BSAProtein Standard I,bovine γ-globulin,1bottle 500-0005500-0006 Protein Assay Dye Reagent Concentrate,450mlbottle500-0007 Protein Standard II,bovine serum albumin,1standard500-0121 RC DC Protein Assay Kit I,bovine γ-globulin500-0122 RC DC Protein Assay Kit II,BSAstandard1.3 试剂盒及其试剂ReadyPrepExtractionKit 163-2100 SequentialTributylphosphine(TBP),200mM,0.6ml 163-2101163-2102 ReadyPrep Sequential Extraction Kit Reagent 1,1vial163-2103 ReadyPrep Sequential Extraction Kit Reagent 2,1vial163-2104 ReadyPrep Sequential Extraction Kit Reagent 3,1vialKit 163-2105 StarterReadyPrep2-DReadyPrep 2-D Starter Kit Rehydration/Sample Buffer 163-2106DTT 163-2107 ReadyPrep Starter Kit Equilibration Buffer I,withBufferIIEquilibration163-2108 ReadyPrepStarterKitIodoacetamide,30g 163-2109E.coli Protein Sample,lyophilized,2.7mg163-2110 ReadyPrep Overlay Agarose,50ml 163-2111 1.4 化学试剂尿素Urea,250g 161-0730 尿素Urea,1kg 161-0731Resin 142-6425 AG 501-X8(D)MixedBedCHAPS,1g 161-0460 CHAPSO,1g 161-0465 Triton X-100,500ml 161-0407 DTT(Dithiothreitol),1g 161-0610 DTT(Dithiothreitol),5g 161-0611Tributylphosphine(TBP),200mM,0.6ml 163-2101 溴酚蓝(Bromophenol Blue),10g 161-0404 矿物油(Mineral Oil),500ml 163-2129 SDS,25g 161-0300 SDS,100g 161-0301 SDS,1kg 161-0302 Tris,100g 161-0715 Tris,500g 161-0716 Tris,1kg 161-0719Iodoacetamide,30g 163-2109 低熔点琼脂糖,25g 161-3111161-0717 甘氨酸,250g甘氨酸,1kg 161-0718甘氨酸,2kg 161-0724丙烯酰胺(Acrylamide),99.9%,100g 161-0100 丙烯酰胺(Acrylamide),99.9%,500g 161-0101 丙烯酰胺(Acrylamide),99.9%,2kg 161-0103 丙烯酰胺(Acrylamide),99.9%,1kg 161-0107 丙烯酰胺(Acrylamide),99.9%,5kg 161-0108 甲叉双丙烯酰胺(Bis),5g161-0200 甲叉双丙烯酰胺(Bis),50g 161-0201161-0202 PDA(Piperazine Diacrylamide),10g161-0203 PDA(Piperazine Diacrylamide),50g过硫酸氨(Ammonium Persulfate),10g 161-0700161-0754 过硫酸氨(Ammonium Persulfate),100gTEMED,5ml 161-0800TEMED,50ml 161-0801 甘油国产或Sigma 硫尿Sigma1.5 蛋白质Marker2-D SDS-PAGE Standards,500μl161-0320161-0303 SDS-PAGE Standards,high range,200μlSDS-PAGE Standards,low range,200μl 161-0304161-0317 SDS-PAGE Standards,broad range,200μlPolypeptide SDS-PAGE Standards,200μl 161-0326 Precision Protein Standards,unstained,1500μl,150 applications 161-0362Precision Protein Standards,prestained,500μl,50 applications 161-0372161-0325 Kaleidoscope Polypeptide Standards,500μlKaleidoscope Prestained Standards,broad range,500μl 161-0324161-0309 Prestained SDS-PAGE Standards,high range,500μlPrestained SDS-PAGE Standards,low range,500μl161-0305 Prestained SDS-PAGE Standards,broad range,500μl 161-0318 IEF Standards,pI range 4.45-9.6,250μl 161-0310 1.6 染色试剂Coomassie Brilliant Blue R-250,10g161-0400161-0406 Coomassie Brilliant Blue G-250,10gIEF Gel Staining Solution,1L 161-0434 Coomassie Brilliant Blue R-250 Staining Solutions Kit 161-0435Coomassie Brilliant Blue R-250 Staining Solutions,1L 161-0436161-0437 Coomassie Brilliant Blue R-250 Staining Solutions,4×1L161-0438 Coomassie Brilliant Blue R-250 Destaining Solutions,1LCoomassie Brilliant Blue R-250 Destaining Solutions,4×1L 161-0439 Bio-Safe Coomassie Stain,1L 161-0786 Bio-Safe Coomassie Stain,5L 161-0787 SYPRO Ruby Protein Gel Stain,200ml 170-3126 SYPRO Ruby Protein Gel Stain,1L 170-3125 SYPRO Ruby Protein Gel Stain,5L 170-3138 Silver Stain Kit 161-0443Kit 161-0450 PlusSilverStain1.7 注意事项1.双向电泳中所用的化学试剂纯度要高,至少为分析级。
biorad序列电泳使用说明书

Sequi-Gen®GTNucleic Acid Electrophoresis Cell Instruction Manual Catalog Numbers165-3860, 165-3861, 165-3862 and 165-3863T able of ContentsPage Warranty Information ........................................................................Inside Front Cover Section 1 General Information (1)1.1 Introduction to Sequi-Gen GT DNA Electrophoresis Cell (1)1.2 Specifications (5)Section 2 Description of Major Parts (6)2.1 Sequi-Gen GT Parts (6)2.2 Gel Reagents and Electrophoresis Buffers (7)Path (8)2.3 ElectricalSection 3 Cleaning and Maintenance (8)3.1 Cleaning and Siliconizing Plates (8)3.2 Cleaning Plastic Parts (9)Section 4 Operating Instruction (10)Assembly (10)4.1 Before4.2 Assembling the Glass Plate Sandwich (10)4.3 Casting the Gel (12)4.4 Preparing for Operations (17)4.5 Loading the Gel (18)Electrophoresis (19)4.6 Gel4.7 Disassembly (20)Section 5 Troubleshooting Guide (22)Troubleshooter (22)5.1 Operational5.2 DNA Sequencing Artifacts (23)Section 6 Equipment and Accessories (26)6.1 Sequi-Gen GT Nucleic Acid Electrophoresis Cells and Accessories (26)Reagents (30)6.2 Electrophoresis6.3 Power Supplies and Slab Gel Dryers (31)6.4 DNA Template Purification, Sequencing, and Cloning Products (31)Handling (32)6.5 LiquidSection 7 Appendix A- Applications (32)7.1 DNA Sequencing Checklist (32)7.2 Standard Gel Protocol (33)7.3 Gel Drying Autoradiography (34)7.4 Applications for Sequi-Gen GT Nucleic Acid Electrophoresis Cell (34)Reading (35)7.5 SuggestedThe precision caster allows quick and easy gel casting without acrylamide spills or waste. By casting the gel with a syringe through the precision caster base, gels can be poured in less than 1 minute. The gel is cast with the glass plate assembly in the horizontal position. Two full-length clamps secure the assembly and allow attachment of the precision caster base to the bottom of the glass plate sandwich. A seal between the caster gasket and the plates is created without tape or grease. The gel is injected from the bottom of the glass plate sandwich (via the injection port of the precision caster base) and moves to the top of the glass plates as a dome-shaped gel front. Acrylamide spills and waste can be eliminated by controlling the flow of the gel front at the top of the glass plates.Modular AssemblyThere are four IPC dimensions to choose from, as shown in Figure 1.1. One universal base functions as the lower buffer chamber for all IPC sizes.21 x 40 cm21 x 50 cm38 x 30 cm38 x 50 cmFig. 1.1. Interchangeable sizes.1.2 SpecificationsGeneral SpecificationsBase footprint16 x 48 cmMaximum unit height65 cm (50 cm cells); 55 cm (40 cm cells); 45 cm (30 cm cells) IPC sizes21 x 40, 21 x 50, 38 x 30 cm and 38 x 50 cm(width x length)Actual gel sizes17 x 40, 17 x 50, 34 x 30 cm, 34 x 50 cmGel thickness range0.25 – 0.75 mmNominal gel volumes (0.25 mm)17 ml (21 x 40 cm); 21 ml (21 x 50 cm); 40 ml (38 x 30 cm);43 ml (38 x 50 cm)Nominal gel volumes (0.40 mm)27 ml (21 x 40 cm); 34 ml (21 x 50 cm); 50 ml (38 x 30 cm);68 ml (38 x 50 cm)Minimum upper buffer volumes500 ml (21 x 40 cm); 575 ml (21 x 50 cm); 650 ml(38 x 30 cm);1,400 ml (38 x 50 cm)Minimum lower buffer volume350 mlMaximum lower buffer volume500 mlElectrical SpecificationsElectrical Safety Certification IEC 1010-1Rated voltage limit3,000 voltsRated power limit100 wattsRated temperature limit60 ˚CElectrical cables Rated to 3,000 volts (VDC)Electrical leads Rated to 3,000 volts (VDC)Banana plugs Rated to 3,000 volts (VDC)•Rinse off all of the detergent with warm water.•Rinse with deionized water.•Wipe the cleaned plate with a large lint free tissue to dry.2.Inspect the plates carefully for pieces of detergent, dried polyacrylamide, or other particles.Rewash if necessary.3.Perform siliconizing under a fume hood, to reduce the hazard from breathing silanizingreagent. Alternatively, several non-toxic, non-corrosive glass plate coating solutions are commercially available. We recommend siliconizing or coating only the outer (long) plate, so that when the plates are separated, the gel sticks to the IPC-bound glass plate.•Use a glass Pasteur pipette to dispense 2 ml of the silanizing reagent onto the front plate. Coat the plate completely and evenly by spreading the silanizing reagent on the plate surface with a large lint free tissue, using a motion that travels from the top to the bottom of the plate.Caution: Do not siliconize the IPC plate unless hexane, heptane, or water is used as a solvent in the silanizing reagent. Other organic solvents will craze or damage the IPC plastic and weaken the adhesive bond.•Never heat an IPC in an oven. Severe damage will result to the adhesive bond. Use siliconizing compounds that react, or cure, at room temperature.Note: If the gels will be fixed or stained, the IPC (short) plate should be siliconized or coated, since its immersion into fixing or staining solutions is not recommended.4.Prior to assembling the plates, apply a small amount of ethanol to each plate and rub todryness with a tissue. Using the same tissue, clean the spacers.3.2 Cleaning Sequi-Gen GT Components1.Rinse the universal base buffer chamber, stabilizer bar, combs, spacers and precisioncaster base, gasket, syringe and tubing assembly with a mild detergent solution in warm water. Use a soft-bristled brush or sponge to remove polyacrylamide gel pieces.Note: Do not snag or break the electrode wire in the universal base while cleaning.2.Rinse thoroughly with warm water and air dry.Compatible Cleaning Agents for Polycarbonate PartsChemically compatible cleaners must be used to ensure long life of parts. These include:•Aqueous solutions of soaps and mild detergents•Organic solvents:•Hexane•Aliphatic hydrocarbons•Alcohols•Methanol•Ethanol•Isopropyl alcohol•Dilute acidsNote on Gel Bubble Formation•The following injection times (from the bottom of IPC to the top) were found to result in bubble-free gels: for 50 cm gels with 0.4 mm spacers, between 40–45 seconds; for50 cm gels with 0.25 spacers, between 50–65 seconds. Injection times of 10 secondsor less can result in bubble formation in the gel.•Bubbles can form at the gel front because of soiled areas or uneven siliconization or coating of the glass plates.•To achieve bubble free gels, thoroughly clean both plates and siliconize the outer glass plate before each use.•If bubbles begin to form at the gel front, hard tapping on top of the IPC assembly (above the bubble formation) while slowly injecting the gel solution should eliminate the bubble. Alternatively, the comb end of the IPC assembly can be momentarily lifted at an angle to facilitate elimination.10.Continue to slowly inject the gel solution until the gel solution emerges a few centimetersfrom the top of the notched (shorter) glass plate (across the entire width of the gel).Important: If pouring a 38 x 50 cm IPC, remove the support that created an incline and lay the unit level on the benchtop (use the Leveling Bubble provided). An additional 2 cm support will be needed to level the IPC assembly. Some users find it convenient to use two1.5 ml tube racks as props.When the gel is past the short plate, lay the syringe on top of IPC assembly until gel polymerization is complete. Do not remove the luer taper from the precision caster base injection port, or the gel solution will drain out of the plates. Do not adjust the syringe plunger after the gel has been cast (Figure 4.8).11.Insert the comb(s) between the plates to the desired depth.•If a sharkstooth comb is used, insert the flat edge of the comb no more than 5 mm past the short glass plate.•Clamp the comb(s) in place with three large metal binder clamps.Fig. 4.8.Syringe position for gel polymerization.•Alternatively, prior to injecting the gel solution, insert the corner of the comb to facilitate comb placement and insertion after gel casting.12.Let the gel polymerize for 30–60 minutes.•After gel polymerization, remove the luer taper from the precision caster base.•The syringe, tubing, and luer taper can be cleaned of any remaining polymerized gel solution by rinsing with hot tap water, followed by a distilled water rinse.13.Remove the precision caster base from the IPC assembly and clean the caster base andgasket of polymerized gel solution with tap water, followed by a distilled water rinse.4.4 Preparing for Operation1.Adhere a gel temperature indicator onto the outside of the outer plate, somewhere near thecenter, to monitor the gel temperature during electrophoresis.•Place the IPC assembly into the universal base, against the back wall, between the alignment tabs.2.Insert the stabilizer bar (Figure 4.9).•The stabilizer bar should slide into place with a snug fit, locking the IPC to the base in a vertical position.•The heads of the screws on the stabilizer bar should push against the front wall of the base to press the IPC clamps against the back wall of the universal base.Note: When first setting up your Sequi-Gen GT cell, adjust the screws on the stabilizerFig. 4.9.Inserting the stabilizer bar into the universal base.3.To avoid buffer spills and cell tipping accidents, adjust the leveling screws on the universalbase, as necessary.•To insure that the unit will not tip over during electrophoresis, make sure the leveling feet threaded rods are at least 1 cm deep into the threaded boss of the base.•At this time, test whether the IPC assembly is properly aligned in the universal base by attaching the top and bottom safety covers. The IPC assembly may have to be shifted to the right or the left to properly attach the safety covers. After this final alignment is complete, remove the safety covers.4.Fill the upper buffer chamber (the IPC) with running buffer (1x TBE) using the flaredportion of the panel as a fill spout.•The level of the buffer should be about 1 cm from the top of the fill spout at all times during the run.•Remove the comb(s) from between the glass plates.•Thoroughly rinse the resulting well(s) or gel front using a syringe with a needle, or disposable plastic transfer pipet (catalog number 223-9911).•If using a sharkstooth comb, insert the comb with the teeth facing the gel front. Lower the comb toward the gel surface until the teeth of the comb just touch the gel surface.5.Fill the lower buffer chamber with 350-500 ml of the running buffer. Refer to Appendix7.1 for running buffer recipes.Caution: Do not fill the lower chamber with more than 500 ml of buffer. The lower buffer chamber holds the entire volume of the upper buffer chamber should a leak develop in the IPC. Buffer levels over 500 ml will not allow the entire volume of the upper buffer chamber to be contained in the universal base.6.Attach the top and bottom safety covers and pre-electrophorese the gel at normal operatingvoltage or power (see Section 4.7), if desired, to increase the gel temperature.•Pre-electrophoresis prior to sample loading will create a uniform gel temperature and bring the gel temperature to the recommended run temperature. This will help eliminate any smile patterns from developing early in the run.Note: Gel electrophoresis buffers can be heated to 50 ˚C in a microwave before adding buffer into the upper buffer chamber. This will reduce the time needed to bring the gel to the appropriate run temperature before sample loading, and will greatly reduce pre-electrophoresis time.Warning: The upper buffer level may drop slightly due to evaporation as the system becomes warmer. Make sure that the upper chamber is always filled with buffer during electrophoresis. Do not allow the buffer level to drop below the level of the notched (shorter) IPC glass plate at any time during electrophoresis, as this may cause arcing and cell damage. Additionally, never allow the gel to exceed 60 °C under any circumstance.This excessive heat may crack the plates or cause the IPC/glass bond to deteriorate.4.5 Loading the Gel1.Turn off the power supply, and remove the top safety cover.•Rinse the well(s) with a syringe with needle, or disposable plastic transfer pipet (catalog number 223-9911), (to remove urea) before applying the samples to the gel.5.Ammonium Persulfate, 25% stock solution: 0.25 g in 1 ml distilled H2O (in a microfugetube). Make fresh daily (see Section 6.2).6. A constant power (or constant voltage) power supply (see Section 6.3).7.Slab gel dryer(see Section 6.3).8.Table top micro-centrifuge9.Gel loading syringe (e.g. Hamilton 701-SN, 28 Gauge, 1.25 inch needle)10.1.5 ml microcentrifuge tubes (see Section 6.5)11.Adjustable pipettors (e.g. Pipetman P-20, P-200, P-1000)12.Balance13.Plastic wrap14.Pipette tips, autoclaved (see Section 6.5)15.Waterbath or Temp-Block at 95 °C.16.X-ray film and cassettes (dark-room facilities)17.Filter Paper (see Section 6.3)18.Siliconizing solution or glass coating solution19.Geiger Counter20.Ice bucket7.2 Standard Gel ProtocolThe following protocol is for a standard 7 M urea, 5% polyacrylamide gel for DNA sequencing. See Section 4 for additional information on gel casting, sample loading, and gel electrophoresis. For ordering information on gel reagent and electrophoresis buffers see Section 6.bine 63 g of urea, 15 ml of 10x TBE, and 25 ml of 30% acrylamide stock solution.Bring the volume to 150 ml with distilled water (low heat may be required to dissolve the urea, but do not boil).2.Filter the solution through a 0.45 micron mesh filter (optional). Then, degas under strongvacuum 5–15 minutes to remove dissolved oxygen.3.Add 150 µl TEMED and 150 µl 25% ammonium persulfate (or one microliter of eachreagent for every milliliter of gel solution) prior to gel casting.4.Cast the gel according to procedures in Section 4.Note: Wedge spacers (see Section 6.1) increase the number of readable bases per lane in a sequencing gel. The use of wedge spacers results in a gel which becomes gradually thicker toward the bottom. As thickness increases, resistance, voltage, and DNA mobility decrease.The resulting gel has bands more closely spaced at the bottom. Wedge spacers allow the use of standard polyacrylamide solution and buffers. No alterations to the gel solution, gel casting or electrophoresis protocols are required to run DNA sequencing wedge gels.7.3 Gel Drying and AutoradiographyThe radiolabeled oligonucleotides may be visualized by a variety of techniques involving autoradiography. For the best resolution and signal intensity, dry DNA sequencing gels with a slab gel dryer.1.Transfer sequencing gels to a fresh sheet of filter paper. Wet the gel slightly by misting thegel with deionized H 2O. Lay the dry filter paper on top of the gel, and press firmly. The gel will stick to the paper. Pick up the gel by lifting the filter paper carefully from one end.2.Cover the sequencing gel with plastic wrap. Smooth out air bubbles and folds by rubbingwith a paper towel, and trim the edges to fit the slab gel dryer.3.Set Model 583 Gel Dryer to sequencing cycle. 30 minutes at 80 °C should suffice fordrying thin low percent gels, if the applied vacuum is above 28 inches of mercury or 125 torr. Refer to the dryer’s instruction manual for details.4.Autoradiograph the gel with high speed X-ray film (such as Kodak XAR) and a suitablefilm cassette. Intensifying screens are optional. If 35S radiolabel is used, the gel can be left on the outer glass plate and fixed in 1 liter of 10% acetic acid, 10% methanol for 15 minutes. This removes hygroscopic urea. The gel may then be dried on filter paper.Removal of plastic wrap before autoradiography is important because 35S is a weak beta emitter. Autoradiography of 35S labeled fragments typically requires 1–3 days. (However,we have found the fixative step unnecessary, even when sequencing with 35S.)7.4 Nucleic Acid Separation Applications for the Sequi-Gen GT Electrophoresis SystemSeveral other nucleic acid separation techniques requiring single nucleotide resolution can be conducted using the Sequi-Gen GT systems. Below is a comprehensive list. Refer to Sambrook, J., Fritsch, E. F., and Maniatas, T., Molecular Cloning, A Laboratory Manual,Second Edition, Cold Spring Harbor Laboratory Press, 1989, or Ausubel, F. M., et al., Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley-Interscience, 1987,for more information and protocols.•Microsatellite Analysis•Single-Strand Conformational Polymorphism (SSCP) studies •Heteroduplex analysis •DNA footprinting •DNA fingerprinting •RNase protection assays •S1 nuclease mapping •Primer extension studies•DNA/Protein binding studies (gel shift assays)•Oligonucleotide analysis7.5 Suggested ReadingBankier, A. T. and Barrell, B. G., Shotgun DNA Sequencing. Techniques in Life Sciences, Vol. B5. Elsevier (1983).Biggin, M. D., Gibson, T. J. and Hong, G. F., Buffer gradient gels and 35S label as an aid to rapid DNA sequence determination, Proc. Natl. Acad. Sci. USA, 80, 3963-3965 (1983).Bishop, M. J., Software for molecular biology. 1. Databases and search programs. Bio Essays1, 25-27. Deininger, P. Approaches to rapid DNA sequence analysis, Anal. Biochem., 135, 247-263 (1983). Garoff, H. and Ansorge, W., Improvements of DNA sequencing gels, Anal. Biochem., 115, 450-457 (1981).Henikoff, S. Unidirectional digestion with exonuclease lll creates targeted breakpoints for DNA sequencing, Gene, 28, 351-359 (1984).Hindley, J., DNA Sequencing, Elsevier Biomed. Press (1983).Lane, D. J., Pace, B., Olsen, G. J., Stahl, D. A., Sogin, M. L. and Pace, N. R., Rapid determination of 16S ribosomal RNA sequences for phylogenetic analysis. Proc. Natl. Acad. Sci. USA, 82, 6955-6959 (1985).Maxam, A. M. and Gilbert, W., Sequencing end-labeled DNA with base-specific chemical cleavages, Methods in Enzymology, 65, 449-580 (1980).Messing, J., New M13 vectors for cloning, Methods in Enzymology – recombinant DNA Techniques, 101, 20-79 (1983).Messing, J., Crea, R. and Seeburg, P. H., A system for shotgun DNAsequencing, Nuc. Acids. Res., 9, 2871-2887 (1981).Ornstein, D. L. and Kashdan, M. A., Sequencing DNA using 35S labeling: A troubleshooting guide, BioTechniques, 3, 476-483 (1985).Sanger, F., Nicklen, S. and Coulson, R., DNA sequencing with chain terminating inhibitors, Proc. Natl. Acad. Sci. USA.,74, 5463-5467 (1977).Schreier, P. H. and Cortese, R. J., A fast and simple method for sequencing DNA cloned in the single-stranded bacteriophage M13,J. Mol. Biol., 169-172 (1979).Staden, R., Automation of the computer handling of gel reading data produced by the shotgun method of DNA sequencing, Nucleic Acids Res., 10, 4731-4751 (1982).Tabor, S. and Richardson, C.C.,Proc. Natl. Acad. Sci. USA, 84, 4767-4771 (1987)Yanisch-Perron, C., Viera, J. and Messing, J., Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors, Gene, 33, 103-119 (1985).。
博乐电泳仪设置使用说明

博乐电泳仪设置使用说明
一、Bio-Rad伯乐电泳仪操作指南:
1、首先用导线将电泳槽的两个电极与电泳仪的直流输出端联接,注意极性不要接反。
2、Bio-Rad伯乐电泳仪电源开关调至关的位置,电压旋钮转到小,根据工作需要选择稳压稳流方式及电压电流范围。
3、接通电源,缓缓旋转电压调节钮直到达到的所需电压为止,设定电泳终止时间,此时电泳即开始进行。
4、Bio-Rad伯乐电泳仪工作完毕后,应将各旋钮、开关旋至零位或关闭状态,并拨出电泳插头。
二、Bio-Rad伯乐电泳仪操作注意事项:
1、Bio-Rad伯乐电泳仪通电进入工作状态后,禁止人体接触电极、电泳物及其它可能带电部分,也不能到电泳槽内取放东西,如需要应先断电,以免触电。
同时要求仪器必须有良好接地端,以防漏电。
2、Bio-Rad伯乐电泳仪通电后,不要临时增加或拨除输出导线插头,以防短路现象发生,虽然仪器内部附设有保险丝,但短路现象仍有可能导致仪器损坏。
3、由于不同介质支持物的电阻值不同,电泳时所通过的电流量也不同,其泳动速度及泳至终点所需时间也不同,故不同介质支持物的电泳不要同时在同一电泳仪上进行。
4、在总电流不*过仪器额定电流时(大电流范围),可以多槽关联使用,但要注意不能*载,否则容易影响仪器寿命。
5、某些特殊情况下需检查仪器电泳输入情况时,允许在稳压状态下空载开机,但在稳流状态下必须先接好负载再开机,否则电压表指针将大幅度跳动,容易造成不必要的人为机器损坏。
6、Bio-Rad伯乐电泳仪使用过程中发现异常现象,如较大噪音、放电或异常气味,须立即切断电源,进行检修,以免发生意外事故。
CFX荧光定量PCR仪操作指南(Bio-rad)

CFX荧光定量PCR仪操作指南(Bio-rad)CFX96 实时荧光定量PCR仪操作流程及注意事项开始运行仪器打开电脑打开定量PCR仪底座开关启动CFX Manager软件放置样品将PCR反应体系加入到0.2ml低缘八联管,盖上管盖;或加入低缘96孔板,用光学级封膜封好、注意,必须带一次性塑料手套,不要让手指接触到反应管表面。
将反应管按顺序放入仪器得加热孔中。
设置程序,运行实验定量PCR软件操作基本步骤为:a、设置热循环程序文件(Protocol Tab) b。
设置反应板文件(Plate Tab)。
C。
点击“StartRun”键,运行程序热循环程序文件(Protocol Tab)设置指南:点击Edit(编辑)或 Create New(创建新程序)。
反应板设置文件(PlateTab)设置指南:选择本次实验所要使用得荧光染料种类;单击样品类型;如要某些反应孔第一荧光染料对应得样品类型为标准品(Standard),点击“DilutionSeries”键可设置其标准品浓度及稀释倍数。
点击“Start Run”键、单击openlid(打开热盖)或Close lid(关闭热盖)放置样品;单击Start Run,保存文件,开始运行程序。
结果分析PCR反应结束后,软件会自动计算标准曲线与Ct值等、如需进行表达量分析、等位基因分析等, 在软件窗口选择相应分析功能。
点击右上方得“Report"键,还可输出结果报告单关闭运行仪器实验结束后取出反应管,顺序关闭CFX Manager软件、定量PC R仪电源,关闭电脑。
注意!CFX仪器上盖部分为全自动控制,在通电状态,严禁任何人为干涉上盖开启或关闭得行为,此类行为会导致上盖故障,危及仪器使用。
CFXManager软件操作快速指南实验操作程序设置图 1显示实验设置窗口中一个预览得程序。
点击CreateNew,打开程序编辑器创建一个新得程序、点击Select Existing,通过浏览器加载一个程序或对其进行编辑。
WB操作规程

WB操作规程一、设备和试剂1.设备①电泳电源Mini-PROTEAN 3 Elecreophoresis Cell,Mini Teans-Blot Module,and PowerPac Basic Power Supply (BIO-RAD,Catalog#165-3323)②电泳仪及附件Mini-PROTEAN 3 Elecreophoresis Cell (BIO-RAD,Catalog#165-3301)③电转仪及附件Mini Teans-Blot Elecreophoresis Transfer Cell (BIO-RAD,Catalog#170- 3930)④NC膜PIERCE,Catalog#88018⑤滤纸Whatman,3MM CHR2.试剂①凝胶试剂试剂名称厂家及规格30%丙烯酰胺溶液上海生工Tris-Base SIGMA10%SDS SIGMA10%过硫酸铵上海生工TEMED 上海生工②常用溶液及缓冲液A.5%积层胶所用溶液B.分离胶溶液C.电泳缓冲液,1L3g Tris-Base、14.4g 甘氨酸、1g SDS,加蒸馏水至IL,pH应该在8.3左右。
也可以制成10×的储存液,在室温下长期保存D.电泳转移缓冲液,1L3g Tris-Base、14.4g 甘氨酸、甲醇200ml, 加蒸馏水至1L, 也可以制成10×的储存液,在湿温下长期保存E.5×样品缓冲液,10ml0.6ml 1mol/L的Tris-HCl(Ph6.8)、5ml 50%的甘油、2ml 10%的SDS、0.5ml 2-巯基乙醇、1ml 1%溴酚蓝,0.9ml的蒸馏水,可在4℃保存数周,或在-20℃保存数月。
③二抗稀释液AP-Anti mouse IgG(Whole molecule),SIGMA,CAT#A-3562;AP-Anti mouse IgGAM,ZYMED,CAT#62-6422一般采取1:3000-1:5000的稀释度,用1%BSA-PBS(含0.05%TWEEN20)稀释④显色底物缓冲液A.HRP标记二抗底物缓冲液DAB KIT(KPL LOT NO Y M107 CAT:54-10-00), Tris Buffer 3滴,DAB Substrate 3滴,Peroxide sol ution 2滴于5ml蒸馏水中,避光,混匀B.AKP标记二抗底物缓冲液AKP缓冲液:0.1mol/L Tris-HCL(PH9.5), 0.1mol/L NaCL, 5mmol/L MgCL2BCIP溶液/NBT溶液:50mg/ml BCIP溶于100%二甲基甲酰胺50mg/ml BCIP溶于70%二甲基甲酰胺⑤其他试剂A.考马斯亮蓝染液,1L考马斯亮蓝R-250 1.0g、甲醇450ml、冰醋酸100mlB.考马斯亮蓝脱色液甲醇100ml、冰醋酸100ml、蒸馏水800ml二、方法1.Bio-Rad微型凝胶电泳模具的安装带上手套将凝胶电泳系统的前后玻璃板,梳子用去污剂洗涤,最后再用去离子水冲洗干净,除去黏附在玻璃板上凝固的胶和其他污垢。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Bio-rad小电泳槽及电泳仪操作规程
一.目的
为规范Bio-rad小电泳槽及电泳仪的基本操作、维护保养、异常处理程序,防止人为操作失误,确保Bio-rad小电泳槽及电泳仪正常运转,特制定本程序。
二.适用范围
本程序适用于Bio-rad小电泳槽及电泳仪。
三.责任
1. 本程序的实施者为Bio-rad小电泳槽及电泳仪操作者,各实验室负责人对
本程序的实施情况进行监督。
2. 日常运行及维护、定期维护、定期点检及保养由Bio-rad小电泳槽及电泳
仪操作者负责。
四.内容
1.灌胶
a、取出灌胶模具,打开封底盘至完全开放的状态,松开两侧螺丝,向外侧打开夹子。
b、取长方形和带凹槽的长玻板各一块,从内到外依次按带凹槽玻板及长方形玻板的顺序排列,形成gel sandwich。
注意正确排列以防漏胶。
c、短玻板朝外将sandwich放入模具中,检测sandwich的底是否紧靠底面。
旋紧螺丝。
将封底盘锁至密闭状态。
d、将模具放在桌面上准备灌胶。
e、根据需要配制一定浓度的胶。
1、分离胶的灌制:用加样枪慢慢将胶加入sandwich中,注意不要有气泡。
其上加0.1%SDS封顶.
2、浓缩胶的灌制:待分离胶凝后弃去SDS,开始灌浓缩胶,最后轻轻插入梳子。
f、待胶聚合后,拔出梳子,用电泳缓冲液清洗加样槽,去除未聚合的胶。
g、
模具放入电泳槽中。
2.电泳
a、加适量的电泳缓冲液
b、准备样品,加样。
c、盖上电泳槽的盖子,注意红色对正极,黑色对负极。
打开电源,开始电泳。
初始电压100V,15分钟后改为120v。
d、直至指示剂距前沿1cm处结束电泳,拔掉电源,小心取出胶,根据需要进行染色或转膜,回收电泳缓冲液(可重复利用3次)。
五.注意事项:
a、每次用完后,立即清洗电泳槽,灌胶模具,玻璃板(先用自来水洗干净,再用蒸馏水冲一遍后放在架子上晾干)。
b、扣紧或打开灌胶模具时要用力均匀,以免压坏玻璃板。
c、盖上电泳槽的盖子时,务必注意正负极不要接反。