3,4-DAA GlpBio
降糖药物GLP1A和DDP-4简介

常见的GLP-1RA
随着相关研究的不断深入,临床应用经验的不断积累,GLP-1RA 在国内外糖尿病 治疗指南中的地位逐渐提升。GLP-1RA 从2009年美国糖尿病学会(ADA)和欧洲糖尿
病研究学会(EASD)首次在指南中推荐纳入治疗流程,目前已成为继二甲双胍之后的
首选药物之一。 GLP-1RA 包括多肽和小分子化合物,在短短的几年中,多肽、血浆白蛋白重组肽、 生物表达重组肽等制剂先后问世,以下GLP-1RA 有些已经在临床应用中,有些仍在发 展临床试验过程中:①艾塞那肽注射液(商品名:百泌达,Byetta);②利拉鲁肽注 射液(商品名:诺和力,Victoza);③艾塞那肽长效制剂(商品名:Bydureon);④ 利西拉来Lixisenatide(商品名(Lyxumia);⑤ 阿必鲁肽(Albiglutide);⑥ 杜拉糖肽
验。
5.孕妇与哺乳期用药 妊娠期妇女使用安全性未知。动物实 验证实其可经乳汁分泌,故哺乳期不 推荐使用。
T1DM 患者在胰岛素基础上加用利拉
鲁肽可减少胰岛素剂量、降低体重、 减少低血糖发生率,但在HbA1c及血
糖变异性方面与安慰剂相比差异无统
计学意义。
二肽激酶抑制剂
胰高血糖素样肽-1(glucagon-like peptide 1),1964年,Elrick等发现 口服葡萄糖刺激胰岛素分泌的量明显大于静脉滴注相同剂量葡萄糖,这种现象 被称为“肠促胰素”效应。人体内主要有两种肠促胰素:葡萄糖依赖性促胰岛 素释放肽(glucose-dependent insulinotropic polypeptide, GIP)和GLP-1。因T2DM 患者的血GIP 水平正常或升高,对β细胞的促胰岛素分泌作用显著降低,且对α
症、免疫等相关:
众多高分文章引用的超敏小鼠总胆汁酸TBA检测试剂盒
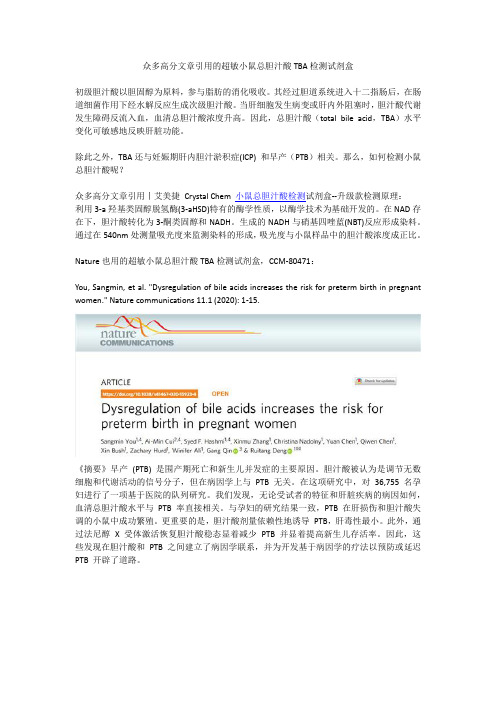
众多高分文章引用的超敏小鼠总胆汁酸TBA检测试剂盒初级胆汁酸以胆固醇为原料,参与脂肪的消化吸收。
其经过胆道系统进入十二指肠后,在肠道细菌作用下经水解反应生成次级胆汁酸。
当肝细胞发生病变或肝内外阻塞时,胆汁酸代谢发生障碍反流入血,血清总胆汁酸浓度升高。
因此,总胆汁酸(total bile acid,TBA)水平变化可敏感地反映肝脏功能。
除此之外,TBA还与妊娠期肝内胆汁淤积症(ICP) 和早产(PTB)相关。
那么,如何检测小鼠总胆汁酸呢?众多高分文章引用丨艾美捷Crystal Chem 小鼠总胆汁酸检测试剂盒--升级款检测原理:利用3-a羟基类固醇脱氢酶(3-aHSD)特有的酶学性质,以酶学技术为基础开发的。
在NAD存在下,胆汁酸转化为3-酮类固醇和NADH。
生成的NADH与硝基四唑蓝(NBT)反应形成染料。
通过在540nm处测量吸光度来监测染料的形成,吸光度与小鼠样品中的胆汁酸浓度成正比。
Nature也用的超敏小鼠总胆汁酸TBA检测试剂盒,CCM-80471:You, Sangmin, et al. "Dysregulation of bile acids increases the risk for preterm birth in pregnant women." Nature communications 11.1 (2020): 1-15.《摘要》早产(PTB) 是围产期死亡和新生儿并发症的主要原因。
胆汁酸被认为是调节无数细胞和代谢活动的信号分子,但在病因学上与PTB 无关。
在这项研究中,对36,755 名孕妇进行了一项基于医院的队列研究。
我们发现,无论受试者的特征和肝脏疾病的病因如何,血清总胆汁酸水平与PTB 率直接相关。
与孕妇的研究结果一致,PTB 在肝损伤和胆汁酸失调的小鼠中成功繁殖。
更重要的是,胆汁酸剂量依赖性地诱导PTB,肝毒性最小。
此外,通过法尼醇X 受体激活恢复胆汁酸稳态显着减少PTB 并显着提高新生儿存活率。
常用多肽缩合试剂[参考内容]
![常用多肽缩合试剂[参考内容]](https://img.taocdn.com/s3/m/ba9dcc56af1ffc4ffe47ace6.png)
HBTU
379.30
6-氯苯并三氮唑-1,1,3,3-
四甲基脲六氟磷酸酯
HCБайду номын сангаасU
413.69
TCTU
355.53
六氟磷酸苯并三唑-1-基-
氧基三吡咯烷基磷
PyBOP
520.40
常用多肽缩合试剂
名称
缩写
分子量
结构
(3H-1,2,3-三唑并[4,5-b]吡啶-3-氧基)三-1-吡咯烷基鏻六氟磷酸盐
PyAOP
521.38
N,N'-二环己基碳二亚胺
DCC
206.33
4-二甲氨基吡啶
DMAP
122.17
1,8-二氮杂双环[5.4.0]十一碳-7-烯
DBU
152.24
1,1’-羰基二咪唑
CDI
162.15
2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯
HATU
380.23
3-羟基-1,2,3-苯并三嗪-4(3H)-酮
O-[(乙氧基羰基)氰基甲胺]-N,N,N',N'-四甲基硫尿四氟硼酸
TOTU
328.1
苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐
BOP
(卡特缩合剂)
442.50
N,N,N',N'-四甲基-O-(3,4-二氢-4-氧代-1,2,3-苯并三嗪-3-基)脲四氟硼酸盐
TDBTU
349.09
常用多肽缩合试剂
名称
缩写
分子量
结构
N,N-二异丙基碳二亚胺
DIC
126.00
羟基苯并三唑
二肽基肽酶4作用原理

二肽基肽酶4作用原理二肽基肽酶4(dipeptidyl peptidase 4,DPP-4)是一种酶类蛋白质,它在人体中起着重要的生理调节作用。
DPP-4主要存在于细胞膜上,特别是肠道上皮细胞、肺细胞和肾小管上皮细胞等组织中。
其作用机制主要包括切割胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)和胰高血糖素(glucagon),从而调节血糖水平。
DPP-4通过切割GLP-1和胰高血糖素的N端二肽基,将其转化为无活性的代谢产物。
GLP-1和胰高血糖素是由肠道中食物摄入引起的胰岛素分泌增加的重要调节因子。
它们通过与胰岛素受体结合,促进胰岛素的释放,从而降低血糖水平。
然而,GLP-1和胰高血糖素的半衰期较短,仅为数分钟,这限制了它们在体内的生物活性。
而DPP-4的存在恰恰是为了调节GLP-1和胰高血糖素的血浆水平,通过降解这两种激素,维持其稳定的浓度。
DPP-4的作用机制可以通过以下步骤来描述:首先,GLP-1和胰高血糖素由肠道内的L细胞分泌出来。
随后,这两种激素通过血液循环输送到胰岛,与胰岛素受体结合,刺激胰岛素的分泌。
同时,DPP-4也存在于胰岛B细胞上,当GLP-1和胰高血糖素进入B细胞时,DPP-4迅速作用于它们的N端二肽基,切割掉这部分肽链,使其失去活性。
这样,GLP-1和胰高血糖素在体内的浓度得到控制,避免过度刺激胰岛素分泌,从而维持血糖水平的稳定。
除了调节GLP-1和胰高血糖素的血浆水平外,DPP-4还参与其他生理过程。
研究表明,DPP-4与免疫调节、细胞增殖和凋亡等过程密切相关。
例如,DPP-4参与调节T细胞的活化和增殖,对免疫系统的正常功能至关重要。
此外,DPP-4还与肿瘤的发生和发展相关,其抑制剂在肿瘤治疗中显示出潜在的抗肿瘤活性。
近年来,针对DPP-4的抑制剂已成为治疗2型糖尿病的重要药物。
通过抑制DPP-4的活性,可以增加GLP-1和胰高血糖素的半衰期,提高其在体内的生物利用度,从而增加胰岛素的分泌和降低血糖水平。
GLPbio产品说明书-SB 431542 (GC11545)

Product Data SheetProduct Name:SB 431542Cat. No.:GC11545Chemical PropertiesCas No.301836-41-9化学名4-[4-(1,3-benzodioxol-5-yl)-5-pyridin-2-yl-1H-imidazol-2-yl]benzamideCanonicalSMILESC1OC2=C(O1)C=C(C=C2)C3=C(NC(=N3)C4=CC=C(C=C4)C(=O)N)C5=CC=CC=N5分子式C22H16N4O3分子量384.39溶解度≥ 19.2mg/mL in DMSO, ≥ 10.06mg/mL in EtOH with ultrasonic储存条件Store at -20°CGeneral tips For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months.Shipping Condition Evaluation sample solution : ship with blue ice All other available size: ship with RT , or blue ice upon request.StructureProtocolKinase experiment [1]:Preparation Method The kinase domain without the GS region was cloned and expressed as a GST fusion protein. Expressing the protein without the GS domain, which has been shown to regulate the kinase activity, creates a constitutively active kinase that is able to phosphorylate GST-Smad3. Test the effects of SB-431542 on ALK5 and ALK4 kinase activity with GST-Smad3 as substrate.Reaction Conditions The Kinase assays were performed with 65 nM GSTALK5 and 184 nM GST-Smad3 in 50 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, 1 mM dithiothreitol, and 3 μM ATP. Reactions were incubated with 0.5 μCi of [33P]γATP for 3 h at 30°C.Applications SB-431542 is a selective ALK5 inhibitor with little activity against p38 MAPK. SB-431542 also inhibits ALK4 activity. Which is consistent with the degree of homology between these kinases, such that ALK4 is the closest related kinase to ALK5. This data clearly demonstrated that SB-431542 is a potent and selective inhibitor of ALK5 and ALK4, with slightly higher selectivity for ALK5.Cell experiment [1]:Cell lines Renal proximal tubule epithelial cells (RPTEC)Product Data SheetPreparation Method Cells were grown in Earle’s minimum essential medium supplemented with 10% fetal calfserum, penicillin (5 units/ml), and streptomycin (5 ng/ml). Cells were serum-starved for 24 hbefore treatment.Reaction Conditions Cells were treated with TGF-β1 (5 ng/ml) plus increasing concentrations of SB-431542 (50,250, 500, and 700 nM).Applications SB-431542 could be used to evaluate whether ALK5 activity is required for TGF-β1-inducedtranslocation of Smad3. SB-431542 at a concentration of 1 μM significantly reduced the TGF-β1-induced nuclear accumulation of Smad proteins. Thus, SB-431542 selectively inhibitsTGF-β1–induced Smad translocation without affecting BMP-induced Smads.Animal experiment [2]:Animal models Male Sprague-Dawley rats aged 5 weeks, weighing 200-220 gPreparation Method Rats lived in air served as control groups, and rats lived in an air condition incubatorcontaining 10% O2 to simulate chronic hypoxia animal model, and served as model groups.Model groups were treated with daily intraperitoneal injections of the SB-431542 for 28days.Dosage form 10 mg/kg; 20 mg/kgApplications SB-431542 inhibited the proliferative activity as a function of exposure time andconcentration. Treated rats with SB-431542 caused more pathological changes in vascularadventitia, and the severity of the changes varied from slight to moderate depending on concentrations. In addition, the pulmonary arteries in the hypoxia-induced model groups hadgreater amounts of collagen fibers than that of the control groups. In comparison, collagenfibers were significantly reduced after treatment with SB-431542 (P < 0.01).References:[1]. Laping NJ, et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002 Jul;62(1):58-64.[2]. Yuan W, et al. SB-431542, a specific inhibitor of the TGF-β type I receptor inhibits hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. Pharmazie. 2016 Feb;71(2):94-100.BackgroundSB-431542, a small molecule inhibitor of the type I TGF-β receptor, blocks intracellular mediators of TGF-1 signaling,which leads to decreased TGF-β1–mediated proliferation, cytokines and collagen expression. In clinical settings,SB-431542 is widely used to treat respiratory asthma, and inhibits proliferation and synthesis of adventitial fibro in the process of pulmonary vascular remodeling.[1]In vitro study indicated that SB-431542 is able to inhibit ALK5 with an IC50 of 94 nM and other type I receptors, such as ALK4. Although SB-431542 inhibited ALK4 with an IC50 of 140 nM. Moreover, SB-431542 inhibited TGF-β1–induced collagen Iα1 and PAI-1 mRNA with IC50 values of 60 and 50 nM, respectively. In addition, SB-431542 inhibited TGF-β1–induced fibronectin mRNA and protein with IC50 values of 62 and 22 nM, respectively. These data demonstrate for the first time that ALK5 activity is required for TGF-β1 regulation of extracellular matrix markers FN, collagen Iα1,and PAI-1 mRNA.[1]In vivo study demonstrated that SB-431542 has the capacity to inhibit TGF-β1-induced gene expression. SB-431542is recognized as a important inhibitor of the TGF-β1 receptors in blocking TGF-β1/Smads signal pathways in vascular remodeling. Moreover, hypoxia-induced vascular remodeling can significantly increase the amount of cytokines and collagen in vascular adventitia. However, after the treatment of SB-431542, attenuation of the fibrosis promoting effects of TGF-β1, including TGF-β1-induced cell proliferation, cell motility, cell migration and cell synthesis were observed. Therefore, it is significant to the identify the potential of SB-431542 for the treatment of hypoxia-induced pulmonary hypertension.[2]References:[1]. Laping NJ, et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novelProduct Data Sheetinhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002 Jul;62(1):58-64.[2]. Yuan W, et al. SB-431542, a specific inhibitor of the TGF-β type I receptor inhibits hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. Pharmazie. 2016 Feb;71(2):94-100.SB-431542 是 I 型 TGF-β 受体的小分子抑制剂,可阻断 TGF-1 信号转导的细胞内介质,从而导致 TGF-β1 介导的增殖、细胞因子和胶原蛋白表达减少。
MicroRNA-34a Suppresses Autophagy in A

MicroRNA-34a Suppresses Autophagy in Alveolar Type II Epithelial Cells in Acute Lung Injury by Inhibiting FoxO3ExpressionLan Song,1,2Fangliang Zhou,1,2Lijuan Cheng,1Mei Hu,1Yingchun He,1Bo Zhang,1Duanfang Liao,2,4and Zhaojun Xu 3,4Abstract —Excessive autophagic activity of alveolar type II epithelial (A T-II)cells is one of the main causes of acute lung injury (ALI);however,the underlying molecular mechanism remains to be determined.The microRNAs (miRNAs)are involved with autophagy in many diseases.The objective of this study was therefore to investigate the relationship between the miRNA expression and the autophagic activity of the A T-II cells in the pathogenesis of ALI and its molecular mechanism.A mouse model of ALI and A T-II cell injury was induced using lipopolysaccharide (LPS)in vivo and in vitro ,and the expression of miR-34a and the autophagy-related proteins LC3II/I and p62were determined.Moreover,the autophagic activity was investigated after miR-34a overexpression and inhibition.The effects of miR-34a on its target gene,FoxO3,in regulating autophagic activity in A T-II cells were also determined.LPS induced autophagic activity and increased the expression of miR-34a in lung tissues and in A T-II cells.The in vitro results showed that the upregulation of miR-34a suppressed,whereas the inhibition of miR-34a promoted,autophagy in A T-II cells.Moreover,miR-34a could directly bind to the 3′-untranslated region of the autophagy-related gene,FoxO3,to decrease its expression.In addition,the knockdown of FoxO3expression inhibited the autophagic activity in A T-II cells.Together,this study suggested that miR-34a might suppress the excessive autophagic activity in A T-II cells via targeting FoxO3to reduce the damage of LPS-induced ALI.KEY WORDS:acute lung injury;alveolar type II epithelial cells;autophagy;miR-34a;FoxO3.INTRODUCTIONAutophagy is an evolutionarily conserved lysosomal degradation pathway [14,15].It has been shown to be both protective and damaging in a variety of different models,suggesting that its role in human diseases is complex.Con-siderable evidence has shown the role of autophagy and selective autophagy in cell survival,cell death,and immune and inflammatory responses related to the pathogenesis of complex lung disease [14].Previous studies have reported that autophagy is an induced reaction in response to various injuries such cigarette smoke,oxidative stress,and treatment with TNF-alpha or lipopolysaccharide (LPS),and different types of cell autophagy may have protective or adverse effects on acute lung injury (ALI)[7].Accumulating evi-dence has reported that the excessive autophagic activityofLan Song and Fangliang Zhou contributed equally to this work.1Department of Biochemistry and Molecular Biology,Hunan University of Chinese Medicine,300Xueshi Road,Hanpu Science and Teaching Park,Changsha,Hunan 410208,China 2Division of Stem Cell Regulation and Application,State Key Laboratory of Chinese Medicine Powder and Medicine Innovation in Hunan (Incu-bation),Hunan University of Chinese Medicine,300Xueshi Road,Hanpu Science and Teaching Park,Changsha,Hunan 410208,China 3Cardiothoracic Surgery of the First Affiliated Hospital,Hunan University of Chinese Medicine,97Shaoshan Road,Changsha,Hunan 41007,China 4To whom correspondence should be addressed to Duanfang Liao at Division of Stem Cell Regulation and Application,State Key Laboratory of Chinese Medicine Powder and Medicine Innovation in Hunan (Incubation),Hunan University of Chinese Medicine,300Xueshi Road,Hanpu Science and Teaching Park,Changsha,Hunan 410208,China.E-mail:dfliao66@;and Zhaojun Xu at Cardiothoracic Surgery of the First Affiliated Hospital,Hunan University of Chinese Medicine,97Shaoshan Road,Changsha,Hunan 41007,China.E-mail:Xuzj1492@0360-3997/17/0000-0001/0#2017Springer Science+Business Media New YorkInflammation (#2017)DOI:10.1007/s10753-017-0537-1alveolar type II epithelial cells plays an important pathogenic role in ALI[7,20],and its inhibition is better to ALI[7,14].Lo et al.reported that autophagy in the septic lung resulting from cecal ligation and puncture(CLP)represent-ed a protective response[12].However,autophagy,by virtue of excessive autophagosome accumulation in alve-olar type II epithelial cells,may play a maladaptive role in the late stages of sepsis,leading to ALI.Two studies[13, 19]independently reported that excessive autophagic ac-tivity of alveolar type II epithelial cells may contribute to the development of ARDS(acute respiratory distress syn-drome)in H5N1influenza patients.Inhibition of autopha-gy could be used as a novel strategy for the treatment of H5N1infection,and studies have suggested that autophagy blocking agents(e.g.,beclin1and3-methyladenine)might be useful for decreasing the incidence of ALI during the infection of H5N1virus[17,19].Previous studies[9,10] also reported that inhalation of nanomaterials,including starburst polyamidoamine dendrimer(PAMAM)and COOH-CNT(a functionalized single-walled carbon nano-tube),induced ALI by deregulating the Akt-TSC2-mTOR signaling pathway in vivo.Additional in vitro studies re-ported that treatment with PAMAM or COOH-CNT result-ed in autophagosome aggregation in alveolar type II epi-thelial cells.The autophagy inhibitor,3-methyladenine, rescued the nanoparticle-induced excessive autophagy and ameliorated ALI in mice.Smoke exposure also caused ALI,and smoke exposure can lead to excessive autophagy in alveolar type II epithelial cells[2].The excessive autophagic activity of alveolar type II epithelial cells could lead to increased secretion of inflam-matory factors,cell death,and various dysfunctions,which resulting in aggravation of ALI.Autophagy inhibitors can reduce alveolar type II epithelial cell autophagic activity and can inhibit the development of ALI.It is therefore important to study the autophagic regulation mechanism of alveolar type II epithelial cells during ALI.MicroRNAs are small non-coding RNAs that nega-tively regulate gene expression by binding to the3′-UTR of their various target mRNAs to promote mRNA degradation or to inhibit translation.Recently,studies to determine the genetic components of ALI/ARDS pathogenesis have in-vestigated the involvement of miRNAs in this process.The microRNA-34a(miR-34a)is a multifunctional regulator involved in cell proliferation,apoptosis,growth,and autoph-agy.It has been reported that miR-34a suppressed autopha-gic activity in angiotensin II-treated cardiomyocytes[8]and tubular epithelial cells during acute kidney injury[11].The miR-34a plays an important role in the develop-ment of the heart and lung in mammals.It has been reported that miR-34a expression was significantly in-creased in neonatal lungs in response to hypoxia[1], bleomycin-induced pulmonary fibrosis[22],and in Staph-ylococcal enterotoxin B-induced ALI[18].A previous study also reported that miR-34a modulated the autophagy activity via the direct inhibition of ATG9A and ATG4B expression[8,24].In this study,we characterized miR-34a expression in ALI mouse lung tissues and in alveolar type II epithelial cells induced by LPS and investigated the effects of miR-34a on alveolar type II epithelial cell autophagy in ALI. The results data showed that miR-34a targeted the3′-UTR sequence of FoxO3mRNA and modulated its expression, suggesting that miR-34a might suppress alveolar type II epithelial cell autophagy by targeting FoxO3. MATERIALS AND METHODSAnimals and the ALI ModelHealthy male C57BL/6mice aged8–10weeks and weighing17.6–25.4g purchased from the Experimental Animal Center of the Hunan University of Chinese Med-icine(Changsha,China)were used for experimentation and allowed to acclimate for3days before experimenta-tion.The animals were fed rodent chow and water ad libitum and were randomly divided into different groups: an ALI group with intratracheal instillation of3mg/kg LPS (Escherichia coli0111:B4,Sigma,St.Louis,Missouri, USA)and a control group with intratracheal instillation of equal volume of normal saline.The mice were anesthe-tized by an intraperitoneal injection of10%chloral hydrate (QingDao YuLong Algae CO.LTD.,QingDao,China)and kept in a supine position while spontaneous breathing was monitored.Mice of ALI group sacrificed at the indicated times(6,12,24h)after injury,and those of control group were sacrificed at24h after intratracheal instillation of normal saline.After the experimental protocol was com-pleted,lung tissue from animals(n=6per time point)was rapidly obtained.ALI induction was verified by patholog-ical examination of the lung.Lung HistopathologyAt necropsy,the left lung was excised and fixed with 4%paraformaldehyde for24h,and then the lung tissue was dehydrated with graded alcohol and embedded in paraffin at52°C.Sections were prepared and stained with hematoxylin and eosin.For evaluation of the severity of lung injury,each lung section was blindly assigned a lung Song,Zhou,Cheng,Hu,He,Zhang,Liao,and Xuinjury score(LIS)by two pathologists using the method described by Nishina et al.[16].Briefly,lung tissue sec-tions were assessed for alveolar congestion,hemorrhage, infiltration or aggregation of neutrophils in the airspace or vessel wall,and thickness of the alveolar wall/hyaline membrane.The degree of lung injury was scored as fol-lows:0,minimum;1,mild;2,moderate;3,severe;and4, maximum damage.For each animal,six high-magnification fields were randomly selected for grading and an average LIS score was calculated.Isolation of Murine Alveolar Type II Epithelial Cells and Induction of Cell InjuryAlveolar type II epithelial cells were isolated at90–95%purity from6-week-old mice following the proce-dure described by Corti and colleagues[3].Briefly,mice were killed,the pulmonary artery was cannulated,and the lungs were perfused with normal saline in situ to flush out blood.The trachea was cannulated,and2ml dispase II(5U/ml in PBS;Becton-Dickinson,San Jose, CA)were injected in the lungs followed by0.3ml warmed low-melting-point agarose(1%in PBS)to pre-vent the isolation of Clara cells and upper airway epi-thelial cells.The lungs were cooled on ice,dissected free,rinsed with saline,and placed in5ml dispase to digest at room temperature for60min with gentle rocking.Pancreatic DNase(0.01%in DMEM;Sigma-Aldrich)was added for the final5min of incubation. Lung tissue was teased apart,and the resulting cell suspension was filtered sequentially through100-,40-, and21-μm sterile nylon meshes.Leukocytes were re-moved by panning with rat polyclonal anti-murine CD45and anti-murine CD16/CD32antibodies(both Becton-Dickinson)for2h at37°C.Nonadherent cells were collected,pelleted by centrifugation,resuspended in normal saline,and counted using a hemocytometer. Purity of isolated AT-II cell preparations was determined by visualization of lamellar bodies in modified Papanicolaou-stained cytospins.For the incubation ex-periments,alveolar type II epithelial cells were induced with LPS(1μg/ml).A dose of LPS was chosen that was consistent with previous reports and with our pilot study [23].Transfection of miR-34a Mimics or InhibitorMiR-34a mimics or a miR-34a inhibitor and their negative control oligonucleotides were obtained from GenePharma(Shanghai,China).The transfection was per-formed using Lipofectamine™2000(Invitrogen,Carlsbad,California,USA)according to the instructions provided by the manufacturer.Briefly,approximately5×105cells were seeded to flasks containing5ml of appropriate complete growth medium and incubated at37°C with5%CO2until the cells were70to80%confluent(24h).After rinsing with serum-free,antibiotic-free medium,cells were transfected with either miR-34a mimics or a miR-34a inhibitor and their negative control oligonucleotides in 20μL lipofectamine,followed by incubation at37°C in a CO2incubator for6h.The transfected cells were resus-pended and cultured in regular culture medium for48–72h before analyses,and cells were treated with LPS(1μg/ml) to induce injury.siRNA TransfectionFoxO3siRNA and control siRNA were purchased from Cell Signaling Technology(Beverly,MAass., USA).Approximately5×104cells were seeded in each well of a24-well microplate and grown for24h to reach 30–50%confluency.The cells were then incubated with a mixture of siRNA and Lipofectamine™2000(Invitrogen) in100μl of serum-free Opti-MEM®according to the manufacturer’s instructions.Medium may be changed after4–6h.Incubate the cells at37°C in a CO2 incubator for48h.The expression of FoxO3was detected using a real-time polymerase chain reaction(RT-PCR)and western blotting.RNA Extraction and RT-PCRTotal RNA was extracted using the TRIzol®reagent (Invitrogen)according to the manufacturer’s instructions. Five micrograms of total RNA was then used as a template to synthesize cDNA using the First Strand Synthesis Kit (Invitrogen).The cDNA from this synthesis was then used in quantitative RT-PCR analyses using the TaqMan system (ABI-Prism7700Sequence Detection System, Biosystems,Bethesda,Maryland,USA)using SYBR®Green dye.The following primer pairs were used:mouse FoxO3;forward,5′-GTGGACCGACTTCCGCTCGC-3′and reverse,5′-GCTTGCCAGGATGGGCGACA-3′; glyceraldehyde-3-phosphate dehydrogenase(GAPDH); forward,5′-TGGTATCGTGGAAGGACTC-3′and re-verse,5′-AGTAGAGGCAGGGATGATG-3′.The RT-PCR data were normalized by measuring the average cycle threshold(Ct)ratios between candidate genes and the control gene,GAPDH.The formula,2Ct(candidate)/ 2Ct(control),was used to calculate the normalized ratios.MiR-34a Suppresses Autophagy in Acute Lung InjuryTaqMan RT-PCR for the Quantitation of miRNATotal RNA was isolated from frozen lung tissue and cell lines using Trizol™(Invitrogen),then reverse transcribed using the Taqman™microRNA reverse transcription kit,and subjected to RT-PCR using the TaqMan™microRNA assay kit(Biosystems)according to the manufacturer’s instructions.The reactions were performed in triplicate using a Stratagene Mx3000 instrument,and the miRNA expression was normalized to U6.Western Blot AnalysisProteins were isolated and separated by12%SDS–PAGE and transferred onto PVDF membranes(Schleicher &Schuell,Germany).The membranes were blocked overnight in phosphate-buffered saline(PBS)containing 10%nonfat dry milk and0.5%Tween-20and then incubated with primary antibodies for2h.Horseradish peroxidase-conjugated IgG was used as secondary anti-bodies.The immunoreactive bands were visualized using diaminobenzidine(DAB;Boster Biological Technology, Wuhan,China),and the protein expression was normalized to GAPDH.The following antibodies were used:rabbit polyclone anti-LC3B(1:1000;Novus Biologicals,Little-ton,Colorado,USA),anti-p62monoclonal antibody(BD Transduction Laboratories,Lake Franklin,New Jersey, USA),rabbit anti-FoxO3polyclonal antibody(Abcam, Cambridge Science Park,UK),goat GAPDH monoclonal antibody(Sigma),HRP-conjugated anti-goat,and anti-rabbit IgG(Boster Biological Technology). Construction of the Plasmid VectorTo determine whether miR-34a directly bound to the 3′-UTR of FoxO3,we utilized a3′-UTR luciferase reporter assay.The sequence of443nucleotides(including the binding sites for miR-34a)of the3′-UTR of FoxO3were amplified by PCR using the following primers:forward, 5′-ACACAAGCTTCTTATCTTGTATTTCCAT-3′and reverse,5′-ACACGAGCTCAGCTGTGAACACCAAC CC-3′and inserted downstream of the luciferase gene in the pLuc luciferase vector(Ambion,Austen,Texas,USA). Site-directed mutagenesis of the miR-34a target site at the 3′-UTR of FoxO3was performed using the QuickChange mutagenesis kit(Stratagene,Heidelberg,Germany).The resulting constructs were sequenced and named pLuc-FoxA1-wt or pLuc-FoxA1-mut.Luciferase AssayFor reporter assays,exponentially growing alveolar type II epithelial cells were cultured in24-well plates and transfected with100ng of pLuc-FoxO3-wt or pLuc-FoxO3-mut and50nM of miR-34a mimic or miR-34a inhibitor using Lipofectamine™2000(Invitrogen).Forty-eight hours after transfection,the cells were harvested and assayed with the Dual-Luciferase Reporter assay kit (Promega,Madison,Wisconsin,USA)according to the manufacturer’s instructions.All transfections were per-formed in triplicate from at least three independent experiments.Statistical AnalysisData in the figures and text were expressed as the mean±standard error of the mean(SEM).Each experiment was performed at least three times,and statistical analyses were performed using one-way analysis of variance.Otherwise,representative data were shown.A value of P<0.05was considered statistically significant.RESULTSLPS Induced ALI in MiceHistological examination by light microscopy revealed that in the control lungs,the alveoli were fully distended and no alveolar wall edema or congestion was found.Occasion-ally,inflammatory cells were scattered throughout the lung. In contrast,in the lung tissue from LPS-treated animals,the histological features were consistent with ALI,including alveolar hemorrhage,destruction of alveolar attachment points,an increased number of interstitial cells,and exten-sive neutrophil infiltration.The LIS significantly increased in the LPS group(Fig.1a).These results showed that LPS successfully induced ALI in mice.LPS Induced Autophagy and miR-34a Expression in Lung Tissue of Mice and in Alveolar Type II Epithelial CellsAccumulating evidence has shown that excessive autophagic activity occurs in alveolar type II epithelial cells during ALI.We therefore characterized the levels of autophagy-related proteins LC3II/Iand p62expres-sion in ALI mouse lung tissues and in LPS-induced alveolar type II epithelial cells.The results showed that the levels of LC3II/I expression were elevated at Song,Zhou,Cheng,Hu,He,Zhang,Liao,and Xu6,12,and 24h in lung tissue of ALI mice and in LPS-induced alveolar type II epithelial cells.In contrast,the levels of p62protein were repressed at 6,12,and 24h after LPS induction (Fig.1b,c).Fig.1.Change of pulmonary histological scores,autophagy,and miR-34a expression in lung tissue of acute lung injury (ALI)mice and alveolar type II epithelial cells induced by lipopolysaccharide (LPS).a The changes of pulmonary histological scores in different groups.b The levels of autophagy-related protein expression were determined by western blotting in lung tissues of an ALI model induced by LPS or in alveolar type II epithelial cells of mice induced by LPS (c )(d )The levels of miR-34a were determined by real-time polymerase chain reaction in lung tissues of an ALI model induced by LPS or in alveolar type II epithelial cells of mice induced by LPS (e ).The relative values of all results were determined and expressed as the mean ±standard error of the mean of three experiments performed in duplicate.*Statistically significant difference versus the control group (Ctrl),p <0.05.MiR-34a Suppresses Autophagy in Acute Lung InjuryA previous study reported that miR-34a is involved in the process of autophagy.We therefore determined the expression of miR-34a in ALI mice and alveolar type II epithelial cells.The results showed that miR-34a expres-sion was significantly increased at6,12,and24h (Fig.1d,e),suggesting that LPS cause autophagy in the lungs of mice and alveolar type II epithelial cells and induce miR-34a expression.The miR-34a Regulated Autophagy in Murine Alveolar Type II Epithelial CellsWe then determined the effect of miR-34a on autoph-agic activity in mouse alveolar type II epithelial cells.The miR-34a expression was upregulated by miR-34a mimcs, while it was downregulated by transfection of a miR-34a inhibitor in alveolar type II epithelial cells(Fig.2a).Trans-fection with miR-34a mimics decreased the levels of LC3 II/Iin alveolar type II epithelial cells treated with LPS at 12h.However,treatment with the miR-34a inhibitor in-creased the levels of LC3II/I in alveolar type II epithelial cells.In contrast,the levels of p62protein were induced in the miR-34a mimic group but suppressed in the miR-34a inhibitor group(Fig.2b).Taken together,the results showed that miR-34a suppressed LPS-induced autophagic activity in alveolar type II epithelial cells.The miR-34a Directly Targeted FoxO3in Murine Al-veolar Type II Epithelial CellsIt has been reported that miRNAs post-transcriptionally reduced the protein expression of specific target mRNAs.Bioinformatic analyses showed that there was one binding site of miR-34a in the3′-UTR region of FoxO3.FoxO3is an important protein in autophagy,so we proposed that the mechanism of miR-34that suppressed autophagy could be partly due to the downregulation of FoxO3during ALI.The role of miR-34a in the regulation of the FoxO3 gene expression was therefore further investigated.To confirm the role of miR-34a in the regulation of FoxO3expression,quantitative RT-PCR and western blotting were performed.The results showed that miR-34a mimics or the miR-34a inhibitor had no significant effect on the mRNA levels of FoxO3when compared with a negative control (Fig.3a).Western blot analyses showed that the miR-34a mimics suppressed FoxO3protein expression,whereas transfection of the miR-34a inhibitor promoted FoxO3 protein expression compared with the negative control (Fig.3b).To further confirm that miR-34a targets FoxO3, the miR-34a binding sequence present at the3′-UTR of the FoxO3mRNA was subcloned downstream of the firefly luciferase reporter gene in pLUC vectors and then co-transfected with either the negative control,miR-34a mimics,or the miR-34a inhibitor into alveolar type II epithelial cells.The relative luciferase activity of the re-porter containing the wild-type3′-UTR of FoxO3was significantly suppressed when the miR-34a mimics were co-transfected,but not when it was co-transfected with a mutant sequence with the miR-34a binding site(Fig.3c). In addition,there was a significant increase inluciferase Fig.2.The miR-34a suppressed autophagic activity in mouse alveolar type II epithelial cells.a Forty-eight hours after alveolar type II epithelial cells were transfected with miR-34a mimics,miR-34a inhibitor,or the negative control,Taqman real-time quantitative polymerase chain reaction analyses were performed to assess miR-34a expression.b Forty-eight hours after transfection with miR-34a mimics,a miR-34a inhibitor,or a negative control,alveolar type II epithelial cells were incubated with LPS, and the autophagy-related protein expression determined by western blot analyses.The relative values of all results were expressed as the mean±standard error of the mean of three experiments performed in duplicate.*A statistically significant difference versus the negative control,p<0.05.Song,Zhou,Cheng,Hu,He,Zhang,Liao,and Xuactivity for the reporter containing the wild-type 3′-UTR of FoxO3when the miR-34a inhibitor was co-transfected,but not when it contained a mutant sequence with a miR-34a binding site.Together,the results showed that miR-34a bound directly to a specific site on the 3′-UTR of FoxO3to regulate its expression.FoxO3Knockdown Inhibited Autophagic Activity in Murine Alveolar Type II Epithelial CellsWe then characterized the effect of FoxO3inhibition on autophagy in alveolar type II epithelial cells.After treating murine alveolar type II epithelial cells with FoxO3siRNA,the FoxO3mRNA and protein expression were significantly suppressed (Fig.4a,b ).Figure 4c shows that FoxO3siRNA treatment significantly decreased the ratio of LC3II/I,whereas it induced p62expression when compared to the control siRNA group when alveolar type II epithelial cells were incubated with LPS for 12h.DISCUSSIONThe role of autophagy involves maintenance of nor-mal cell and tissue homeostasis by elimination ofdamagedFig.3.The miR-34a suppressed FoxO3protein expression in murine alveolar type II epithelial cells.Forty-eight hours after alveolar type II epithelial cells were transfected with miR-34a mimics,a miR-34a inhibitor,or a negative control,Taqman real-time quantitative polymerase chain reaction or western blot analyses were performed to assess FoxO3mRNA (a )and protein (b )expression.*A statistically significant difference versus the negative control,p <0.05.(c )The alveolar type II epithelial cells were co-transfected with miR-34a mimics or a miR-34a inhibitor and pLu-FoxO3.After 48h,the luciferase activity was performed.The values are expressed as the relative luciferase activity after normalization to Renilla luciferase activity.The relative values of all results were determined and expressed as the mean ±SEM of three experiments performed in duplicate.MiR-34a Suppresses Autophagy in Acute Lung Injuryproteins and organelles in cells.However,excessive or deficient autophagy can contribute to disease pathogenesis.In the stage of ALI induced by H5N1influenza,CLP or nanomaterial particles,past studies have reported that al-veolar type II epithelial cell autophagy was excessively activated [12,13,17,19].Further studies confirmed that excessive autophagy activation of alveolar type II epithelial cells was a key feature of aggravated ALI.The present study showed that the ratio of LC3II/Iincreased,whereas the level of p62was reduced in lung tissues of LPS-induced ALI mice and LPS-induced alveolar type II epithelial cells,to simulate alveolar type II epithelial cell injury during ALI.This suggested that the autophagic activity of mouse alveolar type II epithelial cells also increased in LPS-induced ALI.Significant progress has been made towards under-standing the contribution of miRNAs,including miR-34a in autophagy [5].The miR-101inhibits autophagy in the MCF-7cell line [6],and the miRNA-212/132family regu-lates cardiomyocyte autophagy [21].The miR-34a sup-pressed autophagy by suppression of ATG4B in tubular epithelial cells in acute kidney injury [11].AnotherstudyFig.4.Autophagy was suppressed in murine alveolar type II epithelial cells when FoxO3was inhibited by siRNA.a ,b After 48h of transfection with control siRNA or FoxO3siRNA,the real-time polymerase chain reaction or western blotting was used to analyzethe FoxO3mRNA and protein expression in murine alveolar type II epithelial cells.c After 48h of transfection with control siRNA or FoxO3siRNA,alveolar type II epithelial cells were incubated with lipopolysaccharide,and autophagy-related protein expression was determined by western blot analyses.*A statistically significant difference versus the control group,p <0.05.Song,Zhou,Cheng,Hu,He,Zhang,Liao,and Xureported that miR-34a played an important role in the reg-ulation of Ang II-induced cardiomyocyte hypertrophy by the inhibition of A TG9A expression and autophagic activity [8].Previous studies have also reported that miR-34a is also involved in various lung diseases and could regulate senes-cence in alveolar type II epithelial cells of patients with idiopathic pulmonary fibrosis[4].Our study showed a sig-nificant upregulation of miR-34a in LPS-induced mouse lung tissue and alveolar type II epithelial cells.Furthermore, overexpression of miR-34a by miR-34a mimics suppressed LPS-induced autophagy of alveolar type II alveolar epithe-lial cells,but inhibition of the expression of miR-34a by a miR-34a inhibitor significantly increased the LPS-induced autophagy of alveolar type II epithelial cells.These results suggested that inhibiting the excessive autophagic activity of type II alveolar epithelial cells was a mechanism involved in the increased miR-34a expression and protection against ALI.However,the mechanisms involving a miR-34a de-crease in autophagic activity of alveolar type II epithelial cells during ALI still remain poorly understood.Bioinformatic analyses showed that FoxO3,an autophagy-related gene,was a candidate target of miR-34a.Thus,we predicted that miR-34a regulates alveolar type II epithelial cells autophagy by regulation of FoxO3 expression.Thus,the possibility of a regulatory mecha-nism involving miR-34a and FoxO3was further explored. Alveolar type II epithelial cells of mice were isolated and incubated with LPS to simulate alveolar type II epithelial cell injury in ALI,and then transfected with miR-34a mimics or a miR-34a inhibitor.FoxO3expression signifi-cantly decreased posttransfection after treatment with miR-34a mimics compared with a negative control in alveolar type II epithelial cells,while FoxO3expression significant-ly increased posttransfection after treatment with the miR-34a inhibitor.The luciferase reporter assay was then used to characterize the mechanism of miR-34a as a regulator of FoxO3expression during ALI.The results showed that miR-34a suppressed FoxO3protein expression by binding to the3′-UTR region.We therefore concluded that FoxO3 is a target gene of miR-34a.We also transfected FoxO3 siRNA into alveolar type II epithelial cells to show that FoxO3siRNA treatment significantly decreased the au-tophagic activity when alveolar type II epithelial cells were incubated with LPS.The miR-34a suppresses autophagy that might be ben-eficial or prejudiced for pathogenesis,depending on the disorder.However,the function of miR-34a and its targets have not been reported in ALI and alveolar type II epithelial cells.Autophagy can be harmful or beneficial in ALI. Moderate autophagy protects cells against various kinds of injury.However,excessive autophagy,especially for alveo-lar type II epithelial cells,plays an important pathogenic role in ALI,and its inhibition may prevent ALI[7,14,20].In the present study,the autophagy activity of lung tissue increased in an LPS-induced ALI model and in LPS-induced alveolar type II epithelial cells that induced injury to alveolar type II epithelial cells during ALI.The miR-34a was upregulated in ALI,which inhibited LPS-induced autophagy of alveolar type II epithelial cell injury during ALI.In addition,the results showed that targeting of FoxO3,an autophagy-related gene,involved miR-34a,and silencing of FoxO3 expression inhibited LPS-induce autophagy.In conclusion,this study provided the first evidence that miR-34a suppresses the autophagic activity of alveolar type II epithelial cells during LPS-induced ALI by inhibiting FoxO3expression.Because of the important roles of exces-sive autophagic activity of alveolar type II epithelial cells in the pathological process of ALI,we predict that the induc-tion of miR-34a is a protective factor that could be used as a novel therapeutic approach for the treatment of ALI.We therefore propose that the autophagy activity of alveolar type II epithelial cells and the expression of miR-34a increase in LPS-induced ALI.The miR-34a could suppress this excessive autophagic activity of alveolar type II epithelial cells by inhibition of FoxO3expression, resulting in protection against ALI.ACKNOWLEDGEMENTSThis work was supported by funding from the Natural Science Foundation of Hunan Province,China(No. 2014JJ7062),Research Foundation of Traditional Chinese medicine of Hunan Province,China(No.201696),The Open Research Foundation of Key Laboratory of traditional Chinese medicine in Hunan,Hunan University of Chinese Medicine China(No.ZYNK201506),the Research Foun-dation of Education Bureau of Hunan Province,China(No. 13C691),the58batch Postdoctoral Science Foundation funded,China(No.2015M580690),the Research Founda-tion of Stem Cell Regulation and Application,Hunan Uni-versity of Chinese Medicine,China(No.2013GXB02). COMPLIANCE WITH ETHICAL STANDARDSAnimal experimentations were conducted according to the National Institute of Health Guide for the Care and Use of Laboratory Animals and received the approval of the Animal Care and Use Committee of Hunan University of Chinese Medicine,Changsha China.MiR-34a Suppresses Autophagy in Acute Lung Injury。
GLP类似物药物进展

G L P-1类似物药物进展-截止胰高血糖素样肽(glucagon-likepeptide,GLP)是小肠表皮细胞在食物刺激情况下分泌的单肽类肠促胰岛素,包括GLP-1、GLP-2两种类型。
其中GLP-2具有促进小肠生长,抑制细胞凋亡,促进胃排空,增加食欲的药理作用,临床上可用于治疗小肠短小综合症;而GLP-1具有促进胰岛素分泌,保护胰岛β细胞,抑制胰高血糖素分泌,抑制胃排空,降低食欲的药理作用,临床可用于二型糖尿病和肥胖症的治疗。
人体内具有生物活性的GLP-1主要是GLP-1(7-36)酰胺和GLP-1(7-37),天然GLP-1可被二肽基肽酶Ⅳ(dipeptidylpeptidase-Ⅳ,DPP-Ⅳ)迅速水解失活(半衰期小于5min),不具有临床使用价值,因此对GLP-1结构修饰,掩盖DPP-Ⅳ的结合位点,延长半衰期并保证疗效是该类药物研发的主要方向。
一、已上市GLP-1类似物目前已上市的5个GLP-1类似物(表1)包括艾塞那肽(Byetta/Bydureon,byAmylin/Lilly)、利拉鲁肽(Victoza/Saxenda,byNovoNordisk)、利司那肽(Lyxumia,bySanofiAventis/Zealand)、阿必鲁肽(Tanzeum,byGSK)及杜拉鲁肽(Trulicity,byLilly):1.艾塞那肽(Exenatide)艾塞那肽(商品名Byetta)是第一个上市的GLP-1类似物,由Amylin和Lilly公司于1995年开始联合研发,2005年4月获得FDA的批准上市。
艾塞那肽源于从蜥蜴唾液中分离出的GLP-1类似物Exendin-4,与GLP-1大约有53%的同源性。
由于其N端第二位由Gly代替了GLP-1中Ala,不被DPP-Ⅳ降解,而相对天然GLP-1而言具有较长的半衰期和较强的生物活性,临床使用频率为每日2次。
AstraZeneca收购Amylin取得艾塞那肽的全球开发销售权后,开发了其缓释混悬制剂BydureonPen,并于2014年获得FDA批准。
药物GLP认证目录(2022版)

中国药科大学
(新药安全评价
研究中心)
单次和多次给药毒性试验(非啮齿类,灵长类)
GLP21007126
广州医药研究总院有限公司(药物非临床评价研究中心)
1.生殖毒性试验(Ш段)
2.致癌试验
GLP21008127
西咸新区国睿一诺药物安全评价研究有限公司
1.遗传毒性试验(Ames、微核、染色体畸变)
药物GLP认证目录
机 构 名 称
试 验 项 目
认证批件编号
湖南普瑞玛药物研究中心有限公司
1.单次和多次给药毒性试验(啮齿类)
2.单次和多次给药毒性试验(非啮齿类)
3.生殖毒性试验(I段、II段、Ш段)
4.遗传毒性试验(Ames、微核、染色体畸变)
5.局部毒性试验
6.免疫原性试验
安全性药理试验
8.毒代动力学试验
生殖毒性试验(Ш段)
GLP21017136
益诺思生物技术南通有限公司
1.致癌试验
2.免疫原性试验
GLP21020139
上海益诺思生物技术股份有限公司
依赖性试验
GLP22001140
江苏鼎泰药物研究股份有限公司
生殖毒性试验(Ш段)
GLP22002141
1.单次和多次给药毒性试验(非啮齿类)
2.免疫原性试验
3.安全性药理试验
4.依赖性试验
GLP21015134
广州博济医药生物技术股份有限公司
(药物评价中心)
1.单次和多次给药毒性试验(非啮齿类,灵长类)
2.生殖毒性试验(I段、II段)
3.免疫原性试验
GLP21016135
山东省医学科学院药物研究所
(山东省医学科学院药物安全评价中心)
(S)-Crizotinib 1374356-45-2 GlpBio

Product Data SheetProduct Name:(S)-CrizotinibCat. No.:GC38649Chemical PropertiesCas No.1374356-45-2ChemicalNameN/ACanonicalSMILESNC1=NC=C(C2=CN(C3CCNCC3)N=C2)C=C1O[C@H](C4=C(Cl)C=CC(F)=C4Cl)C Formula C21H22Cl2FN5O M.Wt450.34 Solubility Soluble in DMSO Storage Store at -20°CGeneral tips For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months.Shipping Condition Evaluation sample solution : ship with blue ice All other available size: ship with RT , or blue ice upon request.StructureBackground(S)-Crizotinib, a (S)-enantiomer of crizotinib, is a potent and selective MTH1 (mutT homologue) inhibitor with an IC50 of 330 nM. (S)-Crizotinib disrupts nucleotide pool homeostasis via MTH1 inhibition, induces an increase in DNA singleProduct Data Sheetstrand breaks, activates DNA repair in human colon carcinoma cells, and effectively suppresses tumour growth in animal models[1].(S)-crizotinib (0.625-80 μM; 24 hours) decreases the viability of NCI-H460, H1975 and A549 cells with IC50 values of 14.29, 16.54 and 11.25 μM, respectively[2]. (S)-crizotinib (10-30 μM; 24 hours) induces NCI-H460, H1975 and A549 cells apoptosis[2]. (S)-crizotinib (10-30 μM; 24 hours) decreases Bcl-2: Bax ratio. (S)-crizotinib decreases B cell lymphoma 2 (Bcl-2), and Bcl-2 associated protein x (Bax) is either unaltered (H460 cells) or shows an increase(H1975 cells)[2]. (S)-Crizotinib induces apoptosis in human non-small cell lung cancer (NSCLC) cells by activating ROS-dependent ER stress apoptotic pathway independent of mutT homologue (MTH1)[2]. Cell Viability Assay[2] Cell Line: NSCLC cells, NCI-H460, H1975 and A549 cells(S)-crizotinib (7.5 or 15 mg/kg; intraperitoneal injections; once daily for 10 days) results in significant reductions in both tumor volume and tumor weight[2]. Animal Model: Five-week-old, athymic BALB/c nu/nu female mice (17-19 g) with NCI-H460 cells[2][1]. Huber KV, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014 Apr 10;508(7495):222-7. [2]. Dai X, et al. (S)-crizotinib induces apoptosis in human non-small cell lung cancer cells by activating ROS independent of MTH1. J Exp Clin Cancer Res. 2017 Sep 7;36(1):120.。
glp-1诱导表达的原理

GLP-1诱导表达的原理三磷酸鸟苷环化水解酶(GTPCH)Ⅰ是四氢生物蝶呤(BH4)合成的关键限速酶,BH4是内皮一氧化氮合酶(eNOS)的辅助因子。
糖尿病等病理条件下,BH4水平下降促使eNOS脱偶联,导致内皮功能异常。
胰高糖素样肽(GLP-1)可增强内皮eNOS的磷酸化、增加NO、发挥内皮保护作用。
该课题组前期研究显示,同型半胱氨酸可抑制内皮BH4水平、诱导eNOS脱偶联,损害患者冠脉内皮功能。
预实验显示,GLP-1受体激动剂可促进内皮细胞eNOS表达、抑制氧化应激、增加NO。
该研究分别在内皮细胞及apoE敲除小鼠整体水平,探讨GLP-1受体激动剂能否通过上调血管内皮GTPCH1水平、增加BH4、抑制氧化应激、促使eNOS复偶联等机制,发挥血管内皮保护作用,并在临床水平进一步验证。
实验结果显示GLP-1受体激动剂艾塞那肽可以改善内皮细胞氧化损伤,可能是通过激活AMPK途径增加GTPCH1表达,从而上调BH4水平,进而促进eNOS复偶联,NO产生增加,最终达到改善内皮功能的作用。
在ApoE小鼠中持续应用艾塞那肽(8周)能够显著提高主动脉中GTPCH-1的表达水平和BH4的含量,并且使得主动脉eNOS的磷酸化水平相应增高,提高eNOS的活性,使之能够保持并较好地发挥催化生成血管内皮舒张因子NO的功能,最终增加血管内NO的生成同时减少NO的损失,改善血管内皮依赖性舒张功能,从而达到保护内皮功能的作用。
临床研究显示艾塞那肽不仅能降低2型糖尿病患者的血糖,改善胰岛细胞功能,降低患者的体重、体重指数、腹围,调节血脂代谢,还可具有改善血管内皮功能,提高冠脉内血流发挥血管内皮保护作用,减少糖尿病患者心血管不良事件的发生。
GLP-1受体激动剂,通过上调内皮细胞GTPCH1表达、增加BH4水平、促使eNOS复偶联、增加NO,进而改善血管内皮功能;并可通过激活AMPK,反转被抑制的内皮细胞GTPCH1表达,增加BH4水平,促使eNOS复偶联,改善内皮依赖的血管功能,发挥血管保护作用。
3氨基4溴吡唑安全技术说明书MSDS

第一部分化学品及企业标识化学品中文名:3-氨基-4-澳口比噗化学品英文名:3-Amino-4-bromopyrazo1e5-Amino-4-bromo-1H-pyrazo1e CASNo.:16461-94-2分子式:C3H4BrN3产品推荐及限制用途:工业及科研用途。
第二部分危险性概述紧急情况概述吞咽有害。
造成皮肤刺激。
造成严重眼刺激。
可引起呼吸道刺激。
GHS危险性类别急性经口毒性类别4皮肤腐蚀/刺激类别2严重眼损伤/眼刺激类别2特异性靶器官毒性一次接触类别3标签要素:象形图:警示词:警告危险性说明:H302吞咽有害H315造成皮肤刺激H319造成严重眼刺激H335可引起呼吸道刺激防范说明—预防措施:——P264作业后彻底清洗。
——P270使用本产品时不要进食、饮水或吸烟。
-P280戴防护手套/穿防护服/戴防护眼罩/戴防护面具。
——P261避免吸入粉尘/烟/气体/烟雾/蒸气/喷雾。
——P271只能在室外或通风良好处使用。
•事故响应]——P301+P312如误吞咽:如感觉不适,呼叫解毒中心/医生——P33O漱口。
——P302+P352如皮肤沾染:用水充分清洗。
—P332+P313如发生皮肤刺激:求医/就诊。
一一P362+P364脱掉沾染的衣服,清洗后方可重新使用-P305+P351+P338如进入眼睛:用水小心冲洗几分钟。
如戴隐形眼镜并可方便地取出,取出隐形眼镜。
继续冲洗。
一一P337+P313如仍觉眼刺激:求医/就诊。
一一P304+P340如误吸入:将人转移到空气新鲜处,保持呼吸舒适体位。
------ P312如感觉不适,呼叫解毒中心/医生•安全储存:-P403+P233存放在通风良好的地方。
保持容器密闭。
——P405存放处须加锁。
•废弃处置:一一P501按当地法规处置内装物/容器。
物理和化学危险:无资料。
健康危害:吞咽有害。
造成皮肤刺激。
造成严重眼刺激。
可引起呼吸道刺激。
环境危害:无资料。
第三部分成分/组成信息第四部分急救措施急救:吸入:如果吸入,请将患者移到新鲜空气处。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Product Data Sheet
Product Name:3,4-DAA
Cat. No.:GC12624
Chemical Properties
Cas No.
Chemical
Name
2-[3-(3,4-dimethoxy-phenyl)-acryloylamino]-3-hydroxy-benzoic acid
Canonical
SMILES
COC1=C(OC)C=C(/C=C/C(NC2=C(O)C=CC=C2C(O)=O)=O)C=C1
Formula C18H17NO6M.Wt343.3 Solubility Soluble in DMSO Storage Store at -20°C
General tips For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months.
Shipping Condition Evaluation sample solution : ship with blue ice All other available size: ship with RT , or blue ice upon request.
Structure
Background
N-(3,4,-Dimethoxycinnamoyl) anthranilic acid (3,4-DAA) is a synthetic derivative of the tryptophan metabolite anthranilic acid [1].
Product Data Sheet
Degradation of the essential amino acid Trp by indoleamine 2,3-dioxygenase (IDO) plays an important role in immunity. IDO has been implicated in immune modulation through limiting T cell function and engage mechanisms of immune tolerance. Activation of IDO has been observed during tumor development, helping malignant cells escape eradication by the immune system [2].
3,4-DAA suppressed antigen-specific proliferation of MBP Ac1-11 TCR transgenic CD4+ T cells by arrested the cells in G1/S-phase. 3,4-DAA (200 μM) reduced the release of IL-2, IFN-γ, and TNF-α and 3,4-DAA (30 μM)increased the level of IL-4 and IL-10 in splenocytes from MBP Ac1-11 TCR transgenic T cells after antigen stimulation [1]. 3,4-DAA dose-dependently decreased IFNγ–induced cell surface expression of MHC class II and costimulatory molecules and suppressed the expression of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) release from EOC20 cells and phosphorylation of STAT1α induced by IFNγ. In Mice with experimental autoimmune encephalomyelitis, oral administration of 3,4-DAA (300 mg/kg per day) exhibited fewer and milder relapses and less severe disease compared to control animals [1]. In allograft immunorejection model, administration of 3,4-DAA reduced histological severity of allograft immunorejection, decreased serum levels of TNF-α and IFN-γ, and raised serum levels of IL-10 [3].
References:
[1] Platten M, Ho P P, Youssef S, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite[J]. Science, 2005, 310(5749): 850-855.
[2] Hirata F, Ohnishi T, Hayaishi O. Indoleamine 2, 3-Dioxygenase[J]. J. Biol. Chem, 1977, 252: 4637.
[3] Sun Q F, Ding J G, Sheng J F, et al. Novel action of 3, 4‐DAA ameliorating acute liver allograft injury[J]. Cell biochemistry and function, 2011, 29(8): 673-678.。