新型恶唑烷酮介绍
恶唑烷酮类抗菌新药~利奈唑胺

利奈唑胺在体内缓慢代谢为羧酸, 其抗菌作用低。在尿 中排出原型药 $!& , 无活性代谢物 !"& , 粪中排出代谢物 消除半衰期为 ( - ! %"& 。8# 和 8#9 分别为 %’" 和 (" *: 2 *;<, 6 ! - ! 3。 (三)年龄、 性别、 疾病对药代动力学的影响 成人中, 年龄、 性别对药代动力学性质没有影响, 无需调 整剂量。在儿童与青少年中, 各年龄组间药代动力学性质亦 无明显差异
[%’] 感染, 利奈唑胺亦有效 。
在小鼠中性粒细胞减少症模型中, 利奈唑胺对万古霉素 耐药屎肠球菌和氨基糖苷类耐药粪肠球菌所致感染有效。 在治疗粪肠球菌软组织感染时, 利奈唑胺与万古霉素同样有
[%’] 效 。
四、药代动力学
[), %+] (一)代谢与分布
人体药物代谢动力学研究结果显示, 利奈唑胺口服吸收 快速且完全, 在%F+J内 ( ! >;:) 达到峰浓度 ( " >;:) , 其平均生 物利用度为 %’#. 。进食可使利奈唑胺峰浓度降低 +#. , 但 对 &10、 利奈唑胺表观分布容积 ! >;:和生物利用度没有影响,
[*] 球菌均显示了良好的抗菌作用 , 对厌氧菌亦具抗菌活性。
革兰阳性球菌是临床常见致病菌, 且耐药性上升迅速, 甲氧西林耐药葡萄球菌、 糖肽类耐药肠球菌、 青霉素耐药肺 炎链球菌所占比例日益上升, 糖肽类中度敏感葡萄球菌也已 出现。据 %""* 年美国医院感染监测系统资料, 医院内万古
[%] 霉素耐药肠球菌分离比例 - +’. , 重症监护病房 ( /01) 中
恶唑烷酮类抗菌药:利奈唑胺

恶唑烷酮类抗菌药:利奈唑胺
林东昉
【期刊名称】《中国感染与化疗杂志》
【年(卷),期】2001(1)3
【摘要】@@ 革兰阳性球菌是临床常见致病菌,且耐药性上升迅速,甲氧西林耐药葡萄球菌、糖肽类耐药肠球菌、青霉素耐药肺炎链球菌所占比例日益上升,糖肽类中度敏感葡萄球菌也已出现.据 1998年美国医院感染监测系统资料,医院内万古霉素耐药肠球菌分离比例>20%[1] ,重症监护病房(ICU)中甲氧西林耐药葡萄球菌超过50%.而对糖肽类抗生素耐药的肠球菌属,尤其是屎肠球菌 ,可对临床应用抗菌药均出现耐药,感染性疾病的治疗面临严峻的挑战.近年来,人们开发了数个新的抗菌药,用于耐药革兰阳性菌的治疗.利奈唑胺(linezolid)为一类新型抗菌药恶唑烷酮类(oxazolidinones)的第一个用于临床的品种,对各类耐药革兰阳性球菌均具抗菌活性.现对利奈唑胺的抗菌作用、药代动力学、临床应用作一综述.rn一、作用机制【总页数】3页(P184-186)
【作者】林东昉
【作者单位】上海复旦大学附属华山医院抗生素研究所
【正文语种】中文
【中图分类】R978.1
【相关文献】
1.葡萄球菌临床分布特征及对利奈唑胺等新型抗菌药物的耐药性分析 [J], 林少华;骆丰
2.利奈唑胺治疗糖肽类抗菌药物无效的医院获得性MRSA肺炎的病例分析 [J], 龙锐;邱峰;蒙龙;杨佳丹;张成志
3.噁唑烷酮类抗菌药利奈唑胺的临床研究进展 [J], 任少华;秦丽君;胡华成
4.噁唑烷酮类抗菌药利奈唑胺的临床研究进展 [J], 任少华;秦丽君;胡华成
5.新型抗菌药利奈唑胺的临床应用研究进展 [J], 王婷;李树安;张珍明;齐家娟;赵红博;王璇
因版权原因,仅展示原文概要,查看原文内容请购买。
新型恶唑烷酮类抗菌药——利奈唑烷

1 .开 发 概 况
: 唑 烷 酮类 抗 菌 药 于 1 8 首 次 被报 道 。 关 研 究 表 恶 9 7年 有 明 ,此 类 化 合 物在 体 外 对 大多 数 革 兰 氏 阳性 苗 及 在 动 物 感
利 奈 唑 烷体 外 抗 菌 谱 及 其 活 性 大 体 类 似 于 万 古 霉 素 ,
其 实验 中测 得 的 M ( 低 抑 菌 浓 度 ) 纳 如 表 1 J 最 归 。利 奈 唑 烷 也 已被 证 明 对 厌 氧 菌 具 有 活 性 ,其 中 对 脆 弱 拟 杆 菌 的 MI 为 4 g l 但 利 奈 唑 烷 对 革 兰 氏阴 性 菌 基 本 上 没 有 , /m 。 U
染 模 型 中对 具 有耐 药 性 的 葡萄 球 菌 、链 球 菌 和 肠 球 菌 显 现 活性 ,它 们 能在 细 菌生 命 周 期 的一 个 很 早 期 阶 段抑 制 细 苗 蛋 白质 合 成 , 用 机制 独 特 。 外 研 究未 见 与 已有 抗 菌 药 问 作 体 存 在交 叉 耐 药 性 ,且 体 外 使 用 数 种 方 法 均 没 有 获得 耐 药 性 突 变菌 株 , 引起 医药 学 界 的极 大 兴 趣 。 因 当 时所 得 化 合 故 唯 物 毒性 大 而 未 能及 时进 入 临 床 研 究 。 利奈 唑 烷是 由数 种有 前 途 实 验 化合 物 中优 选 得到 的一 个
n ^
: “ 、0 3 -
利 奈 唑 烷 已 经 过 大 量 动 物 和人 体 研 究 , 明其 安 全 性 证 高 并 具 上述 罹 唑 烷酮 类 化 合 物 的各 种 特 性 .已 于 2 O O 0年 4 月 1 8日被 美 国 食 品 药 品 管 理局 ( D F A)批 准 用 于 治 疗 革 兰 氏阳 性 菌 ,包括 耐万 古霉 素 (a e m cn v no yi)粪 肠球 菌 ( a V E) ( 括 血 液 感染 病 例 )和 耐 甲 氧西 林 ( ti ln 包 me el )金 黄 色 酿 h ii 脓葡萄球菌 ( S MR A)所 致 院区 获 得 性 肺 炎 及 并 发 的 皮 肤 和 皮 肤组 织感 染等 。这 是 利 奈 唑 烷 在 世 界 范 围 内首 次 获 得 批 准 , 是 F A4 也 D 0年 来 批 准 的第 一 个 用 于 治 疗 耐 MR A感 染 S 的药物。 2. 菌 活 性 抗
新型恶唑烷酮介绍

第四章新型噁唑烷酮类抗药菌药物多年来随着抗生素在全球的普及和不断被人滥用,无论革兰氏阳性菌或阴性细菌均已出现耐药趋向,革兰氏阳性细菌的耐药问题更为严重。
据世界卫生组织估计,每天大约有5万人死于感染性疾病,感染性疾病从新成为威胁人类健康和社会发展的主要问题。
世界范围内出现的耐甲氧西林的金黄色葡萄球菌(MRSA)和表皮葡萄球菌(MRSE),耐药性肺炎链球菌,多药耐药的结核分支杆菌及耐万古霉素肠球菌(VRE)是当前临床中存在的主要问题。
尤其是耐万古霉素肠球菌(VRE)的出现,突破了严重感染患者治疗的“最后手段”。
面对这种困境,专家们提出“更新战略”,就是用“新一代”的抗生素替代“老”的品种。
世界许多制药公司都在积极寻找能对付耐多种抗生素的耐药细菌的新型药物。
噁唑烷酮就是一类极有发展前景的新型全合成抗菌剂。
Drug Resistance in Hospital-Acquired InfectionsDrug/Pathogen Resistance(%)Vancomycin/enterococci 24.7Methicillin/S.aureus 53.6Methicillin/Coag.-neg.S.aureus 88.2Imipenem/P.aeruginosa 16.4Quinolone/P.aeruginosa 23.03rd-gen.ceph. /P.aeruginosa 20.63rd-gen.ceph. /Enterobacter 33.1SCIENCE 2004,303,1798噁唑烷酮类抗菌药物的发展NSOC l OC H 3OSHO H 2NOOXH A CO C H 3O Dup105 X=S Dup721 X=CNON H A CO ONON H A CONH OOH A CC HNH A CO OFNH A CO NFH OO123451978年,美国杜邦公司科学家报道噁唑烷酮衍生物1在控制细菌引起的植物疾病方面的应用 。
恶唑烷酮类抗菌药

Antibacerial oxazolidinone agents
背景
➢ 由于病原菌对现有抗菌药产生了耐药性,因 此寻找结构新颖、性能独特的新结构类型的 抗菌药物备受关注
➢ 近年来合成抗菌药物的研究热点包括恶唑烷 酮类oxazolidinones等
背景
➢ 早在1987年杜邦公司就合成了恶唑烷酮类化合物 Dup721和Dup,但在I期临床试验中因毒性问题而被终 止
➢ 美国普强公司在Dup721的基础上进行结构改造, 成功开放了两种新的恶唑烷酮类高效抗菌药
✓ 利奈唑酮linezolid,利奈唑胺(2000年,美国上市) ✓ 羟哌恶酮eperezolid,依哌唑胺 ✓ 它们既可以口服,也可以注射
作用机制
➢ 与现有抗菌药不同
➢ 作用于细菌蛋白质合成的最早期阶段
➢ 对革兰阳性菌及耐药肠球菌等的感染均有显 著疗效
➢ 临床用于耐药革兰阳性菌引起的感染性疾病、也 可用于外科感染性疾病的治疗
利奈唑酮Linezolid
➢ 结构
O O HN CH3
N
O
N
O
F
➢ 化学名:
❖ (S)-[N-3-(3’-氟-4‘-吗啉基)苯基-2-氧代-5-恶唑 烷基]甲基乙酰胺
❖ (S)-[N-3-(3’-Fluro-4’-morpholino)phenyl-2oxo-5-oxazoly]methyl acetamide
作用机制:
新型细菌蛋白合成抑制剂,作用于细菌50s核 糖体亚基,不影响肽基转移酶活性
抑制mRNA与核糖体连接,阻止70s起始复合物 的形成,从而抑制细菌蛋白质的合成
O O HN CH3
N
O
N
O
F
Radezolid_新型的恶唑烷酮类抗生素_869884-78-6_Apexbio

细胞和内皮细胞,从而发挥其抗菌作用。 体外:一项研究发现,Radezolid 在所有类型的细胞(人角质形成细胞、内皮细胞、支气管 上皮细胞、成骨细胞、巨噬细胞以及大鼠胚胎成纤维细胞)中具有相似的积累量(~10 倍)。 在所有这些模型中,不论细菌种类、耐药表型或感染的细胞类型,在相等质量浓度下, Radezolid 的抗菌作用均比 Linezolid 高 10 倍。上述数据表明,Radezolid 的胞内积累量在治疗 细胞复发性或持续性感染中起着决定性作用[2]。 体内:接种 24 小时后,50 mg/kg 的 Radezolid 和 Linezolid 表现出相当的细菌负荷减少。组 织药物浓度的曲线下面积(AUC)分析表明,相对于未受感染的大腿组织,Radezolid 在受感 染大腿中的积累量为 2.4 倍。Linezolid 没有积聚于受感染的大腿[3]。 临床试验:Radezolid(INN,RX-1741)由 Rib-X 制药有限公司研发,用于治疗严重的多重耐 药菌感染。Radezolid 已完成了两项 II 期临床试验。上述两项临床试验分别验证了 Radezolid 对无并发症皮肤及皮肤结构感染(uSSSI)和社区获得性肺炎(CAP)的疗效 (/wiki/Radezolid)。
引用文献
1.Michalska K, Gruba E, et al. "Application of spectroscopic methods (FT-IR, Raman, ECD and NMR) in studies of identification and optical purity of radezolid." Spectrochim Acta A Mol Biomol Spectrosc. 2017 Apr 20;183:116-122. PMID:28456082 2.Michalska K, Gruba E, et al. "Enantioselective recognition of radezolid by cyclodextrin modified capillary electrokinetic chromatography and electronic circular dichroism." J Pharm Biomed Anal. 2017 May 30;139:98-108. PMID:28279932
恶唑烷酮类抗菌药研究进展

动物医学进展,2019,40(3):101-105Progress in Veterinary Medicine噁唑烷酮类抗菌药研究进展 收稿日期:2018-03-19 基金项目:国家科技支撑计划项目(2015BAD11B01) 作者简介:李晓婷(1995-),女,安徽宿州人,硕士研究生,主要从事兽医药理学与毒理学研究。
*通讯作者李晓婷1,2,张继瑜1,2*(1.甘肃农业大学动物医学院,甘肃兰州730070;2.中国农业科学院兰州畜牧与兽药研究所,甘肃兰州730050) 摘 要:在世界范围内,细菌耐药性问题日益严重,已严重影响了感染性疾病的治疗。
新型抗耐药菌药物的研究已成为抗菌药物研究的主要方向。
噁唑烷酮类化合物是一类新型的治疗细菌性感染的化学全合成药物,具有抑制多重耐药的革兰阳性菌的功效。
且作用机制独特,不易与其他药物发生交叉耐药性,从而得到了广泛的研究。
在第一个噁唑烷酮类抗菌药物利奈唑胺成功上市后,又有新的化合物进入临床研究,并取得了良好的临床治疗效果。
论文介绍了噁唑烷酮类抗菌药的作用机制、抗菌活性、构效关系及最新的噁唑烷酮类抗菌药的研究,为研发新型噁唑烷酮类抗菌药提供参考。
关键词:噁唑烷酮;作用机制;抗菌活性;构效关系中图分类号:S859.8796文献标识码:A文章编号:1007-5038(2019)03-0101-05 从1928年青霉素被发现以来,抗生素就成为了临床多种疾病治疗常用的药物。
抗生素品种和数量推陈出新的同时,在各种人为和客观因素的影响下,药物选择难度和药物过度、滥用等情况增加,无论革兰阳性菌还是革兰阴性菌均出现了严重的耐药性,此外,细菌通过水平转移获得外源性耐药基因也加快了耐药菌株的产生。
2006年Science发文报道,一株于1930年保存在实验室的金黄色葡萄球菌对目前临床所用抗生素都敏感,而一株从患者身上分离的金黄色葡萄球菌,几乎对所有的抗生素耐药,而这种耐药性在同一细菌内,呈现出对不同类的抗生素的多重耐药机制[1]。
恶唑烷酮类化合物合成的研究

噁唑烷酮类化合物合成的研究
噁唑烷酮类化合物是一类具有广泛生物活性和药用价值的有机化合物,其合成研究一直备受关注。
噁唑烷酮类化合物具有抗菌、抗病毒、抗肿瘤等多种生物活性,因此在药物研发领域具有重要的应用前景。
近年来,许多研究人员致力于开发新的合成方法来合成噁唑烷酮类化合物。
其中,金属催化的合成方法成为研究热点之一。
通过金属催化反应,可以高效地构建噁唑烷酮的骨架,实现对目标化合物的高产率合成。
此外,还有许多基于不同反应机理的合成策略,如环化反应、串联反应等,为噁唑烷酮类化合物的合成提供了多样化的选择。
在合成方法的研究中,不仅要考虑反应的高效性和选择性,还要注重对环境友好性的考量。
因此,绿色合成理念也逐渐成为噁唑烷酮类化合物合成研究的重要方向。
通过开发绿色合成方法,可以减少废弃物的产生,降低对环境的污染,从而实现可持续发展的目标。
总的来说,噁唑烷酮类化合物合成的研究具有重要的科学意义
和应用价值。
随着合成方法的不断完善和绿色合成技术的应用,相信噁唑烷酮类化合物的合成将迎来更加美好的发展前景。
新型恶唑烷酮类化合物5-位侧链的结构改造及抗菌活性

vto a tb ce i la tvte r lmi a iy M e hod Th i e c mp u d r y t e ie n 9 — 1 t p ir n i a t ra ciiis p e i n rl . t s e t l o o n swe e s n h sz d i t se s 2
苯 胺 为 原 料 , 9~1 反 应 合 成 目标化 合 物 ; 用 二 倍 稀 释 法 , 定 目标 化 合 物 的 体 外 抗 菌 活 性 。 结 果 设 计 、 经 2步 采 测 合 成了3 0个 新 化 合 物 , 中 目标 化 合 物 l 其 8个 , 结 构 经 MR 及 M 其 HN S等方 法 确证 , 1 化 合 物 显 示 出 不 同程 度 的抗 1 个
Z AIXi H n,ZHANG o g n Gu — a g, GOU Do g h i L U u — e g, GONG i g n —u , I J np n Pn
( c o l fPh r cui l gn eig,S ey n h r cuia iest,S e y n 0 1 S h o ama e t a o c En iern h n a g P a ma et lUnv r y h n a gJ 0 6,C ia c i 1 hn )
翟 鑫 ,张 国 刚 ,缑 东 辉 ,刘 君 鹏 ,宫 平
( 阳 药 科 大 学 制 药 工 程 学 院 ,辽 宁 沈 阳 10 1 ) 沈 106
摘 要 :目 的
对 嗯 唑 烷 酮 类 化 合 物 5位 侧链 进 行 结 构 修 饰 与 改 造 , 初 步 评 价 其 体 外 抗 菌 活性 。 方 法 以 问 氟 一 并
wih t e sa tng ma e i l3. u r a i n n h i n v to a tb c e i l a t i e r x m i e b s n t h t ri t ra f o o n l e a d t e r i i n i a t r a c i t s we e e a n d y u i g l i r v i M u le — n o r t i i n me h d. Re uls Th ry n w c mp u s we e d s g e nd s n h s z d.i e l r Hi t n b o h d l o ut to s t i t e o o nd r e i n d a y t e ie n wh c i ht e o e i e c mp u d r r p r d a d t e r s r c u e r o f me y H R n i h e g e n n v l tt o l o n s we e p e a e n h i tu t r s we e c n i r db NM ad ES — I MS. El v n c mp u d h we n i a t r a c i i e o a c ra n e t n .a e e o o n s s o d a tb c e i la t t s t e t i x e t mo g t e c m p un s7a. v i n h m o o d
利奈唑胺的药理学特点及临床应用

利奈唑胺的药理学特点及临床应用摘要利奈唑胺是用于临床的第一个噁唑烷酮类抗菌药,治疗耐药革兰阳性菌感染及耐药结核病的疗效显著,但血液和神经系统的不良反应也较突出,且易诱导细菌产生耐药。
因此,临床上必须合理使用利奈唑胺才能充分发挥其优良的抗菌作用、减少不良反应的发生并延缓细菌耐药的产生。
本文介绍利奈唑胺的药效学和药动学特点、着重探讨其临床应用的指征,为临床规范、合理使用利奈唑胺提供参考。
ABSTRACT Linezolid is the first antibacterials of oxazolidinone used in the clinic,which has significant effect in treating drug-resistant Gram-positive bacterial infections and drug-resistant tuberculosis,and also has some obvious adverse reactions in hematologic and nervous system and even induces bacterial resistance easily. Therefore,it must be used in right ways to make full use of its excellent antibacterial activity,and avoid its adverse reaction and delay the generation of bacterial resistance. This article presents the features of pharmacokinetics and pharmacodynamics of linezolid,focuses on the exploration of its suitable indications,and provision of some references for its clinical application.Key words linezolid;pharmacokinetics;pharmacodynamics;clinical application利奈唑胺(linezolid)是一种人工合成的噁唑烷酮类抗菌药,对大多数革兰阳性致病菌都有良好的抗菌活性,与其他抗菌药多无交叉耐药现象,加之组织、体液分布广泛以及给药方法便捷,使得其治疗多重耐药革兰阳性菌感染的有效性和安全性均很好,在临床上受到广泛的关注。
治疗皮肤感染成人患者新型抗菌药物Sivextro
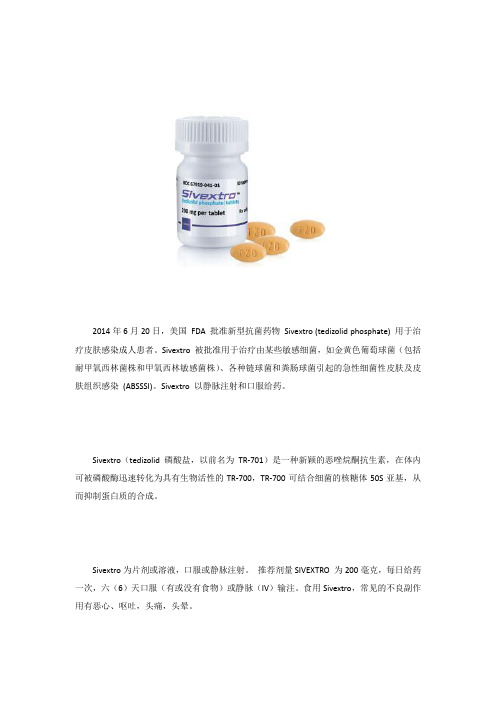
2014年6月20日,美国FDA 批准新型抗菌药物Sivextro (tedizolid phosphate) 用于治疗皮肤感染成人患者。
Sivextro 被批准用于治疗由某些敏感细菌,如金黄色葡萄球菌(包括耐甲氧西林菌株和甲氧西林敏感菌株)、各种链球菌和粪肠球菌引起的急性细菌性皮肤及皮肤组织感染(ABSSSI)。
Sivextro 以静脉注射和口服给药。
Sivextro(tedizolid磷酸盐,以前名为TR-701)是一种新颖的恶唑烷酮抗生素,在体内可被磷酸酶迅速转化为具有生物活性的TR-700,TR-700可结合细菌的核糖体50S亚基,从而抑制蛋白质的合成。
Sivextro为片剂或溶液,口服或静脉注射。
推荐剂量SIVEXTRO 为200毫克,每日给药一次,六(6)天口服(有或没有食物)或静脉(IV)输注。
食用Sivextro,常见的不良副作用有恶心、呕吐,头痛,头晕。
Sivextro(tedizolid phosphate)是的恶唑烷酮。
抗菌活性抗菌剂胺介导的结合导致蛋白质合成的抑制细菌核糖体50S亚基。
Tedizolid抑制细菌蛋白质合成,通过从其他非恶唑烷酮类抗菌药物的作用机制不同;因此,抗菌药物tedizolid和其他类之间的交叉耐药性,是不可能的。
结果体外杀菌研究表明tedizolid是抑菌抗肠球菌,葡萄球菌和链球菌。
Sivextro的安全性和有效性进行的两项临床试验中,与1315名成人ABSSSI评价。
参与者被随机分配接受SIVEXTRO或利奈唑胺,另一种抗菌药物被批准用于治疗ABSSSI。
结果表明,SIVEXTRO为有效治疗ABSSSI利奈唑胺。
Sivextro的FDA的批准是基于两个多中心,随机,双盲,跨国,非劣效性试验。
试验相比SIVEXTRO 200毫克每日一次6天与利奈唑胺600毫克,每12小时一次,10天。
在试验1中,受试者均口服治疗,而实验2科目可以至少一天静脉注射治疗后接受口服治疗。
恶唑烷酮类抗菌药的研究进展

作者简介杨娜,女,硕士生E-mail:ynyangna@ 通讯作者尤启冬,男,博士生导师,研究方向:药物化学E-mail:youqd@收稿日期2011-04-13修回日期2011-05-22*噁唑烷酮类抗菌药的研究进展杨娜,尤启冬*中国药科大学药物化学教研室,南京210009摘要噁唑烷酮类化合物是一类新型的治疗细菌性感染的化学全合成药物,并得到了广泛的研究与发展。
在Linezolid (利奈唑胺)被美国FDA 批准上市后,又有Radezolid (雷得唑来)和Torezolid 进入临床研究。
本文对近年来噁唑烷酮类抗菌化合物的发展进行了综述,并着重介绍了目前在临床研究的两个噁唑烷酮类抗菌药Radezolid 和Torezolid 。
关键词噁唑烷酮;抗菌活性;利奈唑胺;雷得唑来中图分类号R914.5;R978文献标志码A 文章编号1673-7806(2011)04-332-05抗生素和合成抗菌药物是目前人类治疗细菌感染性疾病的首选药物。
在我国,抗菌药物被过度使用甚至滥用的情况已很突出。
据统计,临床上用于预防性抗菌药物处方占抗菌药物总消耗量的50%以上,而其中确实为细菌感染者仅占极少数。
抗生素的过多使用甚至滥用,使得细菌耐药问题日益严重。
其中,革兰阳性菌(G +)的耐药问题尤为严重,例如耐甲氧西林的金黄色葡萄球菌(MRSA )和表皮葡萄球菌(MRSE ),耐青霉素的肺炎链球菌(PRSP )及耐万古霉素的肠球菌(VRE )等,这些耐药菌的出现严重降低了现有药物的疗效,导致患者治疗时间的显著延长和死亡率的提高。
在限制抗菌药物临床使用的同时,开发全新结构和独特作用机制的抗菌药物是解决细菌耐药性问题的根本出路[1-2]。
噁唑烷酮类化合物是一类新型的治疗细菌性感染的抗菌药,可抑制蛋白质合成的起始阶段并很少出现交叉耐药性,由于具有独特的作用机制而备受人们关注。
第一个噁唑烷酮类抗菌剂—Linezolid(利奈唑胺),已于2000年4月在美国批准上市,用于治疗多重耐药G +菌引起的感染[3]。
新型药物恶唑类中间体开发研究可行性报告

新型药物噁唑类中间体开发研究可行性报告郑州大学郑州银通环保技术有限公司2004年8月18日1.概述(R)-N-(3-氟-4-吗啉苯基)-2-氧-噁唑-5-甲基丁酸酯是新一代噁唑酮类抗菌药Linezolid的重要中间体。
Linezolid是一种最新的全合成化学抗菌药物。
Linezolid的化学名称为(R)-N-(3-氟-4-吗啉苯基)-2-氧-噁唑-5-甲基—乙酰胺。
Linezolid是20世纪90年代,由pharmacia∝Upjohn公司,首先研制成功。
并于2000年4月18日在美国已批准上市,Linezolid是第一个应用于临床的新型噁唑烷酮类抗菌药,该药结构和作用机制独特,不易产生耐药性。
美国有关机构进行了2000多种病菌实验与相关性能评价表明:Linezolid具有广谱抗菌、剂量小,毒性小、副作用少。
突出优点是:很少产生较叉耐药性,高度有效,特别是目前现有的抗菌药物不能有效抑制的细菌,如甲氧西林耐药金葡萄球菌(MRSA),耐青霉素肺炎球菌,耐万古霉素肠球菌等具有良好的抗菌效果。
Linezolid从早期来抑制革兰氏阳性菌蛋白质的合成,他束缚了23S粒核糖RNA并阻止70S核糖体的生成。
70S核糖体的生成对RNA变形和蛋白质合成时非常重要的。
该药物在口服1-2小时被快速彻底的吸收,并达到血浆浓度的峰值。
虽然食物可以缩短达到最高浓度的时间,但整个曲线下的持续面积与有无食物无关。
因此它可以随食物或不随食物服用。
临床证明,Linezolid经静脉注射或口服后,对抑制万古霉素的肠霉素(VRE)传染病的彻底治愈占97%,并且对治疗苄甲基噁唑青霉素的黄葡萄糖引起的肺炎,顽固或非顽固的皮肤炎以及皮肤组织的传染病有明显的疗效。
作为第一类阻止早期革兰氏阳性菌蛋白质合成的抗生素。
Linezolid有着广阔的发展前景,Linezolid不仅用于治疗耐多种药物的G+菌和结核杆菌感染。
而且对耐万古酶素肠球菌(VRE)引起的菌血病,耐甲氧西林金黄色葡萄球菌(MRSA)引起的肺炎、皮肤感染,以及耐青霉素肺炎链球菌(PRSP)引起的菌血病有良好的疗效被认为是一种极具应用价值的新型抗菌剂。
利奈唑胺治疗婴幼儿重症肺炎76例分析

利奈唑胺治疗婴幼儿重症肺炎76例分析重症肺炎是严重威胁婴幼儿生命健康的疾病之一,婴幼儿免疫功能低下可能是导致重症肺炎不易控制的重要原因,利奈唑胺(Linezolid)是应用于临床的新型恶烷酮类抗生素,由于该药的作用机制独特,与其他药物无交叉耐药性,在临床对感染的治疗已显示出优势[1]。
尝试在常规治疗的基础上采用利奈唑胺治疗婴幼儿重症肺炎,取得了比较好的效果,现报告如下。
资料与方法2011年1~12月收治婴幼儿重症肺炎患者76例。
其临床表现及实验室检查均符合重症肺炎的诊断标准[2]。
采用单盲、随机对照试验研究,随机分为两组,治疗组38例,男19例,女19例,年龄15天~2岁9个月;对照组38例,男20例,女18例,年龄15天~2岁8个月。
两组患儿均有发热,体温最高39.5℃,咳嗽、喘憋、呼吸困难、烦躁、三凹征、肺部喘鸣和湿啰音。
病程2天~半个月。
血常规:白细胞总数(2.5~23.0)×109/L,淋巴细胞0.4~0.7。
X线表现主要以双肺纹理增重,点片状阴影,肺气肿,其中2例呈节段性肺炎的改变。
治疗方法:两组均按照婴幼儿重症肺炎诊疗常规,采用抗感染,镇静祛痰平喘,吸痰、吸氧、纠正心衰、呼衰、中毒性脑病、中毒性肠麻痹、水、电解质与酸碱平衡失调等并发症及支持治疗等。
治疗组给予利奈唑胺(商品名:斯沃)600mg静滴,12小时/(次·日),疗程7~14天。
对照组给予万古霉素,肾功能无异常患儿给药剂量10mg/kg,每8小时1次。
肾功能异常患儿据肌酐清除率调整剂量。
并监测血万古霉素药物浓度,维持万古霉素血药浓度10~15μg/ml,峰浓度30~40μg/ml。
疗程7~14天。
观察指标:主要观测指标有婴幼儿住院时间、临床有效率、细菌清除率和药物不良反应。
根据(抗菌药物临床研究指导标准):痊愈、显效、有效、无效4级评定。
痊愈、显效、有效合计为总有效,计算临床有效率,判断临床疗效;结合痰培养结果,按病原菌清除、假设清除、未清除、替换和再感染5级评定计算病原菌清除率,判定细菌学疗效;药物主要不良反应有腹泻、肝肾功能损伤、白细胞减少、血小板减少等,统计总例数计算百分比。
吡啶酮酸化学结构

吡啶酮酸化学结构一、概述吡啶酮酸,也被称为恶唑烷酮,是一种重要的化学物质。
它属于吡啶酮家族,是一种具有生物活性的化合物,被广泛应用于药物、农业和工业领域。
吡啶酮酸具有独特的化学结构,使其在多种化学反应中表现出优异的性能。
了解吡啶酮酸的化学结构对于理解其性质、合成方法以及潜在的应用具有重要意义。
二、化学式与分子结构吡啶酮酸的化学式为C3H4N2O2,分子量为100.0752。
其分子结构由以下几个部分组成:一个碳环,环上连接着三个取代基,分别是两个氮原子和一个氧原子。
这三个取代基的排列方式使得吡啶酮酸具有独特的化学性质。
三、性质1.稳定性:吡啶酮酸在常温常压下稳定,但在高温或强酸强碱条件下可能会发生分解。
2.酸碱性:由于吡啶酮酸分子中的氮原子和氧原子,它具有一定的酸性和碱性。
在特定条件下,它可以作为酸或碱进行反应。
3.反应性:吡啶酮酸可以参与多种化学反应,如取代反应、加成反应和氧化还原反应等。
其反应性与其化学结构密切相关。
4.光学与热学性质:吡啶酮酸具有一定的光学活性,并且在特定温度下表现出一定的热学性质。
这些性质与其分子结构紧密相关。
四、合成与生产方法吡啶酮酸的合成方法有多种,以下是其中一种常用的合成方法:首先,将氨基乙酸与氢氧化钠溶液混合,加热至沸腾。
然后,将沸腾的溶液慢慢滴加到乙酐中,同时不断搅拌。
滴加完毕后,继续加热并搅拌一段时间。
最后,将得到的产物进行提纯,即可得到吡啶酮酸。
这种方法具有较高的收率和纯度,适用于大规模生产。
此外,还有其他合成方法如Strecker合成法、Gould-Jacobs合成法等。
不同的合成方法适用于不同的应用场景和需求。
在实际生产中,需要根据具体情况选择合适的合成方法。
吡啶酮酸的生产方法也随着科技的发展而不断改进。
早期的方法通常涉及多步反应和繁琐的纯化过程,导致产率较低且成本较高。
为了提高生产效率和降低成本,研究者们不断探索新的合成路线和改进现有方法。
现代的生产方法通常更加注重绿色化学原则,旨在减少废物产生和降低能耗。
恶唑烷酮结构

恶唑烷酮结构1. 引言恶唑烷酮是一种有机化合物,其分子结构中包含恶唑环和烷基酮基团。
恶唑环是一种五元杂环,其中包含一个氮原子和四个碳原子。
恶唑烷酮具有广泛的应用领域,包括药物化学、材料科学和有机合成等。
2. 恶唑环的特性恶唑环是一种芳香杂环,具有稳定的分子结构和较高的电子密度。
它可以通过多种方法合成,如氧化还原反应、亲核取代反应和金属催化反应等。
恶唑环在天然产物合成中也起到重要作用,许多天然产物中含有恶唑环结构。
3. 烷基酮基团的性质烷基酮基团是由一个碳链与一个羰基结合而成。
它具有极性较强的羰基键,并且可以通过亲核加成反应进行功能化修饰。
烷基酮在药物领域中常用作活性部位或药效团。
4. 恶唑烷酮的合成方法恶唑烷酮可以通过多种方法进行合成。
一种常用的方法是将恶唑环和烷基酮基团进行偶联反应。
这种反应可以使用金属催化剂来促进,如钯、铜等。
另一种方法是通过亲核加成反应,在恶唑环上引入烷基酮基团。
5. 恶唑烷酮的应用领域恶唑烷酮在药物化学中具有广泛的应用。
它可以作为药物分子的核心结构,具有抗菌、抗肿瘤和抗病毒等活性。
此外,恶唑烷酮也可以作为药物合成中的中间体,用于合成更复杂的化合物。
在材料科学领域,恶唑烷酮可以作为有机半导体材料,用于制备有机薄膜晶体管和太阳能电池等器件。
其分子结构稳定且易于修饰,使得其在材料科学中具有广阔的应用前景。
在有机合成领域,恶唑烷酮常被用作功能化合物的合成中间体。
通过对恶唑烷酮的修饰反应,可以引入不同的官能团,从而合成具有特定性质的化合物。
6. 结论恶唑烷酮是一种重要的有机化合物,其分子结构包含恶唑环和烷基酮基团。
恶唑环具有稳定的芳香结构和较高的电子密度,可以通过多种方法进行合成。
烷基酮基团具有极性羰基键,可以进行亲核加成反应。
恶唑烷酮在药物化学、材料科学和有机合成等领域具有广泛应用。
它可以作为药物分子的核心结构或中间体,在材料科学中用于制备器件,在有机合成中用于功能化合物的合成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第四章新型噁唑烷酮类抗药菌药物多年来随着抗生素在全球的普及和不断被人滥用,无论革兰氏阳性菌或阴性细菌均已出现耐药趋向,革兰氏阳性细菌的耐药问题更为严重。
据世界卫生组织估计,每天大约有5万人死于感染性疾病,感染性疾病从新成为威胁人类健康和社会发展的主要问题。
世界范围内出现的耐甲氧西林的金黄色葡萄球菌(MRSA)和表皮葡萄球菌(MRSE),耐药性肺炎链球菌,多药耐药的结核分支杆菌及耐万古霉素肠球菌(VRE)是当前临床中存在的主要问题。
尤其是耐万古霉素肠球菌(VRE)的出现,突破了严重感染患者治疗的“最后手段”。
面对这种困境,专家们提出“更新战略”,就是用“新一代”的抗生素替代“老”的品种。
世界许多制药公司都在积极寻找能对付耐多种抗生素的耐药细菌的新型药物。
噁唑烷酮就是一类极有发展前景的新型全合成抗菌剂。
Drug Resistance in Hospital-Acquired InfectionsDrug/Pathogen Resistance(%)Vancomycin/enterococci 24.7Methicillin/S.aureus 53.6Methicillin/Coag.-neg.S.aureus 88.2Imipenem/P.aeruginosa 16.4Quinolone/P.aeruginosa 23.03rd-gen.ceph. /P.aeruginosa 20.63rd-gen.ceph. /Enterobacter 33.1SCIENCE 2004,303,1798噁唑烷酮类抗菌药物的发展NSOC l OC H 3OSHO H 2NOOXH A CO C H 3O Dup105 X=S Dup721 X=CNON H A CO ONON H A CONH OOH A CC HNH A CO OFNH A CO NFH OO123451978年,美国杜邦公司科学家报道噁唑烷酮衍生物1在控制细菌引起的植物疾病方面的应用 。
随后他们对1进行结构改造得到2即S-6123,S-6123在体内外外对阳性菌和阴性菌表现出中等强度活性。
通过进一步结构优化,1987年,他们发现两个侯选化合物D u P 721和DuP 105。
D u P 721和DuP 105比S-6123抗菌活性有很大提高。
杜邦公司的噁唑烷酮研究工作引起了科学家们极大的兴趣,这是因为:噁唑烷酮的抗菌谱包括了所有重要的革兰氏阳性菌致病菌且和现有抗菌药物没有交叉耐药性,而且在实验室很难诱导产生耐药菌株。
噁唑烷酮是全合成的、具有独特作用机制。
全新结构和独特的作用机制吸引了众多制药公司的关注。
普强,拜尔,和阿特拉斯等公司都参与噁唑烷酮衍生物的研究开发。
普强公司合成了许多并环的化合物,但都没有突破性进展。
1989年杜邦公司因毒性问题停止了他们的噁唑烷酮研究计划。
普强公司则继续深入研究,他们借助喹诺酮类药物结构改造的经验,将哌嗪环,吗啉环和氟原子引入到噁唑烷酮的改造中来,筛选得到了两个高活性噁唑烷酮衍生物:吗啉噁酮(linezolid )和羟哌噁酮(eperezolid) .其中,吗啉噁酮经FDA 批准,2000年首先在美国上市,商品名:Zyvox ,成为第一个获准进入临床应用的噁唑烷酮药物。
NO OFH ACNH A CO NFOH O吗啉噁酮羟哌噁酮二 作用机制噁唑烷酮的作用机制目前还不是十分清楚。
一般认为,噁唑烷酮抑制细菌蛋白质合成的最早期阶段,核糖体50S 亚基是其作用靶位。
通过与靠近30S 界面的50S 亚基结合,以阻止70S 起始复合物的形成。
最近,Patricia Kloss 等人运用耐药变异特性技术,揭示核糖体50S 亚基23S rRNA 的第五中心区是药物作用的基本靶点20。
噁唑烷酮作用机制不同于目前已知的所有抗生素。
三 构效关系噁唑烷酮的基本骨架如下:OA21′2′3′4′5′6′Ⅱ目前合成的噁唑烷酮衍生物大部分都是苯环与3位N 相连。
对于这类衍生物,主要是由环Ⅰ和环Ⅱ两部分构成。
环Ⅰ部分噁唑环的O 以S 或NR (R=H ,Me ,Bu )代替,或2位的羰基以砜基,亚砜基或磷酸甲酯基代替,以及1,2位的开环衍生物,其抗菌活性丧失。
若在环Ⅰ和环Ⅱ之间插入-CO-或-SO 2- ,则活性消失。
3位N 以 -CH 2-或=CH 代替,得到2-四氢呋喃酮及2-二氢呋喃酮化合物23,个别化合物在体外表现出抗菌活性。
5位的立体构型为S 型, R 异构体无效。
2.对于B 基团早期研究表明,乙酰胺基的活性最强。
近年来,B 基团采用五员或六员杂环羟基,或甲氧硫代酰胺基,或硫代乙酰胺基,或二硫代氨基甲酸酯等,也都表现出较好的抗菌活性。
NS NNON FROFNO O FNNHO SFSSNHO XFH 2SHOSS10X=S, O123. 环Ⅱ部分环Ⅱ为苯环时,4-位取代活性最强, 2-位取代活性很弱甚至消失。
当取代基位阻小于乙基时,3-位单取代和4-位单取代活性相当。
3,4-二取代衍生物,当3位为小的取代基(小于Br ),活性与4位单取代相当,3位F 取代有助于增强活性。
2 ,4 ,6 -三取代的衍生物没有活性,这可能与所要求的苯环和噁唑烷酮共平面。
环Ⅱ除苯环外噻吩、吡啶以及苯并呋喃、苯并噻吩、苯并噻唑、苯并吲哚、苯并噻唑酮、苯并二氢喹啉,某些三环稠环均有较好活性。
NH A COSB rNHO SO C H 2C F 3ONOHSOH A CSNSOC H 3OH C O 2C H3ONOOH C O N H 2OOHNNOXOH A CXNN1314151617181920214.A基团A基团变化很大,也是噁唑烷酮衍生物多样性的基础。
A基团可通过C-C键或C-N键和环Ⅱ相连。
都有活性很好的代表化合物。
另外还有以羰基和环Ⅱ相连的,如29-30。
SH C O C H C l 2O OONSH C O C H C l 2OO+_H C O C H 3O NOOHNH C O C H 3OFNH C O C H 3O N NFNH C O C H 3O FNCNH C O C H 3O NFNCH C O C H 3OH NO NSH C O C H 3O ONN S222526272829305、噁唑烷酮和喹诺酮的拼接化合物NOH O 2CF NN HN OO H A cFNOH O 2CFOH A cNOH O 2CFFOH A cH 3C5, 6N2、5、6对耐丙沙星和耐LZ 的阳性菌表现出很好的抗菌活性,但对阴性菌无活性,更多表现为象LZ 类化合物。
NS P A R C E ROO H 3C C O N HFNNFOC O 2HNONS P A R C E RO3K3L3M四、噁唑烷酮类药物的合成噁唑烷酮类药物的5-位必须是S 构型,所以合成的关键是如何建立5-位的手性碳,目前报道的方法都是手性原料开始合成的。
1.5-羟甲基噁唑烷酮的合成P h N H C O 2ROOOHOn-BuLi,THF, -78 CO2.吗啉噁酮的合成FFNO 2NFNH 2OF HN OB nONF OONFOOsO2NF OOA CPd/C, H 22.K O tBuLD A3.2.K 2CO 32.Et 3N , M SClA C 2OTL 37(44) 7937-7940ClcN HNPh H 2COCNFOCH2PhO NPh H 2COCNFO ccNNHNNOp-TsOH/MeOH(EtO )COTL 40(1999),4855-4856 IndiaHOOOA rN H 1.2.(Im)2CO /CH 2Cl 2d il. H ClH A CAr =NH 2NOF NH 2NCBZ NFNH 2NN FHO O钯催化偶合合成噁唑烷酮类化合物 TL 42(2001) 3681-3684BroO Ac OCH3H 3COP d 2d b a 3, B IN A P ,C s 2C O 3, to lu e n e , 100o COAc OCH3H 3COONOAc OODPSO 72%T F A /C H 2C l265%NODPSNO 2NNOHCFOAc OCH3H 3COCl NCOAc OCH3H 3CONBr69%69%69%77%Tetrahedron:Asymmetry 11(2000) 4429-44322HOT rO2T rOO T rNH 3.H 2OT rC l / P y--HOUS6288238H2B2RBRBNCROOHHORB(OH)H2OOCl-一步法合成噁唑烷酮类化合物WO 02/085849NHN OBnOOct-BuOL iNOOFc FX = C l, B r, R S O2OWO 9924393Cl2O OXc 用组合化学的方法合成噁唑烷酮类化合物2clO 2OOHR 1O 2OR 1R 2ONOOR 2R 1R 2N C O /E t3N /rt/3hD B N /C H 2C C l2/rt/2hR 1=C H 2N 3五、吗啉噁酮(linezolid )和羟哌噁酮(eperezolid) 这两个新药候选化合物,它们在完成临床前研究后,结果极其相似,无法判定那一个更好,但在I期临床研究后发现吗啉噁酮人体代谢特性优于羟哌噁酮,吗啉噁酮只需每天服药两次,而羟哌噁酮需要三次。
羟哌噁酮在I期临床研究中被放弃。
吗啉噁酮给药后能够快速和完全的吸收,生物利用度接近100%,既可口服也可注射。
临床上用于皮肤/软组织感染、获得性肺炎及其它革兰氏阳性菌感染。
副作用:吗啉噁酮具有较好的耐受性。
其主要不良反应是肠胃道反应,如恶心和腹泻及舌头变色和口腔念株菌感染。
严重的与药物密切相关的副反应如肝酶升高、房颤或胰腺炎。
最严重的副反应为血小板减少。
六、我们小组的工作我们小组从5年前就开始研究噁唑烷酮类抗菌药物并取得了很好的进展,我们发现的YC-12等化合物体外抗菌活性远远超过2000年上市的吗啉噁酮(linezolid )。
FNOACFNFOOACL in e z o lid /上市A Z D 2563/III 期临床P h a rm a c ia & U p jo nA s tra Z e n e c a合成80多个新化合物Y C -12, Y C -20活性比对照药L Z 强很多表2 YC 12、YC 20、YC 44、和对照药物 对592株临床分离致病菌体外抗菌活性比较表3 YC12、YC20、YC44的MIC、MBC比较接上表:七、发展趋势Linezolid是近30年来出现的第一个全新类型的全合成抗菌剂,作用机制独特。
它既可静脉给药,也可口服给药,口服给药的生物利用度高达100%,万古霉素一般均需静脉给药。