5’ Race protocol (standard rxn)
RACE技术原理

RACE的基本概念 不同厂家RACE试剂盒的介绍 RACE技术的关键环节
存在的问题及解决办法
RACE的基本概念
cDNA末端快速扩增(rapid amplification of cDNA ends, RACE)技术是基于PCR技术由已知的部分
cDNA序列来获得完整cDNA序列的一种方法,
在PCR反应中有足够的全长产物能被探测。
TdT加尾反应及其替代反应
第三,若目的cDNA中含有与多聚尾互补的几个核苷酸同 聚区,则在合成第二条cDNA链时引物的延伸会从内部序 列而不是末端序列开始,产生非全长的第二条cDNA链。
我们所使用Invitron公司的RACE试剂盒,采用PCR介导的
连接反应,在一定程度上避免了以上的问题。
RACE试剂盒中末端转移的是多聚C尾,这样在反转录合 成第一链时5’端多聚G的二级结构影响反转录的彻底进行,
会产生提前终止反应。我们改进了加尾的程序,用TdT酶 加上多聚T尾,结果降低了反转录的难度,两个基因最终 均获得了完整的5’末端。
RACE引物的设计
在实验过程中总结如下经验:
1.
2.
引物不能设计在保守区简并引物区。
2.
反转录提前终止
模板中有特殊的二级结构,反转录提前终止。通过提高 反转录的温度,加大反转录的反应体系以及反转录过程 中一直保持已变性的RNA模板处于50℃以上,避免以解
开二级结构的RNA再恢复原来的结构,以达到5’末端。
提供一种改进的反转录方法
1. 2.
3. 4.
5. 6.
7.
在RNase-free的0.2 mL Eppendorf 管中加入以下成分: Oligo(dT)(0.5μg) 1μL Total RNA(4~5μg) 3μL DEPC-H2O To 25 μL 混匀,70℃保温10min;50℃保温5min;稍微离心一下。 在混合物中,依次加入以下成份: 10X SSⅡ( SSⅢ )Buffer 5.0μL 10mM dNTP mix 1.0μL 0.1M DTT 2.0μL RNase Inhibitor(40U/μL) 1.0μL DEPC-H2O To 24 μL 轻轻混合,在50℃保温5min后,在50℃下将3号管中的混合成分移入 2号管中。 每个反应加入1μL SuperScriptTM Ⅱ(SSⅢ ),轻轻混匀,50℃反应 50min。 70℃放置15min以终止反应。 -20℃保存。
RACE

负链RNA采用PLM-RACE一、样品处理收集新鲜尿囊液,差速离心4000×5min,8000×5min,12 000×5min,取上清冻存备用二、提RNA常规方法,延长作用时间。
每个样品两份(5′和3′)取400μL新鲜尿囊液,加入600μL Trizol,混匀后室温静置10min。
加入200μL氯仿,20μg糖原,振荡混匀,12 000rpm,4℃离心10min。
取500μL上清,加入500μL异丙醇,-20℃静置10min后,12 000rpm,4℃离心10min。
70%酒精漂洗两次,沉淀干燥。
三、3′RACE1、沉淀干燥的RNA,用20μL DEPC水溶解,取出4μL测浓度与纯度2、3′端连接锚定引物(25μL体系)10×T4 RNA Ligase Buffer(A TP free) 2.510mM ATP 10.1%BSA 1 (或1mg/mLBSA 2.5)50 U/μL RNasin 0.510 U/μL T4 RNA Ligase 325μmol/L CL+ 1RNA 16 (14.5)37℃2h 或10℃16~18h,75℃15min3、酒精沉淀,70%酒精洗涤,17μL DEPC水溶解4、连接产物RT-PCR (25μL体系)5×M0-MLV RTase Buffer 5RNasin 0.525μmol/L CL- 1M0-MLV RTase 110mM dNTP 1上一步RNA连接产物16.542℃作用1.5h,-20℃保存5、PCR用反向锚定引物CL-和3SR扩增四、5′RACE1、沉淀干燥的RNA,用34μL DEPC水溶解2、反转录5×M0-MLV RTase Buffer 10RNasin 125μmol/L 5LF 2M0-MLV RTase 1.510mM dNTP 2RNA 33.542℃作用2h3、cDNA纯化或用PCR纯化试剂盒Axygena、50μL(等体积)0.6 mol/L NaOH,60℃20minb、20μL NaAc,80μL DEPC水,500μL无水乙醇,-70℃2hc、12000rpm 4℃30mind、70%酒精洗涤,干燥,溶解4、RNA连接cDNA 3′端单链连接(25μL体系)10×T4 RNA Ligase Buffer(A TP free) 2.510mM ATP 10.1%BSA 1 (或1mg/mLBSA 2.5)10 U/μL T4 RNA Ligase 325μmol/L CL+ 1纯化的cDNA 16.5 (15)37℃2h 或10℃16~18h,75℃15min5、套式PCR第一轮PCR反应引物为CL -和5LF,产物100倍稀释后做第二轮反应的模板。
【良心出品】3,5RACE流程

【良心出品】3,5RACE流程这个文件我看了,其实和普通的cDNA第一链的合成、PCR扩增技术步骤相似!只是引物不同罢了3’端的克隆(3’ RACE)1.2.3.1 特异性引物设计和合成根据测序的Na+/H+逆向转运蛋白基因核心片段设计3’端基因特异引物P3(外侧引物)、P4(巢式引物),分别与GeneRacer TM Kit提供的3’ Primer(3’P)、3’ Nested Primer(3’NP)配对,分别用于外侧PCR扩增和巢式PCR扩增。
P3、P4由上海生工合成。
P3:5’-CTGTATTCTTCTGTGGGATTGTGATG-3’P4:5’-TGGGATGGATGCTTTAGACATTGAG-3’3’P:5’-GCTGTCAACGATACGCTACGTAACG-3’3’NP:5’-CGCTACGTAACGGCATGACAGTG-3’1.2.3.2 cDNA第一链的合成(1)200μl PCR管置于冰上,依次加入下列组分:Total RNA 8μl Oligo dT Primer(0.82μg·μl-1)1μl dNTP Mix (25mmol·L-1)1μl RNase-free ddH2O 3μl (2)轻轻混匀,瞬时离心收集液体。
(3)65℃温育5min,置于冰上冷却1min,瞬时离心收集液体。
(4)于冰上依次加入下列组分:5 First Strand Buffer 4μlDTT(1mol·L-1)1μlRNaseOut?(40U·μl-1)1μl SuperScript? III RT(200U·μl-1)1μl (5)轻轻混匀,瞬时离心收集液体。
(6)50℃温育1h。
(7)70℃温育5min以终止反应,置于冰上冷却2min,瞬时离心收集液体。
(8)加入1μl RNase H (2U·μl-1)。
(9)37℃温育20min,瞬时离心收集液体,即可用于3’外侧PCR扩增或-20℃保存备用。
TAKARA公司的5' full RACE说明书

400 μl 100 μl 100 μl
For Control Reactions(5 次量):
Control HL60 Total RNA(1 μg /μl)*2 5′RACE Control Outer Primer(10 μM)*2 5′RACE Control Inner Primer(10 μM)*2
引物序列(5’→ 3’) CATGGCTACATGCTGACAGCCTA CGCGGATCCACAGCCTACTGATGATCAGTCGATG AGGTAGGTGATGTTCCGAGAGCGT TTGGAGTCGCCCTCAGCAGAGAT
长度 23 mers 34 mers 24 mers 23 mers
团。 3. 使用 T4 RNA Ligase 将 5′RACE Adaptor 连接到去帽后的 mRNA 上。 4. 以上述 3 的mRNA作为模板,使用Reverse Transcriptase M-MLV(RNase H-),Random 9 mers
进行反转录反应合成cDNA。 5. 使用下游外侧特异性引物 GSP1 和 5′RACE Outer Primer 进行 Outer PCR 反应,再使用下游内侧
行 5′RACE 实验。 3. 高特异性的 5′RACE 扩增。套式 PCR 法的使用,大大提高了 5′RACE DNA 片段扩增的特异性。
4. 长片段 5′RACE扩增。试剂盒中使用了本公司特殊改良的M-MLV(RNase H-)反转录酶,反转录性
能良好,使长片段的RT-PCR扩增成为了可能。
●制品内容(10 次量)
以下为使用本试剂盒时的注意事项,使用前请一定认真阅读!
1. 进行 5′RACE 实验时,为提高 RACE 结果的可信度,应同时进行 TAP(-)和 M-MLV(-)的负对 照实验。TAP(-)即:RNA 经去磷酸化反应后不进行 TAP 处理,直接进行 5′RACE Adaptor 的 连接及后续实验,以验证 RNA 去磷酸化是否充分。如果充分,RT-PCR 将不能扩增。但是,真正进 行 TAP(-)负对照实验时,由于不能保证 100%的去磷酸化,往往会有 RT-PCR 扩增结果,但其扩 增片段长度一般不同于目的片段。M-MLV(-)即:5′RACE Adaptor 连接反应后进行 RT 反应时 不添加 M-MLV,以排除基因组 DNA 污染造成的假阳性结果。如果 M-MLV(-)的负对照实验没有 特异性扩增,说明 5′RACE 结果来源于 mRNA。
RACE技术2

原理Figure
三种5’RACE技术的比较与优化 三种5’RACE技术的比较与优化 5’RACE
环化法5’RACE SMART反转录酶法 SMART反转录酶法 5’RACE 末端脱氧核糖核酸 转移酶(TDT酶 转移酶(TDT酶)法 (TDT 5’RACE
三种5’RACE技术的比较与优化 三种5’RACE技术的比较与优化 5’RACE
RACE的优点 RACE的优点
与筛库法相比较,有许多方面的优点: 与筛库法相比较,有许多方面的优点: 此方法是通过PCR技术实现的, PCR技术实现的 此方法是所花费的经费和时间。 节约了实验所花费的经费和时间。 只要引物设计正确, 只要引物设计正确,在初级产物的基础上可以 获得大量的感兴趣基因的全长
RACE 技术
成 员:
董碧秀 古丽扎尔 热依拉
RACE简介 简介
RACE是基于PCR技术基础上由已知的一段 RACE是基于PCR技术基础上由已知的一段 是基于PCR cDNA片段, cDNA片段,通过往两端延伸扩增从而获得 片段 完整的3‘端和5’端的方法 。 完整的3‘端和5’端的方法 3‘端和5’ RACE(rapidends)是通 RACE(rapid-amplification ofcDNA ends)是通 过PCR进行cDNA末端快速克隆的技术。 PCR进行cDNA末端快速克隆的技术。 进行cDNA末端快速克隆的技术
反应中涉及到的一些事项
在进行5 PCR的时候应该使用热 在进行5‘和3’RACE PCR的时候应该使用热 启动。 启动。 中给出了所有引物的相互关系。 表4中给出了所有引物的相互关系。重叠引 物的设计会对全长的产生有帮助。另外, 物的设计会对全长的产生有帮助。另外,重 叠的引物可以为PCR反应提供一个对照。 PCR反应提供一个对照 叠的引物可以为PCR反应提供一个对照。并 不是绝对的要利用设计的引物产生重叠片段。 不是绝对的要利用设计的引物产生重叠片段
RACE原理及应用

RACE的简介目前,全长基因的获得是生物工程及分子生物学研究的一个重点。
尽管已经有多种方法可以获得基因的全长序列,但在很多生物研究中,由于所研究的目的基因丰度较低,从而使得由低丰度mRNA通过转录获得全长cDNA很困难。
近年来发展成熟的cDNA末端快速扩增(RACE)技术为从低丰度转录快速获得全长 cDNA 提供了一个便捷的途径。
cDNA 末端快速扩增 (rapid amplification of cDNA ends,RACE)技术是一种基于mRNA反转录和 PCR技术建立起来的、以部分的已知区域序列为起点,扩增基因转录本的未知区域,从而获得mRNA(cDNA)完整序列的方法。
简单的说就是一种从低丰度转录本中快速增长cDNA5’和cDNA3’末端,进而获得获得全长cDNA简单而有效的方法,该方法具有快捷、方便、高效等优点,可同时获得多个转录本。
因此近年来RACE技术已逐渐取代了经典的cDNA文库筛选技术,成为克隆全长cDNA序列的常用手段。
第二 RACE的原理RACE 是采用PCR 技术由已知的部分cDNA 顺序来扩增出完整cDNA5’和3’末端,是一种简便而有效的方法, 又被称为锚定 PCR (anchoredPCR)和单边PCR(one2side PCR)。
3’RACE的原理一)加入oligo(dT)17和反转录酶对mRNA进行反转录得到(-)cDNA;二)以oligo(dT)l7和一个35bp的接头(dT17-adaptor)为引物,其中在引物的接头中有一在基因组DNA中罕见的限制酶的酶切位点。
这样就在未知cDNA末端接上了一段特殊的接头序列。
再用一个基因特异性引物(3 amp)与少量第一链(-)cDNA退火并延伸,产生互补的第二链(+)cDNA。
三)利用3amp和接头引物进行PCR循环即可扩增得到cDNA双链。
扩增的特异性取决于3amp的碱基只与目的cDNA分子互补.而用接头引物来取代dT17一adaptor则可阻止长(dT)碱基引起的错配。
5'RACE说明

-3-
【使用器具】 尽量使用一次性塑料器材,若用玻璃器材时应在使用前按下列方法进行处理。 (1)用 0.1% DEPC(焦碳酸二乙酯)水溶液在 37℃下处理 12 小时。 (2)然后在 120℃下高压灭菌 30 分钟以除去残留的 DEPC。 RNA 实验用的器具和仪器建议专门使用,不要用于其他实验。
引物序列(5’→ 3’) CATGGCTACATGCTGACAGCCTA CGCGGATCCACAGCCTACTGATGATCAGTCGATG AGGTAGGTGATGTTCCGAGAGCGT TTGGAGTCGCCCTCAGCAGAGAT
长度 23 mers 34 mers 24 mers 23 mers
●保存: -20℃
●试剂盒原理
5′-Full RACE 的扩增原理图
-2-
【操作步骤】 1. 使用 Alkaline Phosphatase(Calf intestine)[简称 CIAP]去掉 Total RNA 中裸露的 5′磷酸基团。 2. 使用 Tobacco Acid Pyrophosphatase [简称 TAP]去掉 mRNA 的 5′帽子结构,保留一个磷酸基
400 μl 100 μl 100 μl
For Control Reactions(5 次量):
Control HL60 Total RNA(1 μg /μl)*2 5′RACE Control Outer Primer(10 μM)*2 5′RACE Control Inner Primer(10 μM)*2
不能形成复杂结构。 4. 引物 3′端的 3~4 个碱基不要与配对引物形成互补序列。
●试剂盒特点
原理
RNA 模板 基因种类 扩增片段大小 反转录酶 扩增特点
5-RACE经验汇总

5 Race(2007-11-18 21:37:08)转载▼分类:核酸技术如果您顺的话,一个礼拜足矣!同意,5'-RACE确实比较难,但也有顺利的时候,我上周做5'-RACE也一次就成功了,我觉得关键还是RNA 的质量,如果RNA模板质量不好,后面做得再认真也是白费!当时要克隆基因的5'GC含量达到了80%,一般的反转录酶和PCR都拿不到,后来加了海藻糖60度反转录一个小时后,用TaKaRa的GC-Buffer和LA-Taq才扩出来,当时就为了这个5端,很是费了力气。
如果RCR产物不是很特异或没有目的条带的话,建议用nest PCR。
在做5‘时,对RNA要求比较高,最好用新鲜的组织提,随即做逆转录,扩增。
做RACE PCR条带单一的不多,尽可能的回收与目的片段大小相近的条带。
克隆的时候,一般kit上建议挑8-10个克隆,多一个,多一份希望,因为RACE,尤其是5’太难了,我作了约6个月。
偶没有买试剂盒,自己合成了3条引物,买了反转录酶。
然后一次就出来了。
RACE的盒子最常用的是CLONTECH和INVITROGEN的,这个方法我也试过很多次了,方法很简单,但是作出来效果非常的不理想。
PCR出来的东西都是杂七杂八的东西,更多的时候是弥散的条带。
SmartRace 既然称之为Smart,因为它的引物能特异的识别5'端帽子位点,因此排除了所有非完全的逆转录。
再加上使用基因特异性引物,使其做出来的可能性更大。
这个方法省钱,但是我不是很看好,祝好运。
另,建议你不要用oligodT进行逆转录,在基因mRNA300bp处设计一条或几条基因特异性逆转录引物更好。
希望你能有好结果。
我刚做过5’、3‘ RACE,我个人的经验如下:1、首先,提的RNA必须完整,最好跑个RNA电泳来验证一下;2、设计引物时要注意:Tm最好大于70度,两引物间要有100-200bp的重叠区;3、用巢氏PCR相对容易出结果;4、如果出现多个条带,要分别验证;5、PCR过程中,我一般分两次,第一次用touchdowm PCR,后产物稀释50倍,作二次,72度尽量长一点,我们一般用2-3min;6、结果分析,最好是重叠区吻合,否则,应重新做。
3GPP 5G基站(BS)R16版本一致性测试英文原版(3GPP TS 38.141-1)

4.2.2
BS type 1-H.................................................................................................................................................. 26
4.3
Base station classes............................................................................................................................................27
1 Scope.......................................................................................................................................................13
All rights reserved. UMTS™ is a Trade Mark of ETSI registered for the benefit of its members 3GPP™ is a Trade Mark of ETSI registered for the benefit of its Members and of the 3GPP Organizational Partners LTE™ is a Trade Mark of ETSI registered for the benefit of its Members and of the 3GPP Organizational Partners GSM® and the GSM logo are registered and owned by the GSM Association
miRNA的确定-3’RACE 5’RACE服务

cDNA末端快速扩增技术(rapid amplification of cDNA ends, RACE)是通过PCR进行cDNA 末端快速克隆的技术,是基于PCR技术由已知的一段cDNA片段,通过两端延伸扩增从而获得完整3’和5’端的方法,无需构建cDNA文库,可以短时间内获得目的基因cDNA序列。
以其简单、快速、廉价等优势而受到越来越多的重视。
主要应用于全长基因获取,转录起始位点的位置、转录调控区域,miRNA靶标验证。
服务内容:
1.克隆获取中间片段,RACE技术获取5’或者3’端序列;从而获得基因全长mRNA序列。
2.PCR产物TA克隆。
3.重组质粒测序。
客户提供
已获得的cDNA片段序列或者提供基因信息。
纯化好的RNA(≥1.5ug)或者提供足够提取1.5ug以上RNA的样品(细胞或者组织)。
我们提供
完整的实验报告(实验步骤、相关的照片等)。
测序的峰图。
序列拼接结果。
克隆的菌液。
RACE原理

5'RACE 及3'RACE的基本原理简介2011-06-20 16:32:24| 分类:分子遗传学| 标签:race|举报|字号订阅RACE 是采用PCR 技术由已知的部分cDNA 顺序来扩增出完整cDNA5’和3’末端,是一种简便而有效的方法, 又被称为锚定PCR (anchoredPCR)和单边PCR(one2side PCR)。
3’RACE的原理一).(Oligo(dT) capture因为Oligo(dT)与mRNA的Poly(A)尾巴可以发生特异性结合,所以用Oligo(dT)特异结合到mRNA上Poly(A)尾端,以此可以特异地将mRNA从总RNA中分离出来。
属于亲和层析技术。
)加入oligo(dT)17和反转录酶对mRNA进行反转录得到(-)cDNA;二)以oligo(dT)l7和一个35bp的接头(dT17-adaptor)为引物,其中在引物的接头中有一在基因组DNA中罕见的限制酶的酶切位点。
这样就在未知cDNA末端接上了一段特殊的接头序列。
再用一个基因特异性引物(3 amp)与少量第一链(-)cDNA退火并延伸,产生互补的第二链(+)cDNA。
三)利用3amp和接头引物进行PCR循环即可扩增得到cDNA双链。
扩增的特异性取决于3amp的碱基只与目的cDNA分子互补.而用接头引物来取代dT17一adaptor则可阻止长(dT)碱基引起的错配。
5’RACE的原理5’RACE与3’ RACE略有不同。
首先,引物多设计了一个用于逆转录的基因特异引物GSP-RT;其次,在酶促反应中增加了逆转录和加尾步骤,即先用GSP-RT逆转录mRNA获得第一链(-)cDNA后,用脱氧核糖核酸末端转移酶和dATP在cDNA5’端加poly(A)尾,再用锚定引物合成第二链(+)cDNA,接下来与3’ RACE过程相同。
用接头引物和位于延伸引物上游的基因特异性引物(5amp)进行PCR扩增。
5’-RACE技术的意义:可以精确定位转录起始位点
5’-RACE技术的意义:可以精确定位转录起始位点。
但是定位转录起始位点又有什么意义呢?以RNA聚合酶Ⅱ为例,启动子区一般位于转录起始位点上游200-300bp的范围内,也即转录起始位点的确定有助于分析基因的转录。
而且有些基因具有多个转录起始位点,通过5’-RACE技术可以精确定位之,进而详细研究基因的调控机制。
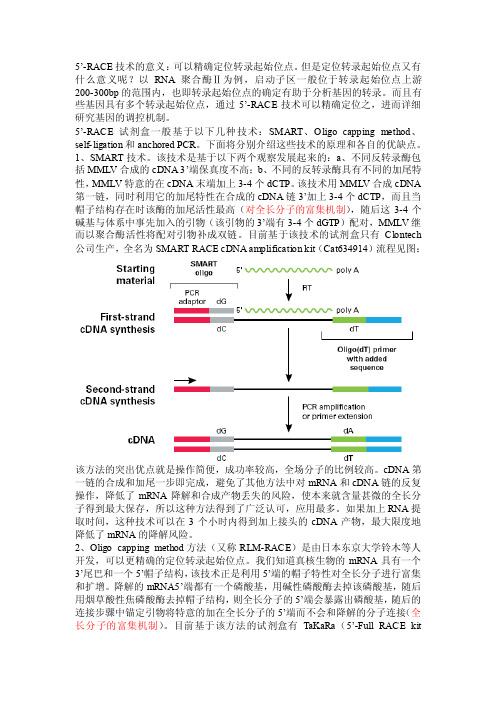
5’-RACE试剂盒一般基于以下几种技术:SMART、Oligo capping method、self-ligation和anchored PCR。
下面将分别介绍这些技术的原理和各自的优缺点。
1、SMART技术。
该技术是基于以下两个观察发展起来的:a、不同反转录酶包括MMLV合成的cDNA 3’端保真度不高;b、不同的反转录酶具有不同的加尾特性,MMLV特意的在cDNA末端加上3-4个dCTP。
该技术用MMLV合成cDNA 第一链,同时利用它的加尾特性在合成的cDNA链3’加上3-4个dCTP,而且当帽子结构存在时该酶的加尾活性最高(对全长分子的富集机制),随后这3-4个碱基与体系中事先加入的引物(该引物的3’端有3-4个dGTP)配对,MMLV继而以聚合酶活性将配对引物补成双链。
目前基于该技术的试剂盒只有Clontech 公司生产,全名为SMART RACE cDNA amplification kit(Cat634914)流程见图:该方法的突出优点就是操作简便,成功率较高,全场分子的比例较高。
cDNA第一链的合成和加尾一步即完成,避免了其他方法中对mRNA和cDNA链的反复操作,降低了mRNA降解和合成产物丢失的风险,使本来就含量甚微的全长分子得到最大保存,所以这种方法得到了广泛认可,应用最多。
如果加上RNA提取时间,这种技术可以在3个小时内得到加上接头的cDNA产物,最大限度地降低了mRNA的降解风险。
2、Oligo capping method方法(又称RLM-RACE)是由日本东京大学铃木等人开发,可以更精确的定位转录起始位点。
RACE技术

RACE技术⽬录1主要分类2发展历史3引物设计4⽐较与优化5实验步骤6PCR扩增7产物验证8克隆和测序9cDNA的获得1主要分类2发展历史3引物设计4⽐较与优化5实验步骤6PCR扩增7产物验证8克隆和测序9cDNA的获得1主要分类编辑本段⼀般分5-和3-RACE两种。
3-RACE较简单,⾸先将mRNA或总RNA⽤PolyT引物反转录,根据⼀般基因具有polyA 尾巴的特点,选⽤特异引物(根据已知序列设计)和PolyT引物PCR即可。
⼤多实验者反映⼀次PCR可以搞定。
5-RACE相对较难,⽬前流⾏⼏种5-RACE。
其⼀为加接头(传统),根据接头引物和⾃⼰设计特异引物PCR,可以设计巢式PCR⼆次扩增。
另外,有利⽤反向PCR技术,连接成环在PCR。
还有,GENE公司⼀种smartRACEPCR,利⽤反转酶末断加C特点,直接加上多G接头,转换模板⽽⽆需⽤连接酶加接头。
2发展历史编辑本段经典的RACE技术是由Frohman等(1988)发明的⼀项技术,主要通过RT-PCR技术由已知部分cDNA序列来得到完整的cDNA5’和3’端,包括单边PCR和锚定PCR。
该技术提出以来经过不断发展和完善,克服了早期技术步骤多、时间长、特异性差的缺点(Frohman等,1995:Schaefer,l995:Chen,1998:Bespalova等,1998:Matz等1999)。
对传统RACE技术的改进主要是引物设计及RT-PCR技术的改进:改进之⼀是利⽤锁定引物((lockdockingprimer)合成第⼀链cDNA,即在oligo(dT)引物的3′端引⼊两个简并的核苷酸【5′-Oligo(dT)16_30MN-3′,M=A/G/C;N=A/G/C/T】,使引物定位在poly(A)尾的起始点,从⽽消除了在合成第⼀条cDNA链时oligo(dT)与poly(A)尾的任何部位的结合所带来的影响;改进之⼆是在5‘端加尾时,采⽤poly(C),⽽不是poly(A);改进之三是采⽤RNaseH⼀莫洛尼⽒⿏⽩⾎病毒(MMLV)反转录酶或选择嗜热DNA聚合酶可能在⾼温h(60度-70度)有效地逆转录mRNA,从⽽消除了5‘端由于⾼CC含量导致的mRNA⼆级结构对逆转录的影响;改进之四是采⽤热启动PCR(hotstartPCR)技术和降落PCR(touchdownPCR)提⾼PCR反应的特异性。
5RACE操作步骤

5RACE操作步骤5'RACE 操作流程一、提总RNA二、合成第一条cDNA链1、加如下组分于一0.5ml离心管(或PCR管)中,混匀Component AmountGSP1...................................................................2.5pmoles (~10 to 25ng) DEPC-treated water.....................sufficient for a final volume of 15.5μl (or sterile, distilled water)2、70°C孵育10min,冰浴1min,短暂离心后依次加入下列组分Component Volume(μl)10X PCR buffer .......................................................................2.525mM MgCl2 ...........................................................................2.510mMdNTP mix (1)0.1 M DTT.................................................................................2.5final volume.............................................................................8.5The final volume of step 1 and 2 is 24μl.3、轻轻混匀,短暂离心,42°C孵育1min4、加1μl Super Script? II RT,轻轻混匀,42°C孵育50min反应体系最终组成如下:20 mM Tris-HCl (pH 8.4)50 mM KCl2.5 mM MgCl210 mM DTT100 nM cDNA primer (GSP1)400 μM each dATP, dCTP, dGTP, dTTP1-5 μg (~40 ng/μl) RNA200 units SuperScript? II RT5、70°C ,15min终止反应6、离心10-20s后将反应体系至于37°C下7、加1μl of RNase mix,轻柔充分混匀,37°C孵育30min8、短暂离心聚集反应液,置冰上三、S.N.A.P柱纯化回收cDNA1、室温下,binding solution平衡柱子2、每份待纯样品分装~100μl DEPC-treated water,65°C预热备用3、加120μl binding solution (6M NaI)于the first strand reaction中4、将cDNA/NaI solution转至S.N.A.P柱,13,000 x g离心20 s5、取出离心柱,将离心液转至一小离心管中保存,知道确保成功回收到cDNA 。
SMARTer-RACE中文说明书

SMARTer® RACE 5’/3’ KitProtocol-At-A-GlanceCode No. 634858/634859使用本Protocol-At-A-Glance前,请阅读试剂盒完整版使用操作手册。
为了方便您的使用,我们提供了该精简版protocol,但不建议第一次操作者使用。
一、引物设计基因特异性引物(GSPs)应符合下列要求:●长度为23–28 nt,以保证特异性退火●GC含量在50–70%之间●Tm ≥65°C;如果Tm大于70°C,这样可以使用touchdown PCR方法,获得最好的结果。
Tm的计算基于基因特异性引物3‘末端而不是整条引物。
●与Universal Primer Mix的3’端不互补。
Long primer = 5’–CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT–3'Short primer = 5’–CTAATACGACTCACTATAGGGC–3’●与目的基因特异性互补●与载体5’端有15个碱基的重叠(例如, 在GSPs序列5’ 端增加序列“GATTACGCCAAGCTT”,细节如下)二、生成RACE-Ready cDNA1.按下列体积配制Buffer Mix用于cDNA合成反应。
为保证体积量充足,请额外多配制1个反应的量。
混合下列试剂,在微型离心机上短暂旋转,室温放置,用于第6步实验。
2.在单独的微量离心管tube中加入下列试剂:*对照实验请加入1 μl的Control Mouse Heart Total RNA(1 μg/μl)3.混合组分,在微型离心机上短暂旋转。
4.72℃孵育3 min,42℃冷却2 min。
冷却后,14,000 xg下离心10 sec,把试剂收集在管底。
NOTE:该步骤可以在thermal cycler中进行。
在孵育tube的同时,可以准备第6步的混合液。
race-pat法

"Race-PAT法" 在生物科学中,特别是分子生物学领域,并不是一个广泛认可的标准术语。
然而,从字面上看,它似乎结合了“Race”和“PAT”两个概念,这可能指的是某种特定的实验方法或技术。
1. Race 通常指的是“Rapid Amplification of cDNA Ends”,即cDNA末端的快速扩增。
这是一种用于
确定mRNA转录本5'和3'末端序列的分子生物学技术。
通过Race,研究人员可以确定基因的转录起始和终止位点,这对于基因的结构和功能研究非常重要。
2. PAT 在生物科学中有多种可能的含义,但没有一个与Race直接相关的标准定义。
它可能指的是某种特
定的分析技术、蛋白质名称的缩写,或者是某种实验方法的简称。
为了明确PAT的具体含义,需要更多的上下文信息。
如果“Race-PAT法”是某种特定的实验方法或技术,那么它可能是结合了Race技术和另一种技术(可能是PAT 代表的技术)的混合方法。
为了获得更准确的解释,建议查阅相关的科学文献或联系使用这一术语的研究人员以获取更详细的信息。
另外,也有可能“Race-PAT法”是一个特定实验室或研究团队内部使用的术语,并没有在更广泛的科学界得到认可或传播。
在这种情况下,最好的办法是直接咨询使用该术语的研究人员或实验室。
Mikroelektronika W5500 ETH Wiz click

23click™BOARD2. Soldering the headers3. Plugging the board inOnce you have soldered the headers your board is ready to be placed into the desired mikroBUS ™ socket. Make sure to align the cut in the lower-right part of the board withthe markings on the silkscreen at themikroBUS ™ socket. If all the pins are aligned correctly, push the board all the way into the socket.Turn the board upward again. Make sure to align the headers so that they are perpendicular to the board, then solder the pins carefully.Turn the board upside down so that the bottom side is facing you upwards. Place shorter pins of the header into the appropriate soldering pads.Before using your click ™ board, make sure to solder 1x8 male headers to both left and right side of the board. Two 1x8 male headers are included with the board in the package.The W5500 module onboard ETH Wiz click ™ employs a variety of solutions to reduce the target MCU’s memory load and enable a stable and secure Internet connection. High speed network communications are realized through a 80 MHz SPI interface. The module has 32KB of internal memory for TX/RX buffers. Reduced power consumption is achieved with Wake on LAN and power down modes . Automatic handshaking, retransmit on collision and automatic rejection of erroneous packets are also supported. The board is ideal for Home network devices and all sorts of embedded servers.1ETH Wiz click ™ carries W5500, a 48-pin, 10/100 BASE-TX standalone Ethernet controller with a hardwired TCP/IP Internet protocol offload engine, along with a standard RJ-45 connector . Wiznet’s W5500 module supports TCP, UDP, IPv4, ICMP, ARP, IGMP, and PPPoE protocols. ETH Wiz click ™ communicates with the target board MCU through mikroBUS ™ RSTn, SCSn, SCLK, MISO, MOSI and INTn lines. The board uses a 3.3V power supply only.ETH WIZ click™1. IntroductionETH WIZ click manualver 1.0101000000762794. Essential featuresTXP AGND AVDD RXN RXP DNC AVDD AGND EXRES1AVDD NCTXN A G N D N C N C P M O D E 0P M O D E 1R S V D R S V D R S V D R S V D R S V D R S T nP M O D E 2INTn MOSI MISO SCLK SCSn XO XI/CLKINGND VDD ACTLED DUPLED LINKLEDN C A G N D A V D D A G N D A V D D V B G A G N D T O C A P A V D D 1V 20R S V D S P D L E DW550048LQFP123456789101112131415161718192021222324484746454443424140393837363534333231302928272625X125MHzR41M R310RR249.9RC1418pFC1518pFC822nFC12100nFGNDGNDGNDGND GNDC194.7uFC1810nFGND TD+CTTD-RD+RD-CTA 2A 1K 1K 2RJ45R149.9RGND GNDGND R1012K4GNDGND R649.9RR749.9RC96.8nFC106.8nF C1610nFVCC_3V3VCC_3V3C2100nFC3100nFC4100nFC5100nFC1100nFC6100nFAVCC_3V3GNDLD2LD3R82K2R92K2AN RST CS SCK MOSI MISO +3.3V GNDPWM INT RX TX SCL SDA +5V GNDMIKROBUS DEVICE CONN.SCSn SCSnMISO MISO MOSI MOSIINTn INTnGNDGNDSCLK SCLK RSTn R S T nVCC_3V3RD-RCT RD+TD-TCT TD+TD+TD-RD-RCT TCT RD+ACTLED LINKLEDACTLEDLINKLEDLD1GNDR112K2VCC_3V3J1J3J2VCC_3V3GNDGNDVCC_3V3VCC_3V3VCC_3V3VCC_3V3C134.7uFR1210KVCC_3V3VCC_3V3VCC_3V3FP1FERRITEAVCC_3V3C204.7uF C21100nFVCC_3V3GNDGND8. Code examplesMikroElektronika offers free tech support (/support) until the end of the product’s lifetime, so if something goes wrong, we’re ready and willing to help!Once you have done all the necessary preparations, it’s time to get your click ™ board up and running. We have provided examples for mikroC ™, mikroBasic ™ and mikroPascal ™ compilers on our Libstock website. Just download them and you are ready to start..com6. DimensionsMikroElektronika assumes no responsibility or liability for any errors or inaccuracies that may appear in the present document. Specification and information contained in the present schematic are subject to change at any time without notice.Copyright © 2015 MikroElektronika.All rights reserved.mmmilsLENGTH 57.152250WIDTH 25.41000HEIGHT*12472.55. Schematic7. Mode selection jumpers9. Support* without headers10. DisclaimerThese three SMD jumpers (zero ohm resistors) are used for specifying the PHY network op-eration mode. For detailed configuration instructions, consult Wiznet’s data sheet for the W5500 module.25.4 m m / 1000 m i l s57.15 mm / 2250 mils。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
5’ Race protocol 5’ Race流程:
5’ Race步骤:
一、RNA提取:
1.收集细胞:
1500rpm常温离心10min收集杂交瘤细胞,细胞状态好,细胞数量为5-10×106。
2.裂解细胞:
生理盐水洗涤一次,1500rpm离心10min将废液丢弃,按照5-10×106/ml的细胞加1ml Trizol混匀后,室温静止5min至细胞完全溶解。
3.萃取RNA:
将上述液体移至经DEPC处理的Eppendorf管中,按照200µl/ml Trizol加入氯仿,充分振荡15s,室温放置2min。
4.分离水相:
12000rpm低温4℃离心15min,吸取水相(上层无色液体)移入另一个DEPC 处理过的Eppendorf管中。
5沉淀RNA:
加入等体积的异丙醇(约500µl/mlTrizol)沉淀10min,或-20℃过夜。
6.洗涤残留的异丙醇:
将废液吸取丢弃,加入预冷的75%乙醇,洗涤5min。
7.计测浓度:
7500rpm低温4℃离心5min,吸出水相,留沉淀晾干(5-10min),用20-30µl DEPC水重悬,55℃-60℃孵育10min(取出1µl用电泳法粗检是,应有三条目的带),测紫外分光光度计A260/280(比值在1.8-2.0为正常范围)每管3µg分装-20保存。
二、RNA的去磷酸化:
1、去磷酸化酶37℃处理1h:
(1)取总量10µg 的总RNA或者250ng的poly(A) RNA,按照下表中配比进行
轻混匀。
(3)37℃孵育1小时。
2、终止去磷酸化反应,酚氯仿抽提,氯仿抽提:
phenol:chloroform的结果要好。
(2)剧烈振荡30s,最高转速(≥10,000g)室温离心5min。
将水相(上层)转移到一个新管中。
(3)加入150µl的氯仿,剧烈振荡30s,最高转速(≥10,000g)室温离心5min。
将水相(上层)转移到一个新管中。
3、150µl的异丙醇冰上沉淀10min,然后用冰浴的70%乙醇洗涤:
(1)往上述溶液中加入150µl的异丙醇,涡旋混匀,冰上静置10min。
(2)最高转速低温4℃离心20min。
用0.5ml冰浴的乙醇洗涤,最高转速低温4℃离心5min。
小心吸取残留的废液,将沉淀的RNA室温风干(大约15min)。
4、Nuclease-free水重悬RNA:
用11µl的Nuclease-free水重悬。
(可以在-20℃中保留1µl CIP处理后的RNA,用于‘minus-TAP’对照反应)
三、5’帽子结构的去除
1、使用TAP在37℃处理1h:
(3)反应结束后保存在-20℃或者进行下一步添加接头。
2、5' RACE Adapter连接:
溶解的沉淀。
由于该buffer中含有ATP,因此不推荐在37℃以上加热融化。
(2)轻轻混匀后短暂离心,37℃孵育1小时。
(3)反应结束后保存在-20℃或者进行下一步反转录。
四、反转录
3、反应结束后保存在-20℃或者进行下一步PCR反应。
五、5' RLM-RACE巢式PCR
1、Outer 5' RLM-RACE PCR:
(购买的PCR酶是Platinum™ Taq DNA (1)在冰上按照下表所示配制PCR体系:
Polymerase High Fidelity)
注意:推荐使用热启动PCR,在冰上配制反应液,将PCR仪预先加热至94℃,
度可以根据自己的引物调整。
2、Inner 5' RLM-RACE PCR:
Acid phenol:chloroform配制:
125:24:1 phenol: chloroform: isoamyl alcohol。