Annex I - EU deviation 8599HS R1
eu gmpannex 17 参数放行+eu gmpannex 19标准样品和留样

ANNEX 17 附件17Parametric Release 参数放行Table of Contents目录1. Principle 原则2. Parametric Release参数放行3. Parametric Release for sterile products. 无菌产品的参数放行4. Glossary术语1. Principle原则1.1 The definition of Parametric Release used in this Annex is based on that proposed by the European Organization for Quality: " A system of release that gives the assurance that the product is of the intended quality based on information collected during the manufacturing process and on the compliance with specific GMP requirements related to Parametric Release."本附件中参数放行的定义,是根据欧洲质量组织提出的:“是根据生产过程中收集的信息和与GMP 中与参数放行相关的要求相符合,保证产品达到预期的质量的放行系统”。
1.2. Parametric release should comply with the basic requirements of GMP, with applicable annexes and the following guidelines.参数放行要符合GMP的基本要求,符合其他相关的附件和以下原则。
2. Parametric release参数放行2.1. It is recognised that a comprehensive set of in-process tests and controls may provide greater assurance of the finished product meeting specification than finished product testing.普遍认为,全面的一套过程控制和检验要比最终产品的检验,能够提供最终产品符合标准的更高的保证。
Diva Decloaker 10X Pretreatment Reagent 说明书

Intended Use:For In Vitro Diagnostic UseHeat induced antigen retrieval of formalin-fixed paraffin-embedded (FFPE) tissues for immunohistochemistry (IHC) procedures. The clinical interpretation of any staining or its absence should be complimented by morphological studies using proper controls and should be evaluated within the context of the patient's clinical history and other diagnostic tests by a qualified pathologist.Summary & Explanation:Diva Decloaker is a heat retrieval solution that is compatible with virtually all antibodies and eliminates the need for multiple buffers including citrate buffer, EDTA or high pH tris buffers. Antibody titers are doubled and tripled when compared to citrate buffer, pH 6.0. Diva Decloaker incorporates Assure™ tech nology, a color-coded high temperatures pH indicator solution. The end-user is assured by visual inspection that the solution is at the correct dilution and pH. This product is specially formulated for superior pH stability at high temperatures and will help prevent the possibility of losing pH sensitive antigens. Diva Decloaker is non-toxic, non-flammable, odorless and sodium azide and thimerosal free.Known Applications:Immunohistochemistry (formalin-fixed paraffin-embedded tissues) Supplied As:100mlDiva Decloaker, 10X concentrate (DV2004LX)500mlDiva Decloaker, 10X concentrate (DV2004MX)Materials and Reagents (Needed But Not Provided): Microscope slides, positively chargedDesert Chamber* (Drying oven)Positive and negative tissue controlsXylene (Could be substituted with xylene substitute*)Ethanol or reagent alcoholDecloaking Chamber* (Pressure cooker)Deionized or distilled waterWash buffer*(TBS/PBS)Enzyme digestion*Avidin-Biotin Blocking Kit*(Labeled Streptavidin Kits Only) Peroxidase block*Protein block*Primary antibody*Negative control reagents*Detection kits*Detection components*Chromogens*Hematoxylin*Bluing reagent*Mounting medium** Biocare Medical Products: Refer to a Biocare Medical catalog for further information regarding catalog number and ordering information. Certain reagents listed above are based on specific application and detection system used. Storage and Stability:Store at room temperature. Do not use after expiration date printed on vial. If reagents are stored under conditions other than those specified in the package insert, they must be verified by the user. Diluted reagents should be used promptly; any remaining reagent should be stored at room temperature.Protocol Recommendations:1. Deparaffinize tissues and hydrate to water. If necessary, block for endogenous peroxidase and wash in DI water.2. Dilute concentrated Diva Decloaker at a ratio of 1:10 (1 ml Diva to 9 ml of deionized water).3. Place slides into 1X retrieval solution in a slide container (e.g. Coplin Jar, Tissue -Tek™ staining dish or metal slide canister).4. Retrieve sections under pressure using Biocare's Decloaking Chamber. Follow the recommendations on the antibody data sheet and Decloaking Chamber User Manual.5. Check solution for appropriate color change. (See Technical Note #1)6. Gently rinse by gradually adding DI water to the solution, then remove slides and rinse with DI water.Technical Notes:1. Concentrated Diva Decloaker is a bright yellow color. RTU or 1X solution is a pale yellow color. When the solution reaches 80-125°C, the solution turns yellow and indicates that the high temperature solution is at correct pH. Should the pH rise above 7.0, the solution turns a fuschia red color. Should the pH drop too low, thesolution turns a pink color.2. If using Biocare’s Desert Chamber Pro (a programmable turbo-action drying oven), dry sections at 25ºC overnight or at 37ºC for 30-60 minutes and then dry slides at 60ºC for 30 minutes.3. Use positive char ged slides (use Biocare’s Kling-On HIER Slides) and cut tissues at 4-5 microns. Do not use any adhesives in the water bath. Poor fixation and processing of tissues will cause tissue sections to fall off the slides, especially fatty tissues such as breast. Tissues should be fixed a minimum of 6-12 hours.4. Protocol time and temperatures for HIER can vary depending on the Decloaking Chamber model used. Please refer to the relevant Decloaking Chamber manual for appropriate protocol times and temperatures.Limitations:The protocols for a specific application can vary. These include, but are not limited to: fixation, heat-retrieval method, incubation times, tissue section thickness and detection kit used. Due to the superior sensitivity of these unique reagents, the recommended incubation times and titers listed are not applicable to other detection systems, asresults may vary. The data sheet recommendations and protocols are based on exclusive use of Biocare products. Ultimately, it is the responsibility of the investigator to determine optimal conditions. The clinical interpretation of any positive or negative staining should be evaluated within the context of clinical presentation, morphology and other histopathological criteria by a qualified pathologist. The clinical interpretation of any positive or negative staining should be complemented by morphological studies using proper positive and negative internal and external controls as well as other diagnostic tests.Catalog Number: DV2004 LX, MX Description: 100, 500 ml, concentrateQuality Control:Refer to CLSI Quality Standards for Design and Implementation of Immunohistochemistry Assays; Approved Guideline-Second edition (I/LA28-A2). CLSI Wayne, PA, USA (). 2011 Precautions:1. This product is not classified as hazardous. The preservative used in this reagent is Proclin 300 and the concentration is less than 0.25%. Overexposure to Proclin 300 can cause skin and eye irritation and irritation to mucous membranes and upper respiratory tract. The concentration of Proclin 300 in this product does not meet the OSHA criteria for a hazardous substance. Wear disposable gloves when handling reagents.2. Specimens, before and after fixation, and all materials exposed to them should be handled as if capable of transmitting infection and disposed of with proper precautions. Never pipette reagents by mouth and avoid contacting the skin and mucous membranes with reagents and specimens. If reagents or specimens come in contact with sensitive areas, wash with copious amounts of water.3. Microbial contamination of reagents may result in an increase in nonspecific staining.4. Incubation times or temperatures other than those specified may give erroneous results. The user must validate any such change.5. Do not use reagent after the expiration date printed on the vial.6. The SDS is available upon request and is located at /.7. Consult OSHA, federal, state or local regulations for disposal of any toxic substances. Proclin is a trademark of Rohm and Haas Company, or of its subsidiaries or affiliates.Troubleshooting:Follow the antibody specific protocol recommendations according to data sheet provided. If atypical results occur, contact Biocare's Technical Support at 1-800-542-2002.。
EDQM实验室GC设备确认 Annex_2_Qualification_of_GC_equipment[1]
![EDQM实验室GC设备确认 Annex_2_Qualification_of_GC_equipment[1]](https://img.taocdn.com/s3/m/345dbfd03186bceb19e8bb9b.png)
OMCL Network of the Council of Europe QUALITY ASSURANCE DOCUMENTPA/PH/OMCL (06) 86 DEFQUALIFICATION OF EQUIPMENTANNEX 2: QUALIFICATION OF GC EQUIPMENTFull document title and reference Qualification of EquipmentAnnex 2: Qualification of GC Equipment PA/PH/OMCL (06) 86 DEFDocument type GuidelineLegislative basis The present document was also accepted by EA asrecommendation document to be used in the context of QualityManagement System audits of OMCLsDate of first adoption May 2006Date of original entryinto forceJune 2006Date of entry into forceof revised documentOctober 2006Previous titles/other references This document replaces part of document PA/PH/OMCL (06) 46 DEFCustodian Organisation The present document was elaborated by the OMCL Network/EDQM of the Council of EuropeConcerned Network GEONANNEX 2 OF THE OMCL NETWORK GUIDELINE“QUALIFICATION OF EQUIPMENT”QUALIFICATION OF GC EQUIPMENTIntroductionThe present document is the second Annex of the core document “Qualification of Equipment”, and it should be used in combination with it when planning, performing and documenting the GC equipment qualification process.The core document contains the general introduction and the Level I and II of qualification, common to all type of instruments, and the present annex contains GC instrument-related recommendations on parameters to be checked and the corresponding typical acceptance limits, as well as practical examples on the methodology that can be used to carry out these checks.The tests proposed in the Level III and IV of qualification are based on an overall approach, in which several parameters are checked at the same time in a combined test procedure, to obtain information on the overall system performance (e.g. peak area precision, retention time precision, temperature programme reproducibility, etc).Nevertheless, it should be noted that it is also acceptable to check these parameters individually by using other well-defined procedures.TABLE IIILevel III. Periodic and motivated instrument checksExamples of requirements for GC instruments with FIDInstrument moduleParameter to be checkedTypical tolerance limits 1.1 Injector leak testPressure drop ≤ 15 kPa within 5 minutes 1.2. Pressure/flow accuracy and stabilityCovered by overall test 1 1.3. Repeatability of injection (overall test 1) - In split mode- In split less modeRSD ≤ 3.0% RSD ≤ 3.0%1.4. Injector temperature accuracy and stability Covered by overall test 2 1. Inlet system1.5. Carry-over (overall test 3) ≤ 0.2%2. Oven2.1. Repeatability of oven temperaturecharacteristicsCovered by overall test 23.1. Linearity (overall test 3) r 2 ≥ 0.9993.2. Constant detector response Covered by overall test 1 or 2 3.3. Noise See Annex I 3. FID detector3.3. DriftSee Annex ITABLE IV Level IV. In-use instrument checksExamples of requirements for GC instruments with FIDParameter to be checked Typical tolerance limits1. System suitability check for the method According to Ph. Eur. or MAH dossier or validated in-house method2. Peak area precision RSD ≤3.0% unless otherwise prescribed*3. Retention time repeatability RSD ≤ 2.0%4. Sensitivity (where relevant, e.g. for related substances tests) According to Ph. Eur. or MAH dossier or validated in-house methodAll parameters given here should be checked when performing analyses under the working conditions for the actual sample determinations. Normally, the test and reference solutions to be prepared for this purpose are given as a part of the method.ANNEX ILevel III. Periodic and motivated instrument checksPractical examples of tests and their associated tolerance limits for several parameters related to the performance of the different modules of a GC are presented below.These examples can be considered by the OMCLs as possible approaches to perform the Level III of the equipment qualification process: “Periodic and motivated instrument checks”.Several tests are proposed to check various parameters at the same time (overall tests). In order to run the tests in a more economical way, other suitable solutions can be used, as for example, the “Grob Test” mixture, available from different suppliers (e.g. Alltech, Sigma, Thames Restek). This commercial solution should be appropriate to the column material used.It is recommended to run the overall tests by using always the same test column, exclusively dedicated to qualification purposes, to guarantee reproducible conditions.1. INLET SYSTEMThe following tests are proposed for the periodic and motivated check of the GC Inlet System.1.1. INJECTOR LEAK TESTMethod:If not otherwise specified by the instrument manufacturer, the leak test is carried out according to the procedure laid down in the instrument manual or by the built in automatic leak check procedure of the instrument.Otherwise use the test described below:Disconnect the column from the injector and close the injector outlet with a sealed cap. Close the septum purge and the bypass.Adjust the flow and pressure controller to the maximal possible value of the pressure gauge. Adjust the flow controller to zero.Read the pressure after 1 minute and record the value.Record the pressure after 5 minutes.Limits:Pressure drop ≤ 15 kPa within 5 minutes.1.2. INLET PRESSURE/FLOW ACCURACY AND STABILITYA direct measurement of these parameters was not deemed practical or necessary, but the optimal conditions of flow/pressure can be verified by the overall test 1.Limits: refer to overall test 1.1.3. REPEATABILITY OF INJECTIONThe verification of this parameter is covered by the overall test 1.This test is to be performed in both split and split less mode.Limits: refer to overall test 1.1.4. INJECTOR TEMPERATURE ACCURACY AND STABILITYDue to the fact that the temperature cannot be reliably measured without opening and modifying the system and due to the difficulties of introducing a probe inside this module, the verification of this parameter is considered to be covered by the overall test 2.Limits: refer to overall test 2.1.5. INJECTOR CARRY OVERAfter having injected the solutions for the linearity test of the FID detector, in increasing order, inject the blank and measure the peaks that correspond to the major peaks (= analytes) in the linearity solutions.The verification of this parameter is covered by the overall test 3.Limits: refer to overall test 3.2. OVEN2.1. REPEATABILITY OF THE OVEN TEMPERATURE CHARACTERISTICSDue to the fact that the temperature cannot be reliably measured without opening and modifying the system conditions and that even when introducing a probe inside the oven, its location would not reflect the real temperature conditions at all points, the verification of this parameter is covered by the overall tests 2A and 2B.Limits: refer to overall test 2.3. FID DETECTORThe following tests are proposed for the periodic and motivated check of the GC FID detector.3.1. FID DETECTOR LINEARITYIncreasing amounts of analyte are injected and a linear response should be obtained.The verification of this parameter is covered by the overall test 3.Limits: refer to overall test 3.3.2. CONSTANT FID DETECTOR RESPONSEThe proper and reproducible functioning of the FID can be demonstrated by checking the peak areas obtained from a pre-defined standard solution.The verification of this parameter is covered by the overall test 1 or 2.Limits: refer to overall test 1 or 2.3.3. FID DETECTOR NOISE AND DRIFTIf the instrument has a built-in automatic system for the verification of the noise and drift, follow the manufacturer’s instructions and apply the defined acceptance criteria. Otherwise, use the test described below:Settings:Column installedSuitable flow, depending on column length/diameterNo injectionOven temperature: 40°CDetector on and heated at working temperature (270-300°C)Method:After stabilisation of the system, record the signal for 15 minutes.Noise: evaluate 10 periods of 1 minute and calculate the mean value.Drift: evaluate the slope of the baseline over the 15 minutes.Limits:The acceptance criteria for these parameters have to be chosen in accordance with the instrument vendor’s instructions and the intended use of the instrument. If no instructions are given, the user has to pre-define these acceptance criteria by taking into account the previous experience and the intended use of the instrument.No fixed values can be pre-defined in this guideline due to the high variety of integration systems used and consequently the acceptance criteria may be expressed in different units (voltage, current, arbitrary units per time).OVERALL TEST 1The overall test 1 covers the following parameters:-Pressure/flow accuracy and stability in the inlet system: Retention time repeatability -Repeatability of injection: peak area precision-In split mode-In split less modeThe test may be combined with overall test 3.Split mode:Test solution:1-octanol in n-hexane 1% (v/v).Settings:Column: SPB-1 (30m x 0.32mm ID x 0.25µm film)Carrier gas: HeVelocity: 25cm/secSplit: 1:100Injection: 1µlInjector temperature: 220°COven temperature: 100°C isothermDetector temperature: 300°CRuntime: 8 minRetention time of 1-octanol: about 5 minSplit less mode:Stock solution: 1-octanol in n-hexane 1% (v/v)Test solution: Dilute 10 ml of the stock solution with n-hexane to 100 ml (corresponds to 1µl/ml of 1-octanol in n-hexane)Settings:Column: SPB-1, 30m, 0.32mm ID, 0.25µm filmCarrier: HeVelocity: 30cm/secSplit less injection: purge valve closed during 2 minInjection: 0.2µl of the test solutionInjector Temperature: 220°COven Temperature: Initial 60°C for 4 min, 15°C/min. up to 135°C, final time 1min Detector temperature: 300°CRuntime: 9.5 minRetention time of 1-octanol: about 8 minMethod:Carry out 6 consecutive injections of the test solution and calculate the RSD of the different peak areas and retention times.Limits:Retention time repeatability: the RSD of the retention times should be ≤ 2.0%Peak area precision (split and split less mode): the RSD of the peak areas should be ≤3.0% OVERALL TEST 2The overall test 2 covers the following parameters:-Injector, oven and detector temperature accuracy and stability: retention time repeatabilityTwo alternative tests are proposed:Overall test 2ATest solution:0.035 ml 1-octanol0.035 ml 2-octanone0.035 ml 2,6-dimethylanilin0.035 ml n-tridecane0.035 ml n-tetradecane35 mg n-eicosanedissolved in 50 ml DichloromethaneSettings:Column: SPB-1 (30m x 0.32mm ID x 0.25µm film)Carrier gas: HeliumVelocity: 25 cm/sSplit: 1:100Injection volume: 1 µlInjector temperature: 220°CDetector: FIDDetector temperature: 300°CGradient programme: 60°C (4 min), 5°C/min, 270°C (3 min)Method:Inject the solution twice and calculate the relative retention times in relation to n-eicosane (RRT = 1)The following table shows the approximately expected relative retention times.Analyte 1-octanol 2-octanone 2,6-dimethylaniline n-tridecane n-tetradecane RRT 0.30 0.22 0.37 0.52 0.60 Limits:The RSD of each RRT from two consecutive injections should be ≤1.0%Overall test 2BTest Solution:1.0% (W/W) n-Nonane and Hexadecane in Tetradecane.Settings:Column: Ultra-1 (25m x 0.32mm ID x 0.52µm film)Injection volume: 1 µlSolvent: TetradecaneOven temperature: 110°CGradient programme: 110°C, 20°C/min, 180°C (final time: 3.5 min)Detector temperature: 250°CInjector temperature: 200°CDetector: FIDFlow rates: Carrier gas (Helium): 2 ± 0.2 ml/minHydrogen: 30 ± 1.0 ml/minAir: 400 ± 20.0 ml/minMakeup (Nitrogen): 28 ± 1.0 ml/minSplit ratio: 15Split vent: 30 ± 3.0 ml/minSeptum purge: 3-5 ml/minMethod:Allow the system to equilibrate.Injection sequence:1)blank (Tetradecane)2) 6 replicates of the test solution. Calculate the mean of the retention times and peakareas and the relative standard deviation of n-Nonane and n-Hexadecane.Limits:Retention time repeatability: RSD of the peak retention times of the 6 replicates ≤ 2% Retention time (Rt) accuracy: for this example, the retention time ranges shown in the table below are proposed. Nevertheless, individual ranges should be predefined by the laboratory depending on the column used (e.g. Rt ± 0.2 min).Compound Rt (min)n-Nonane (C9) 1.3 – 1.7Tetradecane (C14) 4.0 – 4.7Hexadecane (C16) 5.1 – 6.0OVERALL TEST 3This test is a modified version of the overall test 1 to be used for the verification of: -Detector linearity: linearity of the areas recorded-Injector carry-over: area recorded in the blank runIt is described for both split and split less mode and may be combined with overall test 1. Split mode:Test solution: 1-octanol in n-hexane 1% (v/v)Prepare further reference solutions by diluting the test solution as described below. Settings: see overall test 1Injection sequence:5.0 ml of the test solution diluted to 25.0 ml with n-hexane (2 µl/ml): 2 injections10.0 ml of the test solution diluted to 25.0 ml with n-hexane (4 µl/ml): 2 injections15.0 ml of the test solution diluted to 25.0 ml with n-hexane (6 µl/ml): 2 injections20.0 ml of the test solution diluted to 25.0 ml with n-hexane (8 µl/ml): 2 injectionsi f combined with overall test 1 for repeatability: test solution (10 µl/ml): 6 injectionsn-hexane as blank (carry over)PA/PH/OMCL (06) 86 DEF - OMCL Guideline on Qualification of GC equipment (Annex 2) Split less mode:Stock solution: 1-octanol in n-hexane 1% (v/v)Test solution: Dilute 10 ml of the stock solution with n-hexane to 100 ml (corresponds to 1µl/ml of 1-octanol in n-hexane).Prepare further reference solutions by diluting the test solution with n-hexane.Settings: see overall test 1Injection sequence:5.0 ml of the test solution diluted to 25.0 ml with n-hexane (0.2 µl/ml): 2 injections10.0 ml of the test solution diluted to 25.0 ml with n-hexane (0.4 µl/ml): 2 injections15.0 ml of the test solution diluted to 25.0 ml with n-hexane (0.6 µl/ml): 2 injections20.0 ml of the test solution diluted to 25.0 ml with n-hexane (0.8 µl/ml): 2 injectionsif combined with overall test 1 for repeatability: test solution (1 µl/ml): 6 injectionsn-hexane as blank (carry over)Limits:Linearity: coefficient of correlation of the calibration line obtained with the reference solutions and the test solution: r2≥ 0.999.Carry-over: the percentage of the peak area corresponding to the analyte in the blank solution should be ≤ 0.2% of the peak area of this analyte in the chromatogram obtained with the solution with the highest concentration within the sequence.October 2006Page 11 of 11。
欧盟GMP指南:仪器确认_附件4_红外 EDQM_OMCL_Annex4_IR

N.B. This OMCL Quality Management System document is applicable to members of the European OMCL Network only. Other laboratories might use the document on a voluntary basis. However, please note thatGeneral European OMCL Network (GEON) QUALITY MANAGEMENT DOCUMENTPA/PH/OMCL (18) 24 R1QUALIFICATION OF EQUIPMENTQUALIFICATION OF IR SPECTROPHOTOMETERSFull document titleand reference Qualification of EquipmentAnnex 4: Qualification of IR Spectrophotometers PA/PH/OMCL (18) 24 R1 Document typeGuidelineLegislative basisThe present document was also accepted by EA as recommendation document to be used in the context of Quality Management System audits of OMCLs Date of first adoptionMay 2007 Date of original entry into forceJuly 2007 Date of entry into force of revised documentSeptember 2019Previous titles/other references / last valid version This document replaces document PA/PH/OMCL (07) 12 DEF CORR in force since December 2007 Custodian Organisation The present document was elaborated by the OMCL Network / EDQM of the Council of Europe Concerned NetworkGEONANNEX 4 OF THE OMCL NETWORK GUIDELINE“QUALIFICATION OF EQUIPMENT”QUALIFICATION OF IR SPECTROPHOTOMETERSNote: Mandatory requirements in this annex are defined using the terms “shall” or “must”. The use of “should” indicates a recommendation. For these parts of the text other appropriately justified approaches are acceptable. The term “can” indicates a possibility or an example w ith non-binding character.1.INTRODUCTIONThe present document is the 4th Annex of the core document “Qualification of Equipment”, and it shall be used in combination with it when planning, performing and documenting the qualification process of Infrared (IR) spectrophotometers.The core document contains the Introduction and general forms for Levels I (Selection of instruments and suppliers) and II (Installation and release for use) of qualification, which are common to all types of instruments.The present Annex 4 contains a general introduction and requirements for IR spectrophotometers. Level III (Periodic and motivated instrument calibration/checks) and IV (In-use instrument checks) qualifications must be carried out as an ISO 17025 requirement.Requirements and (if applicable) corresponding typical acceptance limits given in bold should be applied; however other appropriately justified approaches are acceptable.Exemplary procedures provided in this document have non-binding character. They can be helpful when carrying out the required qualification. Nevertheless, it is left to the professional judgement and background experience of each OMCL to decide on the most relevant procedures to be undertaken in order to provide evidence that their IR spectrophotometers are working properly and are suitable for their intended use.If the qualification of equipment is done by the manufacturer or an external service provider, it is the responsibility of the OMCL to make sure that this is in line with the requirements set out in this guideline.TABLE ILevel III. Periodic and motivated instrument checksRequirements and related typical acceptance limits are indicated in boldParameter to be checked Typical acceptance limits* Accuracy of wavenumber scale*Refer to Ph. Eur. Chapter 2.2.24.“Control of equipment performance”Spectral Resolution*Refer to Ph. Eur. Chapter 2.2.24.“Control of equipment performance” Detector energy ratio Limit to be set based on OMCLexperience/service provider’s instructionsSignal-to-Noise ratio Limit to be set based on OMCLexperience/service provider’s instructionsZero test Limit to be set based on OMCLexperience/service provider’s instructionsContamination check (only for ATR instruments)Wavenumber (cm-1) Upper limit (A) 3100.0 – 2800.0 0.1 1800.0 – 1600.0 0.1 1400.0 – 1100.0 0.2Throughput check (only for ATR instruments)T min = 80 % (n=3 wavenumbers)(The lower limit of the transmittance for the 3 wavenumbers must be 80 %)TABLE IILevel IV. In-use instrument checksExamples of requirements for IR spectrophotometersParameter to be checked/Typical acceptance limitsSystem suitability check: according to Ph. Eur. Chapter 2.2.24. “Control of equipment performance” or specific Monographs or MAH dossier or validated in-house method2. LEVEL III: examples of periodic and motivated instrument checksThis section contains practical examples of tests and their associated tolerance limits for several parameters related to the performance of an IR spectrophotometer.These examples can be considered by OMCLs as possible approaches to perform Level III of the equipment qualification process: “Periodic and motivated instrument checks”.Note: if available and judged appropriate, the use of the automatic internal calibration function of the instrument is encouraged. Please refer to the manufacturer’s instructions.2.1 DETECTOR ENERGY RATIOMethod:Record the minimum energy ratio value for at least one of the following measurement points and compare it to the vendor’s specifications:-Energy at 3990 cm-1 / energy at 2000 cm-1-Energy at 4000 cm-1 / energy at 2000 cm-1-Energy at 3400 cm-1 / energy at 1300 cm-1-Energy at 2000 cm-1 / energy at 1000 cm-1Energy ratio test specifications vary for each spectrometer configuration.2.2 SIGNAL-TO-NOISE RATIOMethod:Record the maximum noise level for each of the following regions:Peak-to-peak noise between:4050 cm-1 and 3950 cm-12050 cm-1 and 1950 cm-11050 cm-1 and 950 cm-1550 cm-1 and 450 cm-1(systems with DTGS detector only)RMS (root mean square) noise between:4050 cm-1 and 3950 cm-12050 cm-1 and 1950 cm-11050 cm-1 and 950 cm-1550 cm-1 and 450 cm-1(systems with DTGS detector only)Noise level test specifications vary for each spectrometer configuration.2.3 ZERO TESTMethod:When using a polystyrene film of approximately 35 µm in thickness as standard at wavelengths of 2925 cm-1and 700 cm-1, almost complete absorption of the irradiated energy can be observed. With this test, the remaining transmission is measured. As the maximum absorption can be observed at 700 cm-1, a negative value may be observed. The objective of the test is to evaluate whether, despite the fact that there is almost complete absorption, energy is still detectable.Non-valid results are an indication of non-linear behaviour of the detector and the electronic system.2.4 CONTAMINATION TEST (only for Attenuated Total Reflection (ATR) instruments)Note: if an automated system is available, this test can be run more frequently or it can be transferred to Level IV, to be run before each analysis.Method:This test checks the presence of peaks that signal a contamination problem.Use the automated function of the instrument (if available) to perform this test. If not available, a background spectrum should be recorded and compared with the one generated during the instrument qualification or provided by the supplier.2.5 THROUGHPUT CHECK (only for Attenuated Total Reflection (ATR) instruments)Note: if an automated system is available, this test can be run more frequently or it can be transferred to Level IV, to be run before each analysis.Method:This test checks for an unexpected reduction of the transmittance. An instrument-specific automated test can be used.A background spectrum is recorded and the transmittance is measured at 3 wavenumbers e.g. 4000, 2600 and 1000 cm-1. The background spectrum should be compared with the one generated during the instrument qualification or provided by the supplier.3.LEVEL IV: examples of in-use instrument checksThis section contains practical examples of tests and their associated tolerance limits for several parameters related to the performance of an IR spectrophotometer.These examples can be considered by OMCLs as possible approaches to perform Level IV of the equipment qualification process: “In-use instrument checks”.Note: if available and judged appropriate, the use of the automatic internal calibration function of the instrument is encouraged. Please refer to the manufacturer’s instructions.3.1 SYSTEM SUITABILITY TEST OF THE METHODMethod:This test should be performed according to the Ph. Eur. 2.2.24. “Control of equipment performance”, the MAH dossier or a suitably validated in-house method.Note: regeneration or replacement of the desiccant should be done if the system suitability test fails (e.g. by drying it for 8-12h at 250°C, then flushing with N2).REFERENCES(For all references, the latest version applies)1)Ph. Eur. 2.2.24, Absorption spectrophotometry, Infrared.。
annex 15 验证 201504 中英文 完

EUROPEAN COMMISSIONENTERPRISE DIRECTORATE-GENERALSingle market, regulatory environment, industries under vertical legislationPharmaceuticals and cosmeticsBrussels, 30 March 2015EudraLexVolume 4EU Guidelines forGood Manufacturing Practice forMedicinal Products for Human and Veterinary UseAnnex 15: Qualification and ValidationEU GMP附录15:确认和验证Legal basis for publishing the detailed guidelines: Article 47 of Directive 2001/83/EC on the Community code relating to medicinal products for human use and Article 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinal products as laid down in Directive 2003/94/EC for medicinal products for human use and Directive91/412/EEC for veterinary use.公布详细指南的法律依据:指令2001/83/EC第47款关于人药共同体代码,和2001/82/EC第51款关于兽药共同体代码的要求。
2010-26-EU-欧盟新排放指令

DIRECTIVESCOMMISSION DIRECTIVE 2010/26/EUof 31 March 2010amending Directive 97/68/EC of the European Parliament and of the Council on the approximation of the laws of the Member States relating to measures against the emission of gaseous and particulate pollutants from internal combustion engines to be installed in non-road mobile machinery(Text with EEA relevance)THE EUROPEAN COMMISSION, Having regard to the Treaty on the Functioning of the European Union,Having regard to Directive 97/68/EC of 16 December 1997 of the European Parliament and of the Council on the approxi mation of the laws of the Member States relating to measures against the emission of gaseous and particulate pollutants from internal combustion engines to be installed in non-road mobile machinery ( 1 ), and in particular Articles 14 and 14a thereof, Whereas:(1) Article 14a of Directive 97/68/EC sets out the criteria and the procedure for extending the period referred to in Article 9a(7) of that Directive. Studies carried out in accordance with Article 14a of Directive 97/68/EC show that there are substantial technical difficulties to comply with stage II requirements for professional use, multi- positional, hand-held mobile machinery in which engines of classes SH:2 and SH:3 are installed. It is therefore necessary to extend the period referred to in Article 9a(7) until 31 July 2013. (2) Since the amendment of Directive 97/68/EC in 2004, technical progress has been made in the design of diesel engines with a view to make them compliant with the exhaust emission limits for stages IIIB and IV. Electronically controlled engines, largely replacing me- chanically controlled fuel injection and control systems, have been developed. Therefore, the current general type- approval requirements in Annex I to Directive 97/68/EC should be adapted accordingly and general type-approval requirements for stages IIIB and IV should be introduced. (3) Annex II to Directive 97/68/EC specifies the technical details of the information documents that need to be submitted by the manufacturer to the type-approval authority with the application for engine type-approval. The details specified regarding the additional anti- pollution devices are generic and should be adapted to the specific after-treatment systems that need to be used to ensure that engines comply with exhaust emission limit stages IIIB and IV. More detailed information on the after-treatment devices installed on the engines should be submitted to enable type-approval authorities to assess the engine’s capability to comply with stages IIIB and IV.(4) Annex III to Directive 97/68/EC sets out the methodtesting the engines and determining their level of emissions of gaseous and particulate pollutants. The type-approval testing procedure of engines to demon strate compliance with the exhaust emission limits of stage IIIB and IV should ensure that the simultaneous compliance with the gaseous (carbon monoxide, hydro carbons, oxides of nitrogen) and the particulate emission limits is demonstrated. The non-road steady cycle (NRSC) and non-road transient cycle (NRTC) should be adapted accordingly. (5) Point 1.3.2 of Annex III to Directive 97/68/EC foreseesthe modification of the symbols (section 2.18 of Annex I), the test sequence (Annex III) and calculation equations (Appendix III to Annex III), prior to the introduction of the cold/hot composite test sequence. The type approval procedure to demonstrate compliance with the exhaust emission limits of stage IIIB and IV requires the intro duction of a detailed description of the cold start cycle. (6) Section 3.7.1 of Annex III to Directive 97/68/EC sets out the test cycle for the different equipment specifications. The test cycle under point 3.7.1.1 (specification A) needs to be adapted to clarify which engine speed needs to be used in the type approval calculation method. It is also necessary to adapt the reference to the updated version of the international testing standard ISO 8178-4:2007.( 1 ) OJ L 59, 27.2.1998, p. 1.(7) Section 4.5 of Annex III to Directive 97/68/EC outlines the emissions test run. This section needs to be adapted to take account of the cold start cycle. (8) Appendix 3 of Annex III to Directive 97/68/EC sets out the criteria for the data evaluation and calculation of the gaseous emissions and the particulate emissions, for both the NRSC test and the NRTC test set out in Annex III. The type approval of engines in accordance with stage IIIB and IV requires the adaptation of the calculation method for the NRTC test. (9) Annex XIII to Directive 97/68/EC sets out the provisions for engines placed on the market under a ‘flexible scheme’. To ensure a smooth implementation of stage IIIB, an increased use of this flexibility scheme may be needed. Therefore, the adaptation to technical progress to enable the introduction of stage IIIB compliant engines needs to be accompanied by measures to avoid that the use of the flexibility scheme may be hampered by notifi cation requirements which are no longer adapted to the introduction of such engines. The measures should aim at simplifying the notification requirements and the reporting obligations, and at making them more focused and tailored to the need for market surveillance authorities to respond to the increased use of the flexi bility scheme that will result from the introduction of stage IIIB. (10) Since Directive 97/68/EC provides for the type-approval of stage IIIB engines (category L) as from 1 January 2010 it is necessary to provide for the possibility to grant type approval from that date. (11) For reasons of legal certainty this Directive should enter into force as a matter of urgency. (12) The measures provided for in this Directive are in accordance with the opinion of the Committee estab lished in Article 15(1) of Directive 97/68/EC, HAS ADOPTED THIS DIRECTIVE: Article 1 Amendments to Directive 97/68/EC Directive 97/68/EC is amended as follows: 1. in Article 9a(7), the following subparagraph is added: ‘Notwithstanding the first subparagraph, an extension of the derogation period is granted until 31 July 2013, within the category of top handle machines, for professional use, multi- positional, hand-held hedge trimmers and top handle tree service chainsaws in which engines of classes SH:2 and SH:3 are installed.’;2. Annex I is amended in accordance with Annex I to this Directive;3. Annex II is amended in accordance with Annex II to this Directive;4. Annex III is amended in accordance with Annex III to this Directive;5. Annex V is amended in accordance to Annex IV to this Directive;6. Annex XIII is amended in accordance with Annex V to this Directive.Article 2Transitional provisionWith effect from the day following the publication of this Directive in the Official Journal, Member States may grant type-approval in respect of electronically controlled engines which comply with the requirements laid down in Annexes I, II, III, V and XIII to Directive 97/68/EC, as amended by this Directive.Article 3Transposition1. Member States shall bring into force the laws, regulations and administrative provisions necessary to comply with the Directive within 12 months after the publication of the Directive. They shall forthwith communicate to the Commission the text of those provisions.They shall apply those provisions from 31 March 2011.When Member States adopt those provisions, they shall contain a reference to this Directive or be accompanied by such a reference on the occasion of their official publication. Member States shall determine how such reference is to be made.2. Member States shall communicate to the Commission the text of the main provisions of national law which they adopt in the field covered by this Directive.Article 4Entry into forceThis Directive shall enter into force on the day following its publication in the Official Journal of the European Union .Article 5AddresseesThis Directive is addressed to the Member States. Done at Brussels, 31 March 2010. For the Commission The President José Manuel BARROSOANNEX IThe following section 8 is added to Annex I to Directive 97/68/EC:IIIBIVSTAGESANDFOR‘8. TYPEAPPROVALREQUIREMENTS8.1. This section shall apply to the type-approval of electronically controlled engines, which uses electronic control todetermine both the quantity and timing of injecting fuel (hereafter “engine”). This section shall apply irrespective of the technology applied to such engines to comply with the emission limit values set out in sections 4.1.2.5 and 4.1.2.6 of this Annex.8.2. DefinitionsFor the purpose of this section, the following definitions shall apply:8.2.1. “emission control strategy” means a combination of an emission control system with one base emission controlstrategy and with one set of auxiliary emission control strategies, incorporated into the overall design of an engine or non-road mobile machinery into which the engine is installed.8.2.2. “reagent” means any consumable or non-recoverable medium required and used for the effective operation of theexhaust after-treatment system.8.3. Generalrequirements8.3.1. Requirements for base emission control strategy8.3.1.1. The base emission control strategy, activated throughout the speed and torque operating range of the engine,shall be designed as to enable the engine to comply with the provisions of this Directive8.3.1.2. Any base emission control strategy that can distinguish engine operation between a standardised type approvaltest and other operating conditions and subsequently reduce the level of emission control when not operating under conditions substantially included in the type approval procedure is prohibited.8.3.2. Requirements for auxiliary emission control strategy8.3.2.1. An auxiliary emission control strategy may be used by an engine or a non-road mobile machine, provided thatthe auxiliary emission control strategy, when activated, modifies the base emission control strategy in response toa specific set of ambient and/or operating conditions but does not permanently reduce the effectiveness of theemission control system:(a) where the auxiliary emission control strategy is activated during the type approval test, sections 8.3.2.2 and8.3.2.3 shall not apply;(b) where the auxiliary emission control strategy is not activated during the type approval test, it must bedemonstrated that the auxiliary emission control strategy is active only for as long as required for thepurposes identified in section 8.3.2.3.8.3.2.2. The control conditions applicable to this section are all of the following:(a) an altitude not exceeding 1 000 metres (or equivalent atmospheric pressure of 90 kPa);(b) an ambient temperature within the range 275 K to 303 K (2 °C to 30 °C);(c) the engine coolant temperature above 343 K (70 °C).Where the auxiliary emission control strategy is activated when the engine is operating within the control conditions set out in points (a), (b) and (c), the strategy shall only be activated exceptionally.8.3.2.3. An auxiliary emission control strategy may be activated in particular for the following purposes:(a) by onboard signals, for protecting the engine (including air-handling device protection) and/or non-roadmobile machine into which the engine is installed from damage;(b) for operational safety and strategies;(c) for prevention of excessive emissions, during cold start or warming-up, during shut-down;(d) if used to trade-off the control of one regulated pollutant under specific ambient or operating conditions, formaintaining control of all other regulated pollutants, within the emission limit values that are appropriate forthe engine concerned. The purpose is to compensate for naturally occurring phenomena in a manner thatprovides acceptable control of all emission constituents.8.3.2.4. The manufacturer shall demonstrate to the technical service at the time of the type-approval test that theoperation of any auxiliary emission strategy complies with the provisions of section 8.3.2. The demonstration shall consist of an evaluation of the documentation referred to in section 8.3.3.8.3.2.5. Any operation of an auxiliary emission control strategy not compliant with section 8.3.2 is prohibited.8.3.3. Documentation requirements8.3.3.1. The manufacturer shall provide an information folder accompanying the application for type-approval at thetime of submission to the technical service, which ensures access to any element of design and emission control strategy and the means by which the auxiliary strategy directly or indirectly controls the output variables. The information folder shall be made available in two parts:(a) the documentation package, annexed to the application for type-approval, shall include a full overview of theemission control strategy. Evidence shall be provided that all outputs permitted by a matrix, obtained fromthe range of control of the individual unit inputs, have been identified. This evidence shall be attached to theinformation folder as referred to in Annex II;(b) the additional material, presented to the technical service but not annexed to the application for type-approval, shall include all the modified parameters by any auxiliary emission control strategy and theboundary conditions under which this strategy operates and in particular:(i) a description of the control logic and of timing strategies and switch points, during all modes ofoperation for the fuel and other essential systems, resulting in effective emissions control (such asexhaust gas recirculation system (EGR) or reagent dosing);(ii) a justification for the use of any auxiliary emission control strategy applied to the engine, accompanied by material and test data, demonstrating the effect on exhaust emissions. This justification may be basedon test data, sound engineering analysis, or a combination of both;(iii) a detailed description of algorithms or sensors (where applicable) used for identifying, analysing, or diagnosing incorrect operation of the NO x control system;(iv) the tolerance used to satisfy the requirements in section 8.4.7.2, regardless of the used means.8.3.3.2. The additional material referred to in point (b) of section 8.3.3.1 shall be treated as strictly confidential. It shallbe made available to the type-approval authority on request. The type-approval authority shall treat this material as confidential.ofoperationNO x control measures8.4. Requirementstoensurecorrect8.4.1. The manufacturer shall provide information that fully describes the functional operational characteristics of theNO x control measures using the documents set out in section 2 of Appendix 1 to Annex II and in section 2 of Appendix 3 to Annex II.8.4.2. If the emission control system requires a reagent, the characteristics of that reagent, including the type of reagent,information on concentration when the reagent is in solution, operational temperature conditions and reference to international standards for composition and quality must be specified by the manufacturer, in section 2.2.1.13 of Appendix 1 and in section 2.2.1.13 of Appendix 3 to Annex II.8.4.3. The engine emission control strategy shall be operational under all environmental conditions regularly pertainingin the territory of the Community, especially at low ambient temperatures.8.4.4. The manufacturer shall demonstrate that the emission of ammonia during the applicable emission test cycle ofthe type approval procedure, when a reagent is used, does not exceed a mean value of 25 ppm.8.4.5. If separate reagent containers are installed on or connected to a non-road mobile machine, means for taking asample of the reagent inside the containers must be included. The sampling point must be easily accessible without requiring the use of any specialised tool or device.8.4.6. Use and maintenance requirements8.4.6.1. The type approval shall be made conditional, in accordance with Article 4(3), upon providing to each operator ofnon-road mobile machinery written instructions comprising the following:(a) detailed warnings, explaining possible malfunctions generated by incorrect operation, use or maintenance ofthe installed engine, accompanied by respective rectification measures;(b) detailed warnings on the incorrect use of the machine resulting in possible malfunctions of the engine,accompanied by respective rectification measures;(c) information on the correct use of the reagent, accompanied by an instruction on refilling the reagentbetween normal maintenance intervals;(d) a clear warning, that the type-approval certificate, issued for the type of engine concerned, is valid only whenall of the following conditions are met:(i) the engine is operated, used and maintained in accordance with the instructions provided;(ii) prompt action has been taken for rectifying incorrect operation, use or maintenance in accordance with the rectification measures indicated by the warnings referred to in point (a) and (b);(iii) no deliberate misuse of the engine has taken place, in particular deactivating or not maintaining an EGR or reagent dosing system.The instructions shall be written in a clear and non-technical manner using the same language as is used in the operator’s manual on non-road mobile machinery or engine.8.4.7. Reagent control (where applicable)8.4.7.1. The type approval shall be made conditional, in accordance with the provisions of section 3 of Article 4, uponproviding indicators or other appropriate means, according to the configuration of the non-road mobile machinery, informing the operator on:(a) the amount of reagent remaining in the reagent storage container and by an additional specific signal, whenthe remaining reagent is less than 10 % of the full container’s capacity;(b) when the reagent container becomes empty, or almost empty;(c) when the reagent in the storage tank does not comply with the characteristics declared and recorded insection 2.2.1.13 of Appendix 1 and section 2.2.1.13 of Appendix 3 to Annex II, according to the installedmeans of assessment.(d) when the dosing activity of the reagent is interrupted, in cases other than those executed by the engine ECUor the dosing controller, reacting to engine operating conditions where the dosing is not required, providedthat these operating conditions are made available to the type approval authority.8.4.7.2. By the choice of the manufacturer the requirements of reagent compliance with the declared characteristics andthe associated NO x emission tolerance shall be satisfied by one of the following means:(a) direct means, such as the use of a reagent quality sensor.(b) indirect means, such as the use of a NO x sensor in the exhaust to evaluate reagent effectiveness.(c) any other means, provided that its efficacy is at least equal to the one resulting by the use of the means ofpoints (a) or (b) and the main requirements of this section are maintained.’ANNEX IIAnnex II to Directive 97/68/EC is amended as follows:1. Section 2 of Appendix 1 is replaced by the following:POLLUTIONAIRAGAINSTTAKEN‘2. MEASURESyes/no(*)............................................................................................................gases:recyclingcrankcase2.1. Deviceforcoverednotbyheading)ifanother(ifanti-pollutiondevices2.2. Additionalandany,(*)yes/noconverter:2.2.1. Catalytic.......................................................................................................................................................................................2.2.1.1. Make(s):........................................................................................................................................................................................2.2.1.2. Type(s):converterselements................................................................................................................andcatalytic2.2.1.3. Numberofconverter(s):...............................................................................................thecatalyticofandvolume2.2.1.4. Dimensions-........................................................................................................................................................action:ofcatalytic2.2.1.5. Typeprecious........................................................................................................................................metals:of2.2.1.6. Totalchargeconcentration:...........................................................................................................................................................2.2.1.7. Relative.....................................................................................................................................material):and2.2.1.8. Substrate(structure...............................................................................................................................................................................2.2.1.9. Celldensity:2.2.1.10. Type of casing for the catalytic converter(s): .................................................................................................................2.2.1.11. Location of the catalytic converter(s) (place(s) and maximum/minimum distance(s) from engine): ............2.2.1.12. Normal operating range (K): ................................................................................................................................................2.2.1.13. Consumable reagent (where appropriate): .......................................................................................................................2.2.1.13.1. Type and concentration of reagent needed for catalytic action: .............................................................................2.2.1.13.2. Normal operational temperature range of reagent: ......................................................................................................2.2.1.13.3. International standard (where appropriate): ....................................................................................................................2.2.1.14. NO x sensor: yes/no (*)(*)yes/nosensor:2.2.2. Oxygen.......................................................................................................................................................................................2.2.2.1. Make(s):............................................................................................................................................................................................2.2.2.2. Type:.....................................................................................................................................................................................2.2.2.3. Location:(*)yes/noinjection:2.2.3. Airetc.):.........................................................................................................................................pump,2.2.3.1. Type(pulseair,air(*)yes/no2.2.4. EGR:etc.):pressure,........................................................................2.2.4.1. Characteristicspressure/low(cooled/uncooled,high(*)yes/no2.2.5. Particulatetrap:particulate.........................................................................................................thetrap:capacityof2.2.5.1. Dimensionsandparticulatetrap:.........................................................................................................................theandof2.2.5.2. Typedesignengine):..................................................................fromdistance(s)2.2.5.3. Locationand(place(s)maximum/minimumdescriptionand/ordrawing:regeneration,............................................................................ofor2.2.5.4. Methodsystempressure(kPa)and..................................................................................range:2.2.5.5. Normal(K)operatingtemperature(*)yes/nosystems:2.2.6. Otheroperation:...................................................................................................................................................and2.2.6.1. Description___________(*) Strike out what does not apply.’2. Section 2 of Appendix 3 is replaced by the following:POLLUTIONAGAINSTAIRTAKEN‘2. MEASURESyes/no(*)............................................................................................................gases:crankcase2.1. Deviceforrecyclingcoverednotbyheading)ifanotherany,anti-pollutiondevices(ifand2.2. Additional(*)yes/noconverter:2.2.1. Catalytic.......................................................................................................................................................................................2.2.1.1. Make(s):........................................................................................................................................................................................2.2.1.2. Type(s):and................................................................................................................converterselementscatalyticof2.2.1.3. Numberconverter(s):...............................................................................................thecatalyticofandvolume2.2.1.4. Dimensions-........................................................................................................................................................action:ofcatalytic2.2.1.5. Typeprecious........................................................................................................................................metals:of2.2.1.6. Totalchargeconcentration:...........................................................................................................................................................2.2.1.7. Relative.....................................................................................................................................material):and2.2.1.8. Substrate(structure...............................................................................................................................................................................2.2.1.9. Celldensity:2.2.1.10. Type of casing for the catalytic converter(s): .................................................................................................................2.2.1.11. Location of the catalytic converter(s) (place(s) and maximum/minimum distance(s) from engine): ............2.2.1.12. Normal operating range (K) .................................................................................................................................................2.2.1.13. Consumable reagent (where appropriate): .......................................................................................................................2.2.1.13.1. Type and concentration of reagent needed for catalytic action: .............................................................................2.2.1.13.2. Normal operational temperature range of reagent: ......................................................................................................2.2.1.13.3. International standard (where appropriate): ....................................................................................................................2.2.1.14. NO x sensor: yes/no (*)yes/no(*)sensor:2.2.2. Oxygen.......................................................................................................................................................................................2.2.2.1. Make(s):............................................................................................................................................................................................2.2.2.2. Type:.....................................................................................................................................................................................2.2.2.3. Location:(*)yes/noinjection:2.2.3. Airetc.):.........................................................................................................................................pump,2.2.3.1. Type(pulseair,air(*)yes/no2.2.4. EGR:etc.):pressure,........................................................................2.2.4.1. Characteristicspressure/low(cooled/uncooled,high(*)yes/no2.2.5. Particulatetrap:particulate.........................................................................................................thetrap:capacityof2.2.5.1. Dimensionsandparticulatetrap:.........................................................................................................................theandof2.2.5.2. Typedesignengine):..................................................................fromdistance(s)2.2.5.3. Locationand(place(s)maximum/minimumdescriptionand/ordrawing:regeneration,............................................................................ofor2.2.5.4. Methodsystempressure(kPa)and..................................................................................range:2.2.5.5. Normal(K)operatingtemperature(*)yes/nosystems:2.2.6. Otheroperation:...................................................................................................................................................and2.2.6.1. Description___________(*) Strike out what does not apply.’。
(Annex19)totheEUGMPGuide-EuropeanCommission

EUROPEAN COMMISSIONENTERPRISE DIRECTORATE-GENERALSingle market : management & legislation for consumer goodsPharmaceuticals : regulatory framework and market authorisationsBrussels, 23 June 2004Ad Hoc GMP Inspections Services GroupGood Manufacturing PracticeProposed Addition (Annex 19) to the EU GMP GuideTitle:Reference Samples and Retention SamplesAgreed by ad hoc GMP inspectors services group April 2004 Released for public consultation 15 July 2004 Deadline for comments 15 January 2005 Final draft adopted by ad hoc GMP inspectors services groupAdopted by Pharmaceutical CommitteeDate for coming into operationNote:The new annex to the EU GMP Guide provides guidance on the taking and holding of refer-ence samples of starting materials, packaging materials and finished products as well as for retention samples of finished products. The annex provides definitions of the terms "refer-ence sample" and "retention sample", which are often incorrectly considered as synonyms. The guidance is wide ranging in scope and includes the case of multiple manufacturing sites, the position with respect to importers and what should happen when a manufacturing site ceases to operate. Updated guidance is also given on the size of reference samples and a con-sequential amendment will therefore be necessary to Chapter 6 section 14 of the GMP Guide to maintain consistency.PROPOSED ANNEX TO EC GUIDE TO GOOD MANUFACTURING PRACTICE REFERENCE SAMPLES AND RETENTION SAMPLES1. Scope1.1 This Annex to the Guide to Good Manufacturing Practice for Medicinal Products (“the Guide”) gives guidance on the taking and holding of reference samples of starting materials, packag-ing materials or finished products and retention samples of finished products.1.2 The guidance may also be applied to investigational medicinal products, subject to any differ-ence mentioned in Commission Directive 2003/94/EC and any more specific guidance in Annex 13 to the Guide.1.3 This annex also includes guidance on the taking of retention samples for parallel imported / distributed medicinal products.2. Principle2.1 Samples are retained to fulfil two purposes; firstly to provide a sample for analytical testing and secondly to provide a specimen of the fully finished product. Samples may therefore fall into two categories:Reference sample: a sample of a batch of starting material, packaging material or finished product which is stored for the purpose of being analysed should the need arise during the shelf life of the batch concerned. Where stability permits, reference samples from important intermediate stages of manufacture should also be kept. Examples include tablet cores and different stages of coating proc-esses.Retention sample: a sample of a fully packaged unit from a batch of finished product. It is stored for identification purposes. For example, presentation, packaging, labelling, summary of product charac-teristics / patient information leaflet, batch number, expiry date) should the need arise during the shelf life of the batch concerned.For finished products, in many instances the reference and retention samples will be presented identi-cally, i.e. as fully packaged units. In such circumstances, reference and retention samples may be re-garded as interchangeable.2.2 It is necessary for the manufacturer / importer / site of batch release, as appropriate, to keep reference and/or retention samples from each batch of finished product and, for the manufacturer to keep a reference sample from each delivery of a batch of starting material (subject to certain excep-tions – see3.2 below). Each packaging site should keep reference samples of each batch of primary and printed packaging materials.2.3 The reference and/or retention samples serve as a record of the batch of finished product or starting material and can be assessed in the event of, for example, a dosage form quality complaint, a query relating to compliance with the marketing authorisation, a labelling/packaging query, a pharma-covigilance report or a stability query.3. Duration of Storage3.1 Reference and retention samples from each batch of finished product should be retained for at least one year after the expiry date. The reference sample should be contained in its finished primary packaging or in packaging composed of the same material as the primary container in which the prod-uct is marketed (for veterinary medicinal products other than immunologicals, see also Annex 4, para-graphs 8 & 9).3.2 Unless a longer period is required under the law of the Member State of manufacture, samples of starting materials (other than solvents, gases or water used in the manufacturing process) shall be retained for at least two years after the release of product. That period may be shortened if the period of stability of the material, as indicated in the relevant specification, is shorter.4. Size of Reference and Retention Samples4.1 The reference sample should be of sufficient size to permit the carrying out, on two occasions, of the full analytical controls on the batch in accordance with the Marketing Authorisation File which has been assessed and approved by the relevant Competent Authority / Authorities. Any proposed exception to this should be justified to, and agreed with, the relevant competent authority.4.2 Where applicable, national requirements relating to the size of reference samples and, if nec-essary, retention samples, should be followed.4.3 Reference samples should be representative of the batch of starting material or finished prod-uct from which they are taken. Samples should include the most stressed part of a process (e.g. begin-ning or end of a process). Where a batch is packaged in two, or more, distinct packaging operations, at least one retention sample should be taken from each individual packaging operation. Any pro-posed exception to this should be justified to, and agreed with, the relevant competent authority.4.4 It should be ensured that all necessary analytical materials and equipment are still available, or are readily obtainable, in order to carry out all tests given in the specification until one year after ex-piry of the last batch manufactured. This applies also to analytical reference materials used in tests which have been superseded.5. StorageConditions5.1 Storage of reference/retention samples of finished products and reference samples of starting materials should be in accordance with the current version of the Note for Guidance on Declaration of Storage Conditions for Medicinal Products and Active Substances.5.2 Storage conditions should be in accordance with the marketing authorisation (e.g. refriger-ated storage where relevant).Agreements6. Written6.1 Where the marketing authorisation holder is not the same legal entity as the site(s) responsible for batch release within the EEA, the responsibility for taking and storage of reference/retention sam-ples should be defined in a written agreement between the two parties in accordance with Chapter 7 of the EC Guide to Good Manufacturing Practice. This applies also where any manufacturing or batch release activity is carried out at a site other than that with overall responsibility for the batch on the EEA market and the arrangements between each different site for the taking and keeping of reference and retention samples should be defined in a written agreement.6.2 The Qualified Person who releases a batch for sale should ensure that all relevant reference and retention samples are accessible at all reasonable times. Where necessary, the arrangements for such access should be defined in a written agreement.6.3 Where more than one site is involved in the manufacture of a finished product, the availability of written agreements is key to controlling the taking and location of reference and retention samples.7. Reference Samples – General Points7.1 Reference samples are for the purpose of analysis and, therefore, should be conveniently available to a laboratory with validated methodology. For starting materials and packaging materials, used for medicinal products manufactured within the EEA, these are the original site of manufacture and the site(s) of packaging, respectively. For finished products manufactured within the EEA, this is the original site of manufacture.7.2 For finished products manufactured by a third-country manufacturer and where an operational Mutual Recognition Agreement (MRA) is in place, the reference samples may be taken and stored at the third country site of manufacture. This should be covered in a written agreement (as referred to in section 6. above) between the importer/site of batch release and the third country manufacturer.7.3 For finished products manufactured by a third country manufacturer where no MRA is in place, reference samples should be taken and stored at a licensed manufacturer located within the EEA. These samples should be taken in accordance with written agreement(s) between all of the par-ties concerned. The samples should, preferably, be stored at the location where testing on importation has been performed.8. Retention Samples – General Points8.1 A retention sample should represent a batch of finished products as distributed in the EEA and may need to be examined in order to confirm non-technical attributes for compliance with the market-ing authorisation or EU legislation. Therefore, retention samples should in all cases be located within the EEA. These should preferably be stored at the site where the Qualified Person (QP) certifying the finished product batch is located.8.2 In accordance with 8.1 above, where an operational MRA is in place and reference samples are retained at a third country manufacturer (section 7.2 above), separate retention samples should be kept within the EEA.8.3 Retention samples should be stored at the premises of an authorised manufacturer in order to permit ready access by the Competent Authority.8.4 Where more than one manufacturing site within the EEA is involved in the manufacture im-portation/packaging/testing/batch release, as appropriate of a product, the responsibility for taking and storage of retention samples should be defined in a written agreement(s) between the parties con-cerned.9. Reference and Retention Samples for Parallel Imported/Parallel Distributed Products. 9.1 Where the packs are not opened, only the packaging material used needs to be retained, as there is no, or little, risk of product mix up.9.2 Where the packs are opened, for example, to replace the carton or patient information leaflet, then one retention sample, per packaging operation, containing the product should be taken, as there is a risk of product mix-up during the assembly process. It is important to be able to identify quickly who is responsible in the event of a mix-up (original manufacturer or parallel import assembler) as it would affect the extent of any resulting recall.10. Reference and Retention Samples in the Case of Closedown of a Manufacturer10.1 Where a manufacturer closes down and the manufacturing authorisation is surrendered, re-voked, or ceases to exist, it is probable that many unexpired batches of medicinal products manufac-tured by that manufacturer remain on the market. In order for those batches to remain on the market, the manufacturer should make detailed arrangements for transfer of reference and retention samples (and relevant GMP documentation) to an authorised storage site. The manufacturer should satisfy the Competent Authority that the arrangements for storage are satisfactory and that the samples can, if necessary, be readily accessed.10.2 If the manufacturer is not in a position to make the necessary arrangements this may be dele-gated to another manufacturer. The Marketing Authorisation holder (MAH) is responsible for such delegation and for the provision of all necessary information to the Competent Authority. In addition, the MAH should, in relation to the suitability of the proposed arrangements for storage of reference and retention samples, consult with the competent authority of each Member State in which any unex-pired batch has been placed on the market.10.3 These requirements apply also in the event of the closedown of a third country site of manu-facture. In such instances, the importer has a particular responsibility to ensure that satisfactory ar-rangements are put in place and that the competent authority/authorities is/are consulted.--------------------------------------------------------------------------------------------------------------- Consequential amendment to Chapter 6 section 14 of EU GMP Guide6.14 Reference samples from each batch of finished products should be retained till one yearafter the expiry date. Finished products should usually be kept in their final packaging and stored un-der the recommended conditions. Samples of starting materials (other than solvents, gases and water) should be retained for at least two years (1) after the release of the product if their stability allows. This period may be shortened if their stability, as mentioned in the relevant specification, is shorter. Refer-ence samples of materials and products should be of a size sufficient to permit the carrying out, on two occasions, of the full analytical controls on the batch in accordance with the Marketing Authorisation.(1) In Federal Republic of Germany, France, Belgium and Greece, samples of starting materials should be retained for as long as the corresponding finished product.。
ICAO Annex附件版本更新

览表
备注
ICAO Annexes版本更新情
附件代 Annex 号 1 附件英文名称
Personnel Licensing Annex 2 Rules of the Air Annex 3 Meteorological Service for International Air Navigation Annex 4 Aeronautical Charts Annex 5 Units of Measurement to be Used in Air and Ground Operations Annex 6 Operation of Aircraft Part I International Commercial Air Transport – Aeroplanes Part II International General Aviation – Aeroplanes Part III International Operations – Helicopters Annex 7 Aircraft Nationality and Registration Marks Annex 8 Airworthiness of Aircraft Annex 9 Facilitation Annex 10 Aeronautical Telecommunications Volume I Radio Navigation Aids Volume II Communications Procedures Volume III Communications Systems Volume IV Surveillance Radar and Collision Avoidance Systems Volume V Aeronautical Radio Frequency Spectrum Utilization Annex 11 Air Traffic Services Annex 12 Search and Rescue Annex 13 Aircraft Accident and Incident Investigation Annex 14 Aerodrome Volume I Aerodrome Design and Construction Volume II Heliports Annex 15 Aeronautical Information Services Annex 16 Environmental Protection Volume I Environmental Protection – Aircraft Noise Volume II Environmental Protection – Aircraft Engine Emissions Annex 17 Security Annex 18 The Safe Transport of Dangerous Goods by Air Annex 19 Safety Management
博思 3590 系列精密电位器 - 使用手册说明书

Specifi cations are subject to change without notice.Customers should verify actual device performance in their specifi c applications.*RoHS Directive 2002/95/EC Jan. 27, 2003 including annex and RoHS Recast 2011/65/EU June 8, 2011.Stop Strength..............................................................................................................................................................................45 N-cm (64 oz.-in.) minimum Mechanical Angle ...........................................................................................................................................................................................3600 ° +10 °, -0 °Torque (Starting & Running) ................................................................................................................................0.35 N-cm (0.5 oz.-in.) maximum (unsealed) 1.1 N-cm (1.5 oz.-in.) maximum (sealed) Mounting ..............................................................................................................................................................................55-80 N-cm (5-7 lb.-in.) (plastic) 90-113 N-cm (8-10 in.-lb.) (metal)Shaft Runout......................................................................................................................................................................................0.13 mm (0.005 in.) teral Runout ...................................................................................................................................................................................0.20 mm (0.008 in.) T.I.R.Shaft End Play ...................................................................................................................................................................................0.25 mm (0.010 in.) T.I.R.Shaft Radial Play ...............................................................................................................................................................................0.13 mm (0.005 in.) T.I.R.Pilot Diameter Runout .......................................................................................................................................................................0.08 mm (0.003 in.) T.I.R.Backlash ............................................................................................................................................................................................................1.0 ° maximum Weight ........................................................................................................................................................................................................Approximately 19 G Terminals ................................................................................................................................................................................................Solder lugs or PC pins Soldering ConditionManual Soldering...........................................................96.5Sn/3.0Ag/0.5Cu solid wire or no-clean rosin cored wire; 370 °C (700 °F) max. for 3 seconds Wave Soldering ...................................................................................96.5Sn/3.0Ag/0.5Cu solder with no-clean fl ux; 260 °C (500 °F) max. for 5 seconds Wash processes .......................................................................................................................................................................................Not recommended Marking .....................................Manufacturer’s name and part number, resistance value and tolerance, linearity tolerance, wiring diagram, and date code.Ganging (Multiple Section Potentiometers) ......................................................................................................................................................1 cup maximum Hardware ............................................................................................................One lockwasher and one mounting nut is shipped with each potentiometer.NOTE: For Anti-rotation pin add 91 after confi guration dash number. Example: -2 becomes -291 to add AR pin.1At room ambient: +25 °C nominal and 50 % relative humidity nominal, except as noted. 2Consult manufacturer for complete specifi cation details for resistances below 1k ohms.BOLDFACE LISTINGS ARE IN STOCK AND READILY AVAILABLETHROUGH DISTRIBUTION. FOR OTHER OPTIONS CONSULT FACTORY.ROHS IDENTIFIER: L = COMPLIANTRecommended Part Numbers(Printed Circuit)(Solder Lug)(Solder Lug)Resistance (Ω)Resolution (%)3590P-2-102L 3590S-2-102L 3590S-1-102L 1,000.0293590P-2-202L 3590S-2-202L 3590S-1-202L 2,000.0233590P-2-502L 3590S-2-502L 3590S-1-502L 5,000.0253590P-2-103L 3590S-2-103L 3590S-1-103L 10,000.0203590P-2-203L 3590S-2-203L 3590S-1-203L 20,000.0193590P-2-503L 3590S-2-503L 3590S-1-503L 50,000.0133590P-2-104L3590S-2-104L3590S-1-104L100,000.009*Ro H S C O MP L I A N TPanel Thickness Dimensions(For Bushing Mount Only)1.60 +.08/-.03(.063 +.003/-.001)DIA.ANTI-ROTATION PINAnti-rotation pin hole is shown at six o'clockposition for reference only. The actual location isdetermined by the customer's application. Referto the front view of the potentiometer to see thelocation of the optional A/R pin.Panel thickness and hole diameters arerecommended for best fit. However, customersmay adjust the dimensions to suit their specificapplication.Product DimensionsSpecifi cations are subject to change without notice.Customers should verify actual device performance in their specifi c applications.REV. 06/12 MOUNTING SURFACE-2, -4, -6, -8 Confi gurations-1, -3, -5, -7 Confi gurationsRecommended PCB LayoutHOLE DIAMETER5.08(.200)5.08(.200)6.99(.275)SchematicTOLERANCES: EXCEPT WHERE NOTED.508 .127DECIMALS: .XX ±(.02),.XXX ±(.005)FRACTIONS: ±1/64MMDIMENSIONS:(IN.)Shaft & Bushing Confi gurations(Bushing - DxL, Shaft - D):(-1) Plastic Bushing (3/8 ” x 5/16 ”)and Shaft (.2480 + .001, - .002)(-2) Metal Bushing (3/8 ” x 5/16 ”)and Shaft (.2497 + .0000, - .0009)(-3) Sealed, Plastic Bushing (3/8 ” x 5/16 ”)and Shaft (.2480 + .001, - .002)(-4) Sealed, Metal Bushing (3/8 ” x 5/16 ”)and Shaft (.2497 + .0000, - .0009)(-5) Metric, Plastic Bushing (9 mm x 7.94 mm)and Shaft (6 mm + 0, - .076 mm)(-6) Metric, Metal Bushing (9 mm x 7.94 mm)and Shaft (6 mm + 0, - .023 mm)(-7) Metric, Sealed, Plastic Bushing (9 mm x7.94 mm) and Shaft (6 mm + 0, - .076 mm)(-8) Metric, Sealed, Metal Bushing (9 mm x7.94 mm) and Shaft (6 mm + 0, - .023 mm)Terminal Styles“P” Terminal Style“S” Terminal Style。
WHO_TRS_908-Annex9

© World Health OrganizationWHO Technical Report Series, No. 908, 2003Annex 9Guide to good storage practices for pharmaceuticals11.Introduction1252.Glossary1263.Personnel1284.Premises and facilities1285.Storage requirements1316.Returned goods1337.Dispatch and transport1338.Product recall134References134 Bibliography134 Appendix136 Storage and labelling conditions1.IntroductionThis guide is intended for those involved in the storage, transpor-tation and distribution of pharmaceuticals. It is closely linked to other existing guides recommended by the WHO Expert Committee on Specifications for Pharmaceutical Preparations, such as:•Good trade and distribution practice (GTDP) of pharmaceutical starting materials (1);•The stability testing of pharmaceutical products containing well-established drug substances in conventional dosage forms (information given in connection with regulation for marketing authorization) (2);•Good manufacturing practices (GMP) (3);1This guidance has been prepared in close collaboration with the InternationalPharmaceutical Federation (FIP).125•The cold chain, especially for vaccines and biologicals;•The International Pharmacopoeia (4).The objective of this guide is to supplement the above-mentioned documents by describing the special measures considered appropriate for the storage and transportation of pharmaceuticals. However, they may be adapted to meet individual needs where necessary, provided that the desired standards of quality are still achieved.The guidelines are applicable not only to manufacturers of medicinal products but also to pharmaceutical importers, contractors and wholesalers, and community and hospital pharmacies. They should be adjusted in line with the type of activity where the storage of pharma-ceuticals is taking place. National or regional regulations should be followed for all related activities.2.GlossaryThe definitions given below of some of the terms used in this docu-ment take into account the terminology of current regulations and recommendations.active pharmaceutical ingredient (API)Any substance or mixture of substances intended to be used in the manufacture of a pharmaceutical dosage form and that, when used in the production of a drug, becomes an active ingredient of that drug.Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease, or to affect the structure and function of the body.contaminationThe undesired introduction of impurities of a chemical or microbio-logical nature, or of foreign matter, into or onto a starting material, or intermediate or finished product during production, sampling, pack-aging or repackaging, storage or transport.cross-contaminationContamination of a starting material, intermediate product or fin-ished product with another starting material or product during production.excipientA substance, other than the active ingredient, which has been ap-propriately evaluated for safety and is included in a drug delivery system to:126—aid in the processing of the drug delivery system during its manufacture;—protect, support or enhance stability, bioavailability, or patient acceptability;—assist in product identification; or—enhance any other attribute of the overall safety and effectiveness of the drug during storage or use.expiry dateThe date given on the individual container (usually on the label) of a drug product up to and including which the product is expected to remain within specifications, if stored correctly. It is established for each batch by adding the shelf-life to the date of manufacture. labellingThe action involving the selection of the correct label, with the required information, followed by line clearance and application of the label.manufactureAll operations of purchase of materials and products, production, quality control, release, storage and distribution of finished products, and the related controls.materialA general term used to denote starting materials (active pharmaceu-tical ingredients and excipients), reagents, solvents, process aids, intermediates, packaging materials and labelling materials. packaging materialAny material, including printed material, employed in the packaging of a pharmaceutical product, but excluding any outer packaging used for transportation or shipment. Packaging materials are referred to as primary or secondary according to whether or not they are intended to be in direct contact with the product.pharmaceutical productAny medicine intended for human use or veterinary product admin-istered to food-producing animals, presented in its finished dosage form or as a starting material for use in such a dosage form, that is subject to control by pharmaceutical legislation in both the exporting state and the importing state.127productionAll operations involved in the preparation of a pharmaceutical prod-uct, from receipt of materials, through processing, packaging and repackaging, labelling and relabelling, to completion of the finished product.retest dateThe date when a material should be re-examined to ensure that it is still suitable for use.storageThe storing of pharmaceutical products and materials up to their point of use.supplierA person providing pharmaceutical products and materials on re-quest. Suppliers may be agents, brokers, distributors, manufacturers or traders. Where possible, suppliers should be authorized by a com-petent authority.3.Personnel3.1 At each storage site (e.g. that of a manufacturer, distributor,wholesaler, community or hospital pharmacy) there should be an adequate number of qualified personnel to achieve pharmaceutical quality assurance objectives. National regulations on qualifications should be followed.3.2 All personnel should receive proper training in relation to goodstorage practice, regulations, procedures and safety.3.3 All members of staff should be trained in, and observe high levelsof, personal hygiene and sanitation.3.4 Personnel employed in storage areas should wear suitable protec-tive or working garments appropriate for the activities they perform.4.Premises and facilitiesStorage areas4.1 Precautions must be taken to prevent unauthorized persons fromentering storage areas.4.2 Storage areas should be of sufficient capacity to allow the orderlystorage of the various categories of materials and products, namely starting and packaging materials, intermediates, bulk and finished products, products in quarantine, and released, rejected, returned or recalled products.1284.3 Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits. Where special storage conditions are required on the label (e.g. temperature, rela-tive humidity), these should be provided, checked, monitored and recorded. Materials and pharmaceutical products should be stored off the floor and suitably spaced to permit cleaning and inspection. Pallets should be kept in a good state of cleanliness and repair.4.4 Storage areas should be clean, and free from accumulated waste and vermin. A written sanitation programme should be available indicating the frequency of cleaning and the methods to be used to clean the premises and storage areas. There should also be a written programme for pest control. The pest-control agents used should be safe, and there should be no risk of contamination of the materials and pharmaceutical products. There should be appropriate proce-dures for the clean up of any spillage to ensure complete removal of any risk of contamination.4.5 Receiving and dispatch bays should protect materials and prod-ucts from the weather. Reception areas should be designed and equipped to allow containers of incoming materials and pharmaceuti-cal products to be cleaned, if necessary, before storage.4.6 Where quarantine status is ensured by storage in separate areas, these areas must be clearly marked and their access restricted to authorized personnel. Any system replacing physical quarantine should provide equivalent security. For example, computerized sys-tems can be used, provided that they are validated to demonstrate security of access.4.7 There should normally be a separate sampling area for starting materials in a controlled environment. If sampling is performed in the storage area, it should be conducted in such a way as to prevent contamination or cross-contamination. Adequate cleaning proce-dures should be in place for the sampling areas.4.8 Physical or other equivalent validated (e.g. electronic) segregation should be provided for the storage of rejected, expired, recalled or returned materials or products. The materials or products, and areas concerned should be appropriately identified.4.9 Highly active and radioactive materials, narcotics and other hazardous, sensitive and/or dangerous materials and pharmaceutical products, as well as substances presenting special risks of abuse, fire or explosion, (e.g. combustible liquids and solids and pressurized129gases) should be stored in a dedicated area that is subject to appropri-ate additional safety and security measures.4.10 Materials and pharmaceutical products should be handled anddistributed according to GMP as defined in this document.4.11 Materials and pharmaceutical products should be handled andstored in such a manner as to prevent contamination, mix-ups and cross-contamination.4.12 Materials and pharmaceutical products should be stored in con-ditions which assure that their quality is maintained, and stock should be appropriately rotated. The “first expired/first out” (FEFO) prin-ciple should be followed.4.13 Rejected materials and pharmaceutical products should be iden-tified and controlled under a quarantine system designed to prevent their use until a final decision is taken on their fate.4.14 Narcotic drugs should be stored in compliance with internationalconventions, and national laws and regulations on narcotics.4.15 Broken or damaged items should be withdrawn from usablestock and separated.4.16 Storage areas should provide adequate lighting to enable alloperations to be carried out accurately and safely.Storage conditions4.17 Storage conditions for pharmaceutical products and materialsshould be in compliance with the labelling, which is based on the results of stability testing (see Appendix).Monitoring of storage conditions4.18 Recorded temperature monitoring data should be available forreview. The equipment used for monitoring should be checked at suitable predetermined intervals and the results of such checks should be recorded and retained. All monitoring records should be kept for at least the shelf-life of the stored material or product plus 1 year, or as required by national legislation. Temperature mapping should show uniformity of the temperature across the storage facility. It is recommended that temperature monitors be located in areas that are most likely to show fluctuations.4.19 Equipment used for monitoring should also be calibrated atdefined intervals.1305.Storage requirementsDocumentation: written instructions and records5.1 Written instructions and records should be available which docu-ment all activities in the storage areas including the handling of ex-pired stock. These should adequately describe the storage procedures and define the route of materials and pharmaceutical products and information through the organization in the event of a product recall being required.5.2 Permanent information, written or electronic, should exist foreach stored material or product indicating recommended storage con-ditions, any precautions to be observed and retest dates. Pharmaco-poeial requirements and current national regulations concerning labels and containers should be respected at all times.5.3 Records should be kept for each delivery. They should include thedescription of the goods, quality, quantity, supplier, supplier’s batch number, the date of receipt, assigned batch number and the expiry date. Where national regulations prescribe that records must be re-tained for a certain period, this must be observed. (Otherwise such records should be retained for a period equal to the shelf-life of the incoming materials and products, where applicable, plus 1 year).5.4 Comprehensive records should be maintained showing all receiptsand issues of materials and pharmaceutical products according to a specified system, e.g. by batch number.Labelling and containers5.5 All materials and pharmaceutical products should be stored incontainers which do not adversely affect the quality of the materials or products concerned, and which offer adequate protection from external influences. In some circumstances, this could include bacte-rial contamination.5.6 All containers should be clearly labelled with at least the name ofthe material, the batch number, the expiry date or retest date, the specified storage conditions and reference to the pharmacopoeia, where applicable. Unauthorized abbreviations, names or codes should not be used.Receipt of incoming materials and pharmaceutical products5.7 On receipt, each incoming delivery should be checked against therelevant purchase order and each container physically verified, e.g. by the label description, batch number, type of material or pharmaceuti-cal product and quantity.1315.8 The consignment should be examined for uniformity of the con-tainers and, if necessary, should be subdivided according to the supplier’s batch number should the delivery comprise more than one batch.5.9 Each container should be carefully inspected for possible contami-nation, tampering and damage, and any suspect containers or, if necessary, the entire delivery should be quarantined for further investigation.5.10 When required, samples should be taken only by appropriatelytrained and qualified personnel and in strict accordance with written sampling instructions. Containers from which samples have been taken should be labelled accordingly.5.11 Following sampling, the goods should be subject to quarantine.Batch segregation should be maintained during quarantine and all subsequent storage.5.12 Materials and pharmaceutical products should remain in quaran-tine until an authorized release or rejection is obtained.5.13 Measures should be taken to ensure that rejected materials andpharmaceutical products cannot be used. They should be stored sepa-rately from other materials and pharmaceutical products while await-ing destruction or return to the supplier.Stock rotation and control5.14 Periodic stock reconciliation should be performed by comparingthe actual and recorded stocks.5.15 All significant stock discrepancies should be investigated as acheck against inadvertent mix-ups and/or incorrect issue.5.16 In manufacturing facilities, partly used containers of materialsand pharmaceutical products should be securely reclosed and re-sealed to prevent spoilage and/or contamination during subsequent storage. Materials and pharmaceutical products from containers which have been opened or partly used should be used up before those in unopened containers.5.17 Damaged containers should not be issued unless the quality ofthe material has been shown to be unaffected. Where possible, this should be brought to the attention of the person responsible for quality control. Any action taken should be documented.132Control of obsolete and outdated materials and pharmaceuticalproducts5.18 All stocks should be checked regularly for obsolete and out-dated materials and pharmaceutical products. All due precautions should be observed to prevent the issue of outdated materials and pharmaceutical products.6.Returned goods6.1 Returned goods, including recalled goods, should be handledin accordance with approved procedures and records should be maintained.6.2 All returned goods should be placed in quarantine and returned tosaleable stock only after this has been approved by a nominated, responsible person following a satisfactory quality re-evaluation.6.3 Any stock reissued should be so identified and recorded in stockrecords. Pharmaceuticals returned from patients to the pharmacy should not be taken back as stock, but should be destroyed.7.Dispatch and transport7.1 Materials and pharmaceutical products should be transported insuch a way that their integrity is not impaired and that storage condi-tions are maintained.7.2 Special care should be exercised when using dry ice in cold chains.In addition observing to safety precautions, it must be ensured that the materials or product does not come in into contact with dry ice, as this may adversely affect the product quality, e.g. by freezing.7.3 Where appropriate, the use of devices to monitor conditions suchas temperature during transportation is recommended. Monitoring records should be available for review.7.4 The dispatch and transport of materials and pharmaceutical prod-ucts should be carried out only after receipt of a delivery order. The receipt of the delivery order and the dispatch of the goods must be documented.7.5 Dispatch procedures should be established and documented, tak-ing into account the nature of the materials and pharmaceutical prod-ucts concerned and any special precautions that might be required.7.6 The outside container should offer adequate protection from allexternal influences and should be indelibly and clearly labelled.7.7 Records for dispatch should be retained, stating at least:133—the date of dispatch;—the customer’s name and address;—the product description, e.g. name, dosage form and strength (if appropriate), batch number and quantify;—the transport and storage conditions.7.8 All records should be readily accessible and available on request.8.Product recall8.1 There should be a procedure to recall from the market, promptlyand effectively, pharmaceutical products and materials known or sus-pected to be defective.References1.Good trade and distribution practice (GTDP) of pharmaceutical startingmaterials. Geneva, World Health Organization, 2002 (unpublished documentQAS/01.014; available on request from Essential Drugs and Medicines Policy,World Health Organization, 1211 Geneva 27, Switzerland).2.WHO Expert Committee on Specifications for Pharmaceutical Preparations.Thirty-fourth report. Geneva, World Health Organization, 1996 (WHOTechnical Report Series, No. 863).3.Good manufacturing practices for pharmaceutical products. In: Qualityassurance of pharmaceuticals. A compendium of guidelines and relatedmaterials. Volume 2. Good manufacturing practices and inspection. Geneva,World Health Organization, 1999; WHO Expert Committee on Specificationsfor Pharmaceutical Preparations. Thirty-Fifth report. Geneva, World HealthOrganization, 1999 (WHO Technical Report Series, No. 885); WHO ExpertCommittee on Specifications for Pharmaceutical Preparations. Thirty-Sixthreport. Geneva, World Health Organization, 2002 (WHO Technical ReportSeries, No. 902)4.The international pharmacopoeia, 3rd ed. Vol. 1: General methods ofanalysis; Vol. 2: Quality specifications; Vol. 3: Quality specifications; Vol. 4:Tests, methods, and general requirements. Quality specifications forpharmaceutical substances, excipients, and dosage forms; Volume 5: Testand general requirements for dosage forms. Quality specifications forpharmaceutical substances and tablets (in press). Geneva, World HealthOrganization, 1979–2002.BibliographyQuality assurance of pharmaceuticals. A compendium of guidelines and relatedmaterials. Volume 1. Geneva, World Health Organization, 1997.Quality assurance of pharmaceuticals. A compendium of guidelines and relatedmaterials. Volume 2. Good manufacturing practices and inspection. Geneva,World Health Organization, 1999.Good storage practice: Joint report of the Committee for Official Laboratoriesand Medicinal Control Services and the Industrial Pharmacists Section of the 134International Pharmaceutical Federation (FIP). Pharm. Ind., 1980, 42:1082–1085.Management of drug purchasing, storage and distribution. Manual for developing countries. Geneva, World Health Organization, 1992.135AppendixStorage and labelling conditions2Normal storage conditionsStorage in dry, well-ventilated premises at temperatures of 15–25°C or, depending on climatic conditions, up to 30°C. Extraneous odours, other indications of contamination, and intense light must be excluded.Defined storage instructionsDrug products that must be stored under defined conditions require appropriate storage instructions. Unless otherwise specifically stated(e.g. continuous maintenance of cold storage) deviation may be toler-ated only during short-term interruptions, for example, during local transportation.The use of the following labelling instructions are recommended:On the label Means“Do not store over 30°C”from +2°C to +30°C“Do not store over 25°C”from +2°C to +25°C“Do not store over 15°C”from +2°C to +15°C“Do not store over 8°C”from +2°C to +8°C“Do not store below 8°C”from +8°C to +25°C“Protect from moisture”no more than 60% relative humidityin normal storage conditions; to beprovided to the patient in a moisture-resistant container.“Protect from light”to be provided to the patient in alight-resistant container.2The text was adopted by the WHO Expert Committee on Specifications forPharmaceutical Preparations at its 34th meeting (WHO Expert Committee onSpecifications for Pharmaceutical Preparations. Thirty-Fourth report. Geneva, WorldHealth Organization, 1996, Annex 5 (WHO Technical Report Series No. 863).136。
SensiMix

Shipping: On Dry/Blue Ice Catalog Numbers Batch No.: See vial Concentration: See vial QT605-05: 500 x 50 μL reactions: 10 x 1.25 mL Storage and Stability:SensiMix SYBR ®Hi --20 °C upon receipt. Excessive freeze/thawing is not recommended. Expiry:When stored under the recommended conditions and handled correctly, full activity of the kit is retained until the expiry date on the outer box label.Quality Control:SensiMix SYBR ®Hi -ROX Kit and its components are extensively tested for activity,processivity, efficiency, heat activation, sensitivity, absence of nuclease contamination andabsence of nucleic acid contamination prior to release.Safety Precautions:Please refer to the material safety data sheet for further information.Notes:For research or further manufacturing use only. Trademarks:SensiMix, SensiFAST (Bioline Reagents Ltd), SYBR (Molecular Probes), ROX, LightCycler (Roche), StepOne (ABI), RotorGene (Qiagen), LightCycler (Roche).Kit componentsStore at –20 °CThe SensiMix SYBR ® Hi -ROX Kit has been optimized for use in SYBR ® Green -based qPCR on the real -time instruments listed in the following compatibility table, each of these instruments having the capacity to analyze the qPCR data with the passive reference signal either on or off. The kit is also compatible with several instruments that do not require the use of ROX, such as the BMS Mic, Qiagen Rotor -Gene ™ 6000, Bio -Rad CFX96 or Roche LightCycler ® 480.DescriptionThe SensiMix ™ SYBR ® Hi -ROX Kit is a high -performance reagent designed for superior sensitivity and specificity on various real -time instruments. The SensiMix SYBR ® Hi -ROX Kit employs a hot -start DNA polymerase, for high PCR specificity and sensitivity. SensiMix SYBR ® Hi -ROX is inactivated and possesses no polymerase activity during the reaction set -up, preventing non -specific amplification including primer -dimer formation.For ease -of -use and added convenience, SensiMix SYBR ® Hi -ROX is provided as a 2x master mix containing all the components necessary for real -time PCR (qPCR), including the SYBR ® Green I dye, dNTPs, stabilizers and ROX for optional use. As a ready -to -use premix, only primers and template need to be added. Kit compatibilityPrimers: the sequence and concentration of primer as well as the amplicon length can be critical for specific amplification, yield and overall efficiency of any qPCR. We strongly recommend taking the following into consideration when designing and running your PCR reaction:• use primer -design software, such as Primer3 or visual OMP TM (/primer3/ and DNA Software, Inc ; / respectively). Primers should have a melting temperature (Tm) of approximately 60 °C• optimal amplicon length should be 50-150 bp• a final primer concentration of 250 nM is suitable for most PCR conditions, however to determine the optimal concentration we recommend a primer titration in the range of 0.1–1 μM• use equimolar primer concentrations• when amplifying from cDNA use gene -specific primers. If possible use intron -spanning primers to avoid amplification from genomic DNATemplate: it is important that the DNA template is suitable for use in PCR in terms of purity and concentration. Also, the template needs to be devoid of any contaminating PCR inhibitors (e.g. EDTA). The recommended amount of template for PCR is dependent upon the type of DNA used. The following should be considered when using genomic DNA and cDNA templates:• Genomic DNA: use up to 1 μg of complex (e.g. eukaryotic) genomic DNA in a single PCR. We recommend using the ISOLATE Genomic II DNA Mini Kit (BIO -52066) for high yield and purity from both prokaryotic and eukaryotic sources• cDNA: the optimal amount of cDNA to use in a single PCR is dependent upon the copy number of the target gene. We suggest using 100 ng cDNA per reaction, however it may be necessary to vary this amount. To perform a two -step RT -PCR, we recommend using the SensiFAST cDNA Synthesis Kit (BIO -65053) for reverse transcription of the purified RNA. For high yield and purity of RNA, use the ISOLATE II RNA Mini Kit (BIO -52072)General considerationsTo help prevent any carry -over DNA contamination we recommend that separate areas be maintained for PCR set -up, PCR amplification and any post -PCR gel analysis. It is essential that any amplified PCR product should not be opened in the PCR set -up area.Website:/sensimixemail:****************************PI -50231 V11__________________________________________________________________________________________________________________________LICENSING INFORMATION2) Purchase of this product conveys a licence from Life Technologies to use this SYBR ® containing reagent in an end -user RUO assay. Parties wishing to incorporate this SYBR ® containing reagent into a downstream kit, should contact Life Technologies for SYBR ® Licencing information.Website:/sensimixemail:****************************Associated ProductsBioline Reagents Ltd UNITED KINGDOMTel: +44 (0)20 8830 5300 Fax: +44 (0)20 8452 2822Meridian Life Science Inc. USATel: +1 901 382 8716 Fax: +1 901.382.0027Bioline GmbH GERMANYTel: +49(0)3371 60222 00 Fax: +49(0)3371 60222 01Bioline (Aust) Pty. Ltd AUSTRALIATel: +61 (0)2 9209 4180 Fax: +61 (0)2 9209 4763Technical SupportIf the troubleshooting guide does not solve the difficulty you are experiencing, please contact Technical Support with details of reaction setup, cycling conditions and relevant data. Email: ********************************MgCl 2: The MgCl 2 concentration in the 1x reaction mix is 3 mM. In the majority of qPCR conditions this is optimal for both the reverse transcriptase and the hot -start DNA polymerase. If necessary, we suggest titrating the MgCl 2 to a maximum of 5mM.PCR controls: It is important to detect the presence of contaminating DNA that may affect the reliability of the data. Always include a no template control (NTC), replacing the template with PCR -grade water. When performing a two -step RT -qPCR, set -up a no RT control as the NTC for the PCR.ProcedureReaction mix composition: Prepare a PCR master mix. The volumes given below are based on a standard 50 μL final reaction mix and can be scaled accordingly.Optional ROX: The SensiMix ™SYBR Hi -ROX Kit is premixed with ROX (5-carboxy -X -rhodamine, succinymidyl ester), so that where necessary, ROX fluorescence can be optionally detected on certain real -time instruments. If your real -time instrument has the capability of using ROX and you wish to use this option, then this option must be selected by the user in the software.Troubleshooting GuideSuggested thermal cycling conditionsThe PCR conditions described below are suitable for the SensiMix ™ SYBR ® Hi -ROX Kit for the majority of amplicons and real -time PCR instruments. However, the cycling conditions can be varied to suit customer or machine -specific protocols. The critical step of the PCR is the 10 minute initial activation at 95 °C. The detection channel on the real -time instrument should be set to (SYBR ®) Green or FAM.*Non -variable parameterOptional analysis:After the reaction has reached completion refer to the instrument instructions for the option of melt -profile analysis.Website:/sensimixemail:****************************Website:/sensimixemail:****************************Troubleshooting Guide (Continued)。
欧盟ANNEX1Manufact...

(The words that are in the green background are new standards)(绿色背景下的内容为新标准)ANNEX 1MANUFACTURE OF STERILE MEDICINAL PRODUCTS附录1 无菌医药产品的生产Principle总则The manufacture of sterile products is subject to special requirements in order to minimize risks of microbiological contamination, and of particulate and pyrogen contamination. Much depends on the skill, training and attitudes of the personnel involved. Quality Assurance is particularly important, and this type of manufacture must strictly follow carefully established and validated methods of preparation and procedure. Sole reliance for sterility or other quality aspects must not be placed on any terminal process or finished product test.无菌药品的生产,必须符合一些特殊的要求,以防止微生物、微粒和热源的污染。
这很大程度上依赖与工作人员的技术水平、培训和工作态度。
在这方面质量保证显得特别重要,这种类型的生产,必须严格按照完善的和经过验证的生产方法和工作程序。
仅靠产品的最终灭菌和某一方面的质量控制是不允许的。
Annexin V凋亡检测的阳性质控液及其设备制作方法与设计方案

本技术公开了一种Annexin V凋亡检测的阳性质控液及其制备方法,应用于Annexin V或PI/7AAD单染,在正常收集的细胞中加入适量的阳性质控液,冰上孵育30分钟,即可使细胞死亡,再与等量的活细胞混合,可形成比例接近的活细胞和死细胞,Annexin V或PI/7AAD单染时,可检测到比例接近的阳性细胞群和阴性细胞群,可非常方便地用于仪器的补偿调节。
操作简单、重复性好、方便补偿调节、保证结果的可靠性。
权利要求书1.一种Annexin V凋亡检测的阳性质控液,其特征在于,包括以下成分和配方:高氯酸钠,一水,终浓度:0.1M-0.2M;柠檬酸三钠,二水,终浓度:0.05M-0.1M;氯化镁,六水,终浓度:0.05M-0.1M;氯化钙,二水,终浓度:0.05M-0.1M;甘油,终浓度:5%-15%;异丁醇,终浓度:0.5%-1%;甲醛,终浓度:1%-4%;其余为水;阳性质控液的PH为:7.0-7.4。
2.根据权利要求1所述一种Annexin V凋亡检测的阳性质控液,其特征在于,包括以下成分和配方:高氯酸钠,一水,终浓度:0.15M;柠檬酸三钠,二水,终浓度:0.05M;氯化镁,六水,终浓度:0.05M;氯化钙,二水,终浓度:0.05M;甘油,终浓度:10%;异丁醇,终浓度:0.92%;甲醛,终浓度:4.04%;其余为水;阳性质控液的PH为:7。
3.根据权利要求1所述一种Annexin V凋亡检测的阳性质控液的制备方法,其特征在于,包括以下步骤:步骤一:准确称量高氯酸钠,一水、柠檬酸三钠,二水、氯化镁,六水和氯化钙,二水至烧杯中,加入超纯水,搅拌混匀,充分溶解;步骤二:准确量取甘油、异丁醇和甲醛,加入上述溶液中,搅拌混匀,放在通风橱中过夜;步骤三:第二天用盐酸调节溶液pH值至(7.0),定容至配制体积,搅拌混匀。
技术说明书一种Annexin V凋亡检测的阳性质控液及其制备方法技术领域本技术涉及一种Annexin V凋亡检测的阳性质控液及其制备方法。
EU GMP annex 15 验证 201504 中英文 完

EUROPEAN COMMISSIONENTERPRISE DIRECTORATE-GENERALSingle market, regulatory environment, industries under vertical legislationPharmaceuticals and cosmeticsBrussels, 30 March 2015EudraLexVolume 4EU Guidelines forGood Manufacturing Practice forMedicinal Products for Human and Veterinary UseAnnex 15: Qualification and ValidationEU GMP附录15:确认和验证Legal basis for publishing the detailed guidelines: Article 47 of Directive 2001/83/EC on the Community code relating to medicinal products for human use and Article 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinal products as laid down in Directive 2003/94/EC for medicinal products for human use and Directive91/412/EEC for veterinary use.公布详细指南的法律依据:指令2001/83/EC第47款关于人药共同体代码,和2001/82/EC第51款关于兽药共同体代码的要求。
EU-GMP-Part-II-Annexes-15.pdf

EUROPEAN COMMISSIONENTERPRISE DIRECTORATE-GENERALSingle market, regulatory environment, industries under vertical legislationPharmaceuticals and cosmeticsBrussels, July 2001Working Party on Control of Medicines and InspectionsFinal Version of Annex 15 to the EU Guide toGood Manufacturing PracticeTitle: Qualification and validation.First discussion in drafting groupDiscussion at the working Party on Control of Medicines and16 September 1999Inspection for release for consultationPharmaceutical Committee28 September 1999Released for consultation30 October 1999Deadline for comments28 February 2000 Final approval by Inspector’s working party December 2000Pharmaceutical Committee (for information)April 2001 Date for coming into operation September 2001Note that this document is based in the PICS/S recommendations2 Table of ContentsPage1. Qualification and Validation32. Planning for Validation43. Documentation44. Qualification55. Processs Validation.66. Cleaning Validation77. Change Control88. Revalidation99. Glossary 10QUALIFICATION AND VALIDATIONPrinciple1.This Annex describes the principles of qualification and validation which areapplicable to the manufacture of medicinal products. It is a requirement ofGMP that manufacturers identify what validation work is needed to provecontrol of the critical aspects of their particular operations. Significantchanges to the facilities, the equipment and the processes, which may affectthe quality of the product, should be validated. A risk assessment approachshould be used to determine the scope and extent of validation.PLANNING FOR VALIDATION2.All validation activities should be planned. The key elements of a validationprogramme should be clearly defined and documented in a validation masterplan (VMP) or equivalent documents.3.The VMP should be a summary document which is brief, concise and clear.4.The VMP should contain data on at least the following:(a)validation policy;(b)organisational structure of validation activities;(c)summary of facilities, systems, equipment and processes to be validated;(d)documentation format: the format to be used for protocols and reports;(e)planning and scheduling;(f)change control;(g)reference to existing documents.5.In case of large projects, it may be necessary to create separate validationmaster plans.DOCUMENTATION6. A written protocol should be established that specifies how qualification andvalidation will be conducted. The protocol should be reviewed and approved.The protocol should specify critical steps and acceptance criteria.7. A report that cross-references the qualification and/or validation protocolshould be prepared, summarising the results obtained, commenting on anydeviations observed, and drawing the necessary conclusions, including rec-ommending changes necessary to correct deficiencies. Any changes to theplan as defined in the protocol should be documented with appropriate justi-fication.8.After completion of a satisfactory qualification, a formal release for the nextstep in qualification and validation should be made as a written authorisation.QUALIFICATIONDesign qualification9.The first element of the validation of new facilities, systems or equipmentcould be design qualification (DQ).10.The compliance of the design with GMP should be demonstrated anddocumented.Installation qualification11. Installation qualification (IQ) should be performed on new or modifiedfacilities, systems and equipment.12. IQ should include, but not be limited to the following:(a)installation of equipment, piping, services and instrumentation checked tocurrent engineering drawings and specifications;(b)collection and collation of supplier operating and working instructions andmaintenance requirements;(c)calibration requirements;(d)verification of materials of construction.Operational qualification13. Operational qualification (OQ) should follow Installation qualification.14. OQ should include, but not be limited to the following:(a)tests that have been developed from knowledge of processes, systems andequipment;(b)tests to include a condition or a set of conditions encompassing upper andlower operating limits, sometimes referred to as “worst case” conditions.15.The completion of a successful Operational qualification should allow thefinalisation of calibration, operating and cleaning procedures, operatortraining and preventative maintenance requirements. It should permit aformal "release" of the facilities, systems and equipment.Performance qualification16. Performance qualification (PQ) should follow successful completion ofInstallation qualification and Operational qualification.17. PQ should include, but not be limited to the following:(a)tests, using production materials, qualified substitutes or simulated product,that have been developed from knowledge of the process and the facilities,systems or equipment;(b)tests to include a condition or set of conditions encompassing upper andlower operating limits.18. Although PQ is described as a separate activity, it may in some cases beappropriate to perform it in conjunction with OQ.Qualification of established (in-use) facilities, systems and equipment19. Evidence should be available to support and verify the operating parametersand limits for the critical variables of the operating equipment. Additionally,the calibration, cleaning, preventative maintenance, operating procedures andoperator training procedures and records should be documented.PROCESS VALIDATIONGeneral20. The requirements and principles outlined in this chapter are applicable to themanufacture of pharmaceutical dosage forms. They cover the initialvalidation of new processes, subsequent validation of modified processes andre-validation.21. Process validation should normally be completed prior to the distributionand sale of the medicinal product (prospective validation). In exceptionalcircumstances, where this is not possible, it may be necessary to validateprocesses during routine production (concurrent validation). Processes inuse for some time should also be validated (retrospective validation).22. Facilities, systems and equipment to be used should have been qualified andanalytical testing methods should be validated. Staff taking part in the vali-dation work should have been appropriately trained.23. Facilities, systems, equipment and processes should be periodically evalu-ated to verify that they are still operating in a valid manner.Prospective validation24. Prospective validation should include, but not be limited to the following:(a)short description of the process;(b)summary of the critical processing steps to be investigated;(c)list of the equipment/facilities to be used (including measur-ing/monitoring/recording equipment) together with its calibration status(d)finished product specifications for release;(e)list of analytical methods, as appropriate;(f)proposed in-process controls with acceptance criteria;(g)additional testing to be carried out, with acceptance criteria and analyticalvalidation, as appropriate;(h)sampling plan;(i)methods for recording and evaluating results(j)functions and responsibilities;(k)proposed timetable.25. Using this defined process (including specified components) a series ofbatches of the final product may be produced under routine conditions. Intheory the number of process runs carried out and observations made shouldbe sufficient to allow the normal extent of variation and trends to be estab-lished and to provide sufficient data for evaluation. It is generally consid-ered acceptable that three consecutive batches/runs within the finally agreedparameters, would constitute a validation of the process.26. Batches made for process validation should be the same size as the intendedindustrial scale batches.27. If it is intended that validation batches be sold or supplied, the conditionsunder which they are produced should comply fully with the requirements ofGood Manufacturing Practice, including the satisfactory outcome of the vali-dation exercise, and with the marketing authorisation.Concurrent validation28. In exceptional circumstances it may be acceptable not to complete a valida-tion programme before routine production starts.29. The decision to carry out concurrent validation must be justified, docu-mented and approved by authorised personnel.30. Documentation requirements for concurrent validation are the same asspecified for prospective validation.Retrospective validation31. Retrospective validation is only acceptable for well-established processesand will be inappropriate where there have been recent changes in the com-position of the product, operating procedures or equipment.32. Validation of such processes should be based on historical data. The stepsinvolved require the preparation of a specific protocol and the reporting ofthe results of the data review, leading to a conclusion and a recommendation.33. The source of data for this validation should include, but not be limited tobatch processing and packaging records, process control charts, maintenancelog books, records of personnel changes, process capability studies, finishedproduct data, including trend cards and storage stability results.34. Batches selected for retrospective validation should be representative of allbatches made during the review period, including any batches that failed tomeet specifications, and should be sufficient in number to demonstrate proc-ess consistency. Additional testing of retained samples may be needed toobtain the necessary amount or type of data to retrospectively validate theprocess.35. For retrospective validation, generally data from ten to thirty consecutivebatches should be examined to assess process consistency, but fewer batchesmay be examined if justified.CLEANING VALIDATION36. Cleaning validation should be performed in order to confirm the effective-ness of a cleaning procedure. The rationale for selecting limits of carry overof product residues, cleaning agents and microbial contamination should belogically based on the materials involved. The limits should be achievableand verifiable.37. Validated analytical methods having sensitivity to detect residues or con-taminants should be used. The detection limit for each analytical methodshould be sufficiently sensitive to detect the established acceptable level ofthe residue or contaminant.38. Normally only cleaning procedures for product contact surfaces of theequipment need to be validated. Consideration should be given to non-contact parts. The intervals between use and cleaning as well as cleaning andreuse should be validated. Cleaning intervals and methods should be deter-mined.39. For cleaning procedures for products and processes which are similar, it isconsidered acceptable to select a representative range of similar products andprocesses. A single validation study utilising a “worst case” approach canbe carried out which takes account of the critical issues.40. Typically three consecutive applications of the cleaning procedure should beperformed and shown to be successful in order to prove that the method isvalidated.41. "Test until clean". is not considered an appropriate alternative to cleaningvalidation.42. Products which simulate the physicochemical properties of the substances tobe removed may exceptionally be used instead of the substances themselves,where such substances are either toxic or hazardous.CHANGE CONTROL43. Written procedures should be in place to describe the actions to be taken if achange is proposed to a starting material, product component, processequipment, process environment (or site), method of production or testing orany other change that may affect product quality or reproducibility of theprocess. Change control procedures should ensure that sufficient supportingdata are generated to demonstrate that the revised process will result in aproduct of the desired quality, consistent with the approved specifications.44. All changes that may affect product quality or reproducibility of the processshould be formally requested, documented and accepted. The likely impactof the change of facilities, systems and equipment on the product should beevaluated, including risk analysis. The need for, and the extent of, re-qualification and re-validation should be determined.REVALIDATION45. Facilities, systems, equipment and processes, including cleaning, should beperiodically evaluated to confirm that they remain valid. Where no signifi-cant changes have been made to the validated status, a review with evidencethat facilities, systems, equipment and processes meet the prescribed re-quirements fulfils the need for revalidation.GLOSSARYDefinitions of terms relating to qualification and validation which are not given in the glossary of the current EC Guide to GMP, but which are used in this Annex, are given below.Change ControlA formal system by which qualified representatives of appropriate disciplines review proposed or actual changes that might affect the validated status of facilities, systems, equipment or processes. The intent is to determine the need for action that would en-sure and document that the system is maintained in a validated state.Cleaning ValidationCleaning validation is documented evidence that an approved cleaning procedure will provide equipment which is suitable for processing medicinal products.Concurrent ValidationValidation carried out during routine production of products intended for sale.Design qualification (DQ)The documented verification that the proposed design of the facilities, systems and equipment is suitable for the intended purpose.Installation Qualification (IQ)The documented verification that the facilities, systems and equipment, as installed or modified, comply with the approved design and the manufacturer’s recommendations. Operational Qualification (OQ)The documented verification that the facilities, systems and equipment, as installed or modified, perform as intended throughout the anticipated operating ranges. Performance Qualification (PQ)The documented verification that the facilities, systems and equipment, as connected together, can perform effectively and reproducibly, based on the approved process method and product specification.Process ValidationThe documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its prede-termined specifications and quality attributes.11 Prospective ValidationValidation carried out before routine production of products intended for sale. Retrospective ValidationValidation of a process for a product which has been marketed based upon accumulated manufacturing, testing and control batch data.Re-ValidationA repeat of the process validation to provide an assurance that changes in the proc-ess/equipment introduced in accordance with change control procedures do not ad-versely affect process characteristics and product quality.Risk analysisMethod to assess and characterise the critical parameters in the functionality of an equipment or process..Simulated ProductA material that closely approximates the physical and, where practical, the chemical characteristics (e.g. viscosity, particle size, pH etc.) of the product under validation. In many cases, these characteristics may be satisfied by a placebo product batch.SystemA group of equipment with a common purpose.Worst CaseA condition or set of conditions encompassing upper and lower processing limits and circumstances, within standard operating procedures, which pose the greatest chance of product or process failure when compared to ideal conditions. Such conditions do not necessarily induce product or process failure.-------------。
欧盟化妆品26种过敏香料目录
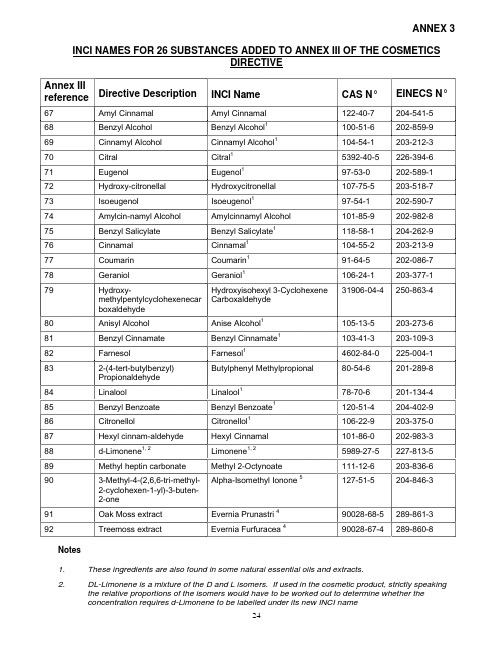
ANNEX 3 INCI NAMES FOR 26 SUBSTANCES ADDED TO ANNEX III OF THE COSMETICS DIRECTIVE Annex III reference67 68 69 70 71 72 73 74 75 76 77 78 79Directive DescriptionAmyl Cinnamal Benzyl Alcohol Cinnamyl Alcohol Citral Eugenol Hydroxy-citronellal Isoeugenol Amylcin-namyl Alcohol Benzyl Salicylate Cinnamal Coumarin Geraniol Hydroxymethylpentylcyclohexenecar boxaldehyde Anisyl Alcohol Benzyl Cinnamate Farnesol 2-(4-tert-butylbenzyl) Propionaldehyde Linalool Benzyl Benzoate Citronellol Hexyl cinnam-aldehyde d-Limonene1, 2INCI NameAmyl Cinnamal Benzyl Alcohol1 1CAS N°122-40-7 100-51-6 104-54-1 5392-40-5 97-53-0 107-75-5 97-54-1 101-85-9 118-58-1 104-55-2 91-64-5 106-24-1 31906-04-4EINECS N°204-541-5 202-859-9 203-212-3 226-394-6 202-589-1 203-518-7 202-590-7 202-982-8 204-262-9 203-213-9 202-086-7 203-377-1 250-863-4Cinnamyl Alcohol1 Citral Eugenol1 Hydroxycitronellal Isoeugenol1 Amylcinnamyl Alcohol Benzyl Salicylate1 Cinnamal1 1Coumarin1 GeraniolHydroxyisohexyl 3-Cyclohexene Carboxaldehyde Anise Alcohol1 Benzyl Cinnamate Farnesol1 180 81 82 83 84 85 86 87 88 89 90105-13-5 103-41-3 4602-84-0 80-54-6 78-70-61203-273-6 203-109-3 225-004-1 201-289-8 201-134-4 204-402-9 203-375-0 202-983-3 227-813-5 203-836-6 204-846-3Butylphenyl Methylpropional Linalool1 Benzyl Benzoate Citronellol1120-51-4 106-22-9 101-86-0 5989-27-5 111-12-65Hexyl Cinnamal Limonene1, 2Methyl heptin carbonate 3-Methyl-4-(2,6,6-tri-methyl2-cyclohexen-1-yl)-3-buten2-one Oak Moss extract Treemoss extract Notes 1. 2.Methyl 2-Octynoate Alpha-Isomethyl Ionone127-51-591 92Evernia Prunastri 4 Evernia Furfuracea 490028-68-5 90028-67-4289-861-3 289-860-8These ingredients are also found in some natural essential oils and extracts. DL-Limonene is a mixture of the D and L isomers. If used in the cosmetic product, strictly speaking the relative proportions of the isomers would have to be worked out to determine whether the concentration requires d-Limonene to be labelled under its new INCI name 24‘Limonene’. In practice, because of the technical difficulty of the analysis, the total level of both isomers will be used to establish whether the threshold is exceeded and labelling is required. 3. The Directive specifies the restrictions for each ingredient as: The presence of the substance must be indicated in the list of ingredients referred to in Article 6(1)(g) when its concentration exceeds: - 0.001 % in leave-on products - 0.01 % in rinse-off products This applies if these ingredients are present in the product for any reason – not just as constituents of fragrances. 4. Evernia Prunastri: as listed in 1996 inventory, we expect this to change to Evernia Prunastri extract in a future update. Evernia Furfuracea: for consistency we have used the format that would have appeared in the 1996 inventory. We expect this to change to Evernia Furfuracea extract in a future update. Alpha-Isomethyl Ionone is the name which appears in the current CTFA On-line listing of the INCI name for 3-Methyl-4-(2,6,6-tri-methyl-2-cyclohexen-1-yl)-3-buten-2-one. Previous hard copy listings omitted ‘iso’ from the name.5.25。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Attachment Form No. ..................... : Attachment Originator ................... : Master Attachment ......................... :
Copyright © 2012 IEC System for Conformity Testing and Certification of Electrical Equipment (IECEE), Geneva, Switzerland. All rights reserved.
Report No.: GZES130900859901
Result – Remark
Verdict P P
The instructions for portable hairdryers shall include the substance of the following: (EN 60335-2-23/A11) - when the hairdryer is used in a bathroom, unplug it after use since the proximity of water presents a hazard even when the hairdryer is switched off (EN 60335-2-23/A11) - for additional protection, the installation of a residual current device (RCD) having a rated residual operating current not exceeding 30 mA is advisable in the electrical circuit supplying the bathroom. Ask your installer for advice. (EN 60335-2-23/A11) The instructions for facial saunas state that after use the appliance should be cleaned to avoid the accumulation of grease and other residues. (EN 60335-2-23/A11) If symbol 60417-5582 (2002-10) is used, together with the prohibition sign, the meaning has to be explained. (EN 60335-2-23/A11) The instructions include the substance of the following: (EN 60335-2-23/A11) WARNING: Do not use this appliance near bathtubs, showers, basins or other vessels containing water. (EN 60335-2-23/A11) 7.12.Z1 The specific instructions related to the safe operation of this appliance is collated together in the front section of the user instructions The height of the characters, measured on the capital letters, is at least 3 mm These instructions are also available in an alternative format, e.g. on a website 7.12.Z101 The specific instructions related to the safe operation of this appliance (as given in 7.12 of this standard) shall be collated together in the front section of the user instructions. The height of the characters, measured on the capital letters, shall be at least 4 mm. (EN 60335-2-23/A11) These instructions shall also be available in an alternative format, e.g. on a website.
— N/A
N/A
N/A
N/A
— P
P
P P P
P
Page 3 of 14 Annex I EN 60335-1, EN 60335-2-23 Clause Requirement – Test These instructions shall also be available in an alternative format, e.g. on a website. The supplying of the instruction in an alternative format is not required for fixed hand dryers for which the operation is simple and clear. 8.1.1 Also test probe 18 of IEC 61032 is applied (EN 60335-2-23/A11) The appliance being in every possible position during the test (EN 60335-2-23/A11) The force on the probe in the straight position is increased to 10 N when probe 18 is used (EN 60335-2-23/A11) When using test probe 18 the appliance is fully assembled as in normal operation without any parts removed, and (EN 60335-2-23/A11) parts intended to be removed for user maintenance are also not removed (EN 60335-2-23/A11) 8.2 Compliance is checked by applying the test probes of IEC 61032 (EN 60335-2-23/A11) For built-in appliances and fixed appliances, the test probe B and probe 18 of EN 61032 are applied only after installation 11.8 In Table 3 delete the row “External enclosure of motor-operated appliancesexcept handles held in normal use". (EN 60335-2-23/A11) Appliances with an automatic cord reel tested with the cord in the most unfavourable position so that the reeling of the wet cord may affect electrical insulation during operation, the cord not being dried before reeling - hand-held appliances are placed on a piece of low density glass-fibre insulation having a coefficient of thermal insulation of approximately 2,5 m² K/W; (EN 60335-2-23/A11) Appliances are fully assembled as in normal operation without any parts removed. (EN 60335-2-23/A11) - a test probe that is similar to test probe B of IEC 61032 but having a circular stop face with a diameter of 50 mm, instead of the non circular face, applied with a force of 5 N and (EN 60335-2-23/A11) - test probe 18 of IEC 61032, applied with a force of 2,5 N. (EN 60335-2-23/A11) 21.101 Appliance is placed on a horizontal surface positioned 700 mm above a rigidly supported hardwood board (EN 60335-2-23/A11 2 of 14 Annex I EN 60335-1, EN 60335-2-23 Clause Requirement – Test - children shall not play with the appliance (EN 60335-1, EN 60335-2-23/A11) - cleaning and user maintenance shall not be made by children without supervision (EN 60335-1, EN 60335-2-23/A11)