Extraction and Purification of Genomic DNA
天津大学制药工程专业 考研《制药工艺学》中英文对译

天津⼤学《制药⼯艺学》中英⽂对译pharmaceutical technology制药⼯工艺学pharmaceutical pipeline制药链pharmacopoeia药典。
Roswell Park Memorial Instirute RPMI good manufacturing practices for drugs GMP制药⾏行行业medicines,drugs药品traditional Chinese medicines中药natural medicines天然药物chemical drugs化学药物biologics,biologic products⽣生物制品generics,generic drugs仿制药物me-too-drug仿制药biosimilars⽣生物类似药biotechnology⽣生物技术.Food and druge administration FDA biotechnology pharmaceutical,biopharmaceutical⽣生物制药nucleotide核苷酸nucleoside核⽢甘enzyme酶enzyme inhibitor酶抑制剂immunomodulator免疫调节剂penicillin⻘青霉素antibody engineering抗体⼯工程inducer诱导剂precursor前体prodrug前药transformation遗传转化.conversion⽣生物转化fermentation发酵.strain breeding菌种选育separation and purification分离纯化和提纯.cell growth phase/fermentationproduct synthesis phase/product secretion phase.Murashige&Skoog MScell autolysis phase/fermentation anaphase.generic通⽤用药物metabolism代谢.substrate培养基质primary/secondary metabolism初级/次级代谢.specific growth rate⽐比⽣生⻓长速率lag/log/decline/stationary/death phase延滞期/对数⽣生⻓长期/减数期/稳定期/死亡期coupling model⽣生⻓长与⽣生产偶联型.PEG聚⼄乙⼆二醇semi-coupling model⽣生⻓长与⽣生产半偶联型.starter culture培养物non-coupling model⽣生⻓长与⽣生产⾮非偶联型.storage保存protoplast fusion原⽣生质体融合.DMSO⼆二甲基亚砜master stock/cell bank MSB/MCB主菌种库.glycerol⽢甘油working stock/cell bank WSB/WCB⼯工作菌种库.Streptomyces链霉菌quality control QC质量量控制.cholramphenicol氯霉素China Center for Type Culture Collection CCTCC中国典型培养物保藏中⼼心China General Microbiological Culture Collection Center CGMCC中国普通微⽣生物保藏管理理中⼼心China Center of Industrial Culture Collection CICC中国⼯工业微⽣生物菌种保藏管理理中⼼心National Center for Medical Culture Collection(Bacteria)CMCC中国医学微⽣生物菌种保藏中⼼心America Type Culture Collection ATCCEuropean Collection of Cell Culture ECACCInstiture for Fermentation,Osaka IFONational Collection of Type Culture mNCTCmedium培养基.carbon source碳源nitrogen source氮源.mineral salt⽆无机盐macroelement⼤大量量元素.trace element microelement微量量元素growth factor⽣生⻓长因⼦子.precursor前体accelerant促进剂.fed medium补料料培养基agar琼脂粉.contaminated microbe杂菌contamination污染.phage噬菌体disinfection消毒.sterilization灭菌pathogen病原微⽣生物.filtration sterilization过滤灭菌filter过滤介质VVM空⽓气流量量(单位时间单位体积内通⼊入的标准状况下的空⽓气体积)primary culture原代培养.passage culture传代培养solid surface culture固体表⾯面培养.liquid submerged culture液体深层培养immobilized culture固定化培养.high cell density culture⾼高密度培养intermittent opration间歇式操作.discontinuous operation不不连续培养semi-continuous operation半连续培养.batch operation分批式操作fed batch operation补料料-分批式(流加)操作.chemostat恒化器器MPa罐压.dissolved oxygen DO溶解氧cell concentration菌体浓度.fermentation heat发酵热production heat产⽣生热.loss heat散失热biological heat⽣生物热.agitation heat搅拌热evaporation heat蒸发热.sensible heat显热radiant heat辐射热.oxygen supply供氧oxygen consumption耗氧.dissolved oxygen coefficient溶解氧系数oxygen transfer rate OTR氧传递速率.oxygen uptake rate OUR摄氧速率ventilation通⽓气.respiratory intensity呼吸强度oxygen saturation concentration氧饱和浓度.respiratory quotient RQ呼吸熵critical oxygen concentration临界氧浓度.fill补料料withdraw放料料.foam泡沫defoaming agent消沫剂.surfactant表⾯面活性剂dispersant分散剂.emulsifier乳化剂inertcarrier惰性载体.antibiotic抗⽣生素carbenicillin羧苄⻘青霉素/⻘青霉素G6-aminopenicillanic acid6-APA6-氨基⻘青霉烷酸.cephalosporin C CPC头孢菌素C erythromycin红霉素.amino acid氨基酸hybridomn杂交瘤.vitamin维⽣生素recombinant DNA technology重组DNA技术.recombinant DNA products rDNA制品plasmid质粒.replicon复制⼦子promoter启动⼦子.terminator终⽌止⼦子multiple cloning site MCS多克隆隆位点.transferability转移性incompatibility不不相容性.cloning vector克隆隆载体expression vector表达载体.shuttle vector穿梭载体intergration vector整合载体.inclusion body包涵体yeast酵⺟母.genetic engineering strain基因⼯工程菌yeast intergration plasmid YIP酵⺟母整合载体yeast episomal plasmid YEP酵⺟母附加载体yeast centromere plasmid YCP酵⺟母着丝粒载体centromere sequence CEN着丝粒序列列autonomously replicating sequences ARS⾃自主复制序列列yeast replicating plasmid YRP酵⺟母复制质粒polymerase chain reaction PCR聚合酶链式反应reverse transcription PCR RT-PCR反转录PCRcomplementary DNA cDNAavian myeloblastosis virus AMV禽源成髓细胞瘤病毒moloney murine leukemia virus MMLV⿏鼠源败⾎血病毒莫勒勒尼株diethyl pyrocarbonate DEPC焦碳酸⼆二⼄乙酯.denaturation变性annelling退⽕火.extension链延伸restriction endonuclease限制性核酸内切酶.ligase连接酶recombinant重组⼦子.interferon IFN⼲干扰素recombinant human interferon rhIFNtricarboxylic acid cycle TCA循环三羧酸循环pentose phosphate pathway PPP磷酸戊糖途径.glycosylation糖基化apoptosis凋亡.diploid cell⼆二倍体primary cell原代细胞.passage cell传代细胞immortal cell永久细胞系.Chinese hamster ovary CHO中国仓⿏鼠卵卵巢DHFR⼆二氢叶酸还原酶.methotrexate MTX甲氨蝶呤baby hamster kidney BHK幼仓⿏鼠肾脏dicistron双顺反⼦子long terminal repeat sequences LTRS逆转录病毒的⻓长末端重复序列列cytomegalovirus CMV⼈人巨噬病毒.ubiquitin泛素蛋⽩白bovine growth hormone.BGH⽜牛⽣生⻓长素.toppoisomerase拓拓扑异构酶internal ribosome entry site IRES核糖体进⼊入位点.serum⾎血清minimum essential medium MEM basal medium Eagle’s BME Dulbecco’s modified Eagle’s medium DMEMGlasgow’s modified Eagle’s medium GMEMJoklik’s Park Memorial Eagle’s medium JMEMRoswell Park Memorial Institute RPMIserum-free medium SEM⽆无⾎血清培养基.buffer solution缓冲液balance saline solution BSS平衡盐溶液monolayer anchorage-dependent culture单层贴壁培养.suspension culture悬浮培养microcarrier微载体.microencapsulation method微囊法phosphonate buffer solution PBS磷酸盐缓冲液.scale-down缩⼩小erythropoietin EPO红细胞⽣生成素.luria bertani LB recombinant human erythropoietin rhEPO重组⼈人红细胞⽣生成素synthon合成⼦子.synthetic equivalent合成等价物protocol solvent质⼦子性溶剂.micronization微晶化catalyst催化剂.phase transfer catalyst相转移催化剂TEBAC三⼄乙基苄基氯化铵Mokosza催化剂TO/CMAC三⾟辛基甲基氯化铵Starks催化剂.Brandstrom催化剂四丁基硫酸氢铵chirality⼿手性.enantiomers对应异构体configuration构型.chiral drug⼿手性药物enantiomeric excesses对映体过量量e.e.%.restrosynthesis追溯求源法resolution拆分.omeprazole奥美拉唑paclitaxel,Taxol紫杉醇.cephalosporin C CPC头孢菌素7-aminocephalosporanic acid7-ACA7-氨基头孢烷酸.cefalexin头孢氨苄tetrahydrofuran THF四氢呋喃.quality by design QbD质量量源于设计process analysis technology PAT过程分析技术quality target product profile QTPP⽬目标产品质量量概况critical material attribute CMA关键物料料属性critical process parameter CPP关键⼯工艺参数normal operation range NOR正常操作区间proven acceptable range PAR可接受的区间critical quality attribute CQA关键质量量属性bioreactor⽣生物反应器器key process parameter KPP重要⼯工艺参数.fermenter发酵罐complete stirred tank reactor CSTR全混流反应器器.yield得率piston fluid reactor PFR平推流反应器器.titer效价stirred tank reactor STR搅拌罐.scale-up放⼤大fixed bed reactor固定化床反应器器.draft tube导流筒packed bed reactor PBR填充床反应器器.bubble column⿎鼓泡塔fluidized bed reactor FBR流化床反应器器.air-lift reactor⽓气升式反应器器disk and turbine impeller涡流式搅拌桨.process validation⼯工艺验证marine style impeller推进式搅拌桨.process design⼯工艺设计process mass intensity PMI过程质量量强度.process qualification⼯工艺确认reaction mass efficiency RME反应质量量效率standard operation procedure SOP标准操作规程.continued process verification⼯工艺核实biochemical oxygen demand BOD⽣生化需氧量量.total nitrogen TN总氮chemical oxygen demand COD化学需氧量量.suspended subatance SS悬浮物mixed liquor suspended solids MLSS混合液悬浮固体total organic carbon TOC总有机碳.sludge volume SV污泥泥沉降⽐比sludge volume index SVI污泥泥指数piping&instrument diagram PID⼯工艺控制流程图。
中国云南及西藏水生环境中的新种——棕孢香港霉(英文)

菌 物 学 报 1275
BAO Dan-Feng et al. /Hongkongmyces brunneisporus sp. nov. (Lindgomycetaceae) from...
摘 要 :在大湄公河次区域的水生真菌调查中Байду номын сангаас从中国云南和西藏的沉水腐木中分离得到4 个菌株。基
于 LSU、SSU、ITS、TEFl-cx和 RPB2 序列进行多基因系统发育分析,表 明 4 个菌株属于菩提科香港霉属真
菌。系统发育分析结果显示4 个菌株聚集在一起,并与泰国香港霉形成姐妹支。基于形态学及分子系统 学研究,将 这 4 个菌株鉴定为新种棕孢香港霉。棕孢香港霉是香港霉属的第二个有性型物种,它因子囊 果的孔口处有棕色至黑色的刚毛,且子囊孢子呈梭形,孢子两端逐渐变窄且钝圆,红棕色至暗棕色,具 有多个隔膜而区别于另一个有性型物种泰国香港霉。本研究提供了该真菌新种的描述及图版并比较了该 种与其他物种的形态差异。 关 键 词 :新 种 ,形 态 学 ,系统 发 育 ,有性 型 ,分类
Supported by the National Natural Science Foundation of China (31860006, 31970021) and Fungal Diversity Conservation and Utilization Innovation Team of Dali University (ZKLX2019213). o Corresponding author. E-mail: suhongyanl6@ Received: 2020-09-23, accepted: 2020-10-29
CTAB结合DNA凝胶回收试剂盒提取食用菌DNA

32生物技术且污染较少[I引。
综合前人研究成果,作者采用间接法提取青贮饲料中的微生物总DNA,使用了CTAB试剂,采用异丙醇沉淀DNA。
吸光度和PcR检测是评价DNA质量常用的手段。
临1,本研究中通过光吸收、琼脂糖电泳及PcR检验,结果表明提取的DNA质量较高。
进一步的DGGE结果表明,所述方法具有较高的涵盖性,对样品中的微生物没有遗漏。
图6目标菌株的扩增结果M:分子量标准;1:添加目标菌株的实验组;2:未添加目标菌株的对照组。
Fig.6A群∞∞egelelectmphon∞isofPCRamplifiedDNAfromlargetstrainM:DNAmarkerD12,000;1:cornstalkwithaddedtariffstrain;2:controlcomslalkwithNolmgetstrain.WhitehouseCA等¨6j通过向处理过的土壤中加入目标菌株Franci.靶llatularensis的方法评价不同商业DNA抽提试剂盒的抽提灵敏度,结果最好的两种试剂盒的极限灵敏度为20—100cfu/g材料。
本文用同样的策略,采用文中所述方法从含有75个L.case/的材料提取的DNA中PCR扩增出了目的片段,灵敏度折合约3cfu/g材料。
YangJin—Long等。
15。
用ERIC—PCR方法评价了从粪便中提取细菌总DNA的方法,本研究中用DGGE法对提取方法进行评价。
发现所述的提取方案能提取出所有的菌株的染色体,没有遗漏。
随着微生物分子生态学的发展,各种提取环境样品总DNA的方法陆续建立起来,但是对多种环境样品而言,没有一种方法能适用于所有的环境样品,每一种样品都需要根据其特有的理化和生物学特性,优化出一种合适的提取DNA的方法‘4'”J。
参考文献:[I]YangHY,WangXF,LiuJB,e1.a1.Effectofwater一.solublecarbo-hydratecontentmsilagefermentationofwheatstl'aw[J].JournalofBi旧-scienceandmomgiaeering.2006,lOI(3):232—237.[2]NisIdnoN,WadaH,瑚idaM,ela1.Micwbialcomls,fermentatian2009年19(1)prt】clucts,andaerobicstabilityofwholecropcornandatotalmixedrationell—siJedwithandwithoutinoeulmion0f厶吲06捌ZmⅢior厶删06∞姚Ⅱbucha硎【JJ.JournalofDairysd蛳蚀,2004,87(8):2563~2570.[3]RossF,De¨aglioF.QualityofsilagesfromItalianfarms∞att8tedbyn幽andindenfityofmicrobialindicators【Jj.JApptM.a删,2007,103(5):1707一1715.[4]JanyJL.BarbierG.Culture—independenti'Ilf,th础qforidentifylngmicrobi—alcommunitiesincheese[J].FoodmiaobioIo科,.2008.25(7):839—848.【5JLeeL,rnnLS.KelleyST.Culture—independentanalysisofbacterialdi—versityiilachild—carefacility[J].BMCMicrobiol,2007.7(27):l—13.[6JAmininAL,WarganegaraFM,AditiawatiP,eta1.Siarpleenrichmentandindependentculturestoexp∞dbacteAalcommunityanalysisfromgedong-songohot@ring[J].JournalofBioscicneeand瑚帕呜.哪州雌,2008,106(2):21l一214.[7]MuyzerG.DGGF¨V,GEamethodforidentifyinggenesfromnaturaleeosys-terns【JJ.Curr0I'illMicrobiol。
微生物资源及开发利用

nature
Vol 467|2 September 10| doi:10.1038 /nature 09354
Bacterial charity work leads to population-wide resistance Henry H. Lee, Michael N. Molla, Charles R. Cantor & James J. Collins
to understand microbial communities. …individual host properties
such未a来s b医od师y m或a可ss依in据de肠x, a型ge的, o区r g划en,de量r 身can打n造ot 病exp患la饮in 食the清o单bserved ente以rot及yp开es处…方,甚至至为抗生素寻求替代品。
Enterotypes of the human gut microbiome
…identify three robust clusters (referred to as enterotypes hereafter)
that人ar类e n肠ot道na微tio生n 物or 系co统nti可ne拥nt 有spe特ci定fic的…分Th类is —ind肠ic型ates further the
othf比a1n2人4th类Eeu基hruo因mpe组aann大gien1nd5ei0vc倍iodmu。apllse…m.eTnth,e…geOnveesre9t,9~%15o0f
times larger the genes are
ba超ct过er9ia9l%, in为d细ica菌tin。g that the entire cohort harbours between
突变检测的方法流程及质控

突变检测的方法流程及质控There are several methods and techniques used for mutation detection, each with its own unique workflow and quality control measures. 突变检测的方法种类繁多,每一种都有其独特的工作流程和质量控制措施。
A common method for mutation detection is Sanger sequencing, which involves amplifying the DNA region of interest, sequencing the amplified DNA, and then analyzing the sequence for mutations. 常见的突变检测方法之一是Sanger测序,其中涉及到扩增感兴趣的DNA区域,对扩增的DNA进行测序,然后分析序列中的突变。
Another widely used method is next-generation sequencing (NGS), which allows for the simultaneous detection of mutations in multiple genes or genomic regions. 另一种广泛使用的方法是下一代测序(NGS),它可以同时检测多个基因或基因组区域中的突变。
Quality control measures for mutation detection methods typically include the use of positive and negative controls to ensure the accuracy and reliability of the results. 突变检测方法的质量控制措施通常包括使用阳性和阴性对照来确保结果的准确性和可靠性。
检测基因组突变比例的方法与流程

检测基因组突变比例的方法与流程英文回答:Methods and Workflow for Detecting Genomic Mutation Rates.Genomic mutation rates are the frequencies at which mutations occur in the genome. Mutations can be caused by various factors, including environmental toxins,replication errors, and DNA damage. Detecting and characterizing genomic mutation rates is essential for understanding genome evolution, identifying disease-causing mutations, and developing targeted therapies.Workflow for Detecting Genomic Mutation Rates.1. Sample Collection and DNA Extraction:Collect high-quality DNA samples from the target organisms or tissues.Extract DNA using appropriate methods, such as phenol-chloroform extraction or silica-based column purification.2. Whole-Genome Sequencing (WGS):Perform WGS to obtain high-coverage sequencing reads that cover the entire genome.Use next-generation sequencing (NGS) platforms, such as Illumina or Ion Torrent, to generate millions of sequencing reads.3. Variant Calling:Align sequencing reads to a reference genome using bioinformatics tools, such as BWA or Burrows-Wheeler Aligner (BWA).Identify sequence variants by comparing aligned reads to the reference genome.Use variant callers, such as SAMtools or GATK, to detect single nucleotide variants (SNVs),insertions/deletions (indels), and copy number variations (CNVs).4. Filtering and Annotation:Filter out false-positive variants using quality control parameters, such as read depth and base quality.Annotate variants with information from databases and tools, such as ClinVar, Ensembl, and dbSNP.This annotation provides insights into the potential functional consequences of mutations.5. Mutation Rate Calculation:Determine the number of mutations per base pair by dividing the total number of mutations by the sequencing coverage.Calculate mutation rates for different genomic regions, such as coding sequences, regulatory elements, or non-coding RNAs.6. Statistical Analysis:Perform statistical analysis to compare mutation rates between different samples, populations, or experimental conditions.Use statistical tests, such as t-tests or ANOVA, to determine significant differences in mutation rates.中文回答:检测基因组突变比例的方法与流程。
_基因组DNA的提取与纯化

文章编号:1000-2286(2004)03-0329-05基因组DNA 的提取与纯化陈桂信1,吕柳新1,赖钟雄2,潘东明1(1.福建农林大学园艺学院,福建福州350002; 2.福建农林大学亚热带果树研究所,福建福州350002)摘要:以 嫩芽为材料,对Dellaporta等用CT AB制备植物基因组DNA的方法进行改良,结果表明,加入质量分数ω=10%PVPP与材料充分研磨,将提取缓冲液的β-巯基乙醇体积分数降低为1%,能有效地去除材料中的多酚类物质;用氯仿/异戊醇(24∶1)抽提裂解液2次所获得的上相,加入1/10体积的65℃的NaCl/CT AB溶液混匀,再用氯仿/异戊醇(24∶1)连续抽提2次,并在DNA粗提液中加入适量高浓度NaCl和无水乙醇沉淀DNA,可以有效地去除多糖的干扰,获得比较纯净的DNA样品;PCR特异性扩增的结果表明,以改良法的DNA为模板可获得PPO基因保守区约600~800bp特异条带,且扩增条带较亮、无引物二聚体出现。
关键词:;基因组DNA;提取;纯化中图分类号:Q813.4 文献标识码:AExtraction and purification of genomic DNA in N ai(Prunus salicina Lindl.var.cordata)CHEN G ui-xin1,LU Liu-xin1,LAI Zhong-xiong2,PAN Dong-ming1 (1.C ollege of H orticulture,FAFU,Fuzhou,Fujian350002,China;2.Institute of Biotechnology in H orticul2 tural Plants,FAFU,Fuzhou,Fujian350002,China) Abstact:Nai’s genom omic DNA from y oung buds was extracted by m odified Dellaporta’method for preparing genomic DNA of plants with Cetyl T riethyl Amm onium Bromide(CT AB).The results showed that polyphenolics were effectively rem oved from materials by blendering10%P oly Vinyl P oly Pyrrilodone(PVPP)to the ground mate2 rials under liquid nitrogen in m ortar and lowering concentration ofβ-mercaptoethanol in extracting buffer to1%; the polysaccharides were effectively rem oved and purer DNA sam ples were obtained by blundering aqueous phase af2 ter extracting lysis s olution twice with24∶1chloroform:is oamyl alcohol with1/10v olume NaCl/CT AB s olution pre2 heated by65℃then successively extracting twice with24∶1chloroform:is oamyl alcohol and adding an appropriate am ount5m ol/L s odium chloride and abs olute ethanol to the rough s olution of DNA.PCR am plication was conducted by using obtained DNA sam ples as tem plates,a blighter600~800bp special band of conserve zone in PPO gene was am plified and no primer dimers emerged.K ey w ords:Nai(Prunus salicina Lindl.var.cordata);genomic DNA;extraction;purification(Prunus salicina Lindl.var.cordata J.Y.Zhang et al)是福建特有的核果类果树,果实桃形李实,核小,肉厚,多汁,质优,深受消费者的青睐,但在果实成熟期和采后贮藏期间,普遍存在果肉的褐变现象[1],导致果实的食用品质和耐贮性下降,甚至丧失其商品价值,给生产带来经济损失。
小花山柰根状茎挥发油和营养成分及其抗植物病原菌活性分析

·2081·小花山柰根状茎挥发油和营养成分及其抗植物病原菌活性分析冯莹1,仇思润2,王张豪2,何宇豪2,黄茵茵3,李逸彤2,李婉琳2,单体江2*(1广东省森林资源保育中心,广东广州510173;2华南农业大学林学与风景园林学院,广东广州510642;3广州医科大学附属口腔医院,广东广州510182)摘要:【目的】阐明小花山柰根状茎挥发油的化学组成和相对含量,明确根状茎中多种营养成分含量,分析根状茎不同提取物的抗菌活性,从而为小花山柰资源的综合开发利用提供理论依据。
【方法】采用水蒸汽蒸馏法提取小花山柰挥发油,通过气相色谱—质谱联用仪(GC-MS )对挥发油进行化学成分分析;采用凯氏定氮法、酸—苯酚比色法和电感耦合等离子体—质谱法(ICP-MS )半定量分析法分别测定小花山柰根状茎中蛋白质、粗多糖和微量元素含量,采用茚三酮柱后衍生离子交换色谱法测定氨基酸组成与含量;进一步采用抑菌圈法和菌丝生长速率法测定小花山柰根状茎不同提取物的抗细菌和抗真菌活性。
【结果】小花山柰根状茎中挥发油得率为0.14‰;从根状茎挥发油中共鉴定出66种成分,占挥发性成分总量的94.51%,主要有烯烃类、醇类、醛类、酮类和酯类,主要成分为冰片(13.02%)、芳樟醇(12.27%)和大根香叶烯D (5.42%)等。
小花山柰根状茎中蛋白质含量为3.83g/100g ,粗多糖含量为3.33g/100g ;16种氨基酸总量为2.94g/100g ,其中精氨酸含量最高(0.61g/100g ),其次是天冬氨酸(0.46g/100g )。
小花山柰根状茎中共检测出28种微量元素,其中钾元素含量最高(3270mg/kg ),镁(813mg/kg )、钙(289mg/kg )、锰(146mg/kg )和铝(135mg/kg )的含量也较高,其他微量元素的含量均在80mg/kg 以下。
小花山柰根状茎乙酸乙酯层提取物的抗细菌活性最强,石油醚层提取物对杜英生假隐丛赤壳菌的抑制活性最强,半最大效应浓度(EC 50)为14.11μg/mL 。
QuickExtract

QuickExtract – Rapid and efficient extraction of PCR-ready genomic DNA from plant and seed samplesMike Freeman MD, Luke Linz PhD; LGC, Biosearch T echnologies, Alexandria, USAIntroductionThe ability to rapidly screen large populations is vital for breeding and characterisation of transgenic plants. Genotyping by methods based on BHQ™ Probes or KASP™ arewidespread but generally require nucleic acid purification. A more cost-effective and less time-consuming approach is needed. The QuickExtract™ Plant DNA Extraction Solution provides a simple, rapid DNA extraction method to prepare genomic DNA for high-throughput processing. It is used for leaf or seed samples. The extracted DNA is ready for amplification-based analyses.The extraction requires less than ten minutes to prepare PCR-quality DNA with two simple heating steps (see Figure 1). The procedure is convenient and can easily be scaled to process hundreds of samples in multi-well robotic automation systems.This application note demonstrates the suitability of using the QuickExtract Plant DNA Extraction Solution for multiple plant species. The requirement for grinding is also investigated. Genotyping results are shown for KASP and BHQ Probes. In addition, the effect of storage on the quality of the extracted DNA was determined.Application noteFigure 1. Overview of the QuickExtract workflow. After the two heating steps, the released DNA can be used directly for amplification-based analysis, stored at +4 °C for four weeks, or transferred to -20 °C for archival purposes.Heat at 65 °C for 8 minutesAdd samplesQuickExtract Plant Solution PCR-ready DNAQuickExtract is part of the Epicentre™ product line, known for its unique genomics kits, enzymes, and reagents which offer high quality and reliable performance.Materials, methods and resultsa) Determination of the impact of grinding Whole seeds from wheat, tomato, pepper and hulled sunflower were ground or incubated whole with QuickExtract Plant DNA Extraction Solution – 100 µL of QuickExtract Solution was added to tomato and pepper seeds, 200 µL was added to wheat seeds, and 300 µL was used for sunflower seeds. The processed samples were diluted 1:4 or 1:16 prior to PCRamplification with KASP on the IntelliQube™. Genotyping results of the four plant species are shown in Figure 2. Grinding had little to no effect on cluster plot analysis for tomato and sunflower seeds. However, grinding was required for amplification of QuickExtracted DNA from wheat seeds and inhibitory for PCR with pepper seeds. The decision whether to grind a seed sample or perform theextraction on whole seeds must be determined empirically.genomic DNA from plant and seed samples4x d i l u t i o n16x d i l u t i o nWheatT omatoSunflowerPepperGround SeedWhole SeedNTCFigure 2. The effect of grinding seed material. Wheat, T omato, Sunflower and Pepper seeds were ground (red circles) or extracted whole (blue circles) with QuickExtract Plant DNA Extraction Solution. Samples were diluted 4-fold (top) or 16-fold (bottom) prior to PCR amplification with KASP on the IntelliQube. Water was used as a negative control (NTC).genomic DNA from plant and seed samplesT able 1. Sample and pre-treatment of validated crops, seeds or leaves, grinding requirement conditions, starting weight and volume of required QuickExtract solution. The guidelines show how much tissue to use, and whether grinding the sample must be determined empirically for each plant species and tissue type. For reference, one punch is 6 mm in diameter. Additional optimisation may be required.b) Evaluation of BHQ and KASP chemistries for eight commercially important crop types QuickExtract Plant DNA Extraction Solution was added to samples of seed or leaf material of eight crop types – Corn, Wheat, Rapeseed, Soy, Tomato, Pepper, Cotton, and Sunflower.Extracts were incubated as in Figure 1,diluted 2- to 8-fold, and PCR amplified on the IntelliQube with KASP and BHQ chemistries. Table 1 lists the crops tested, sample type, grinding requirements, starting weight of the sample and volume of QuickExtract Plant DNA Extraction Solution used.c) Stability of extracted DNA for at least 4 weeks at +4 ˚CWe assessed the stability of the extracted DNA. Corn and tomato leaf samples were processed with the QuickExtract Plant DNA Extraction Solution. The DNA was PCRamplified using two different KASP assays for each crop type. Amplifications were performedimmediately after sample processing. Sample plates were stored at +4 °C. After one month of storage at +4 °C the amplifications were repeated. Figure 4 shows a comparison of the genotyping data, using the original QuickExtract Plant DNA Extraction Solution lysate, and the same QuickExtract lysate one month later. No significant difference in endpoint signal or cluster quality was observed.Figure 3. Cluster plots for KASP and BHQ chemistries. Results are shown using seven different crops – T omato, Rapeseed, Corn, Pepper, Wheat, Sunflower, and Soy. Analyses were performed using KASP and BHQ Probes.T omato RapeseedCornPepperWheatSunflowerSoyB H Q K A S PL e a f L e a fS e e d S e edgenomic DNA from plant and seed samplesO r i g i n a l1 m o n t hCorn assay 1Corn assay 2T omato assay 1Tomato assay 2Figure 4. Stability of DNA stored at 4 ˚C. DNA extracted from corn and tomato with QuickExtract was PCR amplified (original) with two KASP assays and then stored at 4 °C for 1 month. The samples were then again PCR amplified against the same two assays (1 month).ConclusionThe QuickExtract Plant DNA Extraction Solution allows for fast and simple genotyping of plant samples. Tomato and sunflower seeds allow genotyping without grinding. Wheat seeds required grinding for amplification, whereas grinding of pepper seeds was inhibitory for PCR. Leaf material for these crops showed similar results for the seed material (data not shown). We recommend that the necessity or requirement for grinding be determined for each sample type. Also, performing serial dilutions of the QuickExtract extract aftersample processing is advised, for example 1:4, 1:8, 1:16, to determine the optimal amount of processed sample volume for your downstream application. Finally, it is important to emphasise that seeds must be cleaned efficiently ifgenotyping, since interpretation of results may be complicated by any non-plant material adhering to the seed coat.Results show that the QuickExtract Plant DNA Extraction Solution may be used to extract DNA from leaf or seed material from multiple types of plants for endpoint PCR applications. Good genotyping results and clusters, usingKASP and BHQ Probes for different plant species, were generated. Parameters to be considered for each crop and sample (leaf or seed) are: initial sample mass, whether grinding is necessary, volume of the QuickExtract Plant DNA Extraction Solution required and dilution of the QuickExtracted sample to use for the assay.In addition, it was shown that very goodgenotyping results can be achieved even after storing the original lysate four weeks at +4 °C. This application note shows that QuickExtract Plant DNA Extraction Solution provides a fast and simple method to prepare genomic DNA for KASP genotyping, or use of BHQ Probes – all in a single tube, without the use of toxic chemicals, columns or precipitation and resuspension steps. The method isideally suited to high-throughput applications employing a liquid handler combined with the use of a programmable heating block or water bath.genomic DNA from plant and seed samplesIntegrated tools. Accelerated science.For Research Use Only. Not for use in diagnostic procedures.All trademarks and registered trademarks mentioned herein are the property of their respective owners. All other trademarks and registered trademarks are the property of LGC and its subsidiaries. Specifications, terms and pricing are subject to change. Not all products are available in all countries. Please consult your local sales representative for details. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording or any retrieval system, without the written permission of the copyright holder. © LGC Limited, 2019. All rights reserved. GEN/0581/MW/0619@LGCBiosearch。
内生真菌菌核生枝顶孢霉的分离鉴定及生物学特性研究

山东农业大学学报(自然科学版),2023,54(5):641-649VO.54NO.52023 Journal of Shandong Agricultural University(Natural Science Edition)doi:10.3969/j.issn.1000-2324.2023.05.001内生真菌菌核生枝顶孢霉的分离鉴定及生物学特性研究赵晓彤,张凌霄,王桂清*聊城大学农学与农业工程学院,山东聊城252000摘要:为了明确1株来源于国槐枝干内生真菌的分类地位和生物学特性,采用组织和单孢分离法进行了分离纯化,利用显微镜进行了形态鉴定,采用活体接种法进行了致病性鉴定,提取了基因组DNA,通过ITS和18S rDNA序列分析进行了分子生物学鉴定,通过十字交叉法和血球计数法分析了不同生态条件和营养条件下的生长发育状况。
结果表明,该真菌为无性型真菌菌核生枝顶孢霉Acremonium sclerotigenum,这是国内首次从国槐中获得的枝顶孢属非致病性内生真菌;该菌在PDA培养基、温度25~30°C、全光照和酵母浸膏为氮源条件下菌丝生长和产孢能力最优,菌丝生长更适宜以麦芽糖为碳源的中性条件,而产孢更适宜以阿拉伯糖为碳源的偏酸性环境。
该研究为进一步将该A. sclerotigenum开发成新型绿色的微生物制剂、增产菌或生防载体菌奠定了理论基础。
关键词:菌核生枝顶孢霉;形态学鉴定;分子生物学鉴定;生物学特性中图法分类号:S476.1文献标识码:A文章编号:1000-2324(2023)05-0641-09 Isolation Identification and Biological Characteristics of EndophyticFungus Acremonium sclerotigenumZHAO Xiao-tong,ZHANG Ling-xiao,WANG Gui-qing*Agricultural Science and Engineering School/Liaocheng University,Liaocheng252000,ChinaAbstract:In order to clarify the taxonomic status and biological characteristics of one endophytic fungus derived from the stem of Sophora japonica Linn,We used tissue and monospore isolation method for isolation and purification,morphological identification by microscope,pathogenicity dentified by living inoculation,genomic DNA extraction,TS and18S rDNA sequence analysis for molecular biological identification,and clarified the taxonomic status and biological characteristics of an endophytic fungus derived from the stem of Sophora japonica Linn,and analyzed growth and development under different ecological and nutritional conditions by crossing method and blood cell counting method.The results showed that the fungus was Acremonium sclerotigenum,which was the first non-pathogenic endophytic fungus of the genus Acremonium obtained from Sophora japonica Linn in China.This bacterium hyphae growth and spore production capacity were optimal under the conditions of PDA medium,temperature25~30°C,full light and yeast extract as nitrogen source;hyphae growth was more suitable for neutral conditions with maltose as carbon source while spore production was more suitable for acidic environment with arabinose as carbon source.This study laid the theoretical foundation for the further development of A. sclerotigenum into a new type of green microbial preparation,proliferative bacteria or biocontrol vector bacteria. Keywords:Acremonium sclerotigenum;morphological identification;molecular biological identification;biological characteristics目前,在植物病虫害防控过程中主要采用化学防治的措施。
陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析

㊀山东农业科学㊀2023ꎬ55(4):11~23ShandongAgriculturalSciences㊀DOI:10.14083/j.issn.1001-4942.2023.04.002收稿日期:2022-07-19基金项目:山东省农业良种工程项目(2020LZGC002)ꎻ国家自然科学基金项目(31901422)ꎻ山东省农业科学院农业科技创新工程项目(CXGC2021A46)作者简介:陈义珍(1990 )ꎬ女ꎬ助理研究员ꎬ主要从事棉花抗衰老及遗传育种研究ꎮE-mail:chenyizhen0@126.com通信作者:柳展基(1972 )ꎬ男ꎬ研究员ꎬ主要从事棉花遗传育种研究ꎮE-mail:scrcliuzhanji@sina.com陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析陈义珍ꎬ李浩ꎬ傅明川ꎬ王立国ꎬ刘任重ꎬ柳展基(农业部黄淮海棉花遗传改良与栽培生理重点实验室/山东省农业科学院经济作物研究所ꎬ山东济南㊀250100)㊀㊀摘要:膜结合脂肪酸去饱和酶(fattyaciddesaturaseꎬFAD)是生物合成不饱和脂肪酸的关键酶ꎮ本研究以陆地棉基因组数据为基础ꎬ利用生物信息学方法对棉花FAD基因家族进行全基因组鉴定和进化分析ꎮ结果表明ꎬ陆地棉基因组中共含有37个FAD基因ꎬ进化分析发现这些基因分为4个亚家族ꎬ各亚家族成员数量不一ꎬ但相同亚家族成员含有相似的基因结构和保守基序ꎮ共线性分析发现28对复制基因全为片段复制基因ꎬ且都经历了严格的纯化选择作用ꎮ此外ꎬ在陆地棉FAD基因的启动子区域ꎬ发现了丰富的响应植物激素信号和逆境胁迫的顺式作用元件ꎮ转录组分析表明ꎬ陆地棉FAD基因家族响应干旱和盐胁迫ꎬ定量PCR分析表明施加外源褪黑素显著影响了FAD基因的表达ꎮ本研究结果为进一步解析FAD家族基因的功能奠定了基础ꎮ关键词:陆地棉ꎻ膜结合脂肪酸去饱和酶ꎻ全基因组鉴定ꎻ生物信息学分析ꎻ基因表达ꎻ干旱和盐胁迫ꎻ褪黑素中图分类号:S562:Q78㊀㊀文献标识号:A㊀㊀文章编号:1001-4942(2023)04-0011-13Genome ̄WideIdentificationandExpressionAnalysisofMembrane ̄BoundFattyAcidDesaturaseGeneFamilyinUplandCotton(GossypiumhirsutumL.)ChenYizhenꎬLiHaoꎬFuMingchuanꎬWangLiguoꎬLiuRenzhongꎬLiuZhanji(KeyLaboratoryofCottonBreedingandCultivationinHuang ̄Huai ̄HaiPlainꎬMinistryofAgricultureandRuralAffairs/InstituteofIndustrialcropsꎬShandongAcademyofAgriculturalSciencesꎬJinan250100ꎬChina)Abstract㊀Membrane ̄boundfattyaciddesaturases(FADs)arethekeyenzymesforthebiosynthesisofunsaturatedfattyacids.InthisstudyꎬthewholegenomeofFADgenefamilywasidentifiedanditsevolutionwasanalyzedbybioinformaticsmethodbasedonthegenomicdataofuplandcotton(GossypiumhirsutumL.).Theresultsshowedthattherewere37FADgenesintheuplandcottongenome.PhylogeneticanalysesindicatedtheseFADgenesweredividedintofoursubfamiliesꎬandeachcontaineddifferentnumbersofFADgenesꎬbutthemembersinthesamesubfamilycontainedsimilargenestructureandconservedmotifs.Collinearanalysisrevealedthatallthe28pairsofreplicatorswerefragmentreplicatorsandhadundergonerigorouspurificationandselection.Inadditionꎬabundantcis ̄actingelementsrelatedtophytohormonesignalingandstressresponseswereidentifiedinthepromoterregionsofFADgenes.TranscriptomeanalysisshowedthattheFADgenefamilyofuplandcottonrespondedtodroughtandsaltstressꎬandquantitativePCRanalysisshowedthatexogenousmelatoninsignificantlyaffectedtheFADgeneexpression.TheresultsofthisstudylaidafoundationforfurtherfunctionalanalysisofFADfamilygenes.Keywords㊀UplandcottonꎻMembrane ̄boundfattyaciddesaturaseꎻGenome ̄wideidentificationꎻBioin ̄formaticsanalysisꎻGeneexpressionꎻDroughtandsaltstressꎻMelatonin㊀㊀脂肪酸去饱和酶(fattyaciddesaturaseꎬFAD)能够在脂肪酸链的特定位置引入双键ꎬ是产生不饱和脂肪酸的关键酶[1ꎬ2]ꎮ植物中的不饱和脂肪酸主要包括油酸㊁亚油酸㊁亚麻酸和花生四烯酸等ꎬ不仅是储藏油脂的重要组分ꎬ还是植物细胞膜脂的主要成分[3]ꎮFAD包括FAD2㊁FAD3㊁FAD4㊁FAD5㊁FAD6㊁FAD7和FAD8ꎬ其中FAD2和FAD6为ω-6(也称Δ12)去饱和酶ꎬ能够在油酸脂肪酸链的Δ12处引入第二个氢键ꎬ催化油酸转变为亚油酸ꎻFAD3㊁FAD7和FAD8为ω-3(Δ15)去饱和酶ꎬ能够在亚油酸脂肪酸链的Δ15处添加第三个氢键ꎬ进而产生亚麻酸ꎻFAD4㊁FAD5和FAD6的催化底物为棕榈酸[4ꎬ5]ꎮ1992年ꎬArondel等从拟南芥中图位克隆了FAD3基因[6]ꎬ其后FAD基因相继在油菜㊁水稻㊁大豆㊁玉米等作物中被分离[7]ꎮ棉花是世界上最重要的天然纤维作物ꎬ同时也是重要的食用油和蛋白来源[8]ꎮ棉仁中油分含量高达30%~40%ꎬ棉籽油富含多种不饱和脂肪酸ꎬ其中油酸和亚油酸含量约占80%ꎮFAD2是脂肪酸去饱和反应的限速酶ꎬ决定了油脂中油酸和亚油酸的比例和油脂品质ꎬ抑制FAD2基因的表达ꎬ能够降低Δ12-脂肪酸去饱和酶的活性ꎬ致使种子中油酸含量增加ꎮ利用TALENs技术靶向敲除大豆FAD2-1A和FAD2-1B基因ꎬ纯合双基因突变体的油酸含量显著上升ꎬ由20%提高到80%ꎬ而亚油酸含量大幅下降ꎬ由50%降至4%[9ꎬ10]ꎮLiu等利用RNAi技术抑制棉花FAD2-1基因的表达ꎬ获得的棉花高油酸品系High-Ole ̄ic和Mono-Cott的油酸含量分别达到77%和81%[11]ꎮ近年来ꎬ棉花基因组研究进展迅速ꎬ二倍体棉种雷蒙德氏棉(D5)和亚洲棉(A2)以及四倍体棉种陆地棉(AD1)和海岛棉(AD2)的基因组相继被破译[12-15]ꎬ为利用生物信息学在全基因组范围内进行重要基因家族的鉴定和分析提供了可能ꎮ目前ꎬ已知拟南芥基因组中含有17个FAD基因ꎬ水稻中有10个ꎬ油菜中多达84个[16]ꎮ在雷蒙德氏棉基因组中ꎬLiu等鉴定了19个FAD基因[4]ꎮ然而ꎬ陆地棉中FAD基因家族的系统分析尚未见报道ꎮ本研究利用最新发布的陆地棉标准系TM-1的基因组数据[14]ꎬ对膜结合FAD基因家族进行全基因组鉴定ꎬ分析其基因结构㊁染色体定位㊁保守基序㊁基因复制和顺式作用元件ꎬ并分析其在干旱胁迫㊁盐胁迫及施加外源褪黑素下基因的表达模式ꎬ为进一步研究棉花FAD基因家族各成员的功能奠定基础ꎮ1㊀材料与方法1.1㊀供试材料供试陆地棉(GossypiumhirsutumL.)品种为K836ꎬ种子由本实验室保存ꎮ采用1/2Hoagland营养液水培法将棉花幼苗培养至两叶一心ꎮ用100μmol/L褪黑素进行处理ꎬ分别于处理后0㊁3㊁6㊁12h对棉花叶片进行取样ꎬ每个样品3次生物学重复ꎬ置于液氮中冻存备用ꎮ1.2㊀陆地棉FAD基因鉴定与注释陆地棉标准系TM-1的基因组序列数据来自于CottonFGD数据库[17](http://www.cottonfgd.org/)ꎮ利用拟南芥的17个FAD基因的氨基酸序列作为查询序列ꎬ检索棉花基因组蛋白数据库(BlastpꎬEɤe-10)ꎮ同时ꎬ从Pfam数据库(http://pfam.xfam.org/)下载FAD蛋白保守结构域FA_desaturase(PF00487)的隐马尔可夫模型文件ꎬ利用HMMER3.0软件搜索陆地棉的蛋白数据库[18]ꎮ将获得的候选序列提交Pfam和SMART(http://smart.embl-heidelberg.de/)数据库ꎬ确认是否具有FAD蛋白保守结构域ꎮ利用ExPASy(https://web.expasy.org/compute_pi/)分析棉花FAD基因家族成员的分子量和等电点[19]ꎮ利用CELLO(http://cello.life.nctu.edu.tw/)对棉花FAD基因家族成员进行亚细胞定位预测[20]ꎮ1.3㊀陆地棉FAD的系统进化和染色体定位分析利用ClustalW软件对拟南芥和陆地棉FAD蛋白序列进行多重序列比对ꎬ利用MEGA-X软件中的邻接法(neighbor-joiningꎬNJ)构建系统进化树ꎬBootstrap值设置为1000[21]ꎮ根据陆地棉基因组提供的基因注释信息ꎬ获得FAD基因在各染色体上的位置ꎬ利用MapChart2.32软件绘制其染色体分布图[22]ꎮ21山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀1.4㊀陆地棉FAD基因结构、保守基序和上游顺式作用元件分析首先从棉花基因组注释文件(gff文件)获得FAD基因的DNA和cDNA信息ꎬ利用GSDS2.0软件(http://gsds.cbi.pku.edu.cn/)绘制棉花FAD基因家族成员的基因结构图[23]ꎻ利用在线软件MEME(http://meme-suite.org/tools/meme)分析FAD蛋白的保守基序[24]ꎬ基序数值设定为10ꎮ利用陆地棉基因组数据ꎬ提取FAD基因转录起始位点上游1500bp序列ꎬ在PlantCARE数据库(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)中检索ꎬ鉴定上游启动子区域可能存在的顺式作用元件[25]ꎮ1.5㊀陆地棉FAD基因复制分析利用MCScanX软件分析陆地棉基因组内的同源基因[26]ꎬ如比对上的序列长度超过长序列长度的75%且序列的相似性超过75%ꎬ则认为此同源基因为复制基因ꎬ并利用可视化工具Circos(ht ̄tp://circos.ca/)展示复制的同源关系[27ꎬ28]ꎮ采用KaKs_Calculator2.0计算复制基因的Ka(非同义替换率)和Ks(同义替换率)值ꎬ通过Ka/Ks值分析环境选择压力[29]ꎮ1.6㊀RNA-seq分析利用前期试验得到的陆地棉干旱(PEG)和盐胁迫(NaCl)处理3㊁6㊁12㊁24h后的转录组数据(未发表)ꎬ筛选得到FAD基因表达量数据ꎮ利用FPKM(fragmentsperkilobaseofmillionmappedreads)对原始表达数据进行标准化处理ꎬ并利用TBtools软件绘制表达量热图ꎮ以log2(FC)绝对值大于1为差异基因的筛选标准ꎮ1.7㊀基因表达分析采用北京艾德莱生物科技(Aidlab)有限公司植物RNA快速提取试剂盒(RN3802)提取叶片总RNAꎬ分别取1μg总RNA进行反转录ꎬ反应体系和操作程序参照TRUEscriptRTMasterMix(On ̄eStepgDNARemoval)一步法基因组清除反转录试剂盒(PC70)说明ꎮ将反转录产物稀释10倍ꎬ选择Ubiquitin(GenBankNo.AY189972)基因作为内参基因ꎬ进行定量RT-PCRꎬ所用引物见表1ꎬ由北京六合华大基因科技有限公司合成ꎮ选用㊀㊀表1㊀定量PCR引物序列序号基因正向引物(5ᶄ-3ᶄ)反向引物(3ᶄ-5ᶄ)1GH_A01G1380GGGAGCTTTATCGACCGTGGACGAGCCACGTGTATGCGGGTTAT2GH_A01G1382GTTTCCAGCGCTCCGTTTTACGGACCCAAACGCCGGTCAAAATC3GH_A04G1111ACCTTTGGAACAGAAGTCCGGGTCTTTTCGTTCGGTCGGGACAA4GH_A05G0424GCCAAGGTGCCATCTCAGGAAATGGGTTGCTGAGAATCCTTGCC5GH_A05G1248AAACTCTGGCCATCCAGACTCGAAGCCGACACTTTCCCATTGCT6GH_A07G1144CATGGGTTCCGTTGCCTGAGAAACACGGGGAATGCAAATAGGGG7GH_A07G1522AGTGCGGAAGAGCTGAAAGAGCCCAGGGTGATACGCCAGAAAGG8GH_A09G1076GGGACCATGTTTTGGGCTGTCTCGGACATCGGAACCCAAGACTC9GH_A10G2629TCACCATGGTCATGACGAGAAGCATCCCGATCAAGCGTCGTAAGC10GH_A11G0968GGCTTGTCACGGAGTTCTCCAACCCACACGCTATCGCATCTCAA11GH_A11G3588ATGCAGAAGAGCGATCCGTGACTCTTTCGTCAGCTTGCTCCCAC12GH_A12G1258ACCCCACTCTTAAATCTCGCCGAGGTATTGCCAAGCTGTGGTGG13GH_A13G0678CACCAGTACGCCATTACTGCGAAACACGTGGGATGGACCAACTG14GH_A13G2126TATAGGCCACGATTGCGCTCACGTGCCTGTCATGCTTAAACCGC15GH_D01G1464GCGCTCCGTTTTACGCTCATTCACGTTGGAGAGAGCCTGAGGAA16GH_D04G1454ATCATGGCACCCGTTGTCTGAGGGTCGAAGTGCGAACCACTCTT17GH_D05G0426GTTTCCAAGGTTGCCAAGGTGCCTGCAAGGCTGCTGATCGAAGA18GH_D06G1474AGGTGAAGGAGATTGCTCCCGAAGGGCCGATACTCCGATCCATT19GH_D07G1125GAAACTCGACACCAGTACGCGACTTTGCCTGGGCTTCTATGCCA20GH_D07G1526GTATCACCCTGGAACGGCTTGGGTGCCCACACGCTATCACATCT21GH_D08G2811CGACAAGAAAGGACACGGGACACACCATCCCATGCTGATCCCAG22GH_D09G1028ACAGCAACTTGTTTGCTCCCCACTTCTGCTCGTATCCGTGGTGG23GH_D10G2734GAAGCTACCGAAGCAGCAAAGCACACGACATCACCAATGTCACTCA24GH_D11G0999AGGCTTGTCACGGAGTTTTCCACCCACACGCTATCGCATCTCAA25GH_D11G3608TGCAACACACTCACCCTGCATTGCCTTTGTTGCTTCCATTGCGT26UbiquitinAAGACCTACACCAAGCCCAAGACACTCCGCATTAGGACACTC31㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析Aidlab公司的2ˑSybrGreenqPCRMix试剂盒ꎬ反应体系为20μL:2ˑSYBRqPCRMix10μLꎬ基因特异性上下游引物各0.4μL(10μmol/L)ꎬ模板cDNA0.8μLꎬ补ddH2O至20μLꎮ使用Ther ̄moFisherScientificQuantStudio5荧光定量PCR仪ꎬ反应程序:95ħ预变性3minꎻ95ħ15sꎬ60ħ15sꎬ40个循环ꎻ熔点曲线程序为95ħ15sꎬ60ħ1minꎬ95ħ15sꎮ每个样品进行3次重复试验ꎬ采用2-әәCt法计算基因的表达量ꎮ利用Graphpad进行差异显著性分析ꎮ2㊀结果与分析2.1㊀陆地棉FAD基因家族成员的鉴定在陆地棉基因组中共鉴定出37个FAD基因(表2)ꎬ均含有保守的FA_desaturase结构域(PF00487)ꎮFAD蛋白的氨基酸序列长度差异较大ꎬGH_A13G0678最短ꎬ为308个氨基酸ꎬ而GH_A10G2629和GH_D10G2734最长ꎬ均为450个氨基酸ꎻGH_A13G0678的分子量最小ꎬ为35.41kDaꎬGH_A12G1258的分子量最大ꎬ为51.87kDaꎻFAD蛋白的等电点差异较大ꎬGH_D06G1474的等电点最小ꎬ为6.95ꎬ其余FAD蛋白的等电点均在7.29及以上ꎬ为碱性ꎬ以GH_D05G1245的等电点最大(9.37)ꎮ大多数FAD蛋白定位在内质网和质膜上ꎬ少数FAD蛋白位于叶绿体中ꎬ仅GH_D12G1274位于溶酶体中(表2)ꎮ㊀㊀表2㊀陆地棉FAD基因家族基本信息基因染色体定位亚家族外显子数量氨基酸序列长度分子量(kDa)等电点亚细胞定位GH_A01G1380A01:52437258-52438409Omega138344.378.97内质网GH_A01G1382A01:52793676-52794827Omega138344.379.06内质网GH_A01G2453A01:117661885-117664879Omega842749.538.17叶绿体GH_A04G1111A04:76679855-76681857Omega844650.968.47叶绿体GH_A05G0424A05:3932284-3933627Front-end144751.168.43质膜GH_A05G1248A05:11342468-11344453First540546.289.26质膜GH_A06G1435A06:89146264-89147952Sphingolipid233038.407.29质膜GH_A07G1144A07:18014579-18016280Omega837643.508.84内质网GH_A07G1522A07:30432512-30433855Front-end144751.188.36质膜GH_A08G2818A08:124577528-124578871Front-end144751.208.64质膜GH_A09G1076A09:62957866-62959614Omega838845.039.05内质网GH_A10G0260A10:2147390-2149449Sphingolipid233138.628.82质膜GH_A10G2629A10:114411673-114414288Omega845051.198.91叶绿体GH_A11G0968A11:8764840-8766183Front-end144751.388.94质膜GH_A11G3588A11:119680980-119682134Omega138444.258.96内质网GH_A12G1258A12:80872680-80875034Front-end244951.877.98质膜GH_A13G0678A13:14253433-14255056Omega830835.418.69质膜GH_A13G2126A13:105045570-105049386Omega1044551.599.09叶绿体GH_A13G2423A13:108609376-108610533Omega138544.069.09内质网GH_D01G1463D01:30783164-30784315Omega138344.288.94内质网GH_D01G1464D01:30824751-30825902Omega138344.189.04内质网GH_D01G2529D01:64245406-64248408Omega843550.407.42叶绿体GH_D04G1454D04:47832920-47834919Omega844650.728.52叶绿体GH_D05G0426D05:3419790-3421133Front-end144751.208.68质膜GH_D05G1245D06:10327204-10329142First538644.249.37质膜GH_D06G1474D07:44774892-44776562Sphingolipid233038.356.95质膜GH_D07G1125D07:14029866-14031568Omega837643.568.83内质网GH_D07G1526D07:22737990-22739333Front-end144751.198.55质膜GH_D08G2811D08:68604570-68605913Front-end144751.278.61质膜GH_D09G1028D09:35497454-35499216Omega838845.119.05内质网GH_D10G0273D10:2157515-2159474Sphingolipid232337.797.30质膜GH_D10G2734D10:66260614-66263295Omega845051.158.91叶绿体GH_D11G0999D11:8288982-8290325Front-end144751.378.94质膜41山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀㊀㊀表2(续)基因染色体定位亚家族外显子数量氨基酸序列长度分子量(kDa)等电点亚细胞定位GH_D11G3608D11:69715461-69716615Omega138444.279.04内质网GH_D12G1274D12:39933619-39935960Front-end339545.137.30溶酶体GH_D13G2107D13:59015821-59019626Omega1044251.329.17叶绿体GH_D13G2414D13:62456454-62457605Omega138343.898.95内质网2.2㊀陆地棉FAD基因的系统进化和染色体定位分析为了研究陆地棉FAD基因家族成员之间的进化关系ꎬ我们利用陆地棉和拟南芥FAD蛋白序列构建了系统进化树ꎮ按照亲缘关系的远近ꎬ将陆地棉FAD基因家族分为4个亚家族ꎬ分别为First㊁Sphingolipid㊁Front-end和Omega亚家族(图1)ꎮ4个亚家族中的FAD成员数量差异较大ꎬ其中ꎬFirst亚家族中陆地棉FAD成员最少ꎬ仅有2个ꎬ而拟南芥FAD成员多达9个ꎻSphingolipid亚家族中仅含有1个拟南芥FAD成员ꎬ却含有4个陆地棉成员ꎻFront-end亚家族中陆地棉和拟南芥的FAD成员分别为10个和2个ꎻOmega亚家族中陆地棉FAD成员数量最多ꎬ为21个ꎬ拟南芥FAD成员为5个ꎮ图1㊀陆地棉FAD基因家族的系统进化树51㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析㊀㊀根据陆地棉标准系TM-1的基因组注释信息ꎬ将37个FAD基因定位在22条染色体上ꎬ其中A01㊁A13和D01染色体上分别含有3个FAD基因ꎻA05㊁A07㊁A10㊁A11㊁D05㊁D07㊁D10㊁D11和D13染色体上分别含有2个FAD基因ꎻ其余10条染色体上分别含有1个FAD基因(图2)ꎮ图2㊀陆地棉FAD基因家族的染色体分布2.3㊀陆地棉FAD基因结构和保守基序分析陆地棉FAD基因4个亚家族呈现不同的结构特征(图3)ꎮFirst亚家族的FAD成员具有相同的基因结构ꎬ均含有5个外显子ꎻSphingolipid亚家族的FAD基因亦具有相同的基因结构ꎬ均含有2个外显子ꎻFront-end亚家族的基因结构也比较保守ꎬ10个FAD成员中ꎬ8个FAD基因含有1个外显子ꎬ其余2个FAD成员含有2个或3个外显子ꎻOmega亚家族的基因结构存在3种方式ꎬ11个FAD成员含有8个外显子ꎬ2个成员含有10个外显子ꎬ剩余的8个成员含有1个外显子ꎮ利用MEME分析陆地棉FAD蛋白的保守基序ꎬ共发现3个保守组氨酸基序(Histidine-box1㊁Histidine-box2和Histidine-box3)ꎬ但不同亚家族成员的组氨酸基序的序列不同(图4)ꎮOmega亚家族的Histidine-box1为H[D/E]C[G/A]HꎬHis ̄tidine-box2为H[R/D][R/T/I]HHꎬHistidine-box3为H[V/I][I/A/P]HHꎬ尽管中间的氨基酸残基不同ꎬ但两侧均为保守的组氨酸残基ꎮFront-end亚家族的Histidine-box1和Histidine-box2序列十分保守ꎻHistidine-box3为Q[L/I/V]EHHꎬ其起始氨基酸为谷酰胺ꎮSphingolipid亚家族成员的3个组氨酸基序都十分保守ꎬ且基序两侧皆为保守的组氨酸ꎮFirst亚家族Histidine-box3为典型的组氨酸基序ꎬHistidine-box1的最后一个氨基酸残基为亮氨酸ꎬ而Histidine-box2不具有典型的组氨酸基序特征ꎮ2.4㊀陆地棉FAD基因复制分析基因复制在基因家族扩张过程中发挥重要作用ꎮ利用MCScanX对陆地棉基因组中的基因复制事件进行分析ꎬ发现FAD基因家族共存在28对片段复制基因ꎬ无串联复制基因ꎮ其中ꎬ21对基因复制发生在A和D亚基因组之间ꎬ4对发生在A亚基因组之内ꎬ剩余3对发生在D亚基因组之内(图5)ꎬ表明片段复制是棉花FAD基因家族扩张的主要方式ꎮ随后ꎬ利用KaKs_Calculator分析复制基因的Ka/Ks值ꎬ结果显示所有复制基因的Ka/Ks值均小于0.55(表3)ꎮ61山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀图3㊀陆地棉FAD基因结构分析图4㊀陆地棉FAD基因家族的3个保守组氨酸富集区71㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析图5㊀陆地棉FAD基因家族的共线性分析㊀㊀表3㊀陆地棉FAD家族基因的复制分析复制基因对KaKsKa/Ks复制类型GH_A01G1380:GH_D01G14630.0090.0220.413片段复制GH_A04G1111:GH_D04G14540.0110.0560.199片段复制GH_A05G0424:GH_D05G04260.0090.0310.277片段复制GH_A05G1248:GH_D05G12450.0100.0200.517片段复制GH_A06G1435:GH_D06G14740.0080.0400.214片段复制GH_A06G1435:GH_A10G02600.0710.6300.113片段复制GH_A06G1435:GH_D10G02730.0710.6640.106片段复制GH_A07G1144:GH_D07G11250.0100.0320.322片段复制GH_A07G1144:GH_A09G10760.1550.7100.218片段复制GH_A07G1144:GH_D09G10280.1550.7430.208片段复制GH_A07G1522:GH_D07G15260.0010.0370.026片段复制GH_A07G1522:GH_A12G12580.1652.1820.076片段复制GH_A09G1076:GH_D09G10280.0020.0240.093片段复制GH_A09G1076:GH_D07G11250.1450.7560.192片段复制GH_A10G0260:GH_D10G02730.0040.0580.069片段复制GH_A10G2629:GH_D10G27340.0060.0580.100片段复制GH_A10G0260:GH_D06G14740.0750.6700.111片段复制GH_A11G0968:GH_A12G12580.1361.9490.070片段复制GH_A11G0968:GH_D07G15260.0930.8290.112片段复制81山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀㊀㊀表3(续)复制基因对KaKsKa/Ks复制类型GH_A11G0968:GH_D11G09990.0050.0200.237片段复制GH_A11G0968:GH_D12G12740.1421.8910.075片段复制GH_A12G1258:GH_D07G15260.1671.9670.085片段复制GH_A12G1258:GH_D11G09990.1391.9000.073片段复制GH_A12G1258:GH_D12G12740.0160.0840.194片段复制GH_A13G2126:GH_D13G21070.0030.0310.092片段复制GH_D06G1474:GH_D10G02730.0730.7050.103片段复制GH_D07G1125:GH_D09G10280.1450.7920.184片段复制GH_D11G0999:GH_D12G12740.1451.8850.077片段复制2.5㊀陆地棉FAD基因上游顺式作用元件分析利用陆地棉FAD基因转录起始位点上游1500bp序列分析顺式作用元件ꎬ结果发现棉花FAD基因的上游序列除了存在启动子核心元件TATA-box和CAAT-box外ꎬ还存在许多激素和逆境胁迫响应元件(图6)ꎮ其中ꎬ植物激素响应元件有9种ꎬ分别是ABRE㊁AuxRE㊁CGTCA-mo ̄tif㊁TGA-element㊁ERE㊁GARE-motif㊁P-box㊁TCA-element和TGACG-motifꎻABRE㊁ERE和TCA-ele ̄ment分别响应脱落酸㊁乙烯和水杨酸ꎬTGACG-motif和CGTCA-motif为茉莉酸甲酯响应元件ꎬAuxRE和TGA-element为生长素响应元件ꎬGARE-motif和P-box为赤霉素响应元件ꎮ逆境胁迫响应元件有6种ꎬ包括ARE㊁DRE㊁LTR㊁MBS㊁WUN-motif和TC-richꎬ分别是缺氧胁迫㊁冷害㊁低温㊁干旱㊁损伤和防御响应元件ꎮ图6㊀陆地棉FAD基因启动子区域顺式作用元件示意图91㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析2.6㊀陆地棉FAD家族基因在盐胁迫和干旱胁迫下的表达特征为明确FAD基因在盐胁迫和干旱胁迫下的表达模式ꎬ基于转录组数据对37个FAD基因在两种胁迫处理不同时间点陆地棉叶片中的表达量进行聚类分析ꎬ结果如图7所示ꎮ图中log2(FC)代表log2(FoldChange)ꎻNaCl-3h㊁NaCl-6h㊁NaCl-12h㊁NaCl-24h分别代表盐处理3㊁6㊁12㊁24h后的样品ꎻPEG-3h㊁PEG-6h㊁PEG-12h㊁PEG-24h分别代表干旱处理3㊁6㊁12㊁24h后的样品ꎮ图7㊀陆地棉37个FAD基因在盐胁迫和㊀㊀㊀干旱胁迫下的表达分析㊀㊀盐胁迫下ꎬ37个FAD基因呈现不同的表达模式ꎬ其中有28个FAD基因在不同处理时间均有表达ꎬ7个FAD基因未检测到表达信号ꎬ而GH_A06G1435基因仅在处理后6h检测到表达量增加ꎬGH_D01G2529仅在处理后24h检测到表达量增加ꎮ另外ꎬ28个FAD基因呈现不同的表达模式ꎬ17个FAD基因表达量降低ꎬ7个FAD基因表达量增加[log2(FC)>1]ꎬ4个基因的表达量先增后降ꎬ2个基因的表达量先降后增(GH_D08G2811和GH_D11G0999)ꎮ干旱胁迫下ꎬ37个FAD基因中有27个在不同处理时间均有表达ꎬ有6个未检测到表达量变化ꎬ有4个仅在两个处理时间点下表达量增加ꎮ另外ꎬ27个FAD基因中仅GH_D04G1454表达量增加ꎬ17个基因在干旱胁迫下表达量降低ꎬ9个基因仅在一个或两个处理时间(6h或12h)点下表达量增加ꎬ其他时间点的表达量降低ꎮ褪黑素作为一种重要的多效性抗氧化分子ꎬ能帮助植物抵御不利环境的危害ꎬ而且外源褪黑素能显著缓解逆境胁迫引起的伤害[30ꎬ31]ꎮ因此ꎬ本研究基于干旱胁迫和盐胁迫下FAD基因的转录组结果ꎬ选出25个FAD基因(包括Front-end亚家族的8个ꎬOmega亚家族的15个ꎬFirst亚家族的1个ꎬSphingolipid亚家族的1个)ꎬ通过在棉花苗期施加外源褪黑素ꎬ进一步探究FAD基因在外源褪黑素下的表达特征ꎮ定量分析结果显示ꎬ在褪黑素处理后ꎬ1个FAD基因(GH_A01G1380)未检测到表达量的变化ꎬ另外24个FAD基因与对照相比ꎬ有7个基因表达量增加ꎬ17个基因表达量降低ꎬ且上调表达的FAD基因均在处理后6h表达量增加最多ꎮFront-end亚家族8个FAD基因中有6个基因在褪黑素处理后表达量增加ꎬ2个基因表达量降低ꎻOmega亚家族表达的14个FAD基因中仅有1个在处理后表达量增加ꎬ其他基因都呈现表达量降低趋势ꎻFirst㊁Sphingolipid亚家族的各1个基因则均在处理后表现为表达量降低(图8)ꎮ3㊀讨论与结论植物脂肪酸作为储藏油脂和细胞膜脂的重要组分ꎬ既为植物的生长发育提供能量ꎬ也在响应多种逆境胁迫中发挥重要作用[3]ꎮFAD是植物脂肪酸进一步去饱和反应中的关键酶ꎬ目前已在拟南芥㊁水稻㊁花生㊁大豆和油菜等物种中相继完成了全基因组水平上的鉴定和分析[16]ꎮ本研究从陆地棉基因组中鉴定出37个FAD基因ꎬ而二倍体雷蒙德氏棉仅含有19个FAD成员[4]ꎬ是其1.95倍ꎬ造成这种差异的原因可能是棉花进化过程中的异源多倍化现象ꎮ棉属植物进化分析表明ꎬ在170万~190万年前ꎬ陆地棉由A基因组供体亚洲棉和D基因组供体雷蒙德氏棉种间杂交加倍而成[14]ꎮ另外ꎬ陆地棉FAD基因数量远多于模式植物拟南芥和水稻ꎬ分别是其2.18倍和3.70倍ꎮ02山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀柱上∗㊁∗∗分别表示同一基因在不同处理时间与处理0h相比差异显著(P<0.05)㊁极显著(P<0.01)ꎮ图8㊀陆地棉24个FAD基因在褪黑素处理不同时间后的表达分析㊀㊀进化分析将陆地棉和拟南芥的FAD基因分为4个亚家族ꎬ且每个物种的FAD基因在4个亚家族中均有分布ꎬ表明FAD基因的分化可能早于棉花和拟南芥的物种分化ꎮ除了First亚家族ꎬ其他3个亚家族中陆地棉FAD基因的数量远多于拟南芥ꎬ预示Omega㊁Front-end和Sphingolipid亚家族中的陆地棉FAD基因可能在进化过程中发生了物种特异性扩张ꎮ这一结果与之前二倍体雷蒙德氏棉中的报道一致[4]ꎮ基因复制是植物进化的主要动力ꎬ主要包括12㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析串联复制㊁片段复制和全基因组复制[28]ꎮ本研究在陆地棉FAD基因家族中发现了28对复制基因ꎬ来源于4个亚家族ꎬ且全为片段复制ꎬ表明在陆地棉进化过程中FAD基因由于片段复制发生了基因家族扩张ꎮKa/Ks值常用来检验同源基因是否经历了选择作用ꎬKa/Ks>1ꎬ认为有正选择作用ꎻKa/Ks<1ꎬ为纯化或负选择作用ꎻKa/Ks=1ꎬ则认为存在中性选择作用ꎮ本研究中所有复制基因的Ka/Ks值均小于1ꎬ表明陆地棉FAD基因在进化过程中经历了纯化选择作用ꎮ顺式作用元件分析发现ꎬ陆地棉FAD基因的启动子区含有植物激素和逆境胁迫响应元件ꎬ预示其可能在激素信号响应和防御逆境胁迫中起作用ꎮ目前ꎬ已知雷蒙德氏棉中有7个FAD基因受低温诱导表达ꎬ5个FAD基因受低温抑制表达[4]ꎮ部分FAD基因呈现组织特异性表达ꎬ如FAD2-1在发育的种子中特异表达[32]ꎬFAD2-2和FAD2-3在种子发育过程中组成型表达ꎬ在叶片中少量表达[33]ꎮ植物中位于质膜上的脂肪酸大部分是不饱和脂肪酸ꎬ而植物对环境胁迫的抗性与质膜上脂肪酸的不饱和程度密切相关ꎬ膜结合基因对维持植株在多种胁迫下的正常生长起着非常重要的作用ꎮ在拟南芥中ꎬFAD2和FAD6是提高幼苗早期生长耐盐性的重要基因[34ꎬ35]ꎬFAD7基因的反义表达则降低了对盐胁迫和干旱胁迫的耐性[36]ꎮ在番茄中超表达LeFAD3基因增强幼苗早期生长的耐盐性[37]ꎮ为探究陆地棉FAD基因在逆境胁迫下的表达模式ꎬ利用盐胁迫和干旱胁迫的转录组数据对陆地棉37个FAD基因进行分析ꎬ发现70%以上的FAD基因对两种胁迫均有应答ꎬ其中下调表达的FAD基因分别占54%和63%ꎮ值得注意的是ꎬ本研究Front-end亚家族包含的10个基因中除了亚细胞定位预测在溶酶体的GH_D12G1274基因外ꎬ其余9个基因全部定位于质膜ꎬ干旱胁迫后7个基因都是下调表达ꎬGH_A12G1258和GH_D11G0999基因也仅在6h或24h表达量增加ꎬ说明干旱胁迫可能会抑制质膜上FAD基因的表达ꎮ褪黑素在植物非生物胁迫防御系统中发挥重要作用ꎬ且几乎所有非生物胁迫引起的过氧化伤害都能被褪黑素缓解ꎮ褪黑素提高植物对环境胁迫抗性机制主要是通过提高抗氧化物质含量和抗氧化酶活性ꎬ改善光合系统ꎬ调控激素合成和代谢ꎬ调控植物细胞中初级和次级代谢ꎬ刺激细胞合成更多的保护物质等方式[38]ꎮ通过定量PCRꎬ进一步分析了25个FAD基因在施加外源褪黑素后的表达特征ꎬ发现17个基因下调表达ꎬ7个基因上调表达ꎬ1个基因没有表达变化ꎮ值得注意的是ꎬFront-end亚家族中的5个基因在干旱胁迫后下调表达而在褪黑素处理后上调表达ꎬ且在处理后6h表达量显著增加ꎬ它们可作为候选基因用于后续基因功能的进一步探究ꎮ本研究结果可为进一步揭示陆地棉FAD基因逆境响应机理提供参考ꎮ参㊀考㊀文㊀献:[1]㊀戴晓峰ꎬ肖玲ꎬ武玉花ꎬ等.植物脂肪酸去饱和酶及其编码基因研究进展[J].植物学通报ꎬ2007ꎬ24(1):105-113. [2]㊀阮建ꎬ单雷ꎬ李新国ꎬ等.花生FAD基因家族的全基因组鉴定与表达模式分析[J].山东农业科学ꎬ2018ꎬ50(6):1-9. [3]㊀曹福亮ꎬ王欢利ꎬ郁万文ꎬ等.高等植物脂肪酸去饱和酶及其编码基因研究进展[J].南京林业大学学报(自然科学版)ꎬ2012ꎬ36(2):125-132.[4]㊀LiuWꎬLiWꎬHeQꎬetal.Characterizationof19genesenco ̄dingmembrane ̄boundfattyaciddesaturasesandtheirexpres ̄sionprofilesinGossypiumraimondiiunderlowtemperature[J].PLoSONEꎬ2015ꎬ10(4):e0123281.[5]㊀LeeKRꎬLeeYꎬKimEHꎬetal.Functionalidentificationofoleate12 ̄desaturaseandω ̄3fattyaciddesaturasegenesfromPerillafrutescensvar.frutescens[J].PlantCellReportsꎬ2016ꎬ35(12):2523-2537.[6]㊀ArondelVꎬLemieuxBꎬHwangIꎬetal.Map ̄basedcloningofagenecontrollingomega ̄3fattyaciddesaturationinArabidop ̄sis[J].Scienceꎬ1992ꎬ258(5086):1353-1355. [7]㊀杨志刚ꎬ郭子好ꎬ姚琴琴ꎬ等.脂肪酸去饱和酶基因的研究进展[J].生物技术通报ꎬ2013(12):21-26. [8]㊀宋俊乔ꎬ孙培均ꎬ张霞ꎬ等.棉仁高油分材料筛选及其脂肪酸发育分析[J].棉花学报ꎬ2010ꎬ22(4):291-296. [9]㊀HaunWꎬCoffmanAꎬClasenBMꎬetal.Improvedsoybeanoilqualitybytargetedmutagenesisofthefattyaciddesaturase2genefamily[J].PlantBiotechnologyJournalꎬ2014ꎬ12(7):934-940.[10]王福军ꎬ赵开军.基因组编辑技术应用于作物遗传改良的进展与挑战[J].中国农业科学ꎬ2018ꎬ51(1):1-16. [11]LiuQꎬSinghSPꎬGreenAG.High ̄stearicandhigh ̄oleiccot ̄tonseedoilsproducedbyhairpinRNA ̄mediatedpost ̄transcrip ̄tionalgenesilencing[J].PlantPhysiol.ꎬ2002ꎬ129:1732-1743.22山东农业科学㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀第55卷㊀[12]PatersonAHꎬWendelJFꎬGundlachHꎬetal.RepeatedpolyploidizationofGossypiumgeneomesandtheevolutionofspinnablecottonfibres[J].Natureꎬ2012ꎬ492:423-427. [13]LiFꎬFanGꎬWangKꎬetal.Genomesequenceofthecultivat ̄edcottonGossypiumarboretum[J].NatureGeneticsꎬ2014ꎬ46(6):567-572.[14]HuYꎬChenJꎬFangLꎬetal.GossypiumbarbadenseandGos ̄sypiumhirsutumgenomesprovideinsightsintotheoriginande ̄volutionofallotetraploidcotton[J].NatureGeneticsꎬ2019ꎬ51(4):739-748.[15]WangMꎬTuLꎬYuanDꎬetal.ReferencegenomesequencesoftwocultivatedallotetraploidcottonsꎬGossypiumhirsutumandGossypiumbarbadense[J].NatureGeneticsꎬ2019ꎬ51(2):224-229.[16]XuYꎬChenBꎬWangRꎬetal.Genome ̄widesurveyandchar ̄acterizationoffattyaciddesaturasegenefamilyinBrassicana ̄pusanditsparentalspecies[J].AppliedBiochemistryandBio ̄technologyꎬ2018ꎬ184(2):582-598.[17]ZhuTꎬLiangCZꎬMengZGꎬetal.CottonFGD:anintegratedfunctionalgenomicsdatabaseforcotton[J].BMCPlantBiolo ̄gyꎬ2017ꎬ17:101.[18]FinnRDꎬCoggillPꎬEberhardtRYꎬetal.ThePfamproteinfamiliesdatabase:towardsamoresustainablefuture[J].Nu ̄cleicAcidsResearchꎬ2016ꎬ44:D279-D285.[19]ArtimoPꎬJonnalageddaMꎬArnoldKꎬetal.ExPASy:SIBbioinformaticsresourceportal[J].NucleicAcidsResearchꎬ2012ꎬ40:W597-W603.[20]YuCSꎬChenYCꎬLuCHꎬetal.Predictionofproteinsubcel ̄lularlocalization[J].Proteins:StructureꎬFunctionandBioin ̄formaticsꎬ2006ꎬ64(3):643-651.[21]KumarSꎬStecherGꎬLiMꎬetal.MEGAX:molecularevolu ̄tionarygeneticsanalysisacrosscomputingplatforms[J].Molec ̄ularBiologyandEvolutionꎬ2018ꎬ35(6):1547-1549. [22]VoorripsR.MapChart:softwareforthegraphicalpresentationoflinkagemapsandQTLs[J].JournalofHeredityꎬ2002ꎬ93(1):77-78.[23]HuBꎬJinJꎬGuoAYꎬetal.GSDS2.0:anupgradedgenefea ̄turevisualizationserver[J].Bioinformaticsꎬ2015ꎬ31(1):1296-1297.[24]BaileyTLꎬJohnsonJꎬGrantCEꎬetal.TheMEMESuite[J].NucleicAcidsResearchꎬ2015ꎬ43:W39-W49.[25]LescotMꎬDehaisPꎬThijsGꎬetal.PlantCAREꎬadatabaseofplantcis ̄actingregulatoryelementsandaportaltotoolsforinsilicoanalysisofpromotersequences[J].NucleicAcidsRe ̄searchꎬ2002ꎬ30(1):325-327.[26]WangYꎬTangHꎬDeBarryJDꎬetal.MCScanX:atoolkitfordetectionandevolutionaryanalysisofgenesyntenyandcol ̄linearity[J].NucleicAcidsResearchꎬ2012ꎬ40(7):e49. [27]KrzywinskiMꎬScheinJEꎬBirolIꎬetal.Circos:aninforma ̄tionaestheticforcomparativegenomics[J].GenomeResearchꎬ2009ꎬ19(9):1639-1645.[28]LiuZꎬFuMꎬLiHꎬetal.SystematicanalysisofNACtran ̄scriptionfactorsinGossypiumbarbadenseuncoverstheirrolesinresponsetoVerticilliumwilt[J].PeerJꎬ2019ꎬ7:e7995. [29]ZhangZꎬLiJꎬZhaoXQꎬetal.KaKs_Calculator:calculatingKaandKsthroughmodelselectionandmodelaveraging[J].GenomicsProteomicsBioinformaticsꎬ2013ꎬ4(4):259-263. [30]LiHꎬChangJꎬChenHꎬetal.Exogenousmelatoninconferssaltstresstolerancetowatermelonbyimprovingphotosynthesisandredoxhomeostasis[J].FrontiersinPlantScienceꎬ2017ꎬ8:295.[31]LiHꎬGuoYꎬLanZꎬetal.MelatoninantagonizesABAactiontopromoteseedgerminationbyregulatingCa2+effluxandH2O2accumulation[J].PlantScienceꎬ2021ꎬ303:110761. [32]LiuQꎬBrubakerCLꎬGreenAGꎬetal.EvolutionoftheFAD2 ̄1fattyaciddesaturaseintronandthemolecularsystemat ̄icsofGossypium(Malvaceae)[J].AmericanJournalofBota ̄nyꎬ2001ꎬ88(1):92-102.[33]PirtleILꎬKongcharoensuntornWꎬNampaisansukMꎬetal.MolecularcloningandfunctionalexpressionofthegeneforacottonΔ ̄12fattyaciddesaturase(FAD2)[J].BiochimicaetBiophysicaActaꎬ2001ꎬ1522(2):122-129.[34]ZhangJTꎬZhuJQꎬZhuQꎬetal.Fattyaciddesaturase ̄6(Fad6)isrequiredforsalttoleranceinArabidopsisthaliana[J].BiochemicalandBiophysicalResearchCommunicationsꎬ2009ꎬ390(3):469-474.[35]ZhangJꎬLiuHꎬSunJꎬetal.ArabidopsisfattyaciddesaturaseFAD2isrequiredforsalttoleranceduringseedgerminationandearlyseedlinggrowth[J].PLoSONEꎬ2012ꎬ7(1):e30355. [36]ImYJꎬHanOꎬChungGCꎬetal.AntisenseexpressionofanArabidopsisomega ̄3fattyaciddesaturasegenereducessalt/droughttoleranceintransgenictobaccoplants[J].MolecularCellꎬ2002ꎬ13(2):264-271.[37]WangHSꎬYuCꎬTangXFꎬetal.Atomatoendoplasmicre ̄ticulum(ER) ̄typeomega ̄3fattyaciddesaturase(LeFAD3)functionsinearlyseedlingtolerancetosalinitystress[J].PlantCellReportsꎬ2014ꎬ33(1):131-142.[38]ArnaoMBꎬHernández ̄RuizJ.Melatoninagainstenvironmen ̄talplantstressors:areview[J].CurrentProteinandPeptideScienceꎬ2021ꎬ22(5):413-429.32㊀第4期㊀㊀㊀㊀陈义珍ꎬ等:陆地棉膜结合脂肪酸去饱和酶基因家族的全基因组鉴定及表达分析。
生物技术综合实验
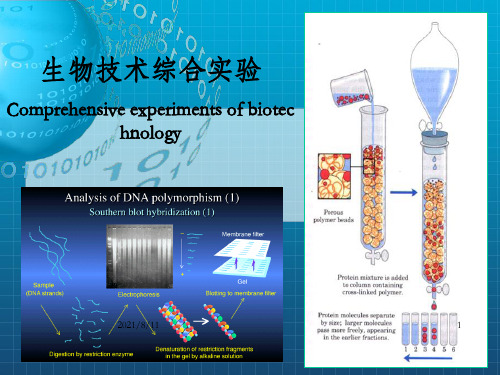
课程简介
现代生物技术综合性实验是以分子生物学为基础的基因克隆 重组技术和生物化学的蛋白质分离、纯化技术为核心的教学, 它是现代生物技术的核心。
主要内容:包括实验理论和实验技术 实验技术-生物技术综合实验,它是集目的基因的制备、克
隆、表达和蛋白质的分离纯化及其活性测定等实验方法和技 术为一体的一门综合性实验课。
chaotropic nature. As with other procedures involving RNA, gloves should be worn at all times to avoid cont
amination
of
samples
with
ribonucleases.
This method describes preparation from small quantities (~ 50mg) of tissue. Appropriate scaling of the volu mes involved can be performed to accomodate larger quantities.
目的:通过本门课程的学习,使学生掌握现代生物工程的上、
下游实验技术,对以基因克隆重组技术为主线的生物实验技
术有一个较全面的了解。
3
教学要求
通过教学,要求学生掌握分子生物学与基因工程的基本理论,巩固所学的理论知识; 使学生了解科研工作的基本思路,学会如何设计实验,如何分析实验,培养分析问题和
解决问题的能力; 培养和训练学生的基本实验技能,培养学生的独立工作能力和创造能力。 掌握基因重组的基本过程,如:核酸DNA、RNA和质粒DNA提取、酶切、连接、转化及
脱氧胆酸钠(DOC)是阴离子去污剂
杨荣武分子生物学

Common sourcethem
Contaminated solutions/buffers
1:1 phenol : chloroform or
25:24:1 phenol : chloroform : isoamyl alcohol
Phenol: denatures proteins, precipitates form at interface between aqueous and organic layer
III. DNA purification • Phenol extraction • Ethanol precipitation
IV. RNA work
What do we need DNA for?
•Detect, enumerate, clone genes •Detect, enumerate species •Detect/sequence specific DNA regions •Create new DNA “constructs” (recombinant DNA
steps
Making and using mRNA (1)
Top 10 sources of RNase contamination (Ambion Scientific website)
1) Ungloved hands 2) Tips and tubes 3) Water and buffers 4) Lab surfaces 5) Endogenous cellular RNases 6) RNA samples 7) Plasmid preps 8) RNA storage (slow action of small amounts of RNAse 9) Chemical nucleases (Mg2+, Ca2+at 80°C for 5’ +) 10) Enzyme preparations
RNAi Core Version 2 103 10 08 池化shRNA文库全基因组RNAi筛选协

Protocol for genome-wide RNAi screening using pooled shRNA library IntroductionGenome-wide RNAi library screen can be performed by two ways, namely, arrayed screen and pooled selection/screen. For arrayed screen, it needs to be assisted by high throughput equipments in order to screen a large number of shRNA constructs. On the other hand, the RNAi pooledselection/screen can be done in a regular lab because the experimental procedures don’t need high throughput instruments for proceeding. The aim of this protocol is to provide a fundamental consideration and guidelines for performing genome-wide RNAi pooled selection/screen.To successfully conduct an RNAi pooled screen/selection, it is extremely important to perform the screen in a condition that most of the transduced cells survived in the selection population receive approximately one copy of shRNA or one lentivirus. Otherwise, potential candidate shRNA mayco-select with other “unwanted shRNA(s)” in the same cell. This unwanted shRNA(s) will lead to serious signal noise or loss of the potential candidates during data analyses.To establish the principle that what MOI shall be used for transduction in order to get suitable condition for the RNAi pooled screen, i.e., most of infected cells received one copy of shRNA or one virus, virologists borrowed the equation of Poisson Distribution to model or to design the experimental condition. The Poisson equation formulates as follow:m n﹒e-mP(n)=n!Wherem is the multiplicity of infection or MOI (the ratio of infectious agent to cell);e is the natural exponent (approximately 2.7183);n is the occurrence of event that virus(s) enters into the cells;P(n) is the probability that a cell will get infected by n viruses.For instance, the probability that a cell got no virus or one virus under the conditions of MOI equals to 0.1, 0.2, 0.3, 0.5, and 1 is calculated according to the formula. In addition, the infected population is derived by the formula of 1-P(0) which is also expressed as P(infected), from where the probability of a cell got infected by greater than or equal to two viruses (≧2) can be derived from the formula, P(infected)-P(1).(If express as percentage, the formula could be expressed as {P(infected)-P(1)/P(infected)}×100%.) Those values calculated are tabulated as follow:The cell numbers required for the RNAi pooled screening are reverse proportion to MOI used. Practically, MOI equals to 0.2 or 0.3 will be recommended to perform large-scale RNAi pooled screening because MOI equals to 0.1 doesn’t reduce the probability of P(n≧2) much but it needs much more cells to reach that condition.Polybrene is a common polycation that increases the infectivity of lentivirus by 3-5 folds (unpublished observation). The cationic polymer enhanced virus adsorption and transduction by neutralizing the charge in between viral envelope and cellular membrane. However, polybrene has potential to facilitate virus aggregation which raises the possibility of multiple virus infection during pooled RNAi screen. This character of aggregation caused by polybrene increases the probability of gaining more than one lentivirus in a cell. Thus, polybrene-assisted transducing cells are not suitable for genome-wide pooled RNAi screen.In addition to these considerations, the following experimental conditions are also important for conducting RNAi pooled screening. They are described as follows:(1)Optimization of puromycin selection conditionTo generate a fully transduced population of cells for analysis, it is important to determine the minimum amount of puromycin required to eliminate untransduced cells. This isaccomplished by performing a puromycin kill curve to determinate the concentration ofpuromycin needed to eliminate the untrasduced cells. For puromycin selection, the minimum antibiotic concentration used is the lowest concentration that kills 100% of untransduced cells and maximal survival transduced cells in 48-72 hours. (Please refer to puromycin kill curve protocol)(2)Re-titrate viral titer in cell used for screeningThe susceptibility of various cell types to virus infection (in this case means VSV-G peudotyped lentivirus) is different. To determine the amounts of lentivirus necessary to achieve the desirable MOI on the recipient cells of interest, re-titration of the pooled lentivirus obtained from the National RNAi Platform is needed. (Please refer to viral titer estimation protocol : RIU method)(3)PolybreneThe use of polybrene (hexadimethrine bromide) during lentivirus transduction is improper while performing a pooled RNAi screen as mentioned above.The protocols described below provide the instructions on how to transduce pooled lentivirus into target cells and guidelines for the isolation of genomic DNA and the preparation of PCR products amplified from genomic DNA for deep sequencing.Protocolsrge-scale infection by pooled shRNA lentivirusesNot all cellular systems can be performed by the method of the RNAi pooled screening.Cell-based systems with the natures of positive or negative selection are able to be conducted by the RNAi pooled screening. Positive selection means that the copy numbers of shRNAcandidates in a particular selection condition are over the non-reactive shRNAs or controlshRNAs. On the other hand, the copy numbers of shRNA candidates in the condition of negative selection will be gradually reduced or even vanished. Usually, the ID numbers of shRNAs being analyzed in the final stage of selection are still huge (almost the same as original input). But the ID numbers of shRNAs can be very few in some positive selection condition, for instanceessential genes required for virus replication or genes involved in apoptosis pathway triggered by some inducers. To get meaningful results, the following protocol uses hTRC1&2 110k pooled lentiviruses (C6-10) as an example to describe. In addition, each shRNA (or correspondingshRNA expressing lentivirus) is duplicate to 250-500 folds (coverage is 250-500) to guarantee that effective shRNA will not lose during transduction and selection.1.Re-titrate the titer of shRNA lentivirus obtained from the National RNAi Platform in therecipient cells of interest using the experimental conditions being performed in your lab, for example spin infection or not. (It is extremely important using the MOI titrated in suchexperimental conditions to perform the RNAi pooled screening.)2.Calculate the cell numbers needed. For example, given MOI = 0.3 and coverage = 250, thenthe cell numbers required are 9.17×107 (110,000×250/0.3).3.Seed cells onto 150 cm2 dish or flask (with a cell density of 60%-80% confluence at thesecond day just before transducing lentivirus). (To reach 60%-80% confluence, the numbers of the cell required are dependent on cell type used.)4.Add lentivirus needed. (Total cell numbers multiply MOI used.) At this stage, you mayperform buck culture or divide into 11 cultures or 3 sub-pools’ viruses as a culture. (There are11 sub-pools of hTRC1&2 pooled lentivirus.)5.Incubate the infected cells at 37o C for 18-24 hours.6.Replace medium with fresh complete culture medium containing optimized concentration ofpuromycin. Return cells to CO2 incubator for selection and change medium (containingpuromycin) every three days.7.Expand and freeze the cells established.II.Library screen/ selection1.Seed shRNA-expressing cells onto 10 cm2 (cells from one sub-pool shRNAs) or 150 cm2(co-culture 3 sub-pools shRNA-expressing cells) dish or other experimental conditions forparticular phenotype selection.2.Subject to phenotype selection (highly depend on experimental designs).3.If the experiment is a long-term selection, keep the cells at an optimal condition (passage thecells regularly). You may take out 2.75×107 to 5.5×107 cells (110,000×250 to 110,000×500 for the compensation of DNA loss during purification) every week (the period is dependent onthe experimental designed)4.In the special positive selection that only a few cells survived, expand the cells to above7.5×106 to 1.5×107 cells for the isolation of genomic DNA.III.Identification of hitsTo identify the potential hits, the ID of shRNAs in selected cells can be determined by deep sequencing or microarray approach. In this protocol, only the method of deep sequencing is mentioned.The genome (with shRNA sequence) of lentivirus integrates into host chromosome after transduction. Thus, shRNA sequence can be passed down to daughter cells during cellproliferation. Therefore, the whole idea of determining shRNA ID is PCR amplification ofshRNA sequence integrated in genomic DNA and followed by sequencing. In general, 180 µg to 200 µg is the minimal amounts of DNA needed for the deconvolution of the ID of the shRNAs in the selected population. The amounts are calculated by the following formula: the numbers of shRNA of the library used × coverage × the mass of single cell. (For instance, when the library of 110K shRNAs is subjected to screening, the amounts of the DNA would be110,000×250×6.6×10-6=182 µg.) However, if only a few cells left after selection, 20 µg to 30 µg of DNA is sufficient to decode the ID of the shRNAs selected.The protocol described here includes: (i) isolation of genomic DNA; (ii) PCR amplification of shRNA; (iii) restriction enzyme digestion and gel purification of the PCR products.It is important to pay attention to note that the PCR products prepared for deep sequencing need that both 5'-ends of PCR products contain phosphor group and 3'-end with OH group. OH group allows the PCR products can be tagged with A nucleotide by taq polymerase and phosphor group allows the PCR products can be ligated to an adaptor (adaptor needs for further PCR amplification of the PCR products for subsequent deep sequencing) by TA pairing method.A.Purification of genomic DNA from selected cells1.Wash cells with ice-cold PBS twice, and then scrap the cells from dish into 15-mlcentrifuge tube. For suspension cells, collect the cells from cultural flask into 15-mlcentrifuge tube and wash the cells twice using step 2 condition.2.Pellet down cells by centrifugation at 1,000-2,000 rpm for 5 min at 4°C.3.Discard the supernatant and resuspend the cells with 3-5 ml TNE or PBS buffer, leavethe tube at RT.{TNE buffer (50 mM Tris-HCl, pH8.0; 1mM EDTA; 150 mM NaCl; 1 mM Na-azide)}4.Add SDS (final concentration: 0.5 %.)5.Mix gently by inverting the tube for several times.6.Add RNaseA (final concentration: 100-200 µg/ml).7.Mix gently by inverting the tube for several times, and then incubate at 37°C for 30min.8.Add proteinase K (final concentration: 100-200 µg/ml).9.Mix gently by inverting the tube for several times, and then incubate at 37°C for O/Nor 52°C for at least 2 hrs.10.Extract once with equal volume of phenol (pH8.0), followed by phenol/chloroformextraction till the interface is clean, finally once with chloroform. (Do NOT vortex!Instead, rotating the mixture on a rotating disc for at least 1 hr or longer.) Centrifuge at4,000 rpm for 15-30 min for separating aqueous and organic phase. (Sorvall super T21, or equivalent ST-H750 rotor.)11.Precipitate DNA by adding 2 volume of absolute ethanol, 1/10 volume of 3M NaOAc(pH5.2), and mix well.12.Transfer DNA aggregates into enppendorf tube and wash DNA twice with 70%alcohol.13.Bring down the DNA by centrifugation at 10,000 rpm for 1 min, and remove residueEtOH as much as possible.14.Air dry.15.Dissolve the DNA in 0.1x TE buffer (pH8.0) with appropriate volume to let theconcentration to be approximately 1 µg/µl. (After completely dissolving, DNAshould be very sticky!!)16.Determine DNA concentration and stock at 4°C ready for use.B.PCR amplification and gel purification5 to 10 µg of PCR products are needed for deep sequencing. To minimize bias causedby PCR amplification, the protocol has been developed/optimized by single round of PCR.The size of the PCR products is 350 bp in length. The regions of amplification andrestriction fragments being purified are depicted in the following diagram:The detailed procedures are described as follows:I.PCR amplification of shRNAs sequence from genomic DNA1.Optimize genomic DNA amounts and PCR cycle (shall be determined for eachbatch of genomic DNA). Typical results are shown in Fig 1 and Fig 2 below:Fig 1. Amounts of genomic DNA used (data from 28 cycles). Titrategenomic DNA amounts used for PCR (per 50 µl reaction). As figureindicated, 2 to 5 µg per reaction in 50 µl is subjected to PCRamplification.Fig 2. Optimization of PCR cycle.To minimize bias caused byover amplification of PCR, the exponential amplification wasdetermined by a kinetic PCR. As indicated, various PCR cycles (23,25, 27, 29, 31) were performed. The reactants were re-addedprimers, dNTPs and enzyme (the same as original amounts) andamplified for one more cycle (24, 26, 28, 30, 32) to enrich theperfect complimentary PCR products (as indicated by the lowerband of lane 5' and others). The upper band of the PCR productswas derived from DNA annealing of different shRNA species. Theresultant annealing products form bubble at the region of shRNAsequences, which lead to slower migration rate, and are resistant toXhoI digestion as indicated by the upper band of the lane of XhoI.“Purified”indicates the DNA fragments purified from gel afterAscI and XhoI digestion.The results show that PCR products of cycle 28 are good quality forNGS in terms of the following two reasons: (i) PCR maintains atexponential stage; (ii) good products for XhoI digestion.2.Set up PCR master mix as follows:Primers used: 5' U6p-TRC1/F: aggcgcgccgagggcctatttcccatg (target TRC1 vector)U6p-TRC2/F: aggcgcgccagagagggcctatttcccatg (target TRC2 vector)LKO1&5/R: tgtggatgaatactgccatttgtctc3.Add the above reagents sequentially into eppendorf tube (except DNA polymerase)and mix thoroughly by vortex.4.Add DNA polymerase after brief spin, and then gentle mix the mixture followed byspin down the mixture.5.Aliquot 50 µl of the mix into six 200 µl PCR tubes.6.Perform the PCR reaction using the following parameters:7.Pool the contents of amplifying products together.8.Analyze by 1-1.5% agarose gel to check the quantity and quality of the PCRproducts.9.Spin column purification to remove primers and dNTPs, and to concentrate PCRproducts to appropriate volume with 0.1x TE buffer. The products are ready forrestriction enzyme digestion.II.Digestion of the PCR products with XhoI and AscI restriction enzymes1.Set up the restriction enzyme reaction to digest PCR products as follows:PCR products ---------------------------------- 650 µl10x FastDigest Buffer ------------------------- 78 µlFastDigest XhoI (進階Cat#: FD0695) ----- 20 µlFastDigest AscI (進階Cat#: FD1894) ---- 20 µl2.Incubate the mixture at 37ºC for 5 hours to overnight.3.Analyze 3 µl of the digested mixture using 1.5% agarose gel electrophoresis toensure the digestion is complete.4.Add dNTP mix (10 mM each) to final concentration of 0.2 mM followed by adding100 units of Klenow fragment of DNA polymerase I.5.Incubate at RT for 30 min to fill-in the protruding ends.6.Concentrate the digestion mixtures by passing them through 6 Qiagen spincolumns (QIAquick Cat. # 14208). This procedure also gets rid of unwanted smallfragments that will interfere the subsequent gel purification.7.Elute the DNA with 60 µl 0.1x TE buffer and pool all eluents together.8.The sample is ready for gel purification.IV.Isolation of digested products from agarose gel1.Cast three sealed wells with 1.5% NuSieve 3:1 agarose (Lonza) containingethidium bromide (0.5 µg/ml) with a volume capacity of 130 µl per sealed well(TAE buffer give rise to better resolution).2.Separate the DNA fragments at 50 volts for above 2 hrs.3.Locate the desired bands with a long-wave (355-360 nm) portable UV lamp.4.Cut out the region of the desired DNA band with a razor blade. Chopped the gelslice into small pieces as much as possible and transfer the sliced gel into a 1.5 mlmicrofuge tube.5.Add 1.5 to 2 volumes of Tris-saturated phenol (pH 8.0) into tube vortex for 30seconds. Incubate the tube at RT for 10 min, then add 20 µg of glycogen (Roche).Freeze the content at -80°C for at least 5 hrs to overnight.6.Centrifuge at 13,000 rpm in a table top centrifuge for 30 to 60 min at 18°C.7.Transfer the upper layer of aqueous phase to a new microfuge tube.8.Extract the aqueous solution once with equal volume of saturated phenol [pH8.0],once with phenol/chloroform, finally once with chloroform. At each step shakes the tube by vortex for 30-60 seconds vigorously and centrifuge at highest speed in atable top centrifuge for 30 min at 4°C.9.Finally, transfer the aqueous phase to a new microfuge tube, add 0.1x volume of 3M sodium acetate (pH 5.2) and 2 volumes of absolute ethanol and put the tube at-20℃for overnight.10.Centrifuge the tube at 13,000 rpm for 10 min at 4℃.11.Wash the pellet with 70% ethanol twice, air dry and dissolve the pellet in 30 µl of0.1x TE. (adjust the DNA concentration to be approximately 200 ng/µl.)12.Determine DNA concentration (usually the ratio of 260/280 shall be in the range of1.85-2.0; and 260/230 shall be greater than 2.3) and check the quality of the DNAby taking out 50-100 ng of DNA and runs the sample in 1.5% agarsoe gel.13.The DNA is ready for deep sequencing.RNAi Core Version 2103/10/08 Links1.Puromycin kill curve protocol.tw/file/protocol/19_PuromycinKillCurve.pdf2.Viral titer estimation protocol: RIU method.tw/file/protocol/4_1_EstimationLentivirusTiterRIUV1.pdf。
黑果枸杞花色苷的提取、纯化及降解动力学研究

连敏,高艺玮,年新,等. 黑果枸杞花色苷的提取、纯化及降解动力学研究[J]. 食品工业科技,2024,45(6):24−31. doi:10.13386/j.issn1002-0306.2023080105LIAN Min, GAO Yiwei, NIAN Xin, et al. Study on Extraction, Purification and Degradation Kinetics of Anthocyanins from Lycium ruthenicum [J]. Science and Technology of Food Industry, 2024, 45(6): 24−31. (in Chinese with English abstract). doi:10.13386/j.issn1002-0306.2023080105· 特邀主编专栏—枸杞、红枣、沙棘等食药同源健康食品研究与开发(客座主编:方海田、田金虎、龚桂萍) ·黑果枸杞花色苷的提取、纯化及降解动力学研究连 敏,高艺玮,年 新,王梦泽*(宁夏大学食品科学与工程学院,宁夏银川 750021)摘 要:以花色苷提取量为主要考察指标,通过单因素和正交试验优化冻干黑果枸杞花色苷提取工艺,并在此条件下研究花色苷纯化工艺及其降解动力学,探讨不同温度、pH 下花色苷提取量的变化。
结果表明,提取最佳工艺条件为:料液比1:25(g :mL )、乙醇浓度60%、pH4、提取时间2 h ,此条件下花色苷提取量达36.507±0.325 mg/g 。
研究显示AB-8大孔树脂纯化黑果枸杞花色苷效果最好,对花色苷吸附量和解吸量的影响效果最佳,其最佳条件为:上样液浓度200 mg/100 g ,解吸乙醇浓度80%,上样流速2 mL/min ,洗脱流速2 mL/min ,上样体积为5 BV ,纯化率为90.02%。
(整理)基因工程学原理专业名词

第十九章基因工程Chapter 19 GeneEngineering第一节绪论Section 1 IntroductionDNA含有4种杂环碱基:嘌呤类有腺嘌呤(A)和鸟嘌呤(G),嘧啶类有胞嘧啶(C)和胸腺嘧啶(T);而在RNA中尿嘧啶(U)替代了结构非常相似的胸腺嘧啶。
In DNA, there are four heterocyclic bases: adenine (A) and guanine (G) are purines; cytosine (C) and thymine (T) are pyrimidines. In RNA, thymine is replaced by the structurally very similar pyrimidine, uracil (U).核苷由碱基共价结合于戊糖分子的1’位而构成。
RNA中的戊糖为核糖,构成核糖核苷或简称核苷;而在DNA中戊糖为2’—脱氧核糖,形成2’—脱氧核糖核苷或简称脱氧核苷。
碱基+糖分子=核苷。
A nucleoside consists of a base covalently bonded to the 1’-position of a pentose sugar molecule. In RNA the sugar is ribose and the compounds are ribonucleosides, or just nucleosides,whereas in DNA it is 2’-deoxyribose,and the nucleosides are named 2’-deoxyribonucleosides,or just deoxynucleosides. Base+sugar=nucleoside.DNA分子通常以双螺旋形式存在。
两条独立的反向平行的单链DNA分子以右手螺旋方式相互缠绕,糖-磷酸骨架在外,靠氢键和堆积力相互配对的碱基在内。
分子生物实验英语

分子生物实验英语作文题目:Exploring the Fascinating World of Molecular Biology through Laboratory ExperimentsMolecular biology experiments provide a fascinating gateway into the intricate inner workings of life at the molecular level. These meticulously designed laboratory exercises allow students and researchers to delve into the complexities of DNA replication, gene expression, protein synthesis, and cellular communication, among other fundamental biological processes.One typical experiment involves the extraction and analysis of DNA from living organisms. Starting with a sample of cells, participants follow a series of steps involving lysis, precipitation, and purification to isolate the genetic material. The thrill of seeing DNA strands precipitate out of solution, often resembling ghostly white threads, is a tangible manifestation of the abstract concepts learned in textbooks. Further techniques like gel electrophoresis and PCR (Polymerase Chain Reaction) enable the visualization and amplification of specific DNA segments, respectively, offering insights into genetic variation and forensic applications.Another captivating experiment revolves around themanipulation of genes through techniques like CRISPR-Cas9 genome editing. By designing guide RNAs to target specific genomic loci and introducing the Cas9 enzyme, scientists can precisely cut and modify DNA sequences. Witnessing the power to alter an organism's genetic blueprint is bothawe-inspiring and thought-provoking, raising ethical discussions while showcasing the immense potential of gene therapy and crop improvement.Protein studies also play a pivotal role in molecular biology labs. Techniques like SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) separate proteins based on their size, allowing for characterization and comparison. Western blotting, another cornerstone method, detects specific proteins within complex mixtures using antibodies, shedding light on protein abundance, modification, and interactions.Moreover, experiments focusing on RNA analysis, such as RT-qPCR (Reverse Transcription Quantitative PCR), help quantify gene expression levels, providing valuable insights into how cells respond to various stimuli or disease states. Additionally, techniques like yeast two-hybrid assays orpull-down assays facilitate the study of protein-proteininteractions, crucial for understanding cellular signaling networks.。
PCR标准方法

PCR标准方法(看不到内容?请点击鼠标右键,选择“刷新”就会出现。
)・Amplification of Genomic DNA by PCR (Robert H. Cruickshank)提供了标准而且详细的方法,包括了一些疑难问题的解答。
・Calculating Concentrations for PCR (Molecular Biology Techniques Manual) 关于如何计算引物和核酸浓度的方法。
・PCR (Fermentas)提供了详细的方法介绍,包括混合液的制备、循环条件等等・PCR (Bowtell Lab)提供了详细的方法介绍……・PCR (Julie B. Wolf,UMBC)阐述了PCR实验的条件。
・PCR (Dr. Chastain)有关的PCR条件和开始反应……・PCR and multiplex PCR guide (Tavi)关于优化PCR方法的详细指导,例如不同试剂的浓度、循环条件等。
・PCR Debugging (Hoshi Lab)提供了一些疑难问题的解决方法。
・PCR Guide (Qiagen)提供了详细的实验指导,包括引物设计(含变性引物的设计)和长期PCR循环的条件……・PCR Guide (Michael Blaber's Lab)对于选择聚合酶、设计引物等的一些源于实验的、有用的指导。
・PCR Manual (BMB)源于实验手册,内容涵盖了PCR方法的详细步骤。
・PCR Program Design (Tavi)提供了一种很有效的方法。
・PCR Trouble Shooting (Alkami Biosystems)非常有用的疑难问题解决方法。
・Standard PCR (Chen)介绍了标准的方法,包括引物的设计、循环的控制等・Standard PCR (Hoshi Lab)提供了一种标准的方法……・Standard PCR (Biotech)提供了详细的实验指导,包括引物制备等等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Experiment 1Extraction and Purification of Genomic DNASafety Guidelines●The temperature of liquid nitrogen is -196ºC, contact with liquid nitrogen willcause frostbite. Wear safety face shield and protective gloves when handling liquid nitrogen.●Phenol and chloroform are both toxic and produce noxious fumes. Work in thefume hood while using phenol:chloroform and chloroform-isoamyl alcohol.Because phenol and chisam (24:1 v/v chloroform:isoamyl alcohol) are solvents that are caustic and will burn skin and clothes, be sure to always wear a lab coat and gloves.I.IntroductionAn organism’s genetic material is contained in its genome encoded in the base sequence of double stranded DNA. It is in this form that genetic information is passed from generation to generation. To facilitate the analysis of genomic DNA, it is first necessary to isolate genomic DNA in a useful (purified and high molecular weight) state. DNA isolation and purification is of crucial importance since pure and high molecular weight DNA is an absolute requirement for the success and reliability of DNA fingerprinting and other molecular biology techniques. Any impurities in the DNA can inhibit enzymatic reactions, electrophoresis, quantification and sequencing. Contamination of genomic DNA with RNA and proteins may exist during the DNA extraction procedures. Therefore, the isolation of genomic DNA from cells or tissue typically involves a sequential process of selective degradations and selective recoveries, such that biomolecules other than DNA are eliminated.Molecular biological studies of plants require high-quality DNA. Plant DNA isolations also face with contaminations of polyphenolic compounds and polysaccharide. Polysaccharide contamination may inhibit most DNA modifying enzymes and measures must be used to prevent polysaccharide contamination during extraction of plant DNA. The CTAB method of DNA extraction is the best method of DNA extraction for obtaining good quality total DNA from many plant species. The method eliminates contaminating polysaacharides by CTAB (cetyltrimethylammonium bromide) precipitation.The preparation of DNA from any cell type involves the same general steps: (i) breaking open the cell (and the nuclear membrane, if applicable), (ii) removingproteins and other cell debris from the nucleic acid and (iii) final purification. If a cell is enclosed by a membrane only, the cell contents can be released by dissolving the membrane with detergent. As the cell membranes dissolve, the cell contents flow out, forming a soup of nucleic acid, dissolved membranes, cell proteins and other cell contents referred to as a cell lysate. Additional treatment is required for cells with walls, such as plant or yeast cells. These treatments can include enzymatic digestion of the cell wall material or physical disruption by means such as blending or grinding. In this experiment, genomic DNA from animal tissues and plant tissues are extracted.II.Materials-Tissue Lysis Buffer at 50°C-CTAB Extraction Buffer at 60°C (For plants containing high po1ysaccharide levels, i.e.glutinous sap, increase the concentration of CTAB to 3% or higher. For plants with high concentrations of phenolic compounds (i.e. oaks, walnuts) add 1% (w/v) polyvinylpyrollidine (PVP-40))-Phenol-chloroform (1:1 v/v) Store at 4°C in a dark glass bottle-Chloroform-isoamyl alcohol (24:1 v/v)-Isopropanol (COLD)-70% (v/v) Ethanol-Autoclaved distilled water-Frozen rat liver tissue-Fresh leaves-Liquid nitrogen-Autoclaved pestle and mortar x 2-Autoclaved spatula x 2- 2 ml microcentrifuge tube x 4- 1.5 ml microcentrifuge tube x 2-Forceps-50°C & 60°C water bath-Vortex-Timer-Ice box-1000-μl, 200-μl and 2-μl pipettes and pipette tips-Wide-bore pipette tips-Safety face shield-Protective gloves-NanoDrop® ND-1000 spectrophotometerTissue Lysis Buffer2X CTAB Extraction BufferIII.ProcedureA.Liver DNA Extraction (~ 1 hr)1.Warm up tissue lysis buffer to 50°C.2.Pre-chill the mortar by pouring liquid nitrogen into it (CAUTION: Liquid N2 isan ASPHYXIANT agent. The temperature of liquid nitrogen is -196ºC, CONTACT WITH LIQUID N2 WILL CAUSE FROSTBITE).3.Place the frozen liver tissue into the pre-chilled mortar by a pre-cooled forceps.Grind the tissue to fine powder with the pre-chilled mortar and pestle. Be sure not to let the liquid nitrogen evaporate completely (if it does, the liver tissue sample will warm and will then bond to the bottom of the mortar and it will be very difficult to remove), liquid N2should be occasionally added to keep the tissue frozen.Continue grinding and adding liquid nitrogen until the tissue is completely pulverized.4.Scrape the frozen powder (~ 20 - 50 mg) into a pre-chilled 2 ml microcentrifugetube with a pre-cooled spatula. Note: The tissues are rich in nuclease, so the frozen powder should not be thawed until disrupted in warm lysis buffer.5.Add 600 μl pre-heated tissue lysis buffer to the tube and mix well by vortex. Note:The vortex/homogenization step is necessary for the effective tissue dispersal.6.Incubate the sample at 50°C for 30 min with occasional gentle swirling.B. Plant DNA Extraction (~1 hr)1.Warm up CTAB extraction buffer to 60°C.2.Wash plant specimens with tap water and then rinse it with distilled waterfollowed by 70% ethanol for 5 min.3.Dry liquid residues with paper towel.4.Peel off any damaged surface with a razor (WEAR GLOVES to avoidcontamination).5.Cut leaves into small pieces.6.Pre-chill the mortar by pouring liquid nitrogen into it.7.Place cut leaves into the pre-chilled mortar, freeze tissue in liquid N2(Note:Avoid pouring liquid N2directly onto sample as this may boil up the sample).Grind the tissue to fine powder with the pre-chilled mortar and pestle. Be sure not to let the liquid nitrogen evaporate completely, liquid N2should be occasionally added to keep the tissue frozen.Continue grinding and adding liquid nitrogen until the tissue is completely pulverized.8.Scrape the frozen powder (~ 0.1 g) into a pre-chilled 2 ml microcentrifuge tubewith a pre-cooled spatula. Note: Plant tissues are rich in nuclease, so the frozen powder should not be thawed until disrupted in warm extraction buffer.9.Add 600 μl pre-heated CTAB extraction buffer to the tube (this should be doneinside the fume hood) and mix well by vortex. Note: The vortex/homogenization step is necessary for the effective tissue dispersal.10.Incubate the sample at 60°C for 30 min with occasional gentle swirling.C. Purification and Quantification of Genomic DNA (~2 hrs)1.Add equal volume (600 μl) of phenol-chloroform (1:1 v/v) to the sample in thefume hood, close the cap and mix the sample by several gentle inversion.2.Centrifuge at 16,000 g at room temperature for 10 min in microcentrifuge.3.Transfer 450 μl upper yellow, aqueous phase with a wide-bore pipette to a new1.5 ml microcentrifuge tube. Note: When transferring the aqueous (upper)phase, it is essential to draw the DNA into the pipette very slowly to avoid disturbing the material at the interface and to minimize hydrodynamic shearingforces.4.Add the same volume (450 μl) of chloroform-isoamyl alcohol (24:1 v/v) to thetube in the fume hood and gently mix the sample by inverting the tube several times. Note: Chloroform-isoamyl alcohol is added to remove traces of phenol.5.Centrifuge at 16,000 g at room temperature for 10 min.6.Carefully transfer 300 μl top, aqueous phase to a new 2 ml microcentrifuge tubewith a wide-bore pipette. Avoid contamination with the interface.7.Precipitate the DNA with 2/3 volume (200 μl) of -20︒C isopropanol (mix gentlyand leave to stand at room temperature for 10 min to precipitate the nucleic acids).8.Spin down the precipitated nucleic acids by centrifuging at maximum speed at4°C for 15 min.9.Discard the supernatant, wash the DNA pellet twice with 1 ml 70% ethanol,centrifuge at maximum speed for 10 min each.10.Discard the supernatant. Air-dry the pellet by concentrator for 3 min (DO NOTOVERDRY genomic DNA) and resuspend the pellet in 100 μl autoclaved distilled water. Dissolve the pellet by gentle pipetting.11.Determine the concentration and purity of the purified DNA with NanoDrop®ND-1000 spectrophotometer by measuring A260 and A280 of the DNA sample with1.5 μl of autoclaved distilled water as blank.Note:Reading “1” at 260 nm or A260 corresponds to 50 μg/ml of double-stranded DNA.A260/A280 ratio of ≥1.75 indicates a good DNA preparation.Results and interpretation:Base on the reading you obtained, calculate the concentration and total amount of each genomic DNA that you isolated. (Show the calculations clearly)Does your DNA appear to be a fairly pure sample? If not, can you speculate on the possible nature of the contaminating material?Questions:1.Some components of tissue lysis buffer and CTAB extraction buffer are different;state the functions of the different components in the buffers.2.Why don’t we use fine-mouth pipette tips in transferring genomic DNA?3.Can you suggest a method that can be used to monitor the integrity and purity ofthe genomic DNA?。