药理学 第三章药动学 重点知识总结
药理学各章重点

药理学第一章一、药理学的性质与任务1.药理学(pharmacology):是研究药物与机体之间相互作用和规律的一门学科。
2.药效动力学(药效学,pharmacodynamics):研究药物对机体的作用及规律。
3.药代动力学(药动学,pharmacokinetics):研究机体对药物的作用及规律。
4.药理学的学科任务:阐明药物作用机制;提高药物疗效;研究开发新药;发现药物新用途;探索细胞生理、生化及病理过程。
第二章药物效应动力学1.药物作用与药物效应:(1)药物作用(drug action)是指药物对机体的间的原发作用。
(2)药物效应(pharmacological effect)是指药物原发作用引起的机体器官原有功能的改变。
2.药物作用的方式:①局部作用:药物无需吸收,而在用药部位直接产生作用。
②全身作用:药物吸收入血循环后分布到机体各组织而发挥作用,也称为吸收作用或系统作用。
Ps:药物不一定要经过吸收才产生全身作用,如iv。
3.药物作用的选择性(selectivity):药物对某些器官或组织有作用或作用强,而对其他器官或组织无作用或作用弱。
选择性分药物对机体组织的选择性和抗菌药对致病菌的选择性即抗菌谱。
4.药物作用的两重性—治疗作用与不良反应:(1)治疗作用(therapeutic effects)凡能达到防治效果的作用称为治疗作用。
①对因治疗(etiological treatment)针对病因的治疗称对因治疗,或称治本,如抗菌药物杀灭致病菌。
②对症治疗(symptomatic treatment)用药目的在于改善症状,称对症治疗,或称治标,包括物理治疗。
③补充治疗(supplementary therapy)也称替代疗法(replacement therapy)用药的目的在于补充营养物质或内源性活性物质的不足。
(2)不良反应(adverse reactions,ADR)与治疗目的无关的,对病人不利的作用。
药理学重点总结终极版

药理学总结第一章绪论药理学是研究药物与机体相互作用及作用规律的学科,既研究药物对机体的作用及作用机制,即药物效应动力学,也研究药物在机体的影响下所发生的变化及其规律,即药物代谢动力学。
第二章药物代谢动力学药物分子通过细胞膜的方式有滤过(水溶性扩散)、简单扩散(脂溶性扩散)和载体转运(包括主动转运和易化扩散)。
绝大多数药物是通过简单扩散的方式通过生物膜。
药物通过细胞膜的速度与可利用的膜面积大小有关。
膜表面大的器官,如肺、小肠,药物通过其细胞膜脂层的速度远比膜表面小的器官(如胃)快。
药物的体内过程:吸收、分布、代谢、排泄;统称为ADME系统。
吸收:药物自用药部位进入血液循环的过程称为吸收。
药物只有经吸收后才能发挥全身作用。
(一)口服大多数药物在胃肠道内是以简单扩散方式被吸收的。
首过消除:从胃肠道吸收入门静脉系统的药物在到达全身血循环前必先通过肝脏,如果肝脏对其代谢能力很强,或由胆汁排泄的量大,则使进入全身血循环内的有效药物量明显减少,这种作用称为首过消除。
(二)吸入(三)局部用药(四)舌下给药(五)注射给药分布:药物一旦被吸收进入血循环内,便可能分布到机体的各个部位和组织。
药物吸收后从血循环到达机体各个部位和组织的过程称为分布。
大多数药物在血浆中均可与血浆蛋白不同程度地结合而形成结合型药物,它与未结合的游离型药物同时存在于血液中,并以一定百分数的结合率而达到平衡。
代谢:体内各种组织对药物的消除,肝是最主要的药物代谢器官排泄:肾是最重要的排泄器官一级消除动力学:是体内药物在单位时间内消除的药物百分率不变,也就是单位时间内消除的药物量与血浆药物浓度成正比,血浆药物浓度高,单位时间内消除的药物多,血浆药物浓度降低时,单位时间内消除的药物也相应降低。
零级消除动力学:是药物在体内以恒定的速率消除,即不论血浆药物浓度高低,单位时间内消除的药物量不变。
药物消除半衰期(t1/2):是血浆药物浓度下降一半所需要的时间。
【药理学】03章 药动学

CYP)酶系,又称肝药酶 (hepatic drug
enzymes)。与药物代谢密切相关的:CYP1A2,
CYP2A6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1, CYP3A4.
家
酶
族
亚
个
家
体
族
药物代谢酶的特性
23
药物转化的两种依赖形式
问题:药物代谢过程中两个时相的主要特点是什么?
两边取对数: pka-pH = lg—[B—H—+]— [B]
则解离度——[BH—+—] = 10 pka -pH [B]
弱碱性药物在pH值低的环境下解离型增加
9
当药物解离50%的时候,
弱酸性药物:
10pH -pka = 1 = 100
pH=pKa
弱碱性药物: 10 pka -pH = 1 = 100
*弱酸性药: HA Ka [H+][A-]
Ka= [HA]
H++A -
两边取对数: pH-pka = lg [A-] [HA]
则解离度 = [A-] = 10pH -pka [HA]
弱酸性药物在pH值增高时解离度 、离子障7 、吸收
Ka
*弱碱性药:BH+
B+H+
[H+][B] Ka =
[BH+]
一、浓-时曲线
血药浓度随时间的推移发生变化
MTC Cmax
ห้องสมุดไป่ตู้
血药浓度(mg/L)
吸 收 分 布 相
潜伏期
Tm ax
持续期
MEC 代谢排泄相
残留期
AUC
时间
速率类型
药理学第三章药动学(考前辅导)

药理学考前辅导要点是我去年的学习笔记,也是今年的我科执业药师考试药理学考前辅导讲稿,肯定有错,请指正!!反冲力2003年1月费时整理,引用者请注明出处。
掌握药物的吸收、分布及其影响因素,P450酶系及其抑制剂和诱导剂,药物排泄途径及其影响肾排泄的因素,血浆蛋白结合率和肝肠循环的概念。
药物代谢动力学,简称为药动学,研究药物体内过程及体内药物浓度随时间变化的规律。
药物在体内分布达到平衡后药理效应强弱与药物血浆浓度成比例。
医生可用药动学规律计算药物剂量以达到所需的血药浓度并掌握药效的强弱久暂。
比单凭经验处方取得较好的疗效。
第一节药物体内过程一、药物的跨膜转运药物在体内的过程:吸收、分布、生物转化、排泄,需进行跨膜转运的过程是吸收、分布、排泄。
1、被动转运(顺梯度转运):药物依赖于膜两侧的浓度差,从高浓度的一侧向低浓度的一侧扩散转运的过程。
多数药物属于被动转运。
(1)特点:不需要载体,不消耗能量,无饱和现象和竞争性抑制。
(2)影响扩散速度的因素:①膜两侧的药物浓度差。
②药物理化性质:分子量小、脂溶性大、极性小、非解离型的药易通过生物膜转运,反之难跨膜转运。
2、主动转运:是一种逆浓度(或电位)差的转运。
特点:需要载体,消耗能量,有饱和现象和竞争性抑制。
二、吸收药物的吸收是指药物进入血液循环的过程。
静脉注射无吸收过程。
吸收速度与程度主要取决于药物的理化性质、剂型、剂量和给药途径。
(一)吸收方式 1.多数药按简单扩散进入(吸收)。
(1)影响扩散速度的因素:1)膜的性质,面积及膜两侧的浓度梯度,2)药物的性质,分子量小的(200D以下),脂溶性大的(油水分布系数大的),极性小的(不易离子化的)药较易通过。
(2)吸收分布排泄的一个可变因素,与环境的酸碱度有关。
(3)离子障现象:非离子型药可自由穿透,而离子型药被限制在膜的一侧。
离子障与吸收有关,可以理解为“酸酸易吸收,酸碱难吸收”。
如弱酸性药在胃液中非离子型多,在胃中即可被吸收。
药理学重点知识归纳吐血

药理学重点知识归纳吐血SANY标准化小组 #QS8QHH-HHGX8Q8-GNHHJ8-HHMHGN#药理学第一章绪论药物:是指可以改变或查明机体的生理功能及病理状态,用于预防、诊断和治疗疾病的物质。
药理学:研究药物与机体(含病原体)相互作用规律的学科第二章药效学药物效应动力学(药效学):是研究药物对机体的作用及作用机制的生物资源科学。
药物作用:是指药物对机体的初始作用,是动因。
药理效应:是药物作用的结果,是机体反应的表现。
治疗效果:也称疗效,是指药物作用的结果有利于改变病人的生理、生化功能或病理过程,使患病的机体恢复正常。
对因治疗:用药目的在于消除原发致病因子,彻底治愈疾病。
对症治疗:用药目的在于改善症状。
药物的不良反应:与用药目的无关,并为病人带来不适或痛苦的反应。
1、副作用:在治疗剂量时出现的与治疗无关的不适反应,可以预知但是难以避免。
2、毒性反应:药物剂量过大或蓄积过多时机体发生的危害性反应,比较严重,可以预知避免。
3、后遗效应:停药后机体血药浓度已降至阈值以下量残存的药理效应。
4、停药反应:突然停药后原有疾病的加剧现象,双称反跳反应。
5、变态反应:机体接受药物刺激后发生的不正常的免疫反应,又称过敏反应。
6、特异性反应:以效应强度为纵坐标,药物剂量或药物浓度为横坐标作图可得量-效曲线。
最小有效量:最低有效浓度,即刚能引起效应的最小药量或最小药物浓度。
最大效应:随着剂量或浓度的增加,效应也增加,当效应增加到一定程度后,若继续增加药物浓度或剂量而效应不再继续增强,这一药理效应的极限称为最大效应,也称效能。
效价强度:能引起等效反应(一般采用50%的效应量)的相对浓度或剂量,其值越小则强度越大。
质反应:药理效应不是随着药物剂量或浓度的增减呈连续性量的变化,而表现为反应性质的变化。
治疗指数:LD50/ED50,治疗指数大的比小的药物安全。
受体:一类介导细胞信号转导的功能蛋白质,能识别周围环境中某种微量化学物质,首先与之结合,并通过中介的信息放大系统,出发后续的生理反应或药理效应。
药理学章节重点知识归纳

药理学章节重点知识归纳第一章绪论1.药理学:是研究药物与机体(包括病原体)相互作用的规律及机制的学科。
2.药效学:研究药物对机体的作用及作用机制。
3.药动学:研究机体对药物的处置。
包括药物在体内过程(吸收、分布、代谢、排泄)及血药浓度随时间而变化的规律。
第二章药物效应动力学(药效学)1、不良反应:(1)副作用:药物在治疗量时出现的与用药目的无关的作用称为副作用。
(2)毒性反应:药物剂量过大或用药时间过长时,药物在体内蓄积过多引起的危害性反应称为毒性反应。
(3)变态反应:药物作为抗原或半抗原,经接触致敏后所引发的病理性免疫反应称为变态反应,又称过敏反应。
常见于过敏体质患者。
如青霉素过敏性休克。
(4)停药反应:长期应用某些药物,突然停药使原有疾病症状重新出现或加剧的现象称停药反应,或称反跳现象。
(5)后遗效应:停药后血药浓度已降至阈浓度以下时残留的药理效应称后遗效应。
后遗效应长短不一。
短的如服用催眠药后,次晨出现的乏力、困倦现象;长的如长期应用肾上腺皮质激素,出现的肾上腺皮质功能低下症状。
(6)续发反应:续发反应是药物的治疗作用引起的不良后果,又称治疗矛盾。
如广谱抗生素。
(7)依赖性:长期应用某些药物后,患者对药物产生主观和客观上连续用药的现象,称为依赖性。
如镇静催眠药和镇痛药。
(8)特异质反应:少数特异体质患者对某些药物产生的反应与常人不同,这种现象称为特异质反应。
如蚕豆病。
2、效能:药物所能产生的最大效应称为该药物的效能。
效能反映了药物内在活性的大小,效能大活性大。
3、效价强度:指能引起等效反应所需要的药物剂量,简称效价。
药物剂量越小,药价的效价越大。
4、评价药物的安全性:治疗指数(TI)可用来评价药物的安全性,是药物的半数致死量(LD50)与半数有效量(ED50)的比值。
这仅用于治疗效应和致死效应的量效曲线平行的药物。
治疗指数越大,药物安全性越高。
两条曲线不平行:LD1/ED99或LD5和ED95之间的距离来评估药物的安全性。
药理学—— 药动学知识点归纳

药理学——药动学知识点归纳一、药物的体内过程药物从进入机体至离开机体,可分为四个过程:简称ADME系统→与膜的转运有关。
(一)药物的跨膜转运:※药物在体内的主要转运方式是:被动转运中的简单扩散!Ⅰ、被动转运——简单扩散1.概念:指药物由浓度高的一侧向浓度低的一侧扩散,以浓度梯度为动力。
2.特点:(1)不消耗能量。
(2)不需要载体。
(3)转运时无饱和现象。
(4)不同药物同时转运时无竞争性抑制现象。
(5)当膜两侧浓度达到平衡时转运即停止。
3.影响简单扩散的药物理化性质(影响跨膜转运的因素)(1)分子量分子量小的药物易扩散。
(2)溶解性脂溶性大,极性小的物质易扩散。
(3)解离性非离子型药物可以自由穿透。
离子障是指离子型药物被限制在膜的一侧的现象。
4.体液pH值对弱酸或弱碱药物的解离的影响:从公式可见,体液pH算数级的变化,会导致解离与不解离药物浓度差的指数级的变化,所以,pH值微小的变动将显著影响药物的解离和转运。
例题:一个pK a=8.4的弱酸性药物在血浆中的解离度为A.10%B.40%C.50%D.60%E.90%『正确答案』A『答案解析』pH对弱酸性药物解离影响的公式为:10 pH-pKa=[解离型]/[非解离型],即解离度为10 7.4-8.4=10-1=0.1。
※总结:体液pH值对药物解离度的影响规律:◇酸性药物在酸性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在碱侧。
◇碱性药物在碱性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在酸侧。
同性相斥、异性相吸或“酸酸碱碱促吸收;酸碱碱酸促排泄”例题:某弱酸性药物pK a=3.4,若已知胃液、血液和碱性尿液的pH 值分别是1.4、7.4和8.4。
问该药物在理论上达到平衡时,哪里的浓度高?A.碱性尿液>血液>胃液B.胃液>血液>碱性尿液C.血液>胃液>碱性尿液D.碱性尿液>胃液>血液E.血液>碱性尿液>胃液『正确答案』A『答案解析』同性相斥、异性相吸。
药学专业知识一_药理学 第三章 药物效力动力学、第四章 影响药物作用的因素_2012年版

A:心率次数
B:死亡个数
C:血压高低的千帕数
D:尿量毫升数
E:体重公斤数
答案:B
2、副作用的产生是由于
A:病人的特异性体质
B:病人的肝肾功能不全
C:病人的遗传变异
D:药物作用的选择性低
E:药物的安全范围小
答案:D
3、药物的安全指数是指
A:ED50/LD50
B:LD5/ED95
C:ED5/LD95
答案:A
6、影响药物效应的因素包括
A:年龄和性别
B:体重
C:给药时间
D:病理状态
E:给药剂量
答案:A,B,C,ຫໍສະໝຸດ ,ED:LD50/ED50
E:LD95/ED50
答案:B
4、β受体阻断剂与利尿剂合用后降压作用大大增强,这种现象称为
A:敏化作用
B:拮抗作用
C:协同作用
D:互补作用
E:相加作用
答案:E
5、连续用药较长时间,药效逐渐减弱,需要增加剂量才能出现药效的现象称为
A:耐受性
B:耐药性
C:成瘾性
D:习惯性
E:快速耐受性
药理学第三章 药动学

门静脉
胆管 肠道
粪便
肝肠循环:部分由胆汁排泄到十二指肠的药物 可在肠道再次被吸收入门静脉进入血液循环
3.肺脏: 某些挥发性药物
4.其他排泄途径: 乳汁、胃液、唾液及汗液。
第二节 速率过程
一、血管外给药的药-时曲线
最低中毒浓度
达峰时间 药峰浓度
血
药
浓 度
吸 收
分
布
相
平衡相
治疗窗
最低有效浓度
消除相
单位时间内用药总量不变,给药间隔时间愈
短,血药浓度的波动愈小,否则反之,但CSS不变, 达CSS时间不变.
问题
某病人病情危急,需立即达到稳 态浓度以控制,应如何给药
加大剂量 缩短给药间隔时间 其它方法
Plasma Drug Concentration
Time
Plasma Drug Concentration
• 血眼屏障:血-房水、血-视网膜。局部用药。 • 胎盘屏障:与一般生物膜无太大区别。孕妇用
药需谨慎。
组织亲和力
• 碘主要集中在甲状腺 • 钙沉积于骨骼 • 汞、砷等重金属多分布在肝、肾 • 硫喷妥钠多分布于脂肪组织 • 四环素可与钙络合沉积于骨骼和牙齿。
(三)生物转化
部位:主要在肝脏,其它如胃肠、肺、
VVdd==33L-5左L右: 主要分布于血液并与血浆蛋白大量结合, 如双香豆素、保泰松。 VVdd==1150L-2左0m右l: 主要分布于细胞外液和血浆,此类药物往 往不易通过细胞膜。如溴化物和碘化物等
VVdd==4400-L左60右ml: 可以分布于细胞内、外液。如利福平、安替比林
VVdd==110000--220000mLl: 特异性分布,可浓集于某些组织,如硫喷 妥钠、131I。
药理学第三章药物代谢动力学

药物 + 蛋白质
复合物
无活性、贮存型、难进入组织
一、与血浆蛋白结合率
特点: ① 差异性。 ② 暂时失活和暂时贮存血液中 。 ③ 可逆性。 ④ 饱和性及竞争性。
由于血浆蛋白总量和结合能力有限,加上结合的非特异性,出现两个问题:①当药物结合达到饱和后,继续增加药量,游离型药物浓度增加,出现药物作用或不良反应增强;
二、细胞膜屏障
血脑屏障(BBB):血管壁与神经胶质细胞形成的血浆与脑细胞外液间的屏障和由脉络膜丛形成的血浆与脑脊液间的屏障。具有保护脑组织生理屏障作用。分子大、极性高的药物不能通过,流脑时SD易通过 。
01
胎盘屏障:胎盘绒毛与子宫血窦间的屏障。几乎所有药物都能穿过胎盘屏障进入胎儿,只是程度和快慢不同。 另外还有血-眼屏障、血-关节囊屏障
分为简单扩散和滤过扩散两种。
特点:(1)药物顺浓度差转运 不耗能 不需要载体 无饱和限速及竞争性抑制
被动转运(下山转运)
添加标题
添加标题
脂溶扩散(lipid diffusion) (简单扩散):大多数药物是通过该方式转运。
影响因素:①膜两侧浓度差:药物在脂质膜的一侧浓度越高,扩散速度越快,当膜两侧浓度相同时,扩散即停止。
代谢的结果:
大多数药物灭活成为无活性的代谢产物; (灭活:药物经转化后活性降低或消失的现象) 少数药物仍有药理活性; (活化:药物经转化后,由无活性转变为有活性的现象。 ) 药物经代谢后水溶性和极性增加。
二、药物转化的酶系统 1、专一性酶:专一性强,主要催化水溶性较大的药物。如AchE、MAO。
2、肝药酶(非专一性酶) 是混合功能氧化酶系统。主要存在于肝细胞内质网上,可促进多种脂溶性药物的转化,其中CYP450酶系统是促进药物转化的主要酶系统。
药理学3药动学

药理学3药动学
生 物 转化 (biotransformation)
又称代谢或药物转化,是指 药物在体内发生的化学结构 改变。转化后的药物活性降 低或失去药理活性,极性增 加,易于排泄。
药理学3药动学
生物转化的部位及其催化酶
•生物转化的主要部位是肝脏。 •生物转化由肝微粒体细胞色
药理学3药动学
药物通过生物膜的转运
• 细胞膜主要由脂类(磷脂、胆固醇与糖脂) 和蛋白质组成。
• 大多数极性(离子化程度较强)药物难于 通过脂质双层,而脂溶性药物可以通透。 小分子药物可从膜孔透过膜。
• 药物通过生物膜的能力主要决定于药物的 脂溶性、解离度及分子量,其转运机制可 分为被动转运和载体转运两大类。
---是指体内药物或其代谢物排出体外 的过程,它与生物转化统称为药物消 除(elimination)。
药理学3药动学
载体转运 ---是指细胞膜上的载体与药物结合,并
载运它到膜另一侧的过程。
• 主动转运:又称“上山”或逆流转运。 特点是:①逆浓度梯度或逆电化学梯度 透过细胞膜 ②细胞膜的载体对药物有特 异的选择性 ③消耗细胞能量 ④以同一载 体转运的两种化合物可出现竞争性抑制 ⑤转运速度有最高限度。
代谢酶的活性,增加自身或其他药物的代谢速率。 苯巴比妥是典型的酶诱导剂。
(2) 酶的抑制:某些化学物质能抑制肝微粒体药物
代谢酶的活性,减慢其他药物的代谢速率。红霉 素是酶抑制剂。
3. 生理因素与营养状态 生物转化有明显的种属差 异、种族差异和个体差异
4. 病理因素
药理学3药动学
排 泄(excretion)
药理学3药动学
pKa值的概念
• pKa值——是弱酸性或弱碱性药物在50% 解离时溶液的pH值。注意:pKa值不是药 物自身的pH值。药物离子化程度受pKa值 及所在溶液的pH值决定。
药理学 第三章 药物效应动力学

➢ 受体 ➢酶 ➢ 离子通道 ➢ 核酸
➢ 载体 ➢ 免疫 ➢ 基因 ➢ 理化反应
一、受体研究的历史
受体的发现可以追溯到20世纪初,1878年,Langley从事药物的某些细胞成分之间相 互作用的研究时,观察到阿托品和毛果芸香碱对猫唾液分泌存在拮抗作用。他认为在 神经末稍或腺体细胞中有一种或一些物质,能分别与该二药形成化合物,而这种化合 物的形成取决于阿托品或毛果芸香碱的相对质量和它们对该物质的亲和力。这是受体 概念的雏形。后来,他在研究菸碱作用时,发现菸碱使鸟类的某些肌肉呈强直性收缩 状态,而且即使切断通向该肌肉的所有神经收缩仍可出现,说明菸碱的作用不是通过 神经。当时认为箭毒必须通过神经末稍才发生麻痹作用,所以他推想菸碱造成的肌肉 收缩作用应不被箭毒拮抗,然而实验结果却表明箭毒能明显拮抗菸碱所引起的肌肉收 缩效应。这说明这两种物质均可直接作用于肌肉细胞,与其中某些成分相结合,他称 这些成分为“接受物质”(receptive-substance)。这些“接受物质”后来被定义 为受体。
二、受体的概念和特性
受体概念
➢ 受体 receptor ➢ 配体 ligand
受体的五大特性
➢ 灵敏性、特异性、饱和性、可逆性、多样性
三 = EC50 亲和力指数:pD2 = -log KD
内在活性 (intrinsic activity) :0≤α≤1
药理学
第三章 药物效应动力学
第一节 药物的基本作用 第二节 药物剂量与效应关系 第三节 药物与受体
掌握 熟悉 了解
1.药理效应、治疗效果、不良反应的基本概念 2.药物量-效曲线的基本参数; 3.受体激动药、拮抗药概念及主要参数含义。
药物安全性评价指标及剂量的概念。
受体类型、细胞内信号转导、受体调节。
药理学 第三章 药动学

上升段:代表药物的吸收与分布。
下降段:代表药物的代谢和排泄(消除)。
药峰时间:血药浓度达到最高浓度的时间。
1)潜伏期:给药后到出现药效的时间,反映 药物的吸收和分布过程。
2)持续期:指药物维持有效浓度的时间,与 药物的吸收和消除速度有关。
3)残留期:指血药浓度降到最低有效浓度 以下至完全消除的时间。
酸性分泌通道 碱性分泌通道 —肾小管重吸收(tubular reabsorption)
尿液的pH影响重吸收
2)胆汁排泄(bile excretion)
分子量大于400-500的化合物主要直接从 胆汁排泄。
★肝肠循环(hepatoenteral circulation) 经胆汁排进小肠的药物部分可再经小肠
2、影响因素:
1)血浆蛋白结合率
结 ① 可逆性暂时的储存库,暂无生
合 理效应,且有饱和作用。
型 药 物
② 不能代谢和具竞争性 ③ 不能排泄,难转运
华法林:抗凝血药 结合99% 游离1%
华法林+保泰松 结合98% 游离2%
2)器官血流量(blood flow of organs)
血流丰富的组织,药物分布快且量多 再分布(redistribution):如硫喷妥钠
(metabolize many drugs)
➢具有个体差异 (have individual variation)
➢具有多种功能 (represent a mixed-function
oxidase system)
➢可以被诱导或抑制(can be induced or
inhibited)
③ 肝药酶诱导剂(enzyme inducer):
药理学:第3章 药物代谢动力学

药物的理化性质决定其固定的pKa值。 。
一个弱酸性药物(布洛芬,pKa=4.4)
胃液 (pH=2.4)
血液
尿液
(pH=7.4) (pH=8.4)
10pH-pKa
10-2
103
104
[离子型][A-] 0.01
1000 10000
[非离子型] [HA] 1
1
1
总量
1.01
1001 10001
胃肠道内影响吸收的因素
(2) 静脉注射给药(Intravenous,iv)
直接将药物注入血管,吸收完全
(3) 肌肉注射和皮下注射 (Intramuscular and subcutaneous injection, im and sc)
被动扩散+过滤,吸收快而全
毛细血管壁孔半径40Å,大多水溶性药 可滤过
肝药酶的特性
1) 选择性低:能催化多种药物转化;
2) 变异性较大:常因遗传、年龄、营养 和疾病等机体状态的影响而存在明显的 个体差异;如CYP450多态性
3) 酶活性易受外界因素影响而出现增强 (酶诱导)或减弱(酶抑制)现象。
肝药酶诱导剂和抑制剂
药酶诱导 (Induction):
苯巴比妥、利福平,环境污染物等
解离性是指水溶性药物在溶液中溶解后 可生成离子型或非离子型。
非离子型药物可自由跨膜转运,易吸收 离子型药物带有正电荷或负电荷不易跨 膜转运,被限制在膜的一侧,形成离子 障(ion trapping)现象。
酸性药 (Acidic drug):
HA H+ + A
碱性药 (Alkaline drug):
logC Time
第四节 药物消除动力学
主管药师专业知识讲义-药理学——第三节 药动学
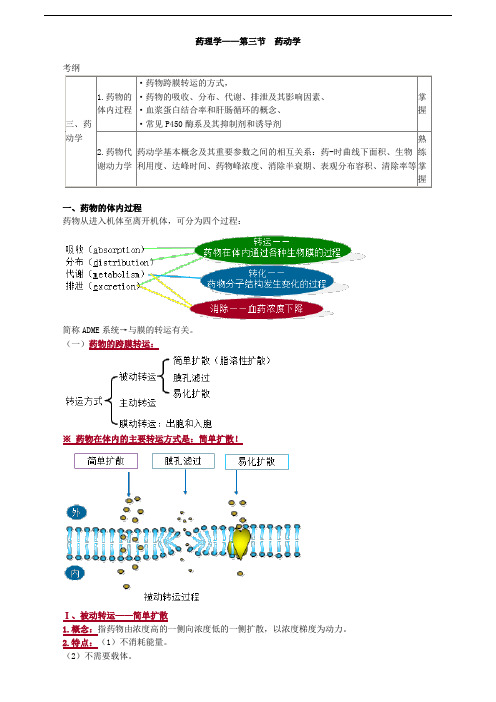
药理学——第三节药动学考纲一、药物的体内过程药物从进入机体至离开机体,可分为四个过程:简称ADME系统→与膜的转运有关。
(一)药物的跨膜转运:※药物在体内的主要转运方式是:简单扩散!Ⅰ、被动转运——简单扩散(3)转运时无饱和现象。
(4)不同药物同时转运时无竞争性抑制现象。
(5)当膜两侧浓度达到平衡时转运即停止。
3.影响简单扩散的药物理化性质(影响跨膜转运的因素)(1)分子量分子量小的药物易扩散。
(2)溶解性脂溶性大,极性小的物质易扩散。
(3)解离性非离子型药物可以自由穿透。
离子障是指离子型药物被限制在膜的一侧的现象。
※药物的解离程度受体液pH值的影响离子型非离子型4.体液pH值对弱酸或弱碱药物的解离的影响:※pK a(解离常数)的含义:>>是指解离和不解离的药物相等时,即药物解离一半时,溶液的pH值。
>>每一种药物都有自己的pK a。
若为弱酸性药物,则:若为弱碱性药物,则:从公式可见,体液pH算数级的变化,会导致解离与不解离药物浓度差的指数级的变化,所以,pH值微小的变动将显著影响药物的解离和转运。
一个pK a=8.4的弱酸性药物在血浆中的解离度为A.10%B.40%C.50%D.60%E.90%『正确答案』A『正确解析』pH对弱酸性药物解离影响的公式为:即:解离度为107.4-8.4=10-1=0.1。
※四两拨千斤:体液pH值对药物解离度的影响规律:◇酸性药物——在酸性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在碱侧。
◇碱性药物——在碱性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在酸侧。
同性相斥、异性相吸或“酸酸碱碱促吸收;酸碱碱酸促排泄”A.碱性尿液>血液>胃液B.胃液>血液>碱性尿液C.血液>胃液>碱性尿液D.碱性尿液>胃液>血液E.血液>碱性尿液>胃液『正确答案』A『正确解析』同性相斥、异性相吸。
在碱性尿液中弱碱性药物A.解离多,重吸收少,排泄快B.解离少,重吸收多,排泄快C.解离多,重吸收多,排泄快D.解离少,重吸收多,排泄慢E.解离多,重吸收少,排泄慢『正确答案』D『正确解析』酸酸碱碱促吸收;酸碱碱酸促排泄。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第三章药动学
药动学:机体对药物的作用。
药物自进入机体到离开机体历经吸收、分布、代谢及排泄过程,这是机体对药物的处置,这些处置可以概括为药物的转运(吸收、分布、排泄)和药物的转化(代谢)
1、吸收(absorption ):是指药物自体外或给药部位经过细胞组成的屏障进入血液循环的过程.
1.2、药物的转运方式:被动转运和主动转运
被动转运:单纯扩散:(脂溶性物质直接溶于膜的类脂相而通过)、
易化扩散:*需特异性载体*顺浓度梯度,不耗能、滤过扩散
主动转运特点:耗能,逆浓度差,需载体参与
影响药物吸收的因素:
(1)、给药途径
静脉>吸入>肌肉(im)>皮下(ih)>舌下>直肠>口服>经皮。
(2)口服给药对药物吸收的影响
首关消除(第一关卡效应或首过消除):有些口服药物首次通过肝脏就发生转化,减少进入体循环量,
(3)血液循环的状态也影响药物的吸收
(4)生物利用度也影响药物的吸收
2、分布:指吸入血液的药物被转运至组织器官的过程。
药物在体内的分布速率主要取决于药物的理化性质,各器官组织的血流量与对药物的通透性,以及药物在组织与血浆的分配比。
影响因素:(1)与血浆蛋白的结合率(2)体内屏障(3)与组织的亲和力(4)组织器官的血流量3、生物转化(代谢):指药物在体内发生的化学过程,这种变化主要是结构的变化,由于结构变化引起
性质变化,以至作用强度的变化。
注意:有少数药不发生化学变化,原型作用,原型排泄,如色甘酸、链霉素等。
1、转化的场所:肝脏微粒体
2、生物转化的类型
第一步:为氧化、还原、水解。
这步反应多数药物灭活,但也有例外(可待因)。
第二步:为结合。
总使药物活性降低或灭活并使极性增加。
影响药物转化的因素
肝脏的功能:肝脏的功能是药物代谢的主要器官,肝脏功能不全时可影响代谢。
药酶诱导剂:某些药物能使肝脏药酶的活性增加或加速其合成。
如:苯巴比妥、水合氯醛、保泰松等可加速其代谢,使药物作用减弱。
药酶抑制剂:凡能抑制药酶活性或减少药酶合成的药物。
如:异烟肼、阿斯匹林等属于这类药。
4、排泄是指药物在体内吸收、分布、或代谢后,最后以原型药或代谢产物通过不同途径排出体外。
(一)肾脏排泄
1、排出方式肾小球滤过(被动),肾小管分泌(主动)
影响肾脏排泄的因素
(1)尿液的PH值,如:弱酸性药物中毒,可用NaHCO3碱化尿液,加速排出(2)竞争性分泌系统(二)、胆汁的分泌与排泄
注意:1、有些药物从胆道排出,有利于治疗胆道病
2、有些药物转化成结合型的药物,随胆汁排出后,在肠腔中水解成游离型,被重吸收,形成肝肠循
环,可延长体内贮存时间。
如洋地黄毒甙。
肝肠循环(hepato-enteral circulation ):有些药物在肝细胞与葡萄糖醛酸等结合后排入胆中,随胆汁到达小肠后被水解,游离药物被重吸收,称为肝肠循环。
药动学的一些基本概念
生物利用度(bioavailability):经过肝脏首过消除过程后能被吸收进入体循环的药物相对量和速度。
(F=A/D)
1、药时曲线:以时间作用横坐标,血药浓度作纵坐标,绘制出一条反映血药浓度随时间动态变化的曲线.
3、药物的消除:指药物浓度在体内逐渐降低的过程,主要与药物的代谢与排泄有关。
恒比(定比)消除:药物在单位时间内消除的比例相等。
恒量(定量)消除:药物在单位时间内所能消除的量相等。
4、半衰期(dimination half-life time):血药浓度下降一半的时间,称半衰期,用t1/2表示。
1、简述药物半衰期(t1/2)的意义。
①确定给药间隔的依据;②可预测连续给药后达到坪值(稳态血药浓度,Css) 的时间。
2、药酶诱导剂有何临床意义?
①药酶诱导剂可使与其同服的药物代谢加速,药效降低,常需增加剂量才能维持疗效。
一旦停用药酶诱导剂,又可使同服的药物浓度过高,药效增强,甚至中毒。
这也是停药敏化现象的原因之一。
②药酶诱导剂还可加速自身代谢,是药物产生耐受性的原因之一。
③利用药酶诱导剂(如苯巴比妥)的酶促作用,可诱导新生儿肝药酶的活性,促进血中游离胆红素与葡萄糖醛酸结合,经胆汁排出,用于预防新生儿“脑核性黄疸”。
3、药物与血浆蛋白结合的意义是什么?
①因药物与血浆蛋白结合后,不易透出血管,故不被转运,不被转化。
同时当游离血药浓度降低时,
又可从血浆蛋白上游离出药物,可见药物血浆蛋白结合型在血液中为一种暂时贮存形式,可延长药物作用的持续时间。
②两种结合率较高的药物合用时,则彼此竞争血浆蛋白,使游离浓度升高,从而使药物的作用和毒性增强。
个体差异(individual variation):同一药物在不同患者不一定能达到相等的血药浓度,相等的血药浓度也不一定达到相同的疗效,这种因人而异的药物反应称为个体差异。
•二、联合用药及药物相互作用
一)联合用药:两种或两种以上的药物,除达到多种治疗目的外都是利用药物间的协同作用增效或利用拮抗作用减少不良反应
协同作用拮抗作用
•(二)相互作用(interaction):不适当的联合用药,使疗效降低或出现毒性反应。
•影响药动学的相互作用
•(1)吸收
•(2)血浆蛋白结合
•(3)肝脏生物转化
•(4)肾脏排泄
•影响药效学的相互作用
•(1)生理性的拮抗或协同
•(2)受体水平的协同与拮抗
•(3)干扰N递质的转运。