重症肌无力指南
最新版《重症肌无力管理国际共识指南:2020更新》

最新版《重症肌无力管理国际共识指南:2020更新》2020年11月3日,神经病学杂志Neurology在线发表了《重症肌无力管理国际共识指南:2020更新》[1],这是继2016年发表首个重症肌无力管理国际共识指南后第一次更新。
让我们一起抢先看一下2020版共识指南的重点内容。
此次共识指南依旧由美国重症肌无力基金会(the Myasthenia Gravis Foundation of America,MGFA)任命的工作组专家来制定,与2016年指南制定时不同,此次工作组增加了一名来自南美的新成员。
新共识指南更新的推荐主题根据2013年以来发表的重症肌无力(myasthenia gravis,MG)治疗研究筛选出来。
工作组专家对各个主题进行了3轮匿名电子邮件投票,每项推荐内容均进行打分,满分9分。
其中,1-3分:不适当;4-6分:不确定;7-9分:适当。
因此这版指南中我们可以看到每项推荐内容后面均附有专家打分的中位数和范围。
本次指南更新的6个主题如下:胸腺切除眼肌型重症肌无力利妥昔单抗甲氨蝶呤伊库丽单抗免疫检查点抑制剂(由于证据质量低,体育锻炼这一主题被排除了)我们将以问答的形式介绍各个主题的新增推荐意见,以便大家理解这些推荐意见背后的临床问题有更多认识和思考。
Q1:非胸腺瘤乙酰胆碱受体(AChR-Ab)阳性的全身型MG患者有必要切除胸腺吗?应该该选择哪种胸腺切除方式?AChR-Ab阴性或者其他抗体阳性的MG患者呢?指南对“胸腺切除”的推荐1a.年龄18-50岁的非胸腺瘤AChR-Ab阳性的全身型MG患者,应当在疾病早期考虑进行胸腺切除来改善临床结局,进而使免疫治疗需求和疾病恶化导致的住院需求最小化。
(中位评分:9,范围:2-9)1b.AChR-Ab阳性的全身型MG患者如果最开始的足量免疫抑制治疗无反应或者发生难以耐受的治疗副反应,强烈考虑胸腺切除术。
(中位评分:9,范围:5-9)2.MG患者进行胸腺切除是择期的,并且应当在患者稳定、安全的时候进行,因为术后的疼痛和机械因素可能限制呼吸功能。
重症肌无力健康教育指南

重症肌无力健康教育指南【教育评估】1、身体状况了解病人有无饮水呛咳、肌肉萎缩、吞咽困难及呼吸困难;了解病人及家属的心理情况,有无焦虑和恐惧;了解病人既往有无住院的经历、家庭经济情况如何。
2、学习要求了解病人、家属的学习能力及对疾病的认识程度;是否了解疾病的原因、诱因、发病特点及发生危象的严重后果;是否清楚主要治疗药物的作用及用药的注意事项,是否掌握病情观察要点、进食技巧及安全防护措施。
【与教育有关的护理问题】1、低效性呼吸形态:与呼吸肌麻痹致呼吸障碍有关2、有误吸的危险:与呼吸肌麻痹、咳嗽反射和呕吐反射减弱、吞咽障碍有关3、不能维持自主呼吸:与呼吸肌疲劳有关4、营养失调:低于机体需要量,与脑神经受损引起的吞咽/咀嚼困难有关5、语言沟通障碍:与言语肌肉的运动失调引起发音不良有关6、焦虑/恐惧:与知识缺乏,担心疾病的预后有关7、活动无耐力:与神经肌肉疾病引起的肌无力、全身疲乏,用力后不适或呼吸困难有关。
8、功能障碍性撤离呼吸机反应:与病人缺乏有关撤机过程的知识、信心不足有关。
【教育目标】教育对象能够:1、简述重症肌无力的表现及治疗、护理的方法。
2、主动配合治疗、护理。
3、演示自我护理技能。
4、复述出院后注意事项。
【教育内容】1、疾病简介:重症肌无力是一种神经肌肉传递障碍的得性自身免疫性疾病。
本病多数病人伴有胸腺增生或胸腺肿瘤,感染、精神创伤、过度劳累、妊娠分娩可诱发或加重病情,临床特征为部分或全身骨骼肌易于疲劳,通常在活动后加重,休息后减轻,晨轻幕重,最常见的首发症状为眼外肌不同程度乏力,其他如咀嚼肌、咽喉舌肌,面肌,四肢肌肉也可受累,分别出现上睑下垂、复视、吞咽困难、发音困难、四肢无力等,如侵犯呼吸肌则出现呼吸困难,称为重症肌无力危象,是致死的主要原因,心肌也可受累,可引起突然死亡。
2、心理指导:本疾病起病隐袭,症状明显,影响日常活动,严重时被迫卧床,因而病人易产生恐惧、焦虑、紧张等悄绪、护士应关心体贴病人、协助生活护理、多与其交谈,向病人和家属讲述本病的起因、过程,鼓励其保持乐观情绪,树立战胜疾病的信心,积极配合治疗和护理。
重症肌无力指南

2.单纤维肌电图(SFEMG):
使用特殊的单纤维针电极通过测定"颤抖"(Jitter)研究神经–肌肉传递功能," 颤抖"通常15~35 μs;超过55 μs为"颤抖增宽",一块肌肉记录20个"颤抖"中 有2个或2个以上大于55 μs则为异常。检测过程中出现阻滞(block)也判定为 异常。SFEMG并非常规的检测手段,但敏感性高。SFEMG不受胆碱酯酶抑 制剂影响。主要用于眼肌型MG或临床怀疑MG但RNS未见异常的患者。
经常从一组肌群无力开始,逐渐累及其他肌群,直到全身肌无力。 部分患者短期内出现全身肌肉收缩无力,甚至发生肌无力危象。
骨骼肌无力表现为波动性和易疲劳性,晨轻暮重,活动后加重、休息后可减轻。
眼外肌无力所致对称或非对称性上睑下垂和(或)双眼复视是MG最常见的首发症 状,见于80%以上的患者[2];还可出现交替性上睑下垂、双侧上睑下垂、眼球活 动障碍等。
④眶内占位病变:眶内肿瘤、脓肿或炎性假瘤等所致,表现为眼外肌麻痹并 伴结膜充血、眼球突出、眼睑水肿。眼眶MRI、CT或超声检查有助于诊断。
⑤Graves眼病:属于自身免疫性甲状腺病,表现为自限性眼外肌无力、眼 睑退缩,不伴眼睑下垂。眼眶CT显示眼外肌肿胀,甲状腺功能亢进或减退 ,抗促甲状腺激素受体抗体阳性或滴度高于界值。
⑥Meige综合征:属于锥体外系疾病,表现为单侧或双侧眼睑痉挛、眼裂变 小,伴有面、下颌和舌肌非节律性强直性痉挛。服用多巴胺受体拮抗剂或局 部注射A型肉毒毒素治疗有效。
2.全身型MG的鉴别诊断:
①吉兰–巴雷综合征:免疫介导的急性炎性周围神经病,表现为弛缓性肢体 肌无力,腱反射减低或消失。肌电图示运动神经传导潜伏期延长、传导速度 减慢、阻滞、异常波形离散等。脑脊液有蛋白–细胞分离现象。
中国重症肌无力诊断和治疗指南

中国重症肌无力诊断和治疗指南一、本文概述《中国重症肌无力诊断和治疗指南》是一份权威的医学指南,旨在为医生和医疗工作者提供关于重症肌无力(Myasthenia Gravis, MG)的标准化诊断和治疗建议。
重症肌无力是一种由自身免疫系统异常引起的神经-肌肉接头传递障碍性疾病,临床表现为部分或全身骨骼肌无力和易疲劳。
本指南的编写目的是确保患者能够得到及时、准确和个性化的医疗服务,改善其生活质量,降低疾病对社会和家庭的影响。
本指南涵盖了重症肌无力的流行病学、发病机制、临床表现、诊断方法、治疗策略以及疾病管理等方面的内容。
通过全面系统地介绍重症肌无力的最新研究进展和临床实践经验,本指南旨在为医生提供科学、规范、实用的治疗建议,以促进重症肌无力患者的康复。
本指南也为政策制定者、医学教育工作者和科研人员提供了有价值的参考信息,有助于推动我国重症肌无力诊疗水平的提高。
本指南的制定遵循了科学、规范、实用的原则,充分借鉴了国内外相关指南和研究成果,结合我国实际情况,形成了具有中国特色的重症肌无力诊断和治疗方案。
我们希望本指南能够为广大医生和医疗工作者提供有益的帮助,共同推动我国重症肌无力诊疗事业的发展。
二、重症肌无力的病因与病理生理机制重症肌无力(Myasthenia Gravis,MG)是一种慢性自身免疫性疾病,主要表现为神经-肌肉接头的传递功能障碍。
其病因复杂,涉及遗传、免疫、环境等多因素。
遗传因素在MG的发病中起着重要作用,许多研究已经证实,MG与主要组织相容性复合体(MHC)基因、乙酰胆碱受体(AChR)基因、补体基因、T细胞受体基因、免疫球蛋白基因以及细胞因子基因等相关。
在病理生理机制方面,MG主要是由于患者体内产生了针对乙酰胆碱受体(AChR)的自身抗体,导致神经-肌肉接头的突触后膜乙酰胆碱受体数量减少或功能受损,进而影响了神经冲动的传递,使骨骼肌发生易疲劳现象。
MG患者体内还可能存在针对肌肉特异性激酶(MuSK)和低密度脂蛋白受体相关蛋白4(LRP4)的自身抗体,这些抗体同样可以影响神经-肌肉接头的功能。
重症肌无力管理国际共识指南2020更新版

重症肌无力管理国际共识指南2020更新版11月3日,神经病学杂志发布了《重症肌无力管理国际共识指南(2020更新版)》,该共识指南基于文献中的最新证据,对2016年《重症肌无力管理国际共识指南》的内容进行了更新。
旨在指导世界各地临床医生的重症肌无力管理策略。
对指南中的要点进行摘译,供大家学习。
2016年《重症肌无力管理国际共识指南》是首个的重症肌无力国际共识指南,发布之前医脉通也对其中的指南要点进行总结。
制定背景2013年10月,美国Myasthenia Gravis基金会任命了一个工作组来制定重症肌无力(MG)的治疗指南,并召集了一个由15名国际专家组成的小组。
RAND / UCLA适当性方法用于制定涉及七个治疗主题的共识性建议。
2019年2月,国际专家小组重新召集,并增加了一名南美代表成员。
该小组对所有先前的建议重新进行了审核,并根据近期文献对需要纳入或更新的主题制定了新的共识建议。
多达三回合的匿名电子邮件投票用于达成共识,并根据专家小组的意见对建议进行了修改。
指南更新的部分包括胸腺切除术;针对利妥昔单抗、依库丽单抗和甲氨蝶呤的使用以及眼型MG的早期免疫抑制和免疫检查点抑制剂治疗相关的MG提出了新的建议。
在本指南共识中,经过三轮投票后未能达成共识的建议被删除。
专家小组以9分制对每项建议进行评分(1-3:不适当,4-6:不确定,7-9:适当),以中位数和范围表示。
共识指南中的主要内容➤胸腺切除术推荐意见: 1a.对于年龄在18至50岁之间,非胸腺性,AChR-Ab+全身性MG患者,应在疾病早期考虑进行胸腺切除术,以改善临床结局并最大程度地降低免疫治疗的需求或住院率(中位数9,范围2-9)。
1b.如果AChR-Ab +全身性MG患者对最初的免疫疗法没有反应或该疗法产生无法忍受的副作用,则应强烈考虑进行胸腺切除术(中位数9,范围5-9)。
2. MG胸腺切除术是择期手术,应在患者稳定且安全(不会因术后疼痛和机械因素限制呼吸功能)的情况下进行(中位数9,范围9)。
重症肌无力指南

中国重症肌无力诊断与治疗指南(2015年版)China guidelines forthe diagnosis and treatment of myasthenia gravis中华医学会神经病学分会神经免疫学组中国免疫学会神经免疫学分会陕西省西安市第四军医大学唐都医院神经内科,李柱一执笔摘要:本次指南简要介绍了重症肌无力(MG)发病机制与诊断要点;重点介绍了MG 治疗方法,方案,特别就是强调了在MG治疗过程中免疫抑制剂得使用方法。
对神经内科临床医生得工作实践具有指导意义。
重症肌无力(myasthenia gravis,MG)就是一种由乙酰胆碱受体(AChR)抗体介导、细胞免疫依赖、补体参与,累及神经肌肉接头突触后膜,引起神经肌肉接头传递障碍,出现骨骼肌收缩无力得获得性自身免疫性疾病。
极少部分MG患者由抗-MuSK(muscle specifickinase )抗体、抗LRP4(low—density lip oproteinreceptor-related protein4)抗体介导。
MG主要临床表现为骨骼肌无力、易疲劳,活动后加重,休息与应用胆碱酯酶抑制剂后症状明显缓解、减轻。
年平均发病率约为8.0—20、0/10万人[1]。
MG在各个年龄阶段均可发病。
在40岁之前,女性发病率高于男性;在40-50岁之间男女发病率相当;在50岁之后,男性发病率略高于女性、一、临床表现与分类1.临床表现全身骨骼肌均可受累。
但在发病早期可单独出现眼外肌、咽喉肌或肢体肌肉无力;颅神经支配得肌肉较脊神经支配得肌肉更易受累。
经常从一组肌群无力开始,逐渐累及到其她肌群,直到全身肌无力。
部分患者短期内出现全身肌肉收缩无力,甚至发生肌无力危象。
骨骼肌无力表现为波动性与易疲劳性,晨轻暮重,活动后加重、休息后可减轻。
眼外肌无力所致对称或非对称性上睑下垂与/或双眼复视就是MG最常见得首发症状,见于80%以上得MG患者[2];还可出现交替性上睑下垂、双侧上睑下垂、眼球活动障碍等、瞳孔大小正常,对光反应正常。
《中国重症肌无力诊断和治疗指南(2020版)》要点

《中国重症肌无力诊断和治疗指南(2020版)》要点重症肌无力(MG)是由自身抗体介导的获得性神经-肌肉接头(NMJ)传递障碍的自身免疫性疾病。
乙酰胆碱受体(AChR)抗体是最常见的致病性抗体;此外,针对突触后膜其他组分,包括肌肉特异性受体酪氨酸激酶(MuSK)、低密度脂蛋白受体相关蛋白4(LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR 功能及NMJ信号传递。
目前,MG的治疗仍以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(IVIG)、血浆置换(PE)以及胸腺切除为主。
1 临床表现、分型及亚组分类1.1 临床表现全身骨骼肌均可受累,表现为波动性无力和易疲劳性,症状呈“晨轻暮重”,活动后加重、休息后可减轻。
眼外肌最易受累,表现为对称或非对称性上睑下垂和/或双眼复视,是MG最常见的首发症状,见于80%以上的MG 患者。
面肌受累可致眼睑闭合无力、鼓腮漏气、鼻唇沟变浅、苦笑或呈肌病面容。
咀嚼肌受累可致咀嚼困难。
咽喉肌受累可出现构音障碍、吞咽困难、鼻音、饮水呛咳及声音嘶哑等。
颈肌受累可出现抬头困难或不能。
肢体无力以近端为著,表现为抬臂、梳头、上楼梯困难,感觉正常。
呼吸肌无力可致呼吸困难。
发病早期可单独出现眼外肌、咽喉肌或肢体肌肉无力;脑神经支配肌肉较脊神经支配肌肉更易受累。
肌无力常从一组肌群开始,逐渐累及到其他肌群,直到全身肌无力。
部分患者短期内病情可出现迅速进展,发生肌无力危象。
1.2 美国重症肌无力基金会(MGFA)临床分型旨在评估疾病严重程度,指导治疗及评估预后(表1)。
1.3 MG亚组分类及临床特点MG临床表现具有极大异质性,以血清抗体及临床特点为基础的亚组分类,对MG个体化治疗及预后评估更具指导意义(表3)。
1.3.1 OMG:MGFA型,可发生于任何年龄阶段。
1.3.2 AChR-全身型MG(GMG):依据发病年龄可分为早发型MG (EOMG)及晚发型MG(LOMG)。
MG2009指南

重症肌无力诊断和治疗指南发表时间:2009-12-14 发表者:李醒亚(访问人次:1373)重症肌无力是乙酰胆碱受体抗体介导,细胞免疫依赖,补体参与的,发生于神经肌接头的自身免疫性疾病。
诊断:(1)横纹肌易疲劳性。
晨轻暮重,活动后肌无力加重,经休息后减轻、缓解。
(2)药理学特征。
胆碱酯酶抑制剂可迅速缓解肌无力症状。
(3)电生理学特征。
低频重复频率刺激可使波幅衰竭50%以上。
(4)血清学特征。
在80-90%的全身型重症肌无力患者血中可检测到特异性的致病因子-乙酰胆碱受体抗体;约在60%左右的单纯眼肌型重症肌无力患者血中可检测到乙酰胆碱受体抗体。
近年来,在乙酰胆碱受体抗体阴性的全身型重症肌无力患者血中检测到抗-MuSK抗体,阳性率约为60%。
(5)影像学特征。
约80%重症肌无力患者伴有胸腺增生;约25%左右的重症肌无力患者同时伴有胸腺瘤;约20-25%胸腺瘤患者出现重症肌无力症状。
重症肌无力治疗:一、不同类型重症肌无力患者的药物选择。
(1)单纯眼肌型重症肌无力患者病初用胆碱酯酶抑制剂即可;如果单用胆碱酯酶抑制剂疗效不佳者可考虑应用糖皮质激素或者甲基强的松龙冲击。
胆碱酯酶抑制剂剂量应个体化。
激素在预防单纯眼肌型重症肌无力向全身型重症肌无力转化中的可能作用。
近年来有文献报道,单纯眼肌型重症肌无力患者早期使用糖皮质激素可预防眼肌型重症肌无力向全身型重症肌无力的转化。
但,直到目前还没有明确证据证明糖皮质激素能够预防眼肌型向全身重症肌无力的转化。
(2)全身型重症肌无力。
一般只用胆碱酯酶抑制剂不足以完全改善症状,需要联合用药治疗。
在应用胆碱酯酶抑制剂的基础上,加用糖皮质激素或者联合应用免疫抑制剂,如硫唑嘌呤、环孢酶素A等。
部分全身型重症肌无力患者需要甲基强的松龙冲击。
在冲击过程中应严密观察病情变化。
部分全身型重症肌无力患者在甲基强的松龙冲击过程中病情一过性加重,甚至需行气管插管或气管切开。
经甲基强的松龙冲击后疗效后仍不佳者,考虑大剂量丙种球蛋白冲击。
全身型重症肌无力诊疗指南(罕见病诊疗指南)

32.全身型重症肌无力概述全身型重症肌无力(generalized myasthenia gravis,GMG)是神经肌肉接头处因自身抗体破坏突触后膜导致神经肌肉接头传递障碍的疾病。
临床表现为骨骼波动性无力、不耐疲劳,活动后加重,休息后减轻。
症状多分布于眼部、球部、四肢肌肉,严重者累及呼吸肌造成呼吸衰竭。
病因和流行病学由特异性抗体介导、细胞和体液免疫参与,累及神经肌肉接头突触后膜,导致神经肌肉接头传递障碍。
患者外周血中可发现抗乙酰胆碱受体(AChR)抗体,、抗骨骼肌特异性酪氨酸受体激酶(MuSK)抗体或脂蛋白4(LRP4)抗体。
例如,MG患者外周血中测得的AChR-Ab,作用于神经肌肉突触后膜的乙酰胆碱受体,起到封闭受体、激活补体、破坏突触后膜等作用,导致神经肌肉接头传递功能障碍,肌肉疲劳无力。
在任何年龄段均可发病。
年平均发病率为8.0/100 000~20.0/100 000人。
在我国有两个发病高峰,40岁以前和60岁以后。
40岁以前女性多于男性,60岁以后男性多于女性。
重症肌无力约有80%合并胸腺异常,20%合并胸腺瘤。
胸腺组织中生发中心存在大量T细胞,刺激外周血中的B细胞活化增殖,生成特异性抗体。
临床表现经常从一组肌群开始,逐渐累及其他肌群,以上睑下垂和复视为最常见的发病症状,占80%。
骨骼肌无力表现为波动性和易疲劳性,晨轻暮重。
根据临床表现,按Osserman分成5型。
Ⅰ型:眼肌型,表现为上睑下垂、复视等。
ⅡA型:轻度全身型,四肢肌群轻度受累。
ⅡB型:中度全身型,四肢肌群中度受累,通常有咀嚼、吞咽和构音障碍。
Ⅲ型:重度激进型,进展快,数周或数月内累及咽喉肌,半年内累及呼吸肌。
Ⅳ型:迟发重度型,缓慢进展,2年内累及呼吸肌。
Ⅴ型:肌萎缩型,起病半年内出现骨骼肌萎缩。
神经科查体可见四肢近端、躯干、颈部、颅面部肌肉无力,不耐疲劳,Jolly试验阳性,腱反射正常,无感觉障碍,无病理征。
部分病人可进展至呼吸衰竭,出现重症肌无力危象。
重症肌无力诊疗结合指南PPT课件

目录
• 重症肌无力概述 • 重症肌无力的诊断 • 重症肌无力的治疗 • 重症肌无力患者的护理与康复 • 重症肌无力诊疗指南的意义与价值
01 重症肌无力概述
定义与特点
定义
重症肌无力是一种由神经-肌肉接头处传递功能障碍所引起的 自身免疫性疾病,临床主要表现为部分或全身骨骼肌无力和 易疲劳,活动后症状加重,经休息后症状减轻。
血液净化疗法
用于治疗重症肌无力危象等严重病情, 通过血液透析、血浆置换等方式清除 体内异常抗体。
04 重症肌无力患者的护理与 康复
日常护理
定期记录病情变化
密切关注患者的病情变化,包括 肌肉无力的程度、范围和进展情 况,以及是否有其他症状的出现。
保持舒适体位
根据患者的具体情况,指导患者保 持舒适的体位,以减轻肌肉疲劳和 不适感。
重复神经电刺激检查
在肌电图检查的基础上,通过重复 神经电刺激检查,观察肌肉反应的 改变,有助于确诊重症肌无力。
抗体检测
检测血清中的乙酰胆碱受体抗 体和肌肉特异性酪氨酸激酶抗 体,有助于确诊重症肌无力。
胸腺影像学检查
胸腺增生或胸腺瘤的存在可能与 重症肌无力的发病有关,因此胸
腺影像学检查有助于诊断。
诊断流程
THANKS FOR WATCHING
感谢您的观看
特点
具有病情波动性、病程长、易复发、治疗难度大等特点。
重症肌无力的分类
01
02
03
眼肌型
主要累及眼肌,表现为上 睑下垂、复视等。
脑干型
累及脑干神经,出现咀嚼、 吞咽困难、声音嘶哑、饮 水呛咳等症状。
全身型
累及全身骨骼肌,出现四 肢无力、抬头困难等症状。
2021重症肌无力管理国际共识指南

2021重症肌无力管理国际共识指南引言:重症肌无力(Myasthenia Gravis,MG)是一种自身免疫性疾病,主要表现为肌力下降,易疲劳以及肌肉无力等症状。
MG的管理一直是临床工作中一个重要的课题,随着对MG病理生理、药物治疗等方面研究的深入,2021年重症肌无力管理国际共识指南对MG的管理进行了全面的更新和修订。
本文将对2021重症肌无力管理国际共识指南进行详细介绍。
一、 MG的病理生理:MG是由于自身免疫系统攻击了神经肌肉接头传导及神经肌肉膜上的抗体,导致肌肉收缩功能异常。
指南强调了MG的早期发病机制和发展过程,包括乙酰胆碱受体(AchR)抗体阳性和MuSK抗体阳性MG的免疫学特点,以及肌肉瘫痪的机制等。
二、 MG的诊断与评估:指南明确了MG的临床表现及诊断标准,其中包括肌无力、疲劳、眼睑下垂等典型症状。
对于可能MG的患者,需要进行详细的身体检查以及相关实验室检查,如抗AchR抗体、MuSK抗体的检测。
此外,指南还介绍了MG的临床分类和MGFA评分的应用,以便评估疾病的程度和监测疾病的进展情况。
三、 MG的治疗策略:指南对MG的治疗策略进行了全面的阐述,包括药物治疗、手术治疗以及其他治疗方法。
药物治疗是MG管理的主要方式,指南详细介绍了抗胆碱酯酶药物、免疫调节药物等的使用和剂量。
此外,对于一些特殊情况的治疗,如妊娠期MG、小儿MG等,指南也提供了相应的指导意见。
四、 MG的监测与随访:MG的治疗是一个长期过程,指南强调了对MG患者的监测与随访的重要性。
此外,针对MG患者手术治疗前后的准备和注意事项,以及其他并发症的管理,指南也进行了详细的介绍。
五、 MG的康复管理:康复管理在MG的治疗中是至关重要的一环。
指南通过介绍运动康复、呼吸康复、言语康复和心理康复等的原则和方法,帮助MG患者更好地进行康复训练和管理。
六、 MG的预后与转归:MG的预后和转归受多种因素影响,包括年龄、发病方式、病程长短等。
重症肌无力症诊断治疗指南

重症肌无力症诊断治疗指南【概述】重症肌无力(MG)是以横纹肌神经肌肉传导障碍为特点的自身免疫性疾92临床诊疗指南胸外科分册病。
MG发病可能是由于机体产生的乙酰胆碱受体抗体(AchRab)作用于横纹肌突触后膜的乙酰胆碱受体(AchR),竞争性抑制乙酰胆碱的作用。
由于突触处传导的安全系数降低而致骨骼肌易疲劳和无力,l晦床表现为活动后横纹肌无力、容易疲劳、休息后可以缓解。
这种疾病并不多见,其发病率为0.5~5/10万人,可以发生于任何年龄,但女性多见于20~30岁,男性多见于50岁以上,女性与男性之比为3:2。
重症肌无力除累及横纹肌外,还存在胸腺的病理改变,观察发现;80%~90%的MG患者胸腺有病理学异常,其中65%~75%的患者伴有胸腺滤泡增生,接近10%的患者伴有胸腺瘤,其他lo%~20%的MG患者表现为胸腺萎缩。
胸腺瘤一般由上皮细胞或淋巴细胞组成,鼯性或恶性主要靠肉眼判断肿瘤是否累及周围组织、胸膜及心包。
胸腺瘤多发现在>30岁的重症肌无力患者,男性居多。
儿童和青少年重症肌无力患者多为胸腺增生。
在光镜下观察到有癌特征的胸腺癌甚为少见。
近年的研究发现正常胸腺内有多种细胞,包括肌样上皮细胞(Myoid cell)、胸腺细胞和上皮细胞等,这些细胞均能表达AchR,结构完全与突触后膜上的AchR相同,但在蛋白水平AchR表达则仅见于胸腺肌样上皮细胞,这种细胞主要位于胸腺髓质和皮髓质交界区,其形态学和生物化学特点是细胞内含有横纹肌肌管。
生理情况下,Ac}水与免疫系统形成免疫耐受,不能诱导机体产生特异性的AchRab,目前尚不知道MC;时如何破坏已经形成的自身耐受,激发自身免疫反应。
胸腺切除术后重症肌无力症状缓解的原因,可能是:①去除了启动自身免疫的胸腺肌样细胞表面新的抗原决定簇;②去除了乙酰胆碱受体致敏的T细胞;③去除了分泌乙酰胆碱受体抗体的B细胞;④去除了与免疫功能障碍有关的其他胸腺因素。
但有些重症肌无力病例胸腺切除效果不显著,其可能原因是:(D胸腺切除不完全.如残留异位胸腺;②神经肌肉接头处的损伤已不可逆;③在胸腺外,位于脾脏和周围淋巴结中的淋巴细胞群仍有类似胸腺的影响。
中国重症肌无力诊断和治疗指南2020(全文版)
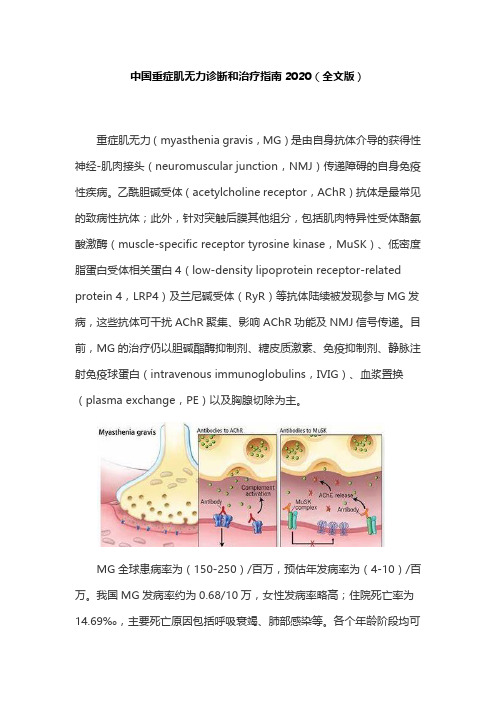
中国重症肌无力诊断和治疗指南2020(全文版)重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经-肌肉接头(neuromuscular junction,NMJ)传递障碍的自身免疫性疾病。
乙酰胆碱受体(acetylcholine receptor,AChR)抗体是最常见的致病性抗体;此外,针对突触后膜其他组分,包括肌肉特异性受体酪氨酸激酶(muscle-specific receptor tyrosine kinase,MuSK)、低密度脂蛋白受体相关蛋白4(low-density lipoprotein receptor-related protein 4,LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR功能及NMJ信号传递。
目前,MG的治疗仍以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(intravenous immunoglobulins,IVIG)、血浆置换(plasma exchange,PE)以及胸腺切除为主。
MG全球患病率为(150-250)/百万,预估年发病率为(4-10)/百万。
我国MG发病率约为0.68/10万,女性发病率略高;住院死亡率为14.69‰,主要死亡原因包括呼吸衰竭、肺部感染等。
各个年龄阶段均可发病,30岁和50岁左右呈现发病双峰,中国儿童及青少年MG(juvenile myasthenia gravis,JMG)患病高达50%,构成第3个发病高峰;JMG 以眼肌型为主,很少向全身型转化。
最新流行病学调查显示,我国70-74岁年龄组为高发人群。
近年来,在MG诊疗方面取得了众多进展,积累了更多循证医学证据。
为此,中国免疫学会神经免疫分会基于近5年国内外文献中的最新证据,参考相关国际指南,反复讨论,在对中国MG诊治指南(2015)更新修订的基础上编写了本指南。
新指南采用MGFA临床分型替代Osserman 分型,旨在对疾病严重程度进行量化评估;提出MG亚组分类,指导精准化治疗;对治疗目标进行了定义;针对胸腺切除,利妥昔单抗、依库珠单抗等生物制剂的应用,眼肌型MG(ocular MG,OMG)早期免疫抑制治疗以及免疫检查点抑制剂(immune check point inhibitors,ICIs)治疗相关MG等方面提出了新的建议。
解放军第309医院重症肌无力治疗指南(

重症肌无力治疗总原则提高神经肌肉接头处传导的安全系数:主要是胆碱酯酶抑制剂;2. 免疫治疗:肾上腺糖皮质激素、免疫球蛋白、血浆交换、免疫抑制剂、胸腺摘除术、胸腺放射治疗,及中药治疗等;3. 避免用乙酰胆碱产生和/或释放的抑制剂。
一、胆碱酯酶抑制剂1.适应证适用于除胆碱能危象以外的所有重症肌无力病人,尤其适用肌无力症状较轻的患者。
2.作用机制该类药能抑制胆碱酯酶对乙酰胆碱的降解,使神经肌肉突触间隙乙酰胆碱量增多,增强与乙酰胆碱受体抗体竞争乙酰胆碱受体的能力,有利于神经-肌肉接头处的传导,而使肌力有所恢复。
3.常用药物新斯的明 (Prostigmine) 包括注射甲基硫酸新斯的明用0.5mg,口服溴化新斯的明15-30mg,3-4次/日。
溴吡啶斯的明 (Pyridostigmine) 60-120 mg;3-4次/日4.副作用主要为胃肠道不良反应,一般用硫酸阿托品、654-2以对抗。
心率过慢,心律不齐,机械性肠梗阻以及哮喘患者均忌用或慎用。
二、肾上腺皮质激素治疗1. 适应症:1、单纯眼型重症肌无力患者。
2、病情恶化又不适于或拒绝做胸腺摘除的重症肌无力患者。
3、胸腺摘除术后的重症肌无力患者。
2. 作用机制(1)免疫抑制抑制乙酰胆碱受体抗体合成,使神经-肌肉接头处突触后膜上的乙酰胆碱受体免受或少受自身免疫攻击所造成的破坏。
也抑制针对骨骼肌其它成分的抗体。
(2)容易化作用使突触前膜易释放乙酰胆碱,使兴奋易于传递。
(3)再生使终板再生,使突触后膜乙酰胆碱受体数目成倍增加。
3. 治疗方法:一般主张自每日大量(强的松60-80,甚至100mg/d)开始,当出现连续好转后逐渐减量,于病情好转后,尽快减量乃至完全停用胆碱酯酶抑制剂。
优点:短期内达满意疗效,减用乃至停用胆碱酯酶抑制剂;明显好转2个月后行胸腺摘除术可减少并发症。
冲击疗法用于危重病例,已经用气管插管及人工呼吸器者。
甲基强的松龙1000mg/d,静脉滴入,连续3天,第4天500mg,连续3天,以后每3天减半量,直到停药。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
A Medical Guide for Patients with Myasthenia Gravis重症肌无力患者指南重症肌无力协会尽最大努力确保其手册中的信息在出版时是完整的和最新的,但是根据英国法律,协会不会对其内容的不准确性或其它可能发生的问题负法律责任每一个重症肌无力病人的情况都不相同,这本手册只能提供我们目前通常的见解,有可能不是唯一的见解。
因此其它人有可能有不同意见重症肌无力及其相关疾病的信息重症肌无力协会(MGA)现在已经更新了关于不同类型的肌无力:重症肌无力,Lambert-Eaton 肌无力综合征和遗传性(先天性)肌无力手册上的资料。
许多患者想充分了解他们所患疾病的本质,这些知识不仅对患者自己有帮助,而且对他们的家人同样有用。
它同样也能使患者容易理解医生是如何在尽力帮助他们。
尽管关于肌无力的相关信息可以在网络上找到,但它并不总是以一个门外汉容易理解的形式叙述的。
我们中有许多同仁为这本新版手册的问世作出了贡献,我们希望这些手册里的信息能容易理解。
但我相信这里仍有改进的空间,因此我们非常感激您的反馈,这将有助于我们继续编写下一版本。
如果您未曾听说过MGA, 对以下信息您也许会有兴趣了解:该协会成立于1968年,1976年独立,1995年开始与一间公司合作。
协会的目标就是为肌无力患者及家人提供关心和支持,促进关于疾病病因和发展更好的治疗手段的研究。
协会希望能与患者/社区成员建立紧密联系,同样希望与医疗人员,研究者建立密切联系。
我们通过当地的分支机构、分部、区域性或国际性的会议(通常有一个专家主讲),或MGA 新闻以及我们的信息手册来建立这些联系。
我们目前的会员接近1,500人。
我们希望您能从中找到有用的信息,请让MGA了解您的意见。
John Newsom-Davis 医学博士主席目录关于这本指南 (5)1 重症肌无力简介 (6)什么是重症肌无力? (8)患上重症肌无力意味着什么? (6)症状是什么样的? (8)什么会导致重症肌无力恶化? (9)谁会患重症肌无力? (9)2 哪里出了问题和为什么会出现问题 (11)正常肌肉的“点火系统”以及它是如何出现问题的 (11)免疫系统以及其如何出现问题的 (13)3 重症肌无力是如何诊断的 (15)4 重症肌无力是如何治疗的 (16)1 一些普通的方法 (16)2 溴吡啶斯的明 (17)3血浆交换和IvIg (18)4胸腺切除术 (19)5长期免疫抑制剂 (19)5麻醉剂与重症肌无力 (22)A 肌肉松弛剂 (23)B 溴化吡啶斯的明 (24)C 激素 (24)D 与麻醉师交谈 (24)E胸腺切除术 (25)6家庭与妇女问题 (25)7前景如何? (26)8历史 (27)9其他类型的肌无力 (28)10牙科与重症肌无力 (31)11眼肌型重症肌无力 (32)附录1对恐慌不安初学者的简单科学知识 (36)附录2术语表 (39)关于这本指南由于每一位重症肌无力患者的经历各不相同,这本指南只能提供一般性的见解。
它只是针对重症肌无力这一我们还不完全了解的复杂疾病的一个方面的认识。
这本指南不仅可以为重症肌无力患者本人提供信息和指导,而且也可以为他们的家人,朋友及任何对其感兴趣的人提供帮助。
这本指南包括了一组到2006年3月为止最新的观点,但它并不是唯一可能的见解。
本卷是关于重症肌无力的介绍。
Lambert-Eaton肌无力综合征和先天性肌无力将在第2卷介绍。
作为一位刚被诊断为重症肌无力的患者,或他的亲戚以及护理他的人,您有可能会发现很多不熟悉的科学和医学术语。
如果是这样,请您在开始阅读本手册前参考附录1,“一些针对恐慌不安初学者的简单科学”(A1页),以及附录2的术语表(A3页)。
本次修订的版本增补了关于牙科(第10章)和眼肌型重症肌无力(第11章)的内容。
1重症肌无力简介什么是重症肌无力?“Myasthenia gravis”,重症肌无力这个名字来自于希腊语(myasthenia:肌无力)和拉丁语(gravis: 严重的)。
幸运的是,肌无力是罕见并且无痛的。
它是由于神经→肌肉传递(我们肌肉的“点火系统”,见第2章)发生障碍所致。
肌无力可有三种不同的表现形式:●重症肌无力(MG)是目前最常见的肌无力。
在这里,免疫攻击仅损害了我们“随意”肌肉的点火系统。
肌无力具有典型的波动性,“易疲劳性”,---你用力越大,无力越重。
所以患者通常晨起最有力,日间活动后肌肉力量逐渐下降。
●先天性肌无力(见于不到二十分之一的肌无力患者)在这里,遗传缺陷导致“点火系统”不能有效地工作。
(见第2卷)●Lambert-Eaton肌无力综合症(LEMS)(约占的肌无力患者的二十分之一)。
类似的免疫攻击不仅损伤了随意肌肉的神经末稍,而且还损伤了“自主”肌肉(例如血管,肠道和膀胱,见第2卷)的神经末梢。
肌肉的无力并不会影响感觉(如触觉或温度觉)或导致疼痛,虽然过度用力可以引起疼痛(常见于颈部或背部)。
对于新病人来说,患上重症肌无力意味着什么我们不希望您看到后面几页后出现恐慌。
从好的方面开始● 重症肌无力症状通常可以得到很好的控制,以至于大多数患者可以度过完整的一生。
● 如果您注定要得一种“自身免疫性”疾病,那么重症肌无力是最具有可治性的疾病之一,它很少伴发疼痛,很少患者有长期不适。
事实上很少有患者死于肌无力。
● 治疗措施随时都在改进,在您的帮助下,我们将不断努力。
● 每一位重症肌无力患者都应该做自己最好的医生,找出自己最佳的方式来控制肌无力症状。
● 您一定不能让重症肌无力控制您的生活。
在另一方面,您需要预先得到些警告:● 您的重症肌无力将会伴随您许多年。
不要坐在那里等着病情缓解;只有二十分之一的重症肌无力患者在一年内没有任何治疗的情况下病情可以得到缓解。
● 您有可能不得不合理安排您的一天,把最重要的活动安排在您最有力的时候。
● 重症肌无力通常会影响到面部的肌肉和声音,因此人们,特别是在第一次见面时,可能通常不会理解您:例如,甚至对您尽力做出的微笑表情产生误解(尽管已经尽力而为,肌无力患者的微笑仍可能看起来像怒吼表情)。
● 您几乎总是需要药物治疗,而且需要忍受它们的不良反应。
对大数人来讲,药物的疗效远大于副作用。
症状是什么样的?在类型和严重程度上,没有两位患者会出现完全相同的症状。
起病可以很突然;也可以很平常,它有可能是逐渐的或隐匿性的,以至于很容易被忽视,或只有在延误后才能确诊。
大多数情况下,它开始于我们时常运用的肌肉—例如,出现眼睑下垂和复视(即影响眼球的活动,但不包括聚焦)。
有时这些眼部肌肉是唯一受累的肌肉。
通常,脸部肌肉的无力会导致“怒吼”样的笑容,或者发音出现鼻音,甚至出现声音减弱,或含混不清,很难让人理解。
重症肌无力经常影响咽喉部、颈部和躯干部的肌肉;有时也影响手部、臂部和腿部的肌肉,导致抓握和行走无力,但这种情况相对少见。
因此,重症肌无力有“从头至脚向下受累”的倾向---离脚越近,症状越轻,肌肉受累的可能性越小。
如果无力影响到吞咽和咀嚼,吃东西可能会很缓慢,会有被食物碎屑噎住或误吸的风险,可能导致肺部感染。
更严重的是,患者可能出现呼吸困难,甚至呈危重状态—因此叫作“重症”:在1930年以前,重症肌无力通常导致很多患者死亡。
如果患者的呼吸困难很严重,将出现“危象”,需要住院进行呼吸机辅助呼吸。
现在,随着治疗水平的显著提高,重症肌无力很少缩短患者的寿命,尽管治疗有一些副作用,大多数患者可以有自主的生活。
重症肌无力症状通常会在患病三至五年内达到顶峰,随后逐渐减轻。
不幸的是,大约每五位患者中仅仅只有一位能永久的“摆脱”重症肌无力(如,达到“缓解”),大多患者需要学会稳定其症状。
什么会导致重症肌无力恶化?许多情况可使无力加重,包括感染(例如感冒,肺炎,或牙龈脓肿),发热,高温,过度用力和情绪紧张。
有些女性患者注意到在月经期,以及怀孕和分娩阶段重症肌无力症状会加重。
甲状腺功能过低和过高都会使重症肌无力加重;使用利尿药物和频繁呕吐导致的盐分丢失,手术或放射治疗的应激也可使重症肌无力加重。
谁会患重症肌无力?我们这一生从摇篮时期起一直到走进坟墓都有可能罹患重症肌无力,无论种族和性别均可受累。
青年女性(10-40岁)更易受累,随着认识的深入,我们发现老年重症肌无力患者的人数,特别是男性患者,似乎正在增加。
“免疫类型”是不会遗传的。
重症肌无力很少出现一个家族内的多个成员受累,尽管也存在某些家族性的危险因素。
例如,当双胞胎中的一位被证实患有重症肌无力时,与其他没有患病的双胞胎个体相比另一位将更易患病,既使如此,风险仍低于五分之一。
没有任何迹象表明重症肌无力将会经由患者而发生传播。
虽然它常在感染后起病,但目前没有任何细菌被重点怀疑与发病相关。
所有的人种均可出现重症肌无力,但疾病的表现形式可以有很大的不同。
例如,仅眼肌受累的重症肌无力(“眼肌型重症肌无力”)在中国和日本青年患者中很常见,尤其多见于10岁以前,而这在欧洲很少见。
患者分型大约有10%的患者为持续的纯眼肌型重症肌无力。
全身型患者中通常有10%患有胸腺肿瘤(胸腺瘤),25%为早发型重症肌无力(40岁以前起病,大多数为妇女),50%为晚发型MG (男性稍多)。
早发型重症肌无力患者看起来胸腺异常较多,但并未发现有胸腺瘤(见第二章)。
最后,在每八位重症肌无力母亲中,大约只有一位母亲的宝宝在出生后的头2至3周可出现无力(见第四章),他们通常对治疗反应较好,并且很快能恢复正常。
为什么只有某些人会得这些疾病呢?关于这点我们知道得很少。
然而,早发型重症肌无力的一个遗传性危险因子也与其他自身免疫性疾病的易患性相关,因此在重症肌无力患者中甲状腺疾病和青年型糖尿病比较多见,与他们有血缘关系的亲属和其他人相比患这两种疾病的也更常见。
但自身免疫性重症肌无力在同一家庭发生两次的机率大约仅为百分之一。
我们怀疑还应该有其他的遗传或外源性危险因子存在,例如感染,但关于这点我们了解得很少。
偶而有些患者服用治疗风湿性关节炎的药物D-青霉胺会出现典型的肌无力抗体和重症肌无力症状,但停药后两者均会消失。
内源性因素包括胸腺肿瘤(“胸腺瘤”),大约存在于十分之一的肌无力患者,以及早发型肌无力患者的胸腺异常(见第二章)。
2哪里出了问题和为什么会出现问题正常肌肉的“点火系统”以及它是如何出现问题的当电冲动从大脑传到神经末梢时,神经末梢释放一种化学递质--乙酰胆碱—“点火钥匙”。
这些递质跨过一个狭窄的间隙,插进了特制的“锁孔”---肌肉表面的乙酰胆碱受体,接着通过复杂的电生理作用机制触发了肌肉的收缩。
多余的乙酰胆碱被一种特殊的蛋白破坏-乙酰胆碱脂酶—它可以使肌肉再次放松。
药物如新斯的明(Neostigmine)和溴吡啶斯的明(Pyrido-stigmine)抑制了乙酰胆碱脂酶,因此递质的传递将持续较长时间,有了更多的触发机会。