药典三部(2015版)-通则-0832水分测定法
2015药典纯化水注射水检验标准
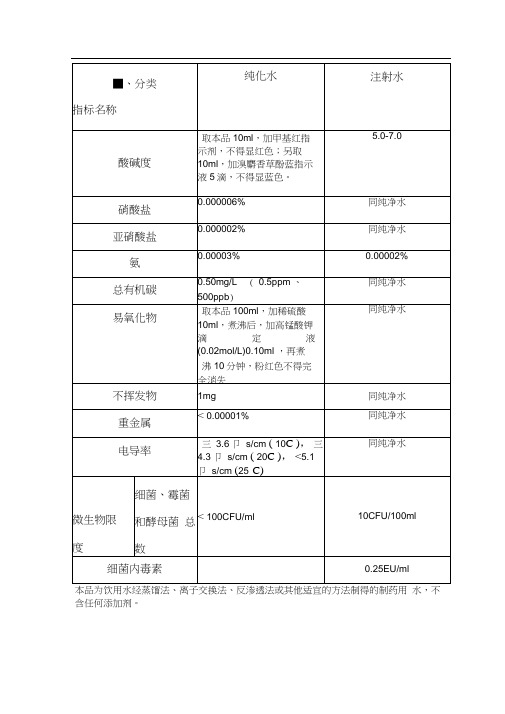
本品为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的制药用水,不含任何添加剂。
纯化水为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的制药用水,不含任何添加剂。
酸碱度:取本品10ml,加甲基红指示液2滴,不得显红色;另取10ml,加溴麝香草酚蓝指示液 5 滴,不得显蓝色;硝酸盐取本品5ml 置试管中,于水浴中冷却,加10%氯化钾溶液0.4ml 与0.1%二苯胺硫酸溶液0.1ml,摇匀,缓缓滴加硫酸5ml,摇匀,将试管于50C 水浴中放置15分钟,溶液产生的蓝色与标准硝酸盐溶液[取硝酸钾0.163g,加水溶解并稀释至100ml,摇匀,精密量取1ml,加水稀释成100ml,再精密量取10ml,加水稀释成100ml,摇匀,即得(每1ml相当于1ugNO3 ]0.3ml,加无硝酸盐的水4.7ml, 用同一方法处理后的颜色比较, 不得更深( 0.000006%);亚硝酸盐取本品10ml,置纳氏管中,对氨基苯磺酰胺的稀盐酸溶液(1 —100) 1ml与盐酸萘乙二胺溶液(0.1 —100)1ml,产生粉红色,与标准亚硝酸盐溶液[取亚硝酸钠0.75g (按干燥品计算),加水溶解,稀释至100ml,摇匀,精密量取1ml,加水稀释成100ml,摇匀,再精密量取1ml,加水稀释成50ml,摇匀,即得(每1ml相当于1ugNO2 ]0.2ml,加无亚硝酸盐的水9.8ml,用同一方法处理后的颜色比较,不得更深( 0.000002%);氨取本品50ml,加碱性碘化汞钾试液2ml,放置15分钟;如显色,与氯化铵溶液(取氨化铵31.5mg,加无氨水适量使溶解并稀释成1000ml) 1.5ml,加无氨水48ml 与碱性碘化汞钾试液2ml 制成的对照液比较,不得更深( 0.00003%);总有机碳不得过0.50mg/L(0.5ppm 、500ppb) (通则0682 2015 版药典第四部P85 )易氧化物取本品100ml,加稀硫酸10ml,煮沸后,加高锰酸钾滴定液(0.02mol/L)0.10ml ,再煮沸10分钟,粉红色不得完全消失;以上总有机碳和易氧化物两项可选做一项不挥发物取本品100ml,置105C恒重的蒸发皿中,在水浴上蒸干,并在105C干燥至恒重,遗留残渣不得过1mg重金属取本品100ml,加水19ml,蒸发至20ml,放冷,加醋酸盐缓冲液(pH3.5) 2ml与水适量使成25ml,加硫代乙酰胺试液2ml,摇匀,放置2分钟, 与标准铅溶液1.0ml加水19ml用同一方法处理后的颜色比较,不得更深(0.000 01%);电导率(10C,三3.6 卩s/cm), (20C,三4.3 卩s/cm), (25C,<5.1 卩s/cm)通则0681 2015 版药典第四部P84);微生物限度取本品,采用薄膜过滤法处理后,依法检查(附录幻J), 细菌、霉菌和酵母菌总数每1ml 不得过100 个(通则1105 2015 版药典第四部P140)。
通则0832水分测定法

创作编号:GB8878185555334563BT9125XW创作者: 凤呜大王*0832水分测定法第一法(费休氏法)1.容量滴定法本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水起定量反应的原理来测定水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。
费休氏试液的制备与标定(1)制备 称取碘(置硫酸干燥器内48小时以上)110g ,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml ,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分浸入的条件下,通入干燥的二氧化硫至重量增加72g ,再加无水甲醇使成1000ml ,密塞,摇匀,在暗处放置24小时。
也可以使用稳定的市售费休氏试液。
市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。
本液应遮光,密封,置阴凉干燥处保存。
临用前应标定滴定度。
(2) 标定 精密称取纯化水10~30mg ,用水分测定仪直接标定;或精密称取纯化水10~30mg ,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分浸入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法(通则0701)等]指示终点;另作空白试验,按下式计算:B-A W F式中 F 为每1ml 费休氏试液相当于水的重量,mg ;W 为称取重蒸馏水的重量,mg ;A 为滴定所消耗费休氏试液的容积,ml ;B 为空白所消耗费休氏试液的容积,ml 。
测定法 精密称取供试品适量(约消耗费休氏试液1~5ml), 除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。
或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另作空白试验,按下式计算:%100W F B A %⨯-=)()供试品中水分含量( 式中 A 为供试品所消耗费休氏试液的容积,ml ;B 为空白所消耗费休氏试液的容积,ml ;F 为每1ml 费休氏试液相当于水的重量,mg ;W 为供试品的重量,mg 。
水分测定法药典分析
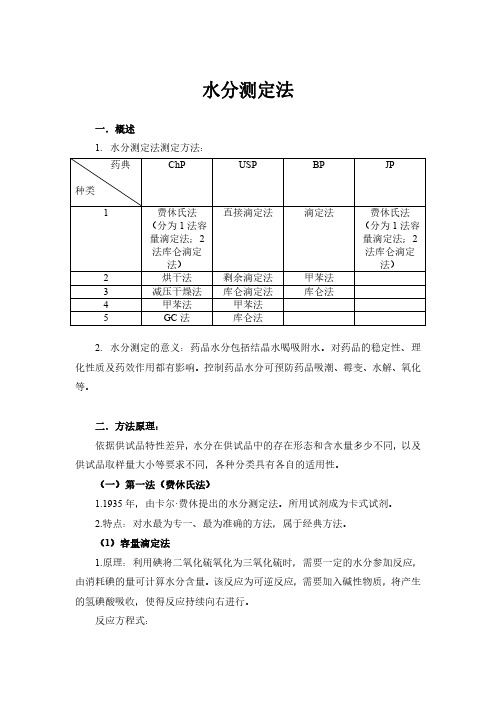
水分测定法一.概述1.水分测定法测定方法:2.水分测定的意义:药品水分包括结晶水喝吸附水。
对药品的稳定性、理化性质及药效作用都有影响。
控制药品水分可预防药品吸潮、霉变、水解、氧化等。
二.方法原理:依据供试品特性差异,水分在供试品中的存在形态和含水量多少不同,以及供试品取样量大小等要求不同,各种分类具有各自的适用性。
(一)第一法(费休氏法)1.1935年,由卡尔·费休提出的水分测定法。
所用试剂成为卡式试剂。
2.特点:对水最为专一、最为准确的方法,属于经典方法。
(1)容量滴定法1.原理:利用碘将二氧化硫氧化为三氧化硫时,需要一定的水分参加反应,由消耗碘的量可计算水分含量。
该反应为可逆反应,需要加入碱性物质,将产生的氢碘酸吸收,使得反应持续向右进行。
反应方程式:I2+SO2+H2O↔2HI+SO32.早期碱性物质采用吡啶吸收,在无水甲醇中,形成稳定的甲基硫酸氢吡啶。
在醇溶液中,碘与水恰好为1:1反应。
无水甲醇既是溶剂又参与反应。
但是吡啶毒性很强,在反应中不直接参与反应,且有很强的致癌性,目前采用咪唑替代。
吡啶只起到调节pH值和缓冲剂的作用。
3.现在卡氏试剂分为两种:无吡啶型和含吡啶型4.典型的醇化剂:无水甲醇或二甘醇乙醚5.常用的碱化剂:吡啶和咪唑。
6.卡氏试剂在使用过程中,滴定度会逐渐变小,由于受到空气中水分的影响。
经试验比较,无吡啶的卡氏试剂稳定性相对更好。
7.仪器装置:容量法卡尔费休水分仪8.测定法:(1)供试品通常采用无水甲醇为溶剂溶解。
(2)滴定终点:①目视法(用于滴定管直接滴定法,颜色由浅黄变为红棕色)应进行溶剂平行空白试验②仪器法(双伏安法)双点位分析法:自动扣除空白和环境影响9.适用性:主要用于化学药品,应用范围广。
可以测定药品中的游离水和结合水;特别适用于遇热易破坏、引湿性强或毒性较大的化学药品。
(2)库仑滴定法1.原理:以卡尔费休反应为基础,采用永停滴定法测定水分。
库仑发碘液不是从滴定管中加入,而是由含有碘离子的阳极电解液电解产生。
中国药典2015年版

中国药典2015年版醋酸曲普瑞林___安奈德乳膏Cusuan Q u’annai d e RugaoT rta n i d i M)量one Acetonide Acetate Cream本品含醋酸曲安奈德(C26H33F07)应为标示量的90。
o%〜110。
0%。
P生状1本品为白色乳膏。
【鉴别】在含量测定项下记录的色谱图中5供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
【检査】应符合乳膏剂项下有关的各项规定(通则0109)。
【含量测定1照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以甲醇-水(60^40)为流动相;检测波长为240nm。
理论板数按醋酸曲安奈德峰计算不低于250(^醋酸曲安奈德峰与内标物质峰的分离度应符合要求。
内标溶液的制备取炔诺酮^加甲醇溶解并稀释制成每l m l中约含0。
15mg的溶液5即得。
测定法取本品适量(约相当于醋酸曲安奈德1。
25mg) 9精密称定9置5O m l量瓶中9加曱醇约3Oml9置80°C水浴中加热2分钟,振摇使醋酸曲安奈德溶解,放冷,精密加内标溶液5m U用甲醇稀释至刻度,摇匀,置冰浴中冷却2小时以上,取出,迅速滤过9取续滤液放至室温,作为供试品溶液,取20p d注入液相色谱仪4己录色谱图;另取醋酸曲安奈德对照品5精密称定,加甲醇溶解并定量稀释制成每l m l中约含C L125m g的溶液,精密量取10m l与内标溶液5m U置50m l量瓶中^兩中醇稀释至刻度9摇匀,同法测定。
按内标法以峰面积计算3卩得。
【类别1同醋酸曲安奈德。
【规格】(l)4g?4mg(2)10g»2,5mg(3)l0g»5mg (4)lOg »40mg【贮藏】密封,在阴凉处保存。
___安奈德注射液Cusuan Q u^annai d e Z hush e y eTriainclnolone Acetonide Acetate Injection本品为醋酸曲安奈德的灭菌混悬液。
《中国药典》2015药典纯化水标准

中国药典2015年版溶液的澄清度与颜色取本品,加水制成每lml中约含阿魏酸钠20m g的溶液,溶液应澄清无色;如显浑浊,与1号池度标准液(通则0902第一法)比较,不得更浓;如显色,与黄色或黄绿色3号标准比色液(通则0901第一法)比较,不得更深。
有关物质避光操作。
取本品,加流动相溶解并稀释制成每l m l中约含0. 7m g的溶液,作为供试品溶液;精密量取lm l,置200m l量瓶中,用流动相稀释至刻度,摇勻,作为对照溶液。
照阿魏酸钠有关物质项下的方法测定。
供试品溶液的色谱图中如有杂质峰,各杂质峰面积的和不得大于对照溶液的主峰面积(0.5%)。
水分取本品,照水分测定法(通则0832第一法1)测定,含水分应为13.0%〜16.0%(供无菌粉末用)或应不超过3.0%(供无菌冻干品用)。
热原取本品,加灭菌注射用水制成每l m l中含阿魏酸钠5m g的溶液,依法检查(通则1142),剂量按家兔体重每l k g 缓慢注射3m l,应符合规定。
无菌照阿魏酸钠项下的方法检査,应符合规定。
其他应符合注射剂项下有关的各项规定(通则0102)。
【含置测定】避光操作。
取装量差异项下的内容物约0.15g,精密称定,加冰醋酸20m l使阿魏酸钠溶解,照阿魏酸钠项下的方法,自“加醋酐3m l”起,依法测定。
每l m l高氣酸滴定液(0.lm o l/L)相当于25.22mg的C10H9N a04•2H20。
【类别】同阿魏酸钠。
【规格】(1)0. lg(2)0. 3g【贮藏】遮光,密封保存。
纯化水ChunhuashuiPurified WaterH2018.02本品为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的制药用水,不含任何添加剂。
【性状】本品为无色的澄清液体;无臭。
【检査】酸碱度取本品10m l,加甲基红指示液2滴,不得显红色;另取10m l,加溴麝香草酚蓝指示液5滴,不得显蓝色。
硝酸盐取本品5m l置试管中,于冰浴中冷却,加10%氣化钾溶液0.4m l与0.1%二苯胺硫酸溶液0.1m l,摇匀,缓缓滴加硫酸5m l,摇勻,将试管于50T:水浴中放置15分钟,溶液产生的蓝色与标准硝酸盐溶液[取硝酸钾0.163g,加水溶解并稀释至100m l,摇匀,精密量取l m l,加水稀释成100m l,再精密量取10m l,加水稀释成100m l,摇匀,即得(每l m l相当于1吨N03)]0.3m l,加无硝酸盐的水4.7m l,用同一方法处理后纯化水的颜色比较,不得更深(0.000006%)。
《中国药典》2015年版通则编码与2010年版附录编码对照表

中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表《中国药典》2015年版通则编码与2010年版附录编码对照表编号通则名称原附录原附录名称0100制剂通则一部工D片剂0101片剂二部I A片剂三部I E片剂一部I u注射剂0102注射剂二部I B注射剂三部I A注射剂一部I L胶囊剂0103胶囊剂二部I E胶囊剂三部I F胶囊剂一部I C颗粒剂0104颗粒剂二部I N颗粒剂三部I J颗粒剂一部I Y眼用制剂0105眼用制剂二部I G眼用制剂三部I c眼用制剂一部I X鼻用制剂0106鼻用制剂二部I R 鼻用制剂三部I L鼻用制剂一部I w栓剂0107栓剂二部I D栓剂三部I B栓剂一部I A丸剂0108丸剂一部I K滴丸剂二部I H丸剂一部I R 软音剂0109软裔剂乳裔剂二部I F软膏剂乳裔剂糊剂三部I G软裔剂、乳裔剂0110糊剂二部I F 软膏剂乳音剂糊剂(指糊剂)0111吸人制剂二部I L气雾剂粉雾剂喷雾剂(指粉雾剂)一部I Z气雾剂喷雾剂(指喷雾剂)0112喷雾剂二部I L气雾剂粉雾剂喷雾剂(指喷雾剂)三部I H喷雾剂一部I Z气雾剂喷雾剂(指气雾剂)0113气雾剂二部I L 气雾剂粉雾剂喷雾剂(指气雾剂)• 425《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称一部I Q凝胶剂0114凝胶剂二部I U凝胶剂三部I M凝胶剂一部I B散剂0115散剂二部I P散剂三部I K散剂一部I H糖浆剂0116糖浆剂二部I K糖浆剂一部I V搽剂洗剂涂膜剂(指搽剂)0117搽剂二部I T搽剂涂剂涂膜剂(指搽剂)二部I T搽剂涂剂涂膜剂(指涂剂)0118涂剂三部I D外用制剂一部I V搽剂洗剂涂膜剂(指涂膜剂)0119涂膜剂二部I T搽剂涂剂涂膜剂(指涂膜剂)一部I N酊剂0120酊剂二部I C酊剂一部I I 贴音剂(指贴剂)0121贴剂二部I V贴剂0122贴裔剂一部I I贴音剂0123口服溶液剂口服混悬剂口服乳剂二部I o口服溶液剂口服混悬剂口服乳剂0124植人剂二部I J植人剂0125膜剂二部I M膜剂0126耳用制剂二部I Q耳用制剂-部I V搽剂洗剂涂膜剂(指洗剂)0127洗剂二部I s洗剂冲洗剂灌肠剂(指洗剂)一部I V搽剂洗剂涂膜剂(指洗剂)0128冲洗剂二部I S洗剂冲洗剂灌肠剂(指冲洗剂〉0129灌肠剂二部I S 洗剂冲洗剂灌肠剂(指灌肠剂)0181合剂一部I J 合剂0182锭剂一部I E锭剂0183煎膏剂(裔滋)一部I F煎裔剂(裔滋)0184胶剂一部I G胶剂0185酒剂—部I M酒剂0186裔药一部I P裔药0187露剂一部I S露剂0188茶剂一部I T茶剂0189流浸裔剂与浸資剂一部I o流浸裔剂与浸裔剂0200其他通则0211药材和饮片取样法一部n a药材和饮片取样法0212药材和饮片检定通则一部n b药材和饮片检定通则0213'炮制通则一部n d炮制通则0251药用辅料二部n药用辅料• 426 •中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表续表.....编号通则名称原附录原附录名称0261制药用水-部m制药用水二部m制药用水0291国家药品标准物质通则新增第二增补本03000301一般鉴别试验一部w一般鉴别试验二部i n一般鉴别试验0400光谱法一部Y分光光度法二部W分光光度法三部n分光光度法0401紫外-可见分光光度法一部V A紫外-可见分光光度法二部IV A 紫外-可见分光光度法三部n a紫外-可见分光光度法0402红外分光光度法一部V c红外分光光度法二部i v c红外分光光度法0405荧光分光光度法二部N E荧光分析法三部n c荧光分析法0406原子吸收分光光度法一部V D原子吸收分光光度法二部IV D原子吸收分光光度法三部n b原子吸收分光光度法0407火焰光度法二部IV F火焰光度法三部n d火焰光度法0411电感耦合等离子体原子发射光谱法一部H E电感耦合等离子体原子发射光谱法0412电感耦合等离子体质谱法一部H D电感耦合等离子体质谱法0421拉曼光谱法二部XK L拉曼光谱法指导原则0431质谱法.二部IX J 质谱法0441核磁共振波谱法二部IX K 核磁共振波谱法0451X射线衍射法二部IX F X射线粉末衍射法0500色谱法0501纸色谱法一部YI A 纸色谱法二部V A纸色谱法三部m a纸色谱法0502薄层色谱法一部VI B 薄层色谱法二部V B薄层色谱法0511柱色谱法一部yi c柱色谱法二部v c柱色谱法0512髙效液相色谱法-部VI D髙效液相色谱法二部V D高效液相色谱法三部冚 B 髙效液相色谱法0513离子色谱法一部YI G离子色谱法二部V J 离子色谱法三部m e离子色谱法• 427《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称0514分子排阻色谱法二部V H 分子排阻色谱法三部m d分子排阻色谱法一部V I E 气相色谱法0521气相色谱法二部V E 气相色谱法三部in c气相色谱法0531超临界流体色谱法新增0532临界点色谱法新增二部V F 电泳法三部W A 醋酸纤维素薄膜电泳法0541电泳法三部IV B 琼脂糖凝胶电泳法三部N C SD&聚丙烯酰胺凝胶电泳法三部IV D 等电聚焦电泳法0542毛细管电泳法一部二部V I F毛细管电泳法V G 毛细管电泳法0600物理常数测定法0601相对密度测定法一部1 A 相对密度测定法二部yi a相对密度测定法0611馏程测定法一部W B 馏程测定法二部V I B馏程测定法0612熔点测定法一部M C 熔点测定法二部*V I C 熔点测定法0613凝点测定法一部1D凝点测定法二部VI D凝点测定法0621旋光度测定法一部1E旋光度测定法二部E旋光度测定法0622折光率测定法一部m f折光率测定法二部V I F折光率测定法一部1G p H值测定法0631p H值测定法二部V I H p H值测定法三部V A p H值测定法一部X I F渗透压摩尔浓度测定法0632渗透压摩尔浓度测定法二部K G 渗透压摩尔浓度测定法三部V H渗透压摩尔浓度测定法0633黏度测定法二部V I G黏度测定法0661热分析法二部1Q 热分析法0681制药用水电导率测定法二部1S制药用水电导率测定法0682制药用水中总有机碳测定法二部1R 制药用水中总有机碳测定法0700其他测定法0701电位滴定法与永停滴定法一部1 A 电位滴定法与永停滴定法二部I A 电位滴定法与永停滴定法%0702非水溶液滴定法一部1B非水溶液滴定法二部M B 非水溶液滴定法• 428中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表续表编号通则名称原附录原附录名称0703氧瓶燃烧法二部1 C 氧瓶燃烧法一部K L氮测定法0704氮测定法二部1D氮测定法三部M l A氮测定法一部K M乙醇量测定法0711乙醇量测定法二部\I E乙醇量测定法0712甲氧基、乙氧基与羟丙氧基测定法二部\l F甲氧基、乙氧基与羟丙氧基测定法一部IX N脂肪与脂肪油測定法0713脂肪与脂肪油测定法二部1H脂肪与脂肪油测定法0721维生素A测定法二部1J 维生素A测定法0722维生素D测定法二部1K维生素D测定法二部1M蛋白质含量测定法0731蛋白质含量测定法三部YI B蛋白质测定法0800限量检査法一部H C氯化物检査法0801氣化物检查法二部1A氯化物检查法0802硫酸盐检查法二部1B硫酸盐检查法0803硫化物检査法二部1 C 硫化物检查法0804硒检查法二部1D砸检查法0805氟检查法二部1E氟检查法二部1F氰化物检查法0806氰化物检查法三部YI X氮化物残留量测定法一部IX D铁盐检查法0807铁盐检查法二部1G铁盐检查法0808铵盐检查法二部1K铵盐检查法一部K E重金属检査法0821重金属检查法二部1H重金属检查法一部IX F砷盐检查法0822砷盐检査法二部1J 砷盐检查法一部K G干燥失重测定法0831干燥失重测定法二部1L干燥失重测定法三部W L干燥失重测定法一部K H水分测定法0832水分测定法二部坩M水分测定法三部\I D水分测定法一部K J 炽灼残渣检查法0841炽灼残渣检查法二部W N炽灼残渣检查法0842易炭化物检查法二部1C) 易炭化物检查法二部1P残留溶剂测定法0861残留溶剂测定法三部VI V残留溶剂测定法0871甲醇量检查法一部IX T甲醇量检查法0872合成多肽中的醋酸测定法二部1N合成多肽中的醋酸测定法429《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称08732-乙基己酸测定法二部1L2-乙基己酸测定法0900特性检査法0901溶液颜色检查法一部H A溶液颜色检查法二部K A溶液颜色检查法0902澄清度检查法二部I X B澄清度检査法一部I X R不溶性微粒检查法0903不溶性微粒检查法二部I X c不溶性微粒检査法三部V I不溶性微粒检査法一部X I c可见异物检查法0904可见异物检査法二部I X H可见异物检查法三部V B 可见异物检査法一部M A崩解时限检查法0921崩解时限检查法二部\A崩解时限检查法三部V C崩解时限检查法一部1B融变时限检查法0922融变时限检査法二部I B融变时限检査法三部V D 融变时限检查法0923片剂脆碎度检查法二部X G片剂脆碎度检査法三部V E 片剂脆碎度检査法0931溶出度与释放度测定法二部X C溶出度测定法二部I D释放度测定法0941含量均匀度检查法二部X E 含量均匀度检查法—部a C最低装量检査法0942最低装量检查法二部\F最低装量检查法三部V F最低装量检查法0951吸人制剂微细粒子空气动力学特性测定法二部吸人气雾剂、吸人粉雾剂、吸入喷雾剂的雾滴(粒)X H分布测定法0952黏附力测定法一部1 E 贴膏剂黏附力测定法二部I J贴剂黏附力测定法0981结晶性检查法二部K D结晶性检查法一部X I B粒度测定法0982粒度和粒度分布测定法二部K E 粒度和粒度分布测定法三部V G粒度测定法0983锥入度测定法二部I K 锥入度测定法1100生物检査法一部I I B无菌检查法1101无菌检查法二部X I H 无菌检查法三部W A无菌检查法非无菌产品微生物限度检査:微生物计数法一部m c微生物限度检查法1105二部H J微生物限度检查法%三部I G微生物限度检査法• 430 •中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表续表4编号通则名称原附录原附录名称非无菌产品微生物限度检查:控制菌检查法一部X I O c微生物限度检査法1106二部X I J微生物限度检査法三部M G微生物限度检查法一部X D I C微生物限度检查法1107非无菌药品微生物限度标准二部X I J微生物限度检査法三部M G微生物限度检査法一部X I D抑菌剂效力检查法指导原则1121抑菌效力检查法二部X I X N抑菌剂效力检査法指导原则三部X I A抑菌剂(防腐剂)效力检查法指导原则一部X f f l E异常毒性检查法1141异常毒性检查法二部X I c异常毒性检査法三部I F异常毒性检査法一部m a热原检査法1142热原检查法二部X I D热原检査法三部M D热原检查法一部X f f l D细菌内毒素检查法1143细菌内毒素检査法二部X I E细菌内毒素检査法三部1E细菌内毒素检查法1144升压物质检查法二部X I F升压物质检查法1145降压物质检查法一部m f降压物质检査法二部X I G降压物质检査法1146组胺类物质检查法新增1147过敏反应检查法一部X I I G过敏反应检査法二部X I K过敏反应检査法1148溶血与凝聚检查法—部X f f l H溶血与凝聚检査法二部X I L溶血与凝聚检查法1200生物活性测定法1201抗生素微生物检定法二部X I A抗生素微生物检定法1202青霉素酶及其活力测定法二部X I B青霉素酶及其活力测定法1205升压素生物测定法二部1A升压素生物测定法1206细胞色素C活力测定法二部I B细胞色素C活力測定法1207玻璃酸酶测定法二部m C玻璃酸酶测定法1208肝素生物测定法二部M D肝素生物测定法1209绒促性素生物测定法二部1E绒促性素生物测定法1210缩宫素生物测定法二部M F缩宫素生物测定法1211胰岛素生物测定法二部M G胰岛素生物测定法1212精蛋白锌胰岛素注射液延缓作用测定法二部I H精蛋白锌胰岛素注射液延缓作用测定法1213硫酸鱼精蛋白生物测定法二部1J硫酸鱼精蛋白生物测定法1214洋地黄生物测定法二部孤K洋地黄生物测定法1215葡萄糖酸锑钠毒力检查法二部I L葡萄糖酸锑钠毒力检查法1216卵泡刺激素生物测定法二部M M卵泡刺激素生物测定法1217黄体生成素生物测定法二部1N黄体生成素生物测定法431《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称1218降钙素生物测定法二部1O降钙素生物测定法1219生长激素生物测定法二部1P 生长激素生物测定法1401放射性药品检定法二部X I放射性药品检定法一部n灭菌法1421灭菌法二部X I灭菌法三部XV 灭菌法1431生物检定统计法二部X W生物检定统计法2000中药其他方法2001显微鉴别法一部n c显微鉴别法2101膨胀度测定法一部IX () 膨胀度测定法2102裔药软化点测定法一部1D裔药软化点测定法2201浸出物测定法一部X A浸出物测定法2202鞣质含量测定法一部X B鞣质含量测定法2203桉油精含量测定法一部X c桉油精含量测定法2204挥发油测定法一部I D挥发油测定法2301杂质检査法一部IX A杂质检查法2302灰分测定法一部K K灰分测定法2303酸败度测定法一部IX P 酸败度测定法2321铅、镉、砷、汞、铜测定法一部IX B 铅、镉、砷、汞、铜测定法2322汞和砷元素形态及其价态测定法新增2331二氧化硫残留量测定法一部IX u二氧化硫残留量测定法2341农药残留量测定法一部IX Q农药残留量测定法2351黄曲霉毒素测定法一部IX V黄曲霉毒素测定法2400注射剂有关物质检查法一部K S 注射剂有关物质检查法3000生物制品相关检査方法3100含量测定法3101固体总量测定法三部1M固体总量测定法3102唾液酸测定法三部yi c唾液酸测定法3103磷测定法三部W A磷测定法3104硫酸铵测定法三部w c硫酸铵测定法3105亚硫酸氢钠测定法三部I E亚硫酸氢钠测定法3106氢氧化铝(或磷酸铝)测定法三部1F氢氧化铝(或磷酸铝)测定法3107氣化钠测定法三部I G氯化钠测定法3108枸橼酸离子测定法三部I H枸橼酸离子测定法3109钾离子测定法三部1I钾离子测定法3110钠离子测定法三部I J 钠离子测定法3111辛酸钠测定法三部V I K辛酸钠测定法3112乙酰色氨酸测定法三部VI W乙酰色氨酸测定法3113苯酚测定法三部V I M苯酚测定法3114间甲酚测定法三部VI N间甲酚测定法3115'硫柳汞测定法三部1B硫柳汞测定法432中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表续表编号通则名称原附录原附录名称对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含三部M l T对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含量测定法3116量测定法3117O-乙酰基测定法三部M l F O乙酰基测定法3118己二酰肼含量测定法三部1K己二酰肼含量测定法3119高分子结合物含量测定法三部L高分子结合物含量测定法3120人血液制品中糖及糖醇测定法三部VI P人血液制品中糖及糖酵测定法3121人血白蛋白多聚体测定法三部yi Q人血白蛋白多聚体测定法人免疫球蛋白类制品Ig G单体加二聚体测三部yi r人免疫球蛋白类制品i g G单体加二聚体测定法3122定法3123人免疫球蛋白中甘氨酸含量测定法三部S 人免疫球蛋白中甘氨酸含量测定法3124重组人粒细胞刺激因子蛋白质含量测定法三部yi u重组人粒细胞刺激因子蛋白质含量测定法3125组胺人免疫球蛋白中游离磷酸组胺测定法三部V I E组胺人免疫球蛋白中游离磷酸组胺测定法3126I g G含量测定法三部H K I g G含量测定法3127单抗分子大小变异体测定法新增3200化学残留物测定法3201乙醇残留量测定法三部Y I D乙醇残留量测定法3202聚乙二醇残留量测定法三部V I G聚乙二醇残留量测定法3203聚山梨酯80残留量测定法三部Y I H聚山梨酯80残留量测定法3204戊二醛残留量测定法三部V I I戊二醛残留量测定法3205磷酸三丁酯残留量测定法三部V I J磷酸三丁酯残留量测定法3206碳二亚胺残留量测定法三部V I Y碳二亚胺残留量测定法3207游离甲醛测定法三部Y I L游离甲醛测定法3208人血白蛋白铝残留量测定法三部1K人血白蛋白铝残留量测定法3209羟胺残留童测定法新增3300微生物检査法3301支原体检査法三部M B支原体检查法3302外源病毒因子检査法三部1C病毒外源因子检査法3303鼠源性病毒检査法三部I H 鼠源性病毒检查法3304S V40核酸序列检査法三部I X H S V40核酸序列检査法3305猴体神经毒力试验三部X I L猴体神经毒力试验血液制品生产用人血浆病毒核酸检测技术新增3306要求3400生物澜定法3401免疫印迹法三部V I A 免疫印迹法3402免疫斑点法三部11 B 免疫斑点法3403免疫双扩散法1三部11 C免疫双扩散法3404免疫电泳法三部1 D 免疫电泳法3405肽图检查法三部1E肽图检查法3406质粒丢失率检査法三部I X G质粒丢失率检査法3407外源性D N A残留量测定法三部K B外源性D N A残留量测定法3408抗生素残留量检查法三部I X A抗生素残留量检查法3409激肽释放酶原激活剂测定法三部I X F激肽释放酶原激活剂测定法433《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称3410抗补体活性测定法三部I X K抗补体活性测定法3411牛血清白蛋白残留量测定法三部1I牛血清白蛋白残留量测定法3412大肠杆菌菌体蛋白质残留量测定法三部I X C大肠杆菌菌体蛋白质残留量测定法3413假单胞菌菌体蛋白质残留量测定法三部I X D假单胞菌菌体蛋白质残留量测定法3414酵母工程菌菌体蛋白质残留量测定法三部K E酵母工程菌菌体蛋白质残留量测定法3415类A血型物质测定法三部K I类A血型物质测定法3416鼠I g G残留量测定法三部I X L鼠I g G残留量测定法3417无细胞百日咳疫苗鉴别试验三部I X s无细胞百日咳疫苗鉴别试验3418抗毒素、抗血清制品鉴别试验三部K T 抗毒素、抗血清制品鉴别试验3419A群脑膜炎球菌多糖分子大小测定法三部1G A群脑膜炎球菌多糖分子大小测定法3420伤寒V i多糖分子大小测定法三部I H 伤寒V i多糖分子大小测定法3421b型流感嗜血杆菌结合疫苗多糖含量测定法三部m J b型流感嗜血杆菌结合疫苗多糖含量测定法3422人凝血酶活性检查法三部I X N人凝血酶活性检查法3423活化的凝血因子活性检查法三部I X0活化的凝血因子活性检查法3424肝素含量测定法三部I X P 肝素含量测定法3425抗A、抗B血凝素测定法三部K J抗A、抗B血凝素测定法3426人红细胞抗体测定法三部I X Q人红细胞抗体测定法3427人血小板抗体测定法三部I X R人血小板抗体测定法3500生物活性/效价测定法3501重组乙型肝炎疫苗(酵母)体外相对效力检査法三部\A重组乙型肝炎疫苗(酵母)体外相对效力检查法3502甲型肝炎灭活疫苗体外相对效力检查法三部I S 甲型肝炎灭活疫苗体外相对效力检査法3503人用狂犬病疫苗效价测定法三部X I A人用狂犬病疫苗效价测定法3504吸附破伤风疫苗效价测定法三部XI B 吸附破伤风疫苗效价测定法3505吸附白喉疫苗效价测定法三部X I C吸附白喉疫苗效价测定法3506类毒素絮状单位测定法三部X I D 类毒素絮状单位测定法3507白喉抗毒素效价测定法三部XI E 白喉抗毒素效价测定法3508破伤风抗毒素效价测定法三部X I F破伤风抗毒素效价测定法3509气性坏疽抗毒素效价测定法三部X I G气性坏疽抗毒素效价测定法3510肉毒抗毒素效价测定法三部X I H 肉毒抗毒素效价测定法3511抗蛇毒血清效价测定法三部X I I抗蛇毒血清效价测定法3512狂大病免疫球蛋白效价测定法三部X I J 狂犬病免疫球蛋白效价测定法3513人免疫球蛋白中白喉抗体效价测定法三部I0人免疫球蛋白中白喉抗体效价测定法3514人免疫球蛋白F c段生物学活性测定法三部I P 人免疫球蛋白F c段生物学活性测定法3515抗人T细胞免疫球蛋白效价测定法(E玫瑰花环形成抑制试验)三部抗人T细胞免疫球蛋白效价测定法(E玫瑰花环形成X Q抑制试验)3516抗人T细胞免疫球蛋白效价测定法(淋巴细胞毒试验)三部抗人T细胞免疫球蛋白效价测定法(淋巴细胞X R毒试验)3517人凝血因子n效价测定法三部x j人凝血因子n效价测定法3518人凝血因子V I[效价测定法三部X K人凝血因子V I I效价测定法3519^人凝血因子I X效价测定法三部X L 人凝血因子K效价测定法3520人凝血因子X效价测定法三部X M人凝血因子X效价测定法• 434 •中国药典2015年版《中国药典》2015年版通则编码与2010年版附录编码对照表续表«编号通则名称原附录原附录名称3521人凝血因子V I E效价测定法三部X N人凝血因子1效价测定法3522重组人促红素体内生物学活性测定法三部I B重组人促红素体内生物学活性测定法3523干扰素生物学活性测定法三部I C 干扰素生物学活性测定法3524重组人白介素-2生物学活性测定法三部I D重组人白介素-2生物学活性测定法3525重组人粒细胞剌激因子生物学活性测定法三部X E重组人粒细胞刺激因子生物学活性测定法重组人粒细胞巨噬细胞剌激因子生物学活三部\F重组人粒细胞巨噬细胞剌激因子生物学活性测定法3526性测定法重组牛碱性成纤维细胞生长因子生物学活三部X G重组牛碱性成纤维细胞生长因子生物学活性测定法3527性测定法3528重组人表皮生长因子生物学活性测定法三部I H重组人表皮生长因子生物学活性测定法3529重组链激酶生物学活性测定法三部I I 重组链激酶生物学活性测定法3530鼠神经生长因子生物学活性测定法新增3531尼妥珠单抗生物学活性测定法新增3532重组人白介素-11生物学活性测定法新增3533A型肉毒毒素效价测定法新增3600特定生物原材料/动物3601无特定病原体鸡胚质量检测要求三部XII A无特定病原体鸡胚质量检测要求m b实验动物微生物学检测要求3602实验动物微生物学检测要求三部m c实验动物寄生虫学检测要求3603实验动物寄生虫学检测要求三部3604新生牛血淸检测要求三部XI D新生牛血清检测要求3605细菌生化反应培养基三部X I V细菌生化反应培养基37003701生物制品国家标准物质目录新增试剂与标准物质8000一部XV A试药8001试药二部XV A试药一部XV B试液8002试液二部I V B试液一部I V c试纸8003试纸二部I V C 试纸一部I V D缓冲液8004缓冲液二部X V D缓冲液一部XV E指示剂与指示液8005指示剂与指示液二部X V E指示剂与指示液一部XV F滴定液8006滴定液二部XV F滴定液8061对照品对照药材对照提取物—部X V G对照品对照药材对照提取物8062标准品与对照品二部I V G 标准品与对照品9000指导原则9001原料药物与制剂稳定性试验指导原则二部XIX c原料药与药物制剂稳定性试验指导原则• 435 •《中国药典》2015年版通则编码与2010年版附录编码对照表中国药典2015年版续表编号通则名称原附录原附录名称9011药物制剂人体生物利用度和生物等效性试验指导原则二部药物制剂人体生物利用度和生物等效性试验指导XIX B原则9012生物样品定量分析方法验证指导原则新增9013缓释、控释和迟释制剂指导原则二部XIX D 缓释、控释和迟释制剂指导原则9014微粒制剂指导原则二部XIX E 微襄、微球与脂质体制剂指导原则9015药品晶型研究及晶型质量控制指导原则新增9101药品质量标准分析方法验证指导原则一部二部X I A 中药质量标准分析方法验证指导原则m A 药品质量标准分析方法验证指导原则9102药品杂质分析指导原则二部XIX F 药品杂质分析指导原则9103药物引湿性试验指导原则二部X K J 药物引湿性试验指导原则9104近红外分光光度法指导原则二部XIX K 近红外分光光度法指导原则9105中药生物活性测定指导原则一部X I C 中药生物活性测定指导原则9106基于基因芯片的药物评价技术与方法指导原则新增9107中药材D N A条形码分子鉴定法指导原则新增一部X I E 药品微生物检验替代方法验证指导原则9201药品微生物检验替代方法验证指导原则二部XIX 0药品微生物检验替代方法验证指导原则三部X I B 药品微生物检验替代方法验证指导原则9202非无菌产品微生物限度检查指导原则一部二部11 F 微生物限度检查法应用指导原则XIX P 微生物限度检查法应用指导原则9203药品微生物实验室质量管理指导原则一部二部11G 药品微生物实验室规范指导原则XIX Q 药品微生物实验室规范指导原则9204微生物鉴定指导原则新增9205药品洁净实验室微生物监测和控制指导原则新增9206无菌检査用隔离系统验证指导原则新增9301注射剂安全性检査法应用指导原则—部二部I I B 中药注射剂安全性检查法应用指导原则XIX M 化学药品注射剂安全性检査法应用指导原则9302中药有害残留物限量制定指导原则新增9303色素测定法指导原则新增9304中药中铝、铬、铁、钡元素测定指导原则新增9305中药中真菌毒素测定指导原则新增9501正电子类放射性药品质量控制指导原则二部M G 正电子类放射性药品质量控制指导原则9502锝[99m T c]放射性药品质量控制指导原则二部XIX H 得[99™Tc]放射性药品质量控制指导原则9601药用辅料功能性指标研究指导原则新增9621药包材通用要求指导原则新增9622药用玻璃材料和容器指导原则新增9901国家药品标准物质制备指导原则新增第二增补本• 436。
15版药典颗粒剂规定

七、颗粒剂的微生物限度应符合要求。
八、根据原料药物和制剂的特性,除来源于动、植物多组分且难以建立测定方法的颗粒剂外,溶出度、释放度、含量均匀度等应符合要求。
九、除另有规定外,颗粒剂应密封,置干燥处贮存,防止受潮。生物制品原液、半成品和成品的生产及质量控制应符合相关品种要求。
除另有规定外,颗粒剂应进行以下相应检查。
【粒度】除另有规定外,照粒度和粒度分布测定法(通则0982第二法双筛分法)测定,不能通过一号筛与能通过五号筛的总和不得超过15%。
【水分】中药颗粒剂照水分测定法(通则0832)测定,除另有规定外,水分不得超过8.0%。
【干燥失重】除另有规定外,化学药品和生物制品颗粒剂照干燥失重测定法(通则0831)测定,于105℃干燥(含糖颗粒应在80℃减压干燥)至恒重,减失重量不得超过2.0%。
泡腾颗粒 系指含有碳酸氢钠和有机酸,遇水可放出大量气体而呈泡腾状的颗粒剂。
泡腾颗粒中的原料药物应是易溶性的,加水产生气泡后应能溶解。有机酸一般用枸橼酸、酒石酸等。
肠溶颗粒 系指采用肠溶材料包裹颗粒或其他适宜方法制成的颗粒剂。肠溶颗粒耐胃酸而在肠液中释放活性成分或控制药物在肠道内定位释放,可防止药物在胃内分解失效,避免对胃的刺激。肠溶颗粒应进行释放度(通则0931)检查。
缓释颗粒 系指在规定的释放介质中缓慢地非恒速释放药物的颗粒剂。
缓释颗粒应符合缓释制剂的有关要求(通则9013),并应进行释放度(通则0931)检查。
控释颗粒 系指在规定的释放介质中缓慢地恒速释放药物的颗粒剂。
控释颗粒应符合控释制剂的有关要求(通则9013),并应进行释放度(通则0931)检查。
颗粒剂在生产与贮藏期间应符合下列规定。
2015版中国药典 工艺用水质要求及检测

Hangzhou Microball Biotech. Co., Ltd.
Microball 2015 all rights reserved
相关的规范性文件、强制性标准规定 6、《体外诊断试剂生产实施细则(试行)》 第二十五条、 工艺用水制水设备应满足水质 要求并通过验证,其制备、储存、输送应能防 止微生物污染和滋生,并不应对产品质量和性 能造成影响。制水设备应定期清洗、消毒、维 护。应配备水质监控的仪器与设备,并定期记 录监控结果。
Hangzhou Microball Biotech. Co., Ltd.
Microball 2015 all rights reserved
相关的规范性文件、强制性标准规定
7、2015中国药典规定,饮用水不能直接用作 制剂的制备或试验用水。纯化水可作为配制普 通药物制剂用的溶剂或试验用水。 注射用水 由纯化水经蒸馏得到。 8、14223.1 《医用输液、输血、注射器具检 验方法第一部分化学分析方法 》本标准中试 验用水若无特殊规定,均应符合roball Biotech. Co., Ltd.
Microball 2015 all rights reserved
相关的规范性文件、强制性标准规定 5《一次性使用麻醉穿刺包生产实施细则》应 建立工艺用水的管理文件,并按相关标准检测 并记录。冷却用水应达到国家饮用水标准,洁 净区工位器具、针的精洗过程应用纯化水清 洗,与药(血)液直接接触部件应用注射用水 清洗。记录检测参数。
Hangzhou Microball Biotech. Co., Ltd.
Microball 2015 all rights reserved
相关的规范性文件、强制性标准规定
药典三部(2015版)-通则-0832水分测定法

0832 水分测定法第一法(费休氏法)1. 容量滴定法本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的原理来测定水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。
费休氏试液的制备与标定(1)制备称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24小时。
也可以使用稳定的市售费休氏试液。
市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。
本试液应遮光,密封,阴凉干燥处保存。
临用前应标定滴定度。
(2)标定精密称取纯化水10~30mg,用水分测定仪直接标定;或精密称取纯化水10~30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法(通则0701)等]指示终点;另做空白试验,按下式计算:F=WA−B式中F为每1ml费休氏试液相当于水的重量,mg;W为称取纯化水的重量,mg;A为滴定所消耗费休氏试液的容积,ml;B为空白所消耗费休氏试液的容积,ml。
测定法精密称取供试品适量(约消耗费休氏试液1~5ml),除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。
或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另做空白试验,按下式计算:×100%供试品中水分含量(%)=(A−B)FW式中A为供试品所消耗费休氏试液的体积,ml;B为空白所消耗费休氏试液的体积,ml;F为每1ml费休氏试液相当于水的重量,mg;W为供试品的重量,mg。
《中国药典》2015年版制剂通则变化比较

中国药典》2015 年版制剂通则变化比较2015 年版《中国药典》无论是在品种收载、标准增修订幅度、检验方法完善、检测限度设定,还是在标准体系的系统完善、质控水平的整体提升都上了一个新的台阶。
通过学习、熟悉2015 年版《中国药典》制剂通则的变化,了解目前我国用药水平、制药水平和监管水平现状,解读未来我国药品行业的趋势。
学习重点:熟悉药典制剂通则,掌握其标准的提升。
第一部分制剂通则比较2015 年版《中国药典》已于2015 年6 月5 日由国家食品药品监督管理总局正式颁布,于2015 年12 月1 日正式实施。
新版药典是国家药品标准的组成部分,是国家药品标准体系的核心。
按照党中央提出的“四个最严”要求,新版药典的制修订始终坚持“科学、先进、实用、规范”的原则,依据试验数据、研究结果、专家评估,体现药典编制的科学性和严谨性,以持续改进提高药品质量。
新版药典的颁布标志着我国用药、制药以及监管水平的全面提升,将促进药品质量的整体提高,对于保障公众用药安全有效意义重大。
《中国药典》2015 年版进一步扩大药品品种的收载和修订,共收载品种5608种。
其中,一部收载品种2598 种,二部收载品种2603 种,三部收载品种137 种。
本版药典是将三部药典的附录合一,加强共性的系统化、完善化及规范化,新版《中国药典》的附录调整为凡例、通则与方法、指导原则、药用辅料等单独成卷,为第四部。
第四部的名称为“《中国药典》2015 年版总则”,包括现有药典一部、二部、三部的附录(现改为“通则”)内容和药用辅料品种正文。
一、《中国药典》2015 年版总则(四部)项目组成:1、前言2、第十届药典委员会委员名单3、目录4、中国药典沿革5、品种及通则变化名单6、凡例(三部合一)7、品名目次(保留笔画索引,品种正文拟改为按拼音排序)8、通则(原药典附录内容):包括导引图、制剂通则、通用方法/检测方法及指导原则9、附表:包括原子量表、国际单位换算表及新旧附录/通则编码对照表10、药用辅料品种正文(原收载于药典二部正文品种第二部分)11、总索引(中英文索引)、修订说明1、使分类更加清晰明确,药典标准更加系统化、规范化将上一版药典中中药、化学药、生物制品三部分别收载的附录(凡例、制剂通则、分析方法、指导原则、药用辅料等)三合一,独立成卷作为第四部。
水分测定法

药物的水分检查1、水分含量的多少,对药物的稳定性、理化性状及生理作用等都可能产生影响。
所以多数原料药及固体制剂的质量标准中都规定有水分检查项。
根据水分子与药物分子之间结合的紧密程度,药物中的水分可以简单的分为结合水和吸附水,但二者之间并没有严格的界限。
2、水分的分析方法很多,有费休氏法、测定法、差示扫描量热法、单晶X-射线衍射法等方法。
其中费休氏法、干燥失重测定法是最为常用的两种方法。
①费休氏法是根据碘和二氧化硫在吡啶和甲醇溶液中与水起定量反应的原理测定水分。
测定时先将样品在规定的溶剂中溶解后滴定,通常情况下样品中所有水分均释放到溶剂中,所以测定结果包括吸附水和结合水两部分。
但应当注意:一些氧化剂如铬酸盐,还原剂如硫代硫酸盐以及其它能与试液生成水的化合物会干扰测定;有些羰基化合物对此法也有干扰:活泼的醛和酮与试剂中的甲醇反应生成缩醛和水,造成检查结果偏高。
②干燥失重测定法测定的是样品在常压或减压的条件下经干燥后所减失的重量,减失部分除有极小量的有机溶剂外主要是水分。
该方法能够准确测定大部分药品的水分含量,但当样品中含有结晶水,尤其是当结晶水与药物分子结合紧密时,测定的结果往往不是样品中水的总量,而只是吸附水。
热重分析法是在程序控制温度下,测量物质重量与温度关系的一种技术。
通常情况下,本法适用于药物结晶水的测定和贵重药物(样品用量较小)或在空气中易氧化药物的干燥失重的分析。
同干燥失重测定法一样,该方法也是利用加热使样品失重,只是利用程序控制温度,优点在于其测定时所需样品的量较小,分析时间较短,数据处理更方便,获得的信息量大。
但当结晶水与药物分子结合的非常紧密时,结晶水在药物熔融分解时才释放,此时的测定结果易造成样品中不含结晶水的假象。
③差示扫描量热法是在程序控制温度下测量输给待测物质与参比物的能量差与温度(或时间)关系的一种技术,当样品中含有结晶水时,其图谱中通常会出现明显的吸热峰。
但在结晶水与样品分子结合紧密时,结晶水的吸热峰有可能与样品熔融吸热峰相重叠。
干燥失重与水分测定的区别

干燥失重与水分测定的区别在药品行业,随着一致性评价的进行,水分的测定已经与我们息息相关了:原料药使用前要测、压片前测一下、WG后要测....那么您是测干燥失重还是测水份呢?《中国药典》(2015版)通则660832记载的是水分测定法,共有五法:费休氏法、烘干法、减压干燥法、甲苯法、气相色谱法;干燥失重记载于通则52,里面只有一种方法,有点类似于水份测定法第二法“烘干法”。
讲到这里,大家应该明白了干燥失重时而等于水份测定、时而不等于水份测定。
一、什么时候等于什么时候不等于?与物料结合的水有自由水和结合水之分:自由水,即以游离的形式存在,可以自由流动;结合水,也称“结晶水”,是结合在化合物中的水分子,它们不属于液态水。
我们常用的乳糖有无水乳糖和一水乳糖,一水乳糖约含5%结合水(一水乳糖分子量为342.3,一水的分子量为18,18/342≈5%),0.2%的自由水,在90℃下干燥只能除去自由水,若温度达到230℃以上则能去除结合水。
水分测定测的水包括自由水和结合水,干燥失重测定的水只是自由水,结合水的失去条件一般较高(如一水乳糖是需达到230℃以上才能去除)。
所以,对于无水乳糖而言,它的干燥失重值=水分测定值;而对于一水乳糖而言,它的干燥失重值≠水分测定值(若温度>230℃则相等)。
值得注意的是,若产品中含有易挥发组分(或残留),干燥失重的测定的结果包括了易挥发组分的含量。
如制剂过程使用50%乙醇造粒,干燥失重测得为结合水+挥发乙醇的总含量。
二、快速水分检测仪快速水分检测仪与卡尔费休水分测定仪现在已经属于我们工作中的辅助伙伴了,可能快速水分检测仪大家更喜欢一点,因为它有两个非常突出的特点:快、使用方便。
快速水分检测仪即红外线快速水分测定仪,采用热解重量原理设计的,是一种新型快速水分检测仪器。
在检测样品重量的同时,红外加热单元和水分蒸发通道快速干燥样品,在干燥过程中,水分仪持续测量并即时显示样品丢失的含水量%,干燥程序完成后,最终测定的水分含量值被锁定显示,从而被读取使用。
(完整版)费休氏水分测定法

费休氏水分测定法1 简述费休氏水分测定法是利用碘在吡啶和甲醇溶液中氧化二氧化硫时需要定量的水参加反应的原理来测定样品中的水分含量,本法可适用任何可溶解于费休氏试液但不与费休氏试液起化学反应的药品的水分测定,故对遇热易破坏的样品仍能用本法测定。
基本反应为I2+SO2+H2O —2HI+SO3上述反应是可逆的,但有吡啶存在时,无水吡啶能定量地吸收HI 和SO3,生成氢碘酸吡啶和亚硫酸吡啶。
C5H5N・|2+C5H5N・SO2+C5H5N+H2O —2C5H5NHI+C5H5NSO3 亚硫酸吡啶亦不稳定,能与水发生副反应,消耗一部分水,因而干扰测定。
C5H5N -SO3+H2O —C5H5NHSO4H加入无水甲醇可使亚硫酸吡啶转变成稳定的甲基硫酸氢吡啶,避免了上述副反应的发生。
C5H5N -SO3+CH3OH —C5H5NHSO4CH3滴定的总反应为C5H5NI2+C5H5NSO2+C5H5N+CH3OH+H2O —2C5H5N -HI+C5H5NHSO4CH3 .............. ⑴由上式可知,吡啶与甲醇不仅作为溶剂,而且参与滴定反应,此外,吡啶还可以与二氧化硫结合降低其蒸气压,使其在溶液中保持比较稳定的浓度。
1.1 容量滴定法根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理;由滴定溶液颜色变化(由淡黄色变为红棕色)或用永停滴定法指示终点;利用纯水首先标定出每1ml费休氏试液相当于水的重量(mg);再根据样品与费休氏试液的反应计算出样品中的水分含量。
1.2 库仑滴定法与容量滴定法相同,库仑滴定法也是根据碘和二氧化硫在吡啶(有些型号仪器改用无臭味的有机胺代替吡啶)和甲醇溶液中能与水起定量反应的原理来进行测定的。
与容量滴定法不同,在库仑滴定法中,碘是由含碘化物的电解液在电解池阳极电解发生碘。
2「—l2+2e (2)只要滴定池中存在水,发生的碘就会按反应(1)进行反应。
当所有的水都反应完毕,阳极电解液中会剩余少许过量的碘。
《中国药典》2015药典纯化水标准
中国药典2015年版溶液的澄清度与颜色取本品,加水制成每lml中约含阿魏酸钠20m g的溶液,溶液应澄清无色;如显浑浊,与1号池度标准液(通则0902第一法)比较,不得更浓;如显色,与黄色或黄绿色3号标准比色液(通则0901第一法)比较,不得更深。
有关物质避光操作。
取本品,加流动相溶解并稀释制成每l m l中约含0. 7m g的溶液,作为供试品溶液;精密量取lm l,置200m l量瓶中,用流动相稀释至刻度,摇勻,作为对照溶液。
照阿魏酸钠有关物质项下的方法测定。
供试品溶液的色谱图中如有杂质峰,各杂质峰面积的和不得大于对照溶液的主峰面积(0.5%)。
水分取本品,照水分测定法(通则0832第一法1)测定,含水分应为13.0%〜16.0%(供无菌粉末用)或应不超过3.0%(供无菌冻干品用)。
热原取本品,加灭菌注射用水制成每l m l中含阿魏酸钠5m g的溶液,依法检查(通则1142),剂量按家兔体重每l k g 缓慢注射3m l,应符合规定。
无菌照阿魏酸钠项下的方法检査,应符合规定。
其他应符合注射剂项下有关的各项规定(通则0102)。
【含置测定】避光操作。
取装量差异项下的内容物约0.15g,精密称定,加冰醋酸20m l使阿魏酸钠溶解,照阿魏酸钠项下的方法,自“加醋酐3m l”起,依法测定。
每l m l高氣酸滴定液(0.lm o l/L)相当于25.22mg的C10H9N a04•2H20。
【类别】同阿魏酸钠。
【规格】(1)0. lg(2)0. 3g【贮藏】遮光,密封保存。
纯化水ChunhuashuiPurified WaterH2018.02本品为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的制药用水,不含任何添加剂。
【性状】本品为无色的澄清液体;无臭。
【检査】酸碱度取本品10m l,加甲基红指示液2滴,不得显红色;另取10m l,加溴麝香草酚蓝指示液5滴,不得显蓝色。
硝酸盐取本品5m l置试管中,于冰浴中冷却,加10%氣化钾溶液0.4m l与0.1%二苯胺硫酸溶液0.1m l,摇匀,缓缓滴加硫酸5m l,摇勻,将试管于50T:水浴中放置15分钟,溶液产生的蓝色与标准硝酸盐溶液[取硝酸钾0.163g,加水溶解并稀释至100m l,摇匀,精密量取l m l,加水稀释成100m l,再精密量取10m l,加水稀释成100m l,摇匀,即得(每l m l相当于1吨N03)]0.3m l,加无硝酸盐的水4.7m l,用同一方法处理后纯化水的颜色比较,不得更深(0.000006%)。
水分测定参考中国药典

水分测定参考中国药典
水分测定的方法参考中国药典2015年版,包括以下几种方法:
1. 重量法:将一定重量的样品在加热器中干燥至恒定重,称取干燥后样品的重量,计算出水分含量。
2. 比重法:将样品与溶剂混合,测得混合物的密度,根据样品的密度和水的密度计算出水分含量。
3. 干燥法:将样品放在高温高湿的环境中,测量样品重量的减少,计算出水分含量。
4. Karl Fischer法:利用Karl Fischer试剂将样品中的水分直接
滴定,并根据滴定量计算出水分含量。
以上方法应根据不同的药物特性和实验条件进行选择。
15版药典上灰分浸出物水分测定方法汇总1

15版药典上灰分浸出物水分测定方法汇总2302灰分测定法i.总灰分测定法测定用的供试品须粉碎,使能通过二号筛,混合均匀后,取供试品2~3g(如须测定酸不溶性灰分,可取供试品3~5g),置炽灼至恒重的坩埚中,称定重量(准确至0. 01g) ,缓缓炽热,注意避免燃烧,至完全炭化时,逐渐升高温度至500~600℃,使完全灰化并至恒重。
根据残渣重量,计算供试品中总灰分的含量(%)。
如供试品不易灰化,可将坩埚放冷,加热水或10%硝酸铵溶液2m1,使残渣湿润,然后置水浴上蒸干,残渣照前法炽灼,至坩埚内容物完全灰化。
2。
酸不溶性灰分测定法取上项所得的灰分,在坩埚中小心加人稀盐酸约10 ml,用表面皿覆盖坩埚,置水浴上加热10分钟,表面皿用热水5ml冲洗,洗液并入坩埚中,用无灰滤纸滤过,坩埚内的残渣用水洗于滤纸上,并洗涤至洗液不显氯化物反应为止。
滤渣连同滤纸移置同一坩埚中,干燥,炽灼至恒重.根据残渣重量,计算供试品中酸不溶性灰分的含量(%)。
稀盐酸取盐酸234m1,加水稀释至1000m1,即得。
马弗炉当马弗炉第一次使用或长期停用后再次使用时,必须进行烘炉。
烘炉的时间应为室温200℃四小时。
200℃至600℃四小时。
使用时,炉温最高不得超过额定温度,以免烧毁电热元件。
禁止向炉内灌注各种液体及易溶解的金属,马弗炉最好在低于最高温度50℃以下工作,此时炉丝有较长的寿命。
空坩埚恒重取洁净坩埚置高温炉内,将坩埚盖斜盖于坩埚上,经加热至700~800℃炽灼约30~60min,停止加热,待高温炉温度冷却至约300℃,取出坩埚,置适宜的干燥器内,盖好坩埚盖,放冷至室温(一般约需60分钟),精密称定坩埚重量(精确至0.1mg)。
再以同样条件重复操作,直至恒重,备用2201浸出物测定法i.水溶性浸出物测定法测定用的供试品需粉碎,使能通过二号筛,并混合均匀。
冷浸法取供试品约4g,精密称定,置250~300ml的锥形瓶中,精密加水100ml,密塞,冷浸,前6小时内时时振摇,再静置18小时,用干燥滤器迅速滤过,精密量取续滤液20ml,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。
药典水分第三法

药典水分第三法检测方法如下:1. 准备足够的供试品,分制备1-20个直径为5.25±0.45毫米的小丸。
把2-3个湿润(小水珠,其表面呈半露状)。
按图四的体积等分装置分样逐粒包裹铝-b砂(所用规格号不宜过小,以免在干燥过程中试样局部过热)。
2. 制备好的供试品应平铺于干燥器内,放置30分钟进行预干燥。
预干燥后试样表面无水珠存在即可进行称量。
3. 将称量后的供试品置于已恒重的称量瓶中,在100-105摄氏度干燥至恒重(一般需要3小时以上)。
取出后置干燥器中,放置至室温后称量。
4. 如此反复直至失重率在0.5%以内。
通常进行恒重操作时,需将供试品置于干燥器中干燥至少三次。
通过以上步骤,可得到干燥后的供试品重量。
接下来进行测定步骤:5. 将已恒重的供试品放在称量瓶中,固定好称量瓶的位置,放在索氏萃取器中。
6. 在索氏萃取器中加入萃取溶剂(丙酮),加热至沸腾,保持30分钟。
提取时间不少于6小时。
提取液应呈无色或微黄色。
7. 提取液冷却后,将提取器下端的阀门关闭,使提取器与浓缩瓶分开,用少量溶剂冲洗浓缩瓶后弃去。
将提取器中的提取液转移至蒸发皿中,用纸筒套在蒸发皿周围进行减压蒸馏(为避免提取液溅出),蒸干溶剂后残留的干燥固体物质重量即为测定结果。
这种方法需要确保实验过程的准确性和规范性,以保证结果的准确性。
水分测定一般多使用电热鼓风干燥箱、真空干燥箱、电子天平等常见的实验设备。
在实验过程中要注意安全,避免发生意外。
以上就是药典水分第三法的检测步骤和注意事项,希望对你有所帮助。
如有更多疑问,可以咨询实验室工作人员或查阅相关文献资料。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
0832 水分测定法
第一法(费休氏法)
1. 容量滴定法
本法是根据碘和二氧化硫在吡啶和甲醇溶液中与水定量反应的
原理来测定水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定应在干燥处进行。
费休氏试液的制备与标定
(1)制备称取碘(置硫酸干燥器内48小时以上)110g,置干燥的具塞锥形瓶(或烧瓶)中,加无水吡啶160ml,注意冷却,振摇至碘全部溶解,加无水甲醇300ml,称定重量,将锥形瓶(或烧瓶)置冰浴中冷却,在避免空气中水分侵入的条件下,通入干燥的二氧化硫至重量增加72g,再加无水甲醇使成1000ml,密塞,摇匀,在暗处放置24小时。
也可以使用稳定的市售费休氏试液。
市售的费休氏试液可以是不含吡啶的其他碱化试剂,或不含甲醇的其他伯醇类等制成;也可以是单一的溶液或由两种溶液临用前混合而成。
本试液应遮光,密封,阴凉干燥处保存。
临用前应标定滴定度。
(2)标定精密称取纯化水10~30mg,用水分测定仪直接标定;或精密称取纯化水10~30mg,置干燥的具塞锥形瓶中,除另有规定外,加无水甲醇适量,在避免空气中水分侵入的条件下,用费休氏试液滴定至溶液由浅黄色变为红棕色,或用电化学方法[如永停滴定法
(通则0701)等]指示终点;另做空白试验,按下式计算:
F=W
A−B
式中F为每1ml费休氏试液相当于水的重量,mg;
W为称取纯化水的重量,mg;
A为滴定所消耗费休氏试液的容积,ml;
B为空白所消耗费休氏试液的容积,ml。
测定法精密称取供试品适量(约消耗费休氏试液1~5ml),除另有规定外,溶剂为无水甲醇,用水分测定仪直接测定。
或精密称取供试品适量,置干燥的具塞锥形瓶中,加溶剂适量,在不断振摇(或搅拌)下用费休氏试液滴定至溶液由浅黄色变为红棕色,或用永停滴定法(通则0701)指示终点;另做空白试验,按下式计算:
×100%
供试品中水分含量(%)=(A−B)F
W
式中A为供试品所消耗费休氏试液的体积,ml;
B为空白所消耗费休氏试液的体积,ml;
F为每1ml费休氏试液相当于水的重量,mg;
W为供试品的重量,mg。
如供试品吸湿性较强,可称取供试品适量置干燥的容器中,密封(可在干燥的隔离箱中操作),精密称定,用干燥的注射器注入适量无水甲醇或其他适宜溶剂,精密称定总重量,振摇使供试品溶解,测定该溶液水分。
洗净并烘干容器,精密称定其重量。
同时测定溶剂的水分。
按下式计算:
供试品中水分含量(%)=(W1−W3)c1−(W1−W2)c2
×100%
W2−W3
式中W1为供试品、溶剂和容器的重量,g;
W2为供试品、容器的重量,g;
W3为容器的重量,g;
c1为供试品溶液的水分含量,g/g;
c2为溶剂的水分含量,g/g。
对热稳定的供试品,亦可将水分测定仪和市售卡氏干燥炉联用测定水分。
即将一定量的供试品在干燥炉或样品瓶中加热,并用干燥气体将蒸发出的水分导入水分测定仪中测定。
2.库仑滴定法
本法仍以卡尔-费休氏(Karl-Fischer)反应为基础,应用永停滴
定法(通则0701)测定水分。
与容量滴定法相比,库仑滴定法中滴
定剂碘不是从滴定管加入,而是由含有碘离子的阳极电解液电解产生。
一旦所有的水被滴定完全,阳极电解液中就会出现少量过量的碘,使铂电极极化而停止碘的产生。
根据法拉第定律,产生碘的量与通过的电量成正比,因此可以通过测量电量总消耗的方法来测定水分总量。
本法主要用于测定含微量水分(0.0001%~0.1%)的供试品,特别适用于测定化学惰性物质如烃类、醇类和酯类中的水分。
所用仪器应干燥,并能避免空气中水分的侵入;测定操作应在干燥处进行。
费休氏试液按卡尔-费休氏库仑滴定仪的要求配制或使用市售
费休氏试液,无需标定滴定度。
测定法于滴定杯加入适量费休氏试液,先将试液和系统中的水分预滴定除去,然后精密量取供试品适量(含水量约为0.5~5mg),
迅速转移至滴定杯中,以永停滴定法(通则0701)指示终点,从仪器显示屏上直接读取供试品中水分的含量,其中每1mg水相当于10.72库仑电量。
第二法(烘干法)
测定法取供试品2~5g,平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm,疏松供试品不超过10mm,精密称定,开启瓶盖在100~105℃干燥5小时,将瓶盖盖好,移置干燥器中,放冷30分钟,精密称定,再在上述温度干燥1小时,放冷,称重,至连续两次称重的差异不超过5mg为止。
根据减失的重量,计算供试品中含水量(%)。
本法适用于不含或少含挥发性成分的药品。
第三法(减压干燥法)
减压干燥器取直径12cm左右的培养皿,加入五氧化二磷干燥剂适量,铺成0.5~1cm的厚度,放入直径30cm的减压干燥器中。
测定法取供试品2~4g,混合均匀,分别取0.5~1g,置已在供试品同样条件下干燥并称重的称量瓶中,精密称定,打开瓶盖,放入上述减压干燥器中,抽气减压至2.67kPa(20mmHg)以下,并持续抽气半小时,室温放置24小时。
在减压干燥器出口连接无水氯化钙干燥管,打开活塞,待内外压一致,关闭活塞,打开干燥器,盖上瓶盖,取出称量瓶迅速精密称定重量,计算供试品中的含水量(%)。
本法适用于含有挥发性成分的贵重药品。
中药测定用的供试品,一般先破碎并需通过二号筛。
第四法(甲苯法)
仪器装置如图。
图中A为500ml的短颈圆底烧瓶;B为水分测定管;C为直形冷凝管,外管长40cm。
使用前,全部仪器应清洁,并置烘箱中烘干。
测定法取供试品适量(约相当于含水量1~4ml),精密称定,置A瓶中,加甲苯约200ml,必要时加入干燥、洁净的无釉小瓷片数片或玻璃珠数粒,连接仪器,自冷凝管顶端加入甲苯至充满B管的狭细部分。
将A瓶置电热套中或用其他适宜方法缓缓加热,待甲苯开始沸腾时,调节温度,使每秒馏出2滴。
待水分完全馏出,即测定管刻度部分的水量不再增加时,将冷凝管内部先用甲苯冲洗,再用饱蘸甲苯的长刷或其他适宜方法,将管壁上附着的甲苯推下,继续蒸馏5分钟,放冷至室温,拆卸装置,如有水黏附在B管的管壁上,可用蘸甲苯的铜丝推下,放置使水分与甲苯完全分离(可加亚甲蓝粉末少量,使水染成蓝色,以便分离观察)。
检读水量,并计算成供试品的含水量(%)。
图甲苯法仪器装置
【附注】(1)测定用的甲苯须先加水少量充分振摇后放置,将水层分离弃去,经蒸馏后使用。
(2)中药测定用的供试品,一般先破碎成直径不超过3mm的颗粒或碎片;直径和长度在3mm以下的可不破碎。
第五法(气相色谱法)
色谱条件与系统适用性试验用直径为0.18~0.25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球作为载体,或采用极性与之相适应的毛细管柱,柱温为140~150℃,热导检测器检测。
注入无水乙醇,照气相色谱法(通则0521)测定,应符合下列要求:
(1)理论板数按水峰计算应大于1000,理论板数按乙醇峰计算应大于150;
(2)水和乙醇两峰的分离度应大于2;
(3)用无水乙醇进样5次,水峰面积的相对标准偏差不得大于3.0%。
对照溶液的制备取纯化水约0.2g,精密称定,置25ml量瓶中,加无水乙醇至刻度,摇匀,即得。
供试品溶液的制备取供试品适量(含水量约0.2g),剪碎或研细,精密称定,置具塞锥形瓶中,精密加入无水乙醇50ml,密塞,混匀,超声处理20分钟,放置12小时,再超声处理20分钟,密塞放置,待澄清后倾取上清液,即得。
测定法取无水乙醇、对照溶液及供试品溶液各1~5μl,注入气相色谱仪,测定,即得。
对照溶液与供试品溶液的配制须用新开启的同一瓶无水乙醇。
用外标法计算供试品中的含水量。
计算时应扣除无水乙醇中的含水量,方法如下:
对照溶液中实际加入的水的峰面积=对照溶液中总水峰面积-K×对照溶液中乙醇峰面积
供试品中水的峰面积=供试品溶液中总水峰面积-K×供试品溶液中乙醇峰面积
K=无水乙醇中水峰面积
无水乙醇中乙醇峰面积。