俄罗斯晶体学所
纪念俄罗斯科学院院士顾哲教授

纪念俄罗斯科学院院士顾哲教授9 |纪念俄罗斯科学院院士顾哲教授*□ 陶 源 周 阔中图分类号:K815 doi:10.19326/ki.2095-9257.2021.01.002*感谢俄罗斯科学院东方所中国部高级研究员C. B.德米特里耶夫(Сергей Викторович Дмитриев)对本文提供的帮助。
2020年7月28日,俄罗斯和波兰汉学界泰斗、历史学博士、教授、俄罗斯科学院东方研究所(Институт востоковедения Российской академии наук)中国部高级研究员斯坦尼斯瓦夫·罗伯特·库切拉(Stanisław Robert Kuczera, Станислав Роберт Кучера,中文名顾哲,后文以中文名指称)逝世。
他是连接俄罗斯汉学与世界汉学的重要纽带,是世界东方学黄金时期传统学术发展中一颗耀眼的明珠。
顾哲1928年5月5日出生于波兰的利沃夫市(Lviv ,1919—1939年属于波兰)。
1947年中学毕业,进入华沙大学东方学学院(Instytut Orientali- styczny Uniwersytetu Warszawskiego )学习,师从波兰著名汉学家夏伯龙(Witold Jabłoński ,1901—1957),并在其指导下完成了毕业论文《道教的基础》(«Основы даоской философии»),1952年毕业并获得东方学哲学硕士学位。
1951年至1953年初在华沙大学(Uniwersytet Warszawski )担任助教。
1953年2月,作为第一批派往中国的西方留学生,顾哲赴北京大学攻读博士学位。
1960年,中国科学院院士张政烺先生(1912—2005)指导顾哲完成了题为《“周礼”之下中国古代社会的阶级结构》(«Классовая структура древнекитайского общества на основе материалов 〈Чжоули〉 »)的博士论文,成为当时唯一一位用汉语完成博士论文的西方留学生。
XRF常用术语解释

术语表吸收边 (1)准确度 (1)老化 (1)分析晶体 (2)分析类型 (2)APC (2)表观浓度 (2)分析方法 (3)分析方法包 (3)应用模板 (3)人工多层晶体 (3)衰减器 (3)背景函数 (4)背景比率法 (4)平衡组分 (5)偏差修正 (5)Bragg 公式 (5)厚样品 (5)校正曲线系数 (6)检查分析 (6)化合物 (6)视野光栏 (6)微分曲线 (7)微分测量 (7)漂移校正 (7)双靶 (8)激发条件 (8)EZ 扫描 (8)固定角度分析 (8)定时分析 (8)流程条 (9)熔剂 (9)FP定量法 (9)全范围定性分析 (10)谱峰的函数分解 (10)玻璃熔片 (11)粒度效应 (11)高次线 (11)识别分析 (12)杂质校正 (12)积分测量 (12)强度积分测量 (12)内标校正 (13)手工输入数据 (13)基体校正 (13)测量光路气氛 (14)矿物效应 (14)净强度 (15)重叠校正 (15)PC (F-PC) (15)PC 芯线清洁 (15)谱峰分解 (15)谱峰搜索 (16)PHA (16)预抽真空 (17)PR 气体 (18)初级束滤光片 (18)定性分析 (18)定量分析 (19)再现性 (19)分辨率 (20)样品膜校正 (20)样品 ID (20)样品ID表 (20)样品模式 (21)样品旋转 (21)SC (21)散射线 (22)真空保护 (22)灵敏度库 (22)灵敏度库分区 (23)灵敏度库样品 (23)准直器 (23)平滑 (23)光谱仪灵敏度系数 (24)SQX 分析 (24)SQX 重叠校正 (24)标准谱形 (24)标准谱分解法 (24)理论基体校正系数 (25)薄膜样品 (25)管电压和管电流 (26)2θ角度 (26)2θ扫描 (26)单位 (26)通用标准样品 (27)X射线强度 (27)X-R 控制 (27)吸收边吸收边是指激发样品中的元素、产生荧光X 射线所需的最小能量。
晶体学基础知识讲义导论X衍射

晶体结构 = 点阵 * 结构基元
点阵点或结点总和称为点阵(lattice),具有平移对称性。
沿着一定方向按某种规则把 结点联结起来,则可以得到 描述各种晶体结构的几何图 象----晶体的空间格子(简 称为晶格)
二 晶 体点 阵
晶体结构最突出的特点是其结构基元(原 子、离子、分子或络合离子)在晶体所占有的 空间中作周期性的排列,构成了晶体点阵结构 图案。点阵总是由为数无限和周围相同点组成。
CsCl的晶胞图
Cs+ Cl-
CsCl晶体结构示意图
CsCl的晶体结构示意图
CsCl的晶胞图 Cl–
Cs+
Cl–
1.1.1 经典晶体学
1669年丹麦学者斯蒂诺,发现了晶面角守恒定律。
1801年法国结晶学家赫羽依,发表了有理指数定律。
1805–1809年间德国学者外斯总结出晶体对称定律。 随后又提出了晶带定律。
1809年乌拉斯顿设计了第一台反射测角仪。 1818–1839年间外斯和英国学者密勒先后创立了用
以表示晶面空间方位的晶面符号。
经典晶体学还包括了对天然矿物物理性质的研究。
1.2.2 近代晶体学
1912年德国科学家劳埃成功发现了X射线对晶体的衍 射现象,具体地证实了晶体结构点阵理论的正确性。
1913年英国晶体学家布拉格父子和俄国晶体学家吴 里弗分别独立地推导出X射线衍射基本公式。 20世纪20年代,完成了收集X射线衍射图谱和推引 空间群方法等工作。
◆ 晶体生长是研究人工培育晶体的方法和规律 ◆ 晶体的几何结构是研究晶体外形的几何理论及内部质
GOST 俄罗斯认证标准目录

GOST俄罗斯标准目录GOST 6999-1968现金出纳机纸GOST 6981-1975工业硫酸亚铁(绿矾)GOST 6979-1954象棋表GOST 6967-1970自动制钉机精度标准GOST 6956-1954管螺纹矫板及板牙GOST 6943.13-1979纺织玻璃纤维材料-硬挺度的测试方法GOST 6943.12-1979纺织玻璃纤维材料-悬垂度的测试方法GOST 6943.11-1979纺织玻璃纤维材料-弯曲硬挺度的测试方法GOST 6943.10-1979纺织玻璃纤维材料-断裂强力和断裂伸长的测试方法GOST 6943.9-1979纺织玻璃纤维材料-动态抗弯性能的测试方法GOST 6943.8-1979纺织玻璃纤维材料-含水量和灼烧损失的测试方法GOST 6943.7-1979纺织玻璃纤维材料-线性尺寸、面积密度和线密度的测定方法GOST 6943.6-1979纺织玻璃纤维材料-密度和难燃性测定方法GOST 6943.5-1979纺织玻璃纤维材料-单丝断裂强度测试方法GOST 6943.4-1979纺织玻璃纤维材料-捻度和捻度平衡指数的测定方法GOST 6943.3-1979纺织玻璃纤维材料-耐磨性测试方法GOST 6943.2-1979纺织玻璃纤维材料-单丝和纤维直径的测试方法GOST 6943.1-1979纺织玻璃纤维材料-线密度测试方法GOST 6943.0-1979纺织玻璃纤维材料-验收规则GOST 6940-1974交换机用白炽灯GOST 6939-1971灌木沼泽犁GOST 6927-1974混凝土外墙装饰技术条件GOST 6926-1975不透光纸GOST 6923-1984工业用辐射温度计一般技术要求GOST 6915-1971间接测量动脉压力的指示式压力仪器GOST 6862-1971永磁合金棒GOST6861-1973彩色书写纸GOST 6860-1968带有电磁式上条机构的汽车钟技术条件GOST 6855-1973框架开榫机主要参数与规格GOST 6854-1977细木工带锯机基本参数与尺寸GOST 6851.3-1977 6MTC-22型铅酸蓄电池技术条件GOST 6851.2-1977 6MTC-9型铅酸蓄电池技术条件GOST 6851.1-1977 3MT-8型铅酸蓄电池技术条件GOST 6851.0-1977摩托车和小型摩托车用铅蓄电池一般技术条件GOST 6850-1976 16mm电影放映机类型、基本参数、技术要求GOST 6826-1978木工四面刨床基本参数与尺寸GOST 6825-1974荧光灯GOST 6810-1974糊墙纸GOST 6809-1970热模锻曲柄压力机基本参数和尺寸GOST 6808-1976空气锤精度标准GOST 6800-1954带有电磁式上条机构的汽车钟技术条件GOST 6787-1980陶瓷地砖技术条件GOST 6787-1969陶瓷地砖GOST 6766-1973剪板机精度标准GOST 6765-1975热轨三层钢板和宽钢带GOST 6756-1957辛香料咸图贡白鲑、索西瓦鲱鱼、欧洲白鲑和高白鲑(桶装)GOST 6749-1974糊墙纸原纸GOST 6746-1975电容量具一般技术条件GOST 6742-1968内封页纸GOST 6727-1980加强钢筋混凝土结构用冷拉低碳钢钢丝GOST 6724-1977拱式和桥式蒸汽空气双作用锻缸精度标准GOST 6723-1972自动切边机精度标准GOST 6722-1975工业过滤纸板GOST 6713-1975桥梁建造用低合金结构钢。
化学文献

【摘要】众所周知,2011年诺贝尔化学奖实至名归地由以色列化学家Daniel Shechtman获奖,获奖理由是“发现准晶体”。
谢赫特曼发现了准晶体,这种材料具有的奇特结构,推翻了晶体学已建立的概念。
许多年以来,凝聚态物理学家们仅仅关心晶态的固体物质。
然而,在过去的几十年,他们逐渐把注意力转向“非晶”材料,如液体或非晶体,这些材料中的原子仅在短程有序,被称为缺少“空间周期性”。
准晶,这种新的结构因为缺少空间周期性而不是晶体,但又不像非晶体,准晶展现了完美的长程有序,这个事实给晶体学界带来了巨大的冲击,它对长程有序与周期性等价的基本概念提出了挑战。
【关键词】准晶的空间结构准晶的应用前景众所周知,2011年诺贝尔化学奖名至实归的由以色列化学家Daniel Shechtman获奖,获奖理由是“发现准晶体” [1]。
想必诺贝尔奖对一个人的重要性不用在此处赘述了吧,那么就请由我为大家简单的介绍一下诺贝尔奖得主与他的研究成果吧一:什么是准晶准晶是一种介于晶体和非晶体之间的固体。
准晶具有完全有序的结构,然而又不具有晶体所应有的平移对称性,因而可以具有晶体所不允许的宏观对称性。
准晶体其实发现的时间并不是很早,与1982年才被发现的,因其具有凸多面体规则外形的,但不同于晶体的固态物质,它们具有晶体物质不具有的五重轴。
目前已知的准晶体都是金属互化物。
2000年以前发现的所有几百种准晶体中至少含有3种金属,如Al65Cu23Fe12,Al70Pd21Mn9等。
但最近发现仅2种金属也可形成准晶体,如Cd57Yb10〔Nature,2000,408:537〕[2]。
由于准晶体的特殊性质,所以长期一来,大多数科学家并不承认其为单独存在的晶体。
2009年,矿物学上的一个发现为准晶是否能在自然条件下形成提供了证据:俄罗斯的一块铝锌铜矿上发现了Al63Cu24Fe13组成的准晶颗粒。
和实验室中合成的一样,这些颗粒的结晶程度都非常好。
第2届国际晶体学和矿物学大会

长过程的影响, 改造了相应生长设备 , 晶体生长中 将
坩锅 的 “ 冷点 ”固定在 中央 , 避免晶体与坩埚的 并 “ 接触 ” 从而在直径为 1 n的坩埚 中生长 了重达 , 4l n 67 的 L O晶体 。 3g B 他们从 B O晶体及熔剂 N 2 B a 0的 基本相关体系和相 图研究 出发 , 确定 了 B O晶体的 B 质量 。此外 , 他们对于布里奇曼法 降法) 生长技术 也作了改进 , 从而以此生长 A G S 红外非线性 晶体 g a2
平台加强与国际、 国内各界的沟通与交流 , 促进经济 社会 发展 , 是重 点需 要解决 的问题 。 本 次研 讨 会 进 一 步 增 进 和加 深 了 国 内外 银 行 业、 企业 和 高校在 支付领 域 的交 流 , 有利 于相 互之 间 . 建立更加密切 的合作 , 有利于支付结算事业的发展 和创新 ,同时也增强了西南财经大学在电子支付研 究领域领头羊的作用 。
其成功经验有利于我们在发展中借鉴 ,尤其是电子 支付 和电子商务 的发展过程是一个循序渐进 的过
程, 需要从进一步健全现有银行体系出发 , 并在此基 础上发 展 电子银 行 的建设 ;然 后再完 善 电子身 份认
足点就是鼓励各种支付工具的推广。 对于高校和电子支付与电子商务领域的研究人 员来说 , 本次研讨会使他们更加深刻地认识到, 应该 明确如何准确地把握和捕捉各种各样 的支付需求 , 在理论研究 、 系统实践等方面不断创新 , 并通过这个
伯利亚分院在非线性光学 晶体生长方面已形成 了特 色, 并具有很大的优势 , 从红外到紫外非线性 晶体生 长方面都取得很大成绩。 这次在会议 匕 AEK l 由 ..0h 【 博 士 报 告 了关 于三 硼 酸锂 (B )和 偏硼 酸 钡 ( LO B— B O 晶体生长的研究 。他们和法国合作 , B) 对于采用 熔盐法生长大尺寸晶体的技术进行 了改进。他们从 温场的对称性 出发 ,研究了温场对于液流及 晶体生
国内外矿物岩石矿床学教材点评分析

矿物学相关教材点评分析赵明教授1、外国教材《Ionic Compounds : Applications of Chemistry to Mineralogy》Applications of Chemistry toMineralogy中译名:离子化合物:矿物应用化学作者:Claude H.Yoder出版单位:A John Wiley & Sons, Inc.出版时间:2006年ISBN-13: 978-0-471-74046-9(pbk.)ISBN-10: 0-471-74046-2(pbk.)这本书是为想要较好地理解化学和矿物学的概念的读者编写的,作者编写此书的目的是希望这本书帮助学地质的人理解化学。
本书论述了矿物的结构和键的基本原理,着重讨论了晶体的结构与晶体的对称和物理性质之间的关系,描述了NaCl、闪锌矿、黄铁矿、方解石萤石、金红石等十多种矿物的结构,分析了影响矿物晶体对称的因素,论述了矿物形态和颜色的成因。
此书对那些从事矿物学工作的人来说具有一定的意义。
本书要求读者必须具备高中以上的化学和矿物学的知识水平。
它主要适合于想要深层次钻研矿物学知识的人使用,同时,也适用于矿物收藏家,因这些收藏家想要更多地了解所收藏的矿物的化学和结晶学的性质。
全书共8章,附录7个,共187页。
各章内容如下:1.键和成分2.离子化合物的结构:紧密堆积3.晶体对称4.简单紧密堆积化合物的结构5.影响单位晶胞对称的因素6.物理性质:形态7.物理性质:颜色8.化学性质本书编写风格独特,以提问和回答问题的活跃方式贯穿全书每个章节,让读者沉思之后,得到结果。
此方式,既能增强读者兴趣,又能提高他们对材料的理解,避免了枯糙乏味,达到了深入浅出的效果。
书中附有精美的矿物彩图,附录中附有周期表、离子、电子等各类化学数据、晶体对称分类,以及参考书目和相关的研究期刊等内容。
读者在使用了此书后将会大有收获的。
为诠释本书,作者还专门编写了非常行之有效的计算机软件程序Crystal-Maker3,并发至网上(网址.),软件Kristall2000程序可用来绘制晶体形状图。
2010诺贝尔物理学奖

2010诺贝尔物理学奖得主:把科研当成快乐的游戏 2010年10月15日 11:16 中国新闻周刊安德烈·海姆和康斯坦丁·诺沃肖洛夫今年的诺贝尔物理学奖可能最具娱乐性:一对师徒用透明胶带在制作铅笔芯的石墨中发现了一种二维平面材料,他们中的一位还曾获得过“搞笑诺贝尔奖”本刊记者/钱炜10月5日,瑞典皇家科学院宣布,将2010年诺贝尔物理学奖授予英国曼彻斯特大学的两位科学家——现年52岁的安德烈·海姆和36岁的康斯坦丁·诺沃肖洛夫,以表彰他们在石墨烯材料方面的卓越研究。
研究:“search”而非“research”石墨烯是怎么被发现的?对此,海姆2008年在接受《科学观察》采访时解释说,除了拥有设备和相关方面的知识,一个重要原因是自己有一种“科研恶习”。
他说,“那段时间里,我关注研究碳纳米管的那拨人,对他们时不时地声称获得这样或那样牛的成果觉得恶心。
我想,我可以做一点不同于碳纳米管的东西,为什么不把碳纳米管剖开呢?于是,就有了后来的研究。
”起初,海姆请实验室新来的一名中国博士生将一块高定向裂解石墨制成薄膜,要求尽可能薄,并给了他一台精巧的抛光机。
三周后,这名博士生拿着培养皿来见海姆,说他成功了。
海姆用显微镜一看,那些石墨碎片估计仍有1000层左右。
海姆希望他能将石墨碎片研磨得更薄一些,但这名博士生最后说:“如果你这么聪明,就自己试试。
”于是这成了一个转折点,海姆决定自己来试试,他就用透明胶带来做这件事。
如今,海姆所用的方法,被业界戏称为“透明胶带技术”。
由于层间的作用力非常弱,石墨很容易剥落脱离。
将石墨放在透明胶带上,反复撕拉 10~20下左右,就获得了10层左右的石墨——这正是海姆当初的实验,他们并没有直接获得石墨烯,但10层左右的石墨就已表现出了足够特殊的物理性能。
海姆曾用磁性克服重力,让一只青蛙漂浮在半空中,因此获得了2000年的“搞笑诺贝尔奖”。
诺贝尔基金会也形容这对师徒“把科学研究当成快乐的游戏”。
晶体结构与晶体化学晶体几何学理论基础

1.1.2 空间点阵
在图3.1的单位平移中,有两个最短的矢量,如图3.2所示。原点的选择是任意 的,任何图案的平移对称都可从图形的一点开始描述。如将图案抽象成一个点, 通过上述的一套平移对称操作即可得到一套平面上点的集合,称为网格或二维 点阵(图3.3)。在空间三维情况下,称作空间格子或空间点阵,点阵中的每个 点称为结点或点阵点。
晶体几何学理论基础
对称性是一种规律的重复,具有变化中的不变性,是自 然科学中一个重要的基本概念。晶体就是指原子或分子 在空间按一定规律重复排列构成的固体物质。晶体结构 的基本特征是其中的质点在三维空间作规律的重复排列。 晶体结构研究的就是揭示晶体内部原子和分子在空间排 列上的对称规律,这种规律只有在晶体结构中每个原子 在空间相对位置揭示出来时才能得到完整证明。
基本图案可以先旋转后反伸,也可以先反伸后旋转。其中1相当于i(反伸中心), 2相当于m)(对称面),3相当于3次轴加反伸中心,6相当于3次轴加对称面, 因此只有4是具有多利意义的旋转反伸轴。
2.点群 2.1 点对称要素 晶体外形上可能出现的对称要素称为点对称要素,包括对称中心、对称面、旋转轴 及旋转反伸轴。这些对称要素的特点是在进行对称操作过程中至少有一点是不动的。 二维空间的对称要素有:旋转点,2、3、4、6次轴;反映线,m。 三维空间的对称要素:旋转轴,2、3、4、6次轴;反伸(对称)中心,i;镜(对称) 面,m;旋转倒反轴,1、2、3、4、6。
1、对称操作 晶体学中的对称图形是通过对称操作来表征的。 对称操作 周期平移对称操作(晶体中) 有公度的
无公度的 准周期平移对称操作(准晶体中) 严格自相似准周期
点对称操作
旋转 反映 反伸
统计自相似准周期
1.1 平移
金兹堡朗道方程

金兹堡朗道方程
金兹堡-朗方程是一种非线性偏微分方程,是一类描述超导现象的非线性抛物型方程组.它是由Ginzburg和Landau在Landau二级相变理论的基础上,综合了超导体的电动力学、量子力学和热力学性质,提出的一个描述超导的唯象模型1937年,俄罗斯的列夫•达维多维奇•朗道(LevDavidovich Landau)提出了外磁场下超导中间态的结构模型,用超导体正常态层与超导态层交替共存的分层结构揭示超导现象的本质。
1950年,俄罗斯的维塔利•金兹堡(Vitaly Ginzburg)与朗道合作,在朗道提出的二级相变理论基础上提出了一个从宏观角度描述超导现象的数学模型(金兹堡-朗道方程,也称G-L方程):超导电子并非单独存在,电子之间可能有关联的最长距离称为它们的相干长度(受到外界扰动后超导恢复所需的空间尺度)。
由G-L方程可推导出超导体在外界磁场强度接近超导体的临界磁场强度(能破坏超导态的磁场强度)时的临界行为:外界磁场并非完全不能进入超导体,而是穿透进入超导体表面,足够强的外磁场则可进入超导体内破坏超导态而恢复成正常态。
超导性的唯象理论,是结合了超导体的电动力学、量子力学和热力学特性,为超导相变给予热力学解释而提出的。
此理论可以由Bcs理论得到,它能相当好地描述第Ⅱ类超导体的磁学性能。
酚与氯化胆碱形成低共熔溶剂

Separation and Purification Technology 63(2008)710–715Contents lists available at ScienceDirectSeparation and PurificationTechnologyj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /s e p p urShort communicationHighly efficient extraction of phenols and aromatic amines into novel ionic liquids incorporating quaternary ammonium cationVladimir M.Egorov,Svetlana V.Smirnova,Igor V.Pletnev ∗Department of Chemistry,M.V.Lomonosov Moscow State University,1/3Leninskiye gory,119992Moscow,Russiaa r t i c l e i n f o Article history:Received 13May 2008Received in revised form 26June 2008Accepted 28June 2008Keywords:Ionic liquidsLiquid–liquid extraction PhenolsAromatic aminesa b s t r a c tTwo novel hydrophobic room temperature ionic liquids (RTIL)incorporating quaternary ammonium cations:tetrahexylammonium dihexylsulfosuccinate (THADHSS)and trioctylmethylammonium salicylate (TOMAS)have been obtained.The mutual solubility of the both RTILs with water and their Reichardt’s polarity index have been measured.The extraction of aromatic amines and phenols into novel RTILs and two 1-alkyl-3-methylimidazolium bis (trifluoromethanesulfonyl)amides has been studied.The depen-dences of RTIL–water distribution ratio for the studied liquids on the phase volume ratio,the time of phase contact,pH value of aqueous solutions and the solute concentration have been obtained.In some cases,the solute distribution ratios for ammonium-based RTILs are as high as n ×104that is much greater than for imidazolium ones.Notably,unlike the case of imidazolium-based RTILs,the quantitative extraction into ammonium RTILs is achieved even at the phase volume ratio V RTIL :V w =1:20.©2008Elsevier B.V.All rights reserved.1.IntroductionRoom temperature ionic liquids (RTIL),which are organic or organoelement salts liquid at room temperature,have nowadays a growing diversity of applications in chemistry and technology [1,2].The advantages of room temperature ionic liquids include wide liq-uid range,high heat capacity,high thermal and chemical stability [3,4].As opposed to conventional organic solvents,most RTILs are non-flammable,have low vapor pressure and good electrochemical properties (electric conductivity,wide electrochemical window)[2–5].It is important that modification of cationic or anionic parts of RTIL may enable a fine-tuning of physical and chemical properties [6,7].Room temperature ionic liquids is an attractive alternative to conventional organic solvents in organic synthesis [1],catalysis [8],biopolymers processing [9–11],electrochemistry and electro-analysis [5,12–14],biochemistry [15],analytical chemistry [16].Furthermore,RTILs have a considerable potential as extraction sol-vents and may serve as a key to the design of more environmentally benign separation processes.However,the most abundant 1,3-dialkylimidazolium ionic liq-uids have some limitations on use,the most important being their high cost.That is why the search for new cheaper ionic liquids with improved characteristics is of current interest.As was indicated∗Corresponding author.Tel.:+74959395464;fax:+74959394675.E-mail address:pletnev@analyt.chem.msu.ru (I.V.Pletnev).elsewhere [17,18],some quaternary ammonium or phosphonium-based RTILs look the promising alternative:they are relatively cheap and easy to synthesize;some of them are water-immiscible and suitable for solvent extraction.A large number of publications devoted to liquid–liquid sep-arations with the use of RTILs have appeared within last years.The applications include extraction of simple substituted arenes [19,20],alcohols [21],carboxylic acids [22,23],and metal ions [24,25];aromatics/aliphatics separations [26,27]and fuel desul-furization [28,29].Extraction of various biological substances is another popular subject [30–32].It is worthy of mention that the majority of papers employ only imidazolium-based RTILs as extrac-tion solvents.Gathering a large array of experimental data on the extraction of representative solutes into different RTILs may give a useful information about the influence of solute/solvent structure on the partitioning of organic compounds.Phenols and aromatic amines may well serve as such representative solutes,since a wide vari-ety of substituted compounds are readily available to examine the role of structural details.Additionally,the data on the extraction of phenols have been previously reported for dialkylimidazolium hexafluorophosphate RTILs (our work [33]),and the extraction of phenols and amines has been reported for tetrafluoroborate ones [34].We report herein on the synthesis and properties of two novel hydrophobic RTILs incorporating quaternary ammo-nium cations,tetrahexylammonium dihexylsulfosuccinate (THADHSS)and trioctylmethylammonium salicylate (TOMAS).1383-5866/$–see front matter ©2008Elsevier B.V.All rights reserved.doi:10.1016/j.seppur.2008.06.024V.M.Egorov et al./Separation and Purification Technology63(2008)710–715711These two RTILs along with two imidazolium-based RTILs,1-hexyl-3-methylimidazolium and1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amides(HMImTf2N and DMImTf2N, respectively),have been tested as extraction solvents for recovery of aromatic amines and phenols from aqueous solution.2.Experimental2.1.ReagentsTetrahexylammonium bromide(Aldrich,99%),Aliquat®336 (trioctylmethylammonium chloride,Aldrich),sodium dihexylsul-fosuccinate(Technolog Ltd.,Russia),sodium salicylate(Panreac, RFE,USP,BP,Ph.Eur.),and2,6-diphenyl-4-(2,4,6-triphenylpyridin-1-yl)-phenolate monohydrate(Reichardt’s dye,Sigma–Aldrich, 70%)were used as received.1-Hexyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide(99%)was purchased at Sol-vent Innovation GmbH,Germany.1-Decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide was synthesized in the Dr.A.V.Yatsenko’s group at the Department of Chemistry of the MSU.The studied solutes,phenol,4-nitrophenol,2,4-dinitrophenol,2,6-dinitrophenol,picric acid,1-naphthol,2-naphthol,aniline hydrochloride,p-toluidine,3-nitroaniline,and tryptamine hydrochloride were of reagent grade purity.Potas-sium ferrocyanide,4-aminoantipyrine,amidopyrine(analytical reagent grade),and iron(III)sulfate(reagent grade)were used for spectrophotometrical measurements.Nitric and sulfuric acids (0.1and3M),potassium hydroxide(0.1and3M),phosphate(pH 6.86)and borate(pH9.18)buffer solutions were used for pH anic solvents,acetone(reagent grade),acetoni-trile(HPLC grade),chloroform(reagent grade)were used without additional purification.Water was freshly distilled before use.2.2.Preparation of quaternary ammonium RTIL2.2.1.Tetrahexylammonium dihexylsulfosuccinate19.42g(0.05mol)of sodium dihexylsulfosuccinate(NaDHSS) was dissolved in80ml of water at50◦C upon shaking.After complete dissolution of NaDHSS,equimolar amount(21.73g)of tetrahexylammonium bromide was added to the solution.The mixture was stirred for20min forming two phases where upper phase is RTIL.Aqueous phase was separated and RTIL phase was repeatedly rinsed with triple volume of fresh water10times upon vigorous shaking.The presence of NaBr in wash water was mon-itored by reaction with silver nitrate.After that the upper phase was collected and centrifuged for several hours to settle the emul-sion of water.A clear,colorless,viscous liquid was obtained.1H NMR(500MHz,CDCl3,␦/ppm relative to TMS):0.88(t;18H),1.33 (m;36H),1.55(m;12H),3.2(m;12H),4.1(m;3H).13C NMR (126MHz,DMSO-d6,␦/ppm relative to TMS):13.64,13.69,13.71 (CH3);20.91,21.76(C*H2CH3);21.87,24.82,24.85,25.34,27.95, 27.99,30.47,30.75,30.82(various CH2CH2);34.01(OOCC*H2CH);57.63(CHSO3);61.30(CH2N);63.89(CH2O);168.31,170.97(COOR).2.2.2.Trioctylmethylammonium salicylate40.42g(0.1mol)of trioctylmethylammonium chloride (Aliquat®336)was dissolved in200ml of acetone,and equal molar amount of sodium salicylate(16.01g)was added to the solution.The mixture was shaken for5h and left overnight.After that the precipitate wasfiltered off and acetone was evaporated fromfiltrate using rotary evaporator.The obtained RTIL was then rinsed with distilled water10times,and the upper RTIL phase was then centrifuged for several hours to settle the emulsion of water.Elemental analysis of the product yielded zero inorganic ash content.A clear,slightly yellowish,viscous liquid was obtained.1H NMR(500MHz,CDCl3,␦/ppm relative to TMS):0.88(t;9H),1.22(m;30H),1.53(m;6H),2.98(s;3H),3.13(m;6H),6.86(t;1H),6.92(d;1H),7.38(t;1H),7.88(d;1H),15.6(s;1H).13C NMR (126MHz,DMSO-d6,␦/ppm relative to TMS):13.80(C*H3C);21.93 (C*H2CH3);21.25,25.68,28.27,28.32,31.04(various CH2CH2);47.41(CH3N);60.50(CH2N);115.35,115.62,120.73,129.73,130.88 (aromatic C);163.17(COH);171.00(COO).1H and13C NMR data were recorded with NMR spectrometer DRX500(Bruker,Germany).2.3.Polarity measurementsFor solvatochromic polarity measurements a pinch of Reichardt’s dye on the tip of a spatula was added to3ml of studied solvent in a glass test-tube.If necessary,the mixture was ultrasonicated to completely dissolve the dye.After that the UV–vis spectrum of the solution was measured relative to distilled water(SF103spectrophotometer,Akvilon,Russia).The Dimroth–Reichardt polarity parameter E T(30)was calculated using the following equation:E T(30),kcal mol−1=28591max(1) where max is a maximum absorbance wavelength[35].2.4.Solubility measurementsThe solubility of THADHSS in water was measured conductimet-rically.A solution of THADHSS was prepared by dissolving a known amount of THADHSS in0.5L of deionized water.Then a series of cal-ibration solutions was made by dilution of the above solution,and their conductivity and the conductivity of the saturated THADHSS solution were measured.The solubility of TOMAS in water was determined spectrofluo-rometrically by salicylate( ex=305nm, em=405nm).The measurement of water content in water-saturated quater-nary ammonium RTILs was made using coulometric Karl Fischer titrator“Expert-007”(Econiks-Expert,Russia).2.5.Extraction procedureThe extraction was carried out in10ml polypropylene cen-trifuge test-tubes at ambient temperature(20±3◦C).The proper volumes of RTIL and pH-adjusted aqueous solution of studied com-pound were placed in a test-tube and shaken for the time necessary for extraction equilibrium to be achieved.Unless otherwise men-tioned,the phase volume ratio V IL:V w was1:3for imidazolium and 1:20for quaternary ammonium RTILs.After the necessary shaking time had elapsed,the systems with quaternary ammonium RTIL were centrifuged for2min(the centrifugation is not imperative but desirable as quaternary ammonium RTILs tend to adhere onto walls of test-tube after shaking).After that the necessary volume of aqueous phase was taken,and pH value was measured(pH-meter pH-410;combined glass microelectrode ESLK-13.7,Akvilon,Rus-sia).Finally,the determination of the solute in the aqueous phase was performed.The recovery(R,%)and the distribution ratio(D)of a solute were calculated using the following equations:R(%)=1−C wC0w100(2) D=C oC w=R100−RV wV o(3) where C0w and C w are the initial and equilibrium concentrations of the studied solute in aqueous phase,respectively(mol L−1),V w and712V.M.Egorov et al./Separation and Purification Technology63(2008)710–715V o denote the volumes of aqueous and RTIL phases,respectively (ml).The studied solutes were monitored spectrophotometrically or spectrofluorometrically[36,37].Spectroscopic measurements were performed using spectrophotometer UV-2201or spectrofluorime-ter RF-5301PC(Shimadzu,Japan),quartz cells.Concentrations of nitrocompounds were determined by their own absorbance after adding3M KOH to pH11–12.The absorbance was measured at400nm(4-nitrophenol),359nm(2,4-dinitrophenol),360nm (2,6-dinitrophenol),356nm(picric acid),365nm(3-nitroaniline). For spectrophotometrical determination of phenol and naphthols, 1.0ml of borate buffer solution(pH9.18),0.1ml of2%wt aque-ous K3Fe(CN)6,and0.1ml of2wt%aqueous4-aminoantipyrine were added to2.5ml of the studied solution.After5min,the absorbance was measured at510nm.For determination of aromatic amines,1ml of phosphate buffer solution,0.3ml of0.1M aqueous amidopyrine and1ml of2wt%K3Fe(CN)6were added to2.5ml of amine solution.After20min,the absorbance was measured at535nm.Tryptamine was determined spectrofluorimetrically at ex=279nm( em=359nm).Spectrophotometrical determination of salicylate in aqueous solutions after contact with TOMAS was car-ried out using freshly prepared5×10−3mol L−1(Fe3+)solution of iron(III)sulfate;2ml of5×10−3mol L−1Fe3+solution were added to2ml of salicylate solution(pH2–3);the absorbance was mea-sured at525nm[36].3.Results and discussion3.1.Properties of the quaternary ammonium RTILsTOMAS and THADHSS are clear liquids with densities slightly less than1g cm−3(0.945and0.975,respectively).Freezing points of the both RTILs are below−10◦C.The liquids are immiscible with water.1H and13C NMR spectra confirmed the identity of the obtained RTILs,and the molar cation–anion ratio calculated from1H NMR data was exactly1:1for the both RTILs.Solubility of THADHSS in water was found to be (8.6±0.2)×10−5mol L−1.Solubility of TOMAS is(2.0±0.2)×10−4mol L−1.These values are up to two orders of magnitude lower than that of common hydrophobic imidazolium-based RTILs[38].This significantly decreases a possible RTIL loss during extraction.The obtained RTILs are hydrolytically stable at pH2–13.Solubility of water in THADHSS and TOMAS is ca.5and7wt%, respectively(Karl Fischer titration).After shaking the RTILs with water,no emulsification was observed in both aqueous and RTIL phases.3.2.Polarity of THADHSS and TOMASPolarity is an important property of a solvent,which affects dif-ferent types of interactions between solvent and solute molecules. There exist several experimental methods for quantitative eval-uation of polarity:inverse gas chromatography,kinetic method, refractive index measurement[39].One of the most popular approaches refers to solvatochromism measured for a specific probe molecule,in particular,Reichardt’s betaine dye.The maxi-mum absorbance wavelength of this dye lies in visible spectrum and strongly depends on the nature of a solvent.In the present study we have measured the Dimroth–Reichardt’s polarity E T(30)of the novel RTILs,and three commonly used organic solvents(acetone,acetonitrile,and chloroform).The results along with literature data are shown in Table1.Table1max and E T(30)values for several solventsSolvent max(nm)E T(30)(kcal mol−1)Acetone67842.2Acetonitrile63045.4Chloroform69841.0 Dichloromethane[40]70040.8HMImTf2N[41]55251.8BMImPF6[40]a54552.5THABzO[35]b65143.9Ethanol[40]54652.4THADHSS(satd.with H2O)61546.5TOMAS(satd.with H2O)59348.2a1-Butyl-3-methylimidazolium hexafluorophosphate.b Tetrahexylammonium benzoate.The E T(30)values for THADHSS and TOMAS are close to each other and comparable with those for the other known ammonium-based RTILs[35].As compared to imidazolium-based RTILs(typical E T(30)values are in between49and60[35]),the novel ionic liquids are less polar.All the measurements and literature data are given for room temperature.It is important to mention that the E T(30)values were measured for water-saturated THADHSS and TOMAS,as they are directly related to the polarity of solvents in extraction conditions.On the basis of polarity measurements one may conclude that the novel RTILs would have solvation/extraction proper-ties different from both conventional extraction solvents and imidazolium-based ionic liquids.As is shown below(see extrac-tion data in Section3.4),there exists no quantitative relationship between E T(30)and extraction efficiency for corresponding RTIL. However,one may note that the lower E T(30)value the more effi-cient is extraction,in general.3.3.Extraction studies3.3.1.Optimization of extraction conditionsThe influence of phase contact time,phase volume ratio and concentration of an inorganic electrolyte(NaCl)on the distribution ratio has been studied on the example of2,4-dinitrophenol.The appropriate values of thefirst two parameters are shown in Table2.As can be seen from Table2,the time of phase contact,which is necessary to achieve extraction equilibrium,is less for the qua-ternary ammonium RTILs.Previously our group has shown that the optimal phase contact time for extraction into imidazolium-based ionic liquids weakly depends on the structure of extracted polar aromatic compound[33].That is why,and also for conditions uni-fication,in all the further experiments the phase contact time for all systems was15min.Like for the other previously studied imidazolium-based RTILs[33],the distribution ratio of2,4-dinitrophenol into both HMImTf2N and DMImTf2N dramatically decreases with increasing phase volume ratio.The3:1phase volume ratio has been chosen as optimal in order to maintain an acceptable distribution coefficientTable2Phase contact time and phase volume ratio for extraction of2,4-dinitrophenol (5×10−4mol L−1;pH2)RTIL Phase contacttime(min)Phase volumeratio(V w:V RTIL) THADHSS520:1TOMAS1020:1HMImTf2N153:1DMImTf2N153:1V.M.Egorov et al./Separation and Purification Technology 63(2008)710–715713and at the same time to minimize the quantity of RTIL,which is necessary for an extraction run.On the contrary,in the case of quaternary ammonium RTILs,the distribution ratio of 2,4-dinitrophenol weakly depends on the phase volume ratio up to 50:1.The value 20:1has been chosen for further experiments with the both quaternary ammonium RTILs in order to decrease the consumption of RTIL while sustaining a good recovery.The concentration of introduced inorganic electrolyte (NaCl)from 5×10−4to 0.5mol L −1has practically no effect on the dis-tribution ratio of 2,4-dinitrophenol.3.3.2.The effect of pH on the recovery of aromatic compoundsAll the studied solutes can be divided into two types based on their acid-base behaviour:phenols and aromatic amines.3.3.2.1.Extraction of phenols.The pH dependence of the distribu-tion ratio of unsubstituted phenol for all studied RTILs is shown in Fig.1.As can be seen,the maximal distribution ratio is observed at acidic pH,where the molecular form of phenol is predomi-nant.Upon the increase in pH the recovery of phenol into the imidazolium-based RTILs dramatically decreases,and at pH >12becomes negligible (R <10%).This is a typical behaviour for parti-tioning of phenol into imidazolium RTILs [33,34].The extraction ability of HMImTf 2N is close to that of DMImTf 2N (though less hydrophobic HMImTf 2N has a small advantage).The distribution ratios of phenol for the ammonium-based RTILs are approximately 0.5and 1order of magnitude (TOMAS and THADHSS,respectively)higher than for the imidazolium-based RTILs.The observed pH dependence of extraction clearly shows that phenol is preferably partitioned into all RTILs in undissociated (molecular)form.However,even at pH 12the recovery of phenol into the ammonium RTILs remains rather significant (R ∼40–50%,see Fig.1).In the case of nitrophenols pH value has small effect on the recovery into quaternary ammonium RTILs,whereas pH depen-dences of distribution ratio of nitrophenols into imdazolium-based RTILs have a trend similar to that for phenol.The aforementioned results clearly point to the possibility of phenolates extraction (anion-exchange)into the quaternary ammonium RTILs,in addi-tion to extraction of a neutral solute.Naturally,the contribution of ion-exchange recovery of ionized phenols should be higher at pH >pK a of solute.Expectedly,the efficiency of anion-exchange extraction into the quaternary ammonium RTILs is higher for more hydrophobic nitrophenols than for phenol.The similarbehaviourFig.1.The effect of pH on distribution ratio of phenol (1×10−4mol L −1)into differ-entRTILs.Fig.2.The increase of salicylate concentrations in aqueous phase after extraction at various pH (solid line)in comparison to calculated initial concentration of 2,4-dinitrophenolate (C total =5×10−4mol L −1;dashed line)in aqueous phase.has been previously observed for the partitioning of picric acid between water and BMImPF 6[33].In order to confirm a contribution of anion exchange to extraction,the concentration of salicylate in aqueous phase after extraction of 2,4-dinitrophenol (5×10−4mol L −1)into TOMAS was monitored.Evidently,anion-exchange extraction of dinitrophe-nolate should be accompanied by a release of stoichiometric quantity of RTIL anion,salicylate,to an aqueous phase.The measured concentration of salicylate after extraction at differ-ent pH was compared with the concentration of salicylate in solutions,which had also been in contact with TOMAS but did not contain 2,4-dinitrophenol (pH values of these solu-tion pairs were approximately the same).Corresponding plot is presented in Fig.2(concentration of 2,4-dinitrophenolate in aqueous phase is calculated with the use of literature pK a ,see Table 3).As can be seen,the increase in aqueous salicylate concentra-tion is higher at higher pH;at pH >7it is nearly equal to the total concentration of 2,4-dinitrophenol.Note that at pH >72,4-dinitrophenol exists mostly as anion,and its recovery into TOMAS is ca.100%.In other words,the increase of aqueous salicylate concen-tration exactly matches the concentration of extracted solute.This proves that the anion exchange mechanism is operative at pH >pK a (solute).3.3.2.2.Extraction of aromatic amines.The extraction of four aro-matic amines into RTILs has been investigated.Fig.3presents theTable 3Extraction of the studied phenols and aromatic amines into quaternary ammonium RTILs (V w :V RTIL =20:1)SolutepK a [42]log D log P OW [43,44]THADHSSTOMAS Phenol10.0 2.5 2.1 1.464-Nitrophenol 7.14 3.6 3.4 1.912,4-Dinitrophenol 4.08 4.1 3.5 1.672,6-Dinitrophenol 4.15 4.0 3.6 1.372,4,6-Trinitrophenol 0.69 3.9 3.8 1.331-Naphthol 9.85 3.8 3.4 2.852-Naphthol 9.63 3.7 3.2 2.70Aniline4.63 1.9 1.80.903-Nitroaniline 2.47 2.3 2.3 1.37p-Toluidine5.07 2.0 2.0 1.39Tryptamine10.23.52.61.55714V.M.Egorov et al./Separation and Purification Technology 63(2008)710–715Fig.3.The effect of pH on distribution ratio of aniline (1×10−4mol L −1)into differ-ent RTILs.pH dependence of the distribution ratio of aniline for all studied RTILs.Three of the studied amines incorporate NH 2group bonded to an aromatic ring,and one (tryptamine)has an aliphatic NH 2group and indole aromatic ring.The pH dependence of extraction of aniline into imidazolium-based RTILs is typical for amines [33,34].Noteworthy,a less hydrophobic HMImTf 2N is a better extraction solvent for aniline than DMImTf 2N.The extraction of aniline into THADHSS and TOMAS is much better than into imidazolium-based RTILs.The characteristic trend of pH dependence for the extraction of aniline,p-toluidine,and m-nitroaniline into the novel RTILs is observed.Tryptamine is efficiently extracted into both THADHSS and TOMAS,but at the optimal pH the distribution ratio of tryptamine for THADHSS is approximately one order of magnitude higher than for TOMAS.For all the ionic liquids studied,the pH profile of aniline extrac-tion is similar to that common for molecular solvents (i.e.efficient extraction takes place at pH >pK a of amine),which is indicative of neutral solute recovery.Unlike phenols,aromatic amines are poorly extracted in ionizedform.parison of distribution data for RTIL/water and 1-octanol/water systems (the trendline is shown for HMImTf 2N).parison of distribution ratios of several compounds for THADHSS and TOMAS (see also Table 3).parative analysis of the extraction resultsThe results obtained in the present work show that the newly obtained quaternary ammonium-based RTILs are much more effi-cient extraction solvents towards the studied aromatic compounds than common imidazolium-based RTILs.The extraction data for non-nitrated phenols and aromatic amines show that the maximal recovery is achieved for molecular forms of these compounds,the decrease in recovery closely corresponding to the respective pK a values.The data on partitioning of the studied phenols and aro-matic amines into quaternary ammonium RTILs are summarized in Table 3.The partitioning of neutral substituted aromatic molecules into imidazolium-based RTILs is often attributed to specific –inter-actions between imidazolium ring and aromatic ring of extracted compound [19,45]or to the ability of imidazolic proton at C2to form hydrogen bonds [46].However,in the case of THADHSS and TOMAS such interactions are unlikely.We suggest that dispersive interactions of solute molecules with cation of RTIL may be a driv-ing force for the preferential partitioning of phenols and aromatic amines into quaternary ammonium RTILs.It is interesting to correlate distribution ratio of organic com-pounds with 1-octanol/water partition coefficients,log P OW .Fig.4shows plot of logarithm of the maximal distribution ratio for the studied compounds (log D )into different RTILs vs.log P OW [43,44].As is clearly seen,the extraction ability of the imidazolium-based RTILs is inferior to the extraction ability of the novel quaternary ammonium RTILs and,in some cases,to that of 1-octanol.Noteworthy,there is a correlation between log P OW and log D for imidazolium-based RTILs (see a trendline in Fig.4).This corresponds well to the literature data [19,33].However,for the novel RTILs a log D –log P OW correlation is poor.This may be because of difference in extraction mechanisms between imidazolium and quaternary ammonium RTILs.At the same time,there exists a good correlation between log D obtained for the same solutes with use of THADHSS and TOMAS (Fig.5).This suggests that solvation/extraction patterns for the two ammonium RTILs be similar (note that,in general,THADHSS is more efficient extraction solvent than TOMAS).4.ConclusionsTwo novel quaternary ammonium based room temperature ionic liquids (THADHSS and TOMAS)have been synthesized andV.M.Egorov et al./Separation and Purification Technology63(2008)710–715715characterized.The Dimroth–Reichardt’s polarities of the novel RTILs,E T(30),are higher than those for the studied molecular sol-vents,but less than for imidazolium ionic liquids.Extraction of11aromatic compounds(phenols and aro-matic amines)into two imidazolium-based RTILs(HMImTf2N and DMImTf2N)and into the novel RTILs has been studied.The best recovery into the novel RTILs has been observed for nitrophenols and naphthols.The recovery of the aforementioned compounds is high(>50%at V IL:V w=1:20)in the whole studied pH range. For phenols at pH>pK a(solute)the contribution of ion-exchange mechanism to partitioning has been observed.In the case of imidazolium-based RTILs the best recovery is achieved for molec-ular form of solutes,the recovery of ionic forms being poor.It has been demonstrated that the extraction abilities of THADHSS and TOMAS well correlate with each other.Generally, THADHSS is more efficient extraction solvent towards the studied compounds than TOMAS.At the same time,it has been shown that the novel RTILs are more efficient extraction solvents than common imidazolium-based RTILs.AcknowledgementsWe are grateful to Dr.A.V.Yatsenko for providing a sample of DMImTf2N.Thanks are also due to Prof.S.I.Petrov for performing Karl Fischer titration,and to M.Khrenova for THADHSS solubil-ity measurements.This work was partially funded by the Russian Foundation for Basic Research(project N05-03-32976)and INTAS (project N05-1000008-8020).References[1]P.Wasserscheid,T.Welton(Eds.),Ionic Liquids in Synthesis,Wiley-VCHVerlagGmbH&CoKgaA,2002.[2]I.V.Pletnev,S.V.Smirnova,K.S.Khachatryan,V.V.Zernov,Russ.Chem.J.58(2004)51–57.[3]T.Welton,Chem.Rev.99(1999)2071–2083.[4]H.L.Ngo,K.Le Compte,L.Hargens,A.B.McEwen,Thermochim.Acta357(2000)97–102.[5]M.C.Buzzeo,R.G.Evans,pton,ChemPhysChem.5(2004)1106–1120.[6]J.G.Huddleston,A.E.Visser,W.M.Reichert,H.D.Willauer,G.A.Broker,R.D.Rogers,Green Chem.3(2001)156–164.[7]J.D.Holbrey,K.R.Seddon,J.Chem.Soc.,Dalton Trans.(1999)2133–2134.[8]T.Welton,Coord.Chem.Rev.248(2004)2459–2477.[9]R.P.Swatloski,S.K.Spear,J.D.Holbrey,R.D.Rogers,J.Am.Chem.Soc.124(2002)4974–4975.[10]V.M.Egorov,S.V.Smirnova,A.A.Formanovsky,I.V.Pletnev,Yu.A.Zolotov,Anal.Bioanal.Chem.387(2007)2263–2269.[11]H.Xie,S.Zhang,S.Li,Green Chem.8(2006)630–633.[12]J.D.Wadhawan,U.Schroder,A.Neudeck,et al.,J.Electroanal.Chem.493(2000)75–83.[13]C.Villagrán,L.Aldous,pton,et al.,J.Electroanal.Chem.588(2006)27–31.[14]N.V.Shvedene,D.V.Chernyshov,M.G.Khrenova,A.A.Formanovsky,V.E.Baulin,I.V.Pletnev,Electroanalysis18(2006)1416–1421.[15]S.Park,R.J.Kazlauskas,Curr.Opin.Biotechnol.14(2003)432–437.[16]M.Koel,Crit.Rev.Anal.Chem.35(2005)177–192.[17]N.Nishi,T.Kawakami,F.Shigematsu,M.Yamamoto,T.Kakiuchi,Green Chem.8(2006)349–355.[18]J.-P.Mikkola,P.Virtanena,R.Sjöholm,Green Chem.8(2006)250–255.[19]J.G.Huddleston,H.D.Willauer,R.P.Swatloski,A.E.Visser,R.D.Rogers,Chem.Commun.(1998)1765–1766.[20]E.Bekou,D.D.Dionysiou,R.-Y.Qian,G.D.Botsaris,ACS Symp.Ser.856(2003)544–560.[21]A.Fadeev,M.Meagher,mun.(2001)295–296.[22]J.Marták,ˇS.Schlosser,Chem.Pap.60(2006)395–398.[23]S.Carda-Broch,A.Berthod,D.W.Armstrong,Anal.Bioanal.Chem.375(2003)191–199.[24]S.Dai,Y.H.Ju,C.E.Barnes,J.Chem.Soc.,Dalton Trans.(1999)1201–1202.[25]A.E.Visser,R.P.Swatloski,W.M.Reichert,et al.,mun.(2001)135–136.[26]M.L.Dietz,J.A.Dzielawa,szak,B.A.Young,M.P.Jensen,Green Chem.5(2003)682–685.[27]A.Arce,M.J.Earle,H.Rodríguez,K.R.Seddon,Green Chem.9(2007)70–74.[28]W.-H.Lo,H.-Y.Yang,G.-T.Wei,Green Chem.5(2003)639–642.[29]C.Huang,B.Chen,J.Zhang,Z.Liu,Y.Li,Energy Fuels18(2004)1862–1864.[30]S.V.Smirnova,I.I.Torocheshnikova, A.A.Formanovsky,I.V.Pletnev,Anal.Bioanal.Chem.378(2004)1369–1375.[31]C.He,S.Li,H.Liu,K.Li,F.Liu,J.Chromatogr.A.1082(2005)143–149.[32]J.-H.Wang,D.-H.Cheng,X.-W.Chen,Z.Du,Z.-L.Fang,Anal.Chem.79(2007)620–625.[33]K.S.Khachatryan,S.V.Smirnova,I.I.Torocheshnikova,N.V.Shvedene,A.A.For-manovsky,I.V.Pletnev,Anal.Bioanal.Chem.381(2005)464–470.[34]B.Y.Pei,J.Wang,K.Wu,Y.Zhao,J.Fan,Z.Phys.Chem.221(2007)825–835.[35]C.Reichardt,Green Chem.7(2005)339–351.[36]I.M.Korenman,Photometricheskiy analiz.Metody opredeleniya organich-eskikh soedineniy(Photometric Analysis.Methods for Determination of Organic Compounds),Khimiya,Moscow,1970(in Russian).[37]R.E.Galian,A.V.Veglia,R.H.de Rossi,Analyst123(1998)1587–1591.[38]N.V.Shvedene,S.V.Borovskaya,V.V.Sviridov,E.R.Ismailova,I.V.Pletnev,Anal.Bioanal.Chem.381(2005)427–430.[39]C.Reichardt,Solvents and Solvent Effects in Organic Chemistry,Wiley-VCH,Weinheim,2003.[40]K.A.Fletcher,I.A.Storey,A.E.Hendricks,Sh.Pandey,S.Pandey,Green Chem.3(2001)210–215.[41]B.R.Mellein,S.N.V.K.Aki,dewski,J.F.Brennecke,J.Phys.Chem.B111(2007)131–138.[42]E.P.Serjeant,B.Dempsey,Ionization Constants of Organic Acids in AqueousSolution,Pergamon,Oxford,1979.[43]NCI database:http://129.43.27.140/ncidb2/.[44]Ya.I.Korenman,Koeffitsienty raspredeleniya organicheskikh soedineniy(Dis-tribution Coefficients of Organic Compounds),Voronezh University Press, Voronezh,Russia,1992(in Russian).[45]J.Liu,Y.Chi,J.Peng,et al.,J.Chem.Eng.Data49(2004)1422–1424.[46]J.F.Huang,P.Y.Chen,I.W.Sun,S.P.Wang,Inorg.Chim.Acta320(2001)7–11.。
俄罗斯的矿产资源
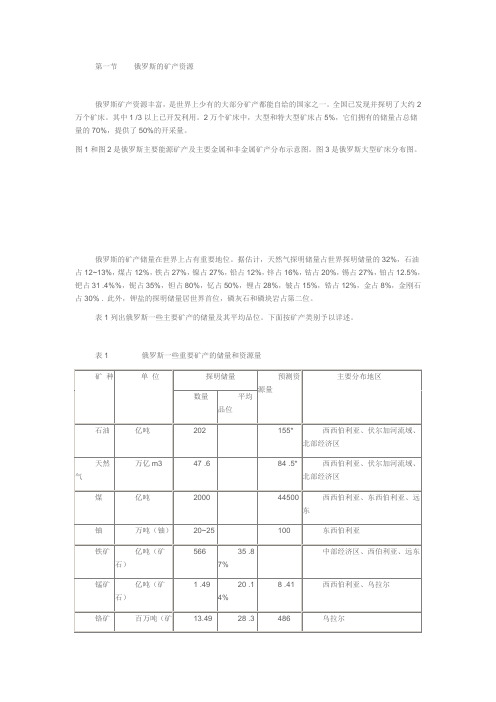
第一节俄罗斯的矿产资源俄罗斯矿产资源丰富,是世界上少有的大部分矿产都能自给的国家之一。
全国已发现并探明了大约2万个矿床。
其中1 /3以上已开发利用。
2万个矿床中,大型和特大型矿床占5%,它们拥有的储量占总储量的70%,提供了50%的开采量。
图1和图2是俄罗斯主要能源矿产及主要金属和非金属矿产分布示意图。
图3是俄罗斯大型矿床分布图。
俄罗斯的矿产储量在世界上占有重要地位。
据估计,天然气探明储量占世界探明储量的32%,石油占12~13%,煤占12%,铁占27%,镍占27%,铅占12%,锌占16%,钴占20%,锡占27%,铂占12.5%,钯占31 .4%%,铌占35%,钽占80%,钇占50%,锂占28%,铍占15%,锆占12%,金占8%,金刚石占30% . 此外,钾盐的探明储量居世界首位,磷灰石和磷块岩占第二位。
表1列出俄罗斯一些主要矿产的储量及其平均品位。
下面按矿产类别予以详述。
表1 俄罗斯一些重要矿产的储量和资源量一.能源矿产俄罗斯能源矿产(石油、天然气、煤、铀)虽然在各个经济区都有探明储量,但是主要还是集中在东部地区--西西伯利亚、东西伯利亚和远东(表2)。
表2 能源矿产资源在俄罗斯各经济区的分布资料来源:Бюллетенъ 《Исполъзование и охрана природных ресурсов России》2001 .1~21)石油截至1998年1月1日,计算有储量的油气矿床共有2152个,其中大约1700个是石油矿床,开采的有1159个。
石油储量据报导为202亿吨,主要集中在107个大型矿床中,它们的储量占俄罗斯石油总储量的69%。
石油矿床在37个联邦主体中都有分布,但主要还是集中在西西伯利亚(汉特曼西自治州和雅马尔-涅涅茨自治州),乌拉尔-伏尔加河中下游(鞑靼斯坦共和国、巴什科尔托斯坦共和国、乌德穆尔特共和国、彼尔姆共和国、萨马尔共和国和奥伦堡等州),以及欧洲北部、科米共和国、涅涅茨自治区等。
俄罗斯固体矿产资源储量管理体制及其启示

俄罗斯固体矿产资源储量管理体制及其启示史瑾瑾,原振雷,张博(中国自然资源经济研究院,北京101149)摘 要:俄罗斯是资源大国,具有成熟的固体矿产资源储量管理体制。
本文通过查阅俄罗斯官方网站信息,对其矿产资源储量管理机构及职能、矿产资源勘查开发中有关矿产资源储量管理的规定以及固体矿产资源储量和资源分类等进行了简要梳理总结。
俄罗斯矿产资源储量管理机构职能分工和管理权限清晰明确,矿产勘查开采中实行资源储量的动态管理,联邦内运行两套矿产资源储量分类体系但服务目标不同,对进一步完善我国固体矿产资源储量管理制度具有重要的借鉴意义。
关键词:俄罗斯 固体矿产 储量管理俄罗斯国土面积居世界第一,拥有丰富的矿产资源,是全球唯一能在矿产资源上自给自足的国家,其石油、天然气、镍、铁、金刚石和铜的储量位居世界前列。
在矿业全球化的大趋势下,近年来俄罗斯采取一系列具体措施积极完善国内储量管理制度及分类体系。
目前我国正处于矿产资源储量管理职能改革的关键阶段,正确认识俄罗斯矿产资源储量管理情况,对进一步完善我国的矿产资源储量管理制度具有重要的借鉴意义。
1 俄罗斯矿产资源储量管理机构及职能1.1 俄罗斯自然资源和生态部及其下设机构俄罗斯自然资源和生态部成立于2008年,体现了国际上自然资源与生态环境一体化管理的发展趋势,是大部制的典型代表,是主要负责制定国家自然资源研究、使用、恢复和保护政策、标准、规范和法律法规的国家执行机构[1]。
根据《俄罗斯联邦地下资源法》,其在矿产资源储量方面的职能是对矿产资源储量及预测性矿产资源进行分类定级,建立各级统一的矿产地质信息库,对已经探明的矿产资源储量的国家级评审鉴定以及对国家矿产资源储量平衡表进行编制管理等。
其下设机构联邦地下资源开发署是负责固体矿产资源储量管理的主要机构,职能包括将新探明的矿产资源储量及时列入国家的储量平衡表,将已消耗的矿产储量及时地从储量平衡表中移出,保持国家储量表的动态平衡和资源家底的清晰[2]。
走进制造“宇宙中最重原子”的俄罗斯工厂

走进制造“宇宙中最重原子”的俄罗斯工厂作者:暂无来源:《读报参考》 2019年第10期杜布纳小城坐落在莫斯科以北靠近伏尔加河的茂密松林中。
离市中心不远,有一条草木繁盛的大街,布满苏联时期的建筑物。
但在这片建筑群中,有一个地方正在发生一些开创性的事情。
新元素补充元素周期表一个巨大的混凝土车间里,戴着安全帽的工人们正忙着组装世界上最强大的研究机器之一。
在我观看时,站在我身边的是世界上唯一健在的用其名命名化学元素的人。
尤里·奥加涅相——118 号化学元素先生——几乎是慈爱地凝视着车间中央那个4 米宽的金属圆盘。
这是一台机器的首批部件之一,而这台机器不久将开始产生出化学元素,但这可不是普通的化学元素,这些元素将是超重元素。
通过第一次制造出数量足以供研究之用的这类奇异的原子,奥加涅相和他的机器应该能够回答有关我们的宇宙是如何形成的一些重大问题,或许还可能给我们提供一个惊人的能量来源。
他甚至可能会推翻支撑元素周期表本身的某些规则。
现代元素周期表的诞生可以追溯到另一个俄罗斯城市——圣彼得堡。
正是在那里,一位名叫德米特里·门捷列夫的科学顾问帮助人们穿透150年前的化学乱象,发明了化学元素周期表。
有了门捷列夫的周期表,化学结构开始变得更加可以理解。
这个俄罗斯人根据原子量把已知的63种元素编制成表。
我们现在知道,原子量是由原子核中的质子和中子的数量决定的。
随着时间的推移,化学家发现了更多的元素,填补了门捷列夫在元素周期表上留下的一些空白。
到了1940年代,我们已经制造出第一批合成元素,比如锝。
但是锝太不稳定,无法在地球上大量存在。
然而,我们通过向原子核中填充更多质子和中子,来不停地试图制造新的元素。
为此,我们需要一台粒子加速器。
这类工作在杜布纳那片建筑群里已经开展很多年。
那里就是联合原子核研究所。
这家机构成立的目的就是同瑞士日内瓦附近的欧洲核子研究中心粒子物理实验室进行竞争。
联合原子核研究所最重要的部分是弗廖罗夫核反应实验室。
ReaxFF_A_Reactive_Force_Field_for_Hydrocarbons

ReaxFF:A Reactive Force Field for HydrocarbonsAdri C.T.van Duin,†,|Siddharth Dasgupta,‡Francois Lorant,§and William A.Goddard III*,‡Department of Fossil Fuels and En V ironmental Geochemistry,Drummond Building,Uni V ersity of Newcastle,Newcastle upon Tyne NE17RU,United Kingdom,Materials and Process Simulation Center,Beckman Institute(139-74),Di V ision of Chemistry and Chemical Engineering,California Institute of Technology,Pasadena,California91125,and Institut Franc¸ais du Pe`trole,Geology and Geochemistry Research Di V ision,1-4A V enuede Bois-Preau,92852Rueil-Malmaison,FranceRecei V ed:December4,2000;In Final Form:March30,2001To make practical the molecular dynamics simulation of large scale reactive chemical systems(1000s ofatoms),we developed ReaxFF,a force field for reactive systems.ReaxFF uses a general relationship betweenbond distance and bond order on one hand and between bond order and bond energy on the other hand thatleads to proper dissociation of bonds to separated atoms.Other valence terms present in the force field(angleand torsion)are defined in terms of the same bond orders so that all these terms go to zero smoothly as bondsbreak.In addition,ReaxFF has Coulomb and Morse(van der Waals)potentials to describe nonbond interactionsbetween all atoms(no exclusions).These nonbond interactions are shielded at short range so that the Coulomband van der Waals interactions become constant as R ij f0.We report here the ReaxFF for hydrocarbons.The parameters were derived from quantum chemical calculations on bond dissociation and reactions of smallmolecules plus heat of formation and geometry data for a number of stable hydrocarbon compounds.We findthat the ReaxFF provides a good description of these data.Generally,the results are of an accuracy similaror better than PM3,while ReaxFF is about100times faster.In turn,the PM3is about100times faster thanthe QC calculations.Thus,with ReaxFF we hope to be able to study complex reactions in hydrocarbons.1.IntroductionThe accuracy and speed of modern quantum chemistry(QC) methods allow the geometries,energies,and vibrational energies to be predicted quite accurately for small molecules.However, QC is not yet practical for studying the dynamic properties of larger molecules and solids.Consequently,it is useful to have accurate force fields(FF)to quickly evaluate the forces and other dynamical properties such as the effects of mechanical shock waves or diffusion of small molecules in polymer and mesoporous zeolites.Indeed,for hydrocarbons a number of FF, particularly MM3,1-3provide accurate predictions of geometries, conformational energy differences,and heats of formation. Generic FF such as DREIDING4and UFF5allow predictions for broad classes of compounds,particularly when coupled to charge equilibration6(Q Eq)or other methods for predicting charges.However,in general,these force fields do not describe chemical reactivity.An exception is the Brenner potential,7 which leads to accurate geometries for ground states of hydrocarbons,but is formulated in such a way that it can describe bond breaking.Unfortunately,the Brenner formalism does not include the van der Waals and Coulomb interactions that are very important in predicting the structures and properties of many systems.In addition,the actual potential curves for bond breaking and reactions are often quite poorly described with the Brenner potential.Generalizations of the Brenner FF have included such nonbonded forces,8but without repairing the fundamental problems in the shapes of the dissociation and reactive potential curves.Two other bond-order-dependent force field methods are noteworthy.The Bond Energy Bond Order(BEBO)method was proposed by Johnston9,10based on Pauling’s relation between bond length and bond order.11The fundamental assumption behind this method is that the path of lowest energy on going from reactant to product is one that conserves total bond order. Originally used for the H+H2reaction surface,it is a very good approximation to more complicated empirical forms such as LEPS surface.12-13While it has recently been extended to more complex reactions,14-15it remains mainly useful for H atom transfer reactions in a linear collision geometry.The VALBOND method formulated by Landis and colleagues is based on the strength functions of hybrid orbitals.The motivation comes from the need to fit large distortions in the softer angle terms of valence force fields,as well as describing multiple equilibrium angles in transition metal complexes(e.g., the90°and180°angles in square planar complexes).Assuming that different ligand atoms,lone pairs,and radical electrons have implicit preference for p-character,VALBOND uses Lewis structure-based allocations to assign hybridizations and the geometries are obtained by minimizing defects in the hybrid orbitals.For a simple force field,it does remarkably well on structures and vibrational frequencies for a wide range of small molecules and transition metal complexes.16-18These methods, however,do not fully address the need to have full chemistry of the breaking and forming bonds,in addition to a proper description of the fully bonded equilibrium geometry of complex molecules.In this paper,we develop a general bond-order-dependent potential in which the van der Waals and Coulomb forces are*Author to whom correspondence should be addressed.E-mail:wag@.†University of Newcastle.‡California Institute of Technology.§Institut Franc¸ais du Pe`trole,Geology and Geochemistry ResearchDivision.|E-mail: A.C.T.van-Duin@.9396J.Phys.Chem.A2001,105,9396-940910.1021/jp004368u CCC:$20.00©2001American Chemical SocietyPublished on Web09/22/2001included from the beginning and the dissociation and reaction curves are derived from QC calculations.In spirit it is derived from the central force concept used earlier by spectroscopists 19but abandoned because a single harmonic potential between all atom pairs was inadequate for complex molecules.We have kept the central force formalism,where all atom pairs have nonbonded interactions,because it dissociates smoothly,but add local perturbations (bond,angle,torsion,etc.)to describe the complex molecules more accurately.We have attempted to obtain accurate descriptions of quantum phenomena such as resonance,unsaturated valences in radical systems,and chemical reactions.While the current work is restricted to hydrocarbons this approach is easily extended to any molecular system of any class of compounds.In a future paper we will report on our extension to CHNO-systems.Section 2describes the general form of the reactive force field (denoted ReaxFF)and the procedure for optimizing the parameters.Section 3presents the results for a number of systems.Section 4discusses these results,and Section 5presents the conclusions.2.Force FieldSimilar to empirical nonreactive force fields,the reactive force field divides the system energy up into various partial energy contributions,as demonstrated by eq 1.The potential energy functions associated with each of these partial energy contributions are described below.Tables 1-6list the parameters used in these potential functions.2.1.Bond Order and Bond Energy.A fundamental as-sumption of ReaxFF is that the bond order BO ′i j between a pair of atoms can be obtained directly from the interatomic distance r ij as given in eq 2and plotted in Figure 1.Equation 2consists of three exponential terms:(1)the sigma bond (p bo,1and p bo,2)which is unity below ∼1.5Åbut negligible above ∼2.5Å;(2)the first pi bond (p bo,3and p b0,4)which is unity below ∼1.2Åand negligible above ∼1.75Å,and (3)the second pi bond (p bo.5and p bo,6)which is unity below ∼1.0Åand negligible above ∼1.4Å.This leads to a carbon -carbon bond with a maximum bond order of 3.For carbon -hydrogen and hydrogen -hydrogen bonds,only the sigma-bond contribution is considered,leading to a maximum bond order of 1The bond orders BO ′i j are corrected for overcoordination and for residual 1-3bond orders in valence angles using the scheme described in eqs 3a -f.While the 1-3bond order correction,described in eqs 3e and 3f,is applied to all the bonds in the molecule,the overcoordination correction (eqs 3b -d)is only applied to bonds containing two carbon atoms.The final bond orders in the molecule are obtained by multiplying the bond orders from Equation 2by the correction factors from eq 3.Figure 2shows the effects of eqs 3a -f on the bond orders in an ethane molecule in which the C -C bond length is reduced from its equilibrium values of about 1.53Åto 1.0Å.This creates overcoordination on both the carbon and the hydrogen atoms,as the sum of bond orders around all atoms exceeds their valences (4for carbon and 1for hydrogen).As Figure (2)shows,application of Equations (3a -f)removes all of the 1-3bond orders,correcting the overcoordination on the hydrogen atoms,and,in addition,corrects most of the additional overcoordinationTABLE 1:General Parametersparameter value descriptionequation λ150.0overcoordination bond order correction 3c λ215.61overcoordination bond order correction 3d λ3 5.021-3bond order correction 3e,f λ418.321-3bond order correction 3e,f λ58.321-3bond order correction 3e,f λ6-8.90overcoordination energy 6λ7 1.94undercoordination energy 7a λ8-3.47undercoordination energy 7a λ9 5.79undercoordination energy 7b λ1012.38undercoordination energy 7b λ11 1.49valence angle energy 8b λ12 1.28valence angle energy 8b λ13 6.30valence angle energy 8c λ14 2.72valence angle energy 8c λ1533.87valence angle energy 8c λ16 6.70valence angle energy 8d λ17 1.06valence angle energy 8d λ18 2.04valence angle energy 8d λ1936.0penalty energy 9a λ207.98penalty energy 9a λ210.40penalty energy 9b λ22 4.00penalty energy 9b λ23 3.17torsion energy 10b λ2410.00torsion energy 10c λ250.90torsion energy 10c λ26-1.14conjugation energy 11a λ27 2.17conjugation energy 11b λ281.69van der Waals energy12bE system )E bond +E over +E under +E val +E pen +E tors +E conj +E vdWaals +E Coulomb (1)TABLE 2:Atom Parameters As Used in Equations 2,6,7,12,13,and 14abond radiiunder/over coordinationCoulomb parameters heat increments units r o År o,πÅr o,ππÅp over kcal/molp under kcal/molηEV EV γÅI kcal/mol C 1.399 1.266 1.23652.229.47.41 4.120.69218.6H0.656-117.59.142.260.3754.3ar o (ij ))1/2[r o (i )+r o (j)].Figure 1.Interatomic distance dependency of the carbon -carbon bond order.BO ′ij )exp [p bo,1‚(r ijr o)p bo,2]+exp [p bo,3‚(r ijπr o)p bo,4]+exp [p bo,5‚(r ijππr o)p bo,6](2)ReaxFF:A Reactive Force Field for HydrocarbonsJ.Phys.Chem.A,Vol.105,No.41,20019397on the carbon atoms.Val i in eqs 3a -3f is the valency of atom i (Val i )4for carbon,Val i )1for hydrogen).∆′i is the degree of deviation of the sum of the uncorrected bond orders around an atomic centerfrom its valency Val i ,as described in eq 4.Equation 5is used to calculate the bond energies from the corrected bond order BO ij .2.2.Atom Under-/Overcoordination.From the valencetheory of bonding we know that the total bond order of C should not exceed 4and that of H should not exceed 1,except in hypervalent cases.However,as Figure 2shows,even after correction of the original bond orders BO ′i j a degree of overcoordination may remain in the molecule.To handle this we have added an overcoordination penalty term to the force field.2.2.1.O V er-Coordination.For an overcoordinated atom (∆i >0),eq 6imposes an energy penalty on the system.The form of eq 6,ensures that E over will quickly vanish to zero for under-coordinated systems (∆i <0).∆i is calculated using eq 4,using the corrected bond orders BO ij from eq 3instead of the uncorrected bond orders from eq 2.2.2.2.Under-Coordination.For an under-coordinated atom(∆i <0),we want to take into account the energy contribution for the resonance of the π-electron between attached under-coordinated atomic centers.This is done by eqs 7a,b where E under is only important if the bonds between under-coordinated atom i and its under-coordinated neighbors j partly have π-bond character (BO ij,π>0as calculated from the last two terms of eq 2).2.3.Valence Angle Terms.Just as for bond terms,it is important that the energy contribution from valence angle terms goes to zero as the bond orders in the valence angle goes to zero.Equations 8a -d are used to calculate the valence angle energy contribution.We use the bond-order-dependent form in eq 8a to calculate energy associated with deviations in valence angle Θijk from its equilibrium value Θo .The f 7(BO )term as described in eq 8b ensures that the valence angle energy contribution disappears smoothly during bond dissociation.Equation 8c deals with the effects of over/undercoordination in central atom j on the valence angle energy.The equilibrium angle Θo for Θijk depends on the sum of π-bond orders (SBO)around the central atom j as described in eq 8d.Thus,theTABLE 3:van der Waals Parameters Used in Equation 12aatom units r vdW Å kcal/mol R γw ÅC 3.9120.086210.71 1.41H3.6490.019410.065.36aArithmetic combination rules are used for all van der Waals parameters.TABLE 4:Bond Parameters (D e in kcal/mol)As Used in Equations 2and 3bond D e p be,1p be,2p bo,1p bo,2p bo,3p bo,4p bo,5p bo,6C -C 145.20.3180.65-0.097 6.38-0.269.37-0.39116.87C -H 183.8-0.45412.80-0.0137.65H -H168.4-0.31010.25-0.0165.98Figure 2.(a)Effect of the bond order correction in eq 2on the C -C and C -H bond orders in an ethane molecule in which the C -C bond is shortened to 1.0Åwith the rest of the geometry fixed.(b)Effects of shortening of the C -C bond length in ethane to 1.0Åon the relaxed C -H bond lengths as calculated by DFT and ReaxFF.Equilibrium C -C and C -H bond lengths are in italics and brackets.BO ij )BO ′ij ‚f 1(∆′i ,∆′j )‚f 4(∆′i ,BO ′ij )‚f 5(∆′j ,BO ′ij )(3a)f 1(∆i ,∆j ))12‚(Val i +f 2(∆′i ,∆′j )Val i +f 2(∆′i ,∆′j )+f 3(∆′i ,∆′j )+Val j +f 2(∆′i ,∆′j )Val j +f 2(∆′i ,∆′j )+f 3(∆′i ,∆′j ))(3b)f 2(∆′i ,∆′j ))exp(-λ1‚∆′i )+exp(-λ1‚∆′j )(3c)f 3(∆′i ,∆′j ))1λ2‚ln {12‚[exp(-λ2‚∆′i )+exp(-λ2‚∆′j )]}(3d)f 4(∆′i ,BO ′ij ))11+exp(-λ3‚(λ4‚BO ′ij ‚BO ′ij -∆′i )+λ5)(3e)f 5(∆′j ,BO ′ij ))11+exp(-λ3‚(λ4‚BO ′ij ‚BO ′ij -∆′i )+λ5)(3f)∆′i )∑j )1n bondBO ′ij -Val i(4)E bond )-D e ‚BO ij ‚exp[p be,1(1-BO ij p be,1)](5)E over )p over ‚∆i ‚(11+exp(λ6‚∆i ))(6)E under )-p under ‚1-exp(λ7‚∆i )1+exp(-λ8‚∆i )‚f 6(BO ij ,π,∆j )(7a)f 6(BO ij ,π,∆j ))11+λ9‚exp(λ10‚∑j )1neighbors(i )∆j ‚BO ij ,π)(7b)9398J.Phys.Chem.A,Vol.105,No.41,2001van Duin et al.equilibrium angle changes from around 109.47°for sp 3hybrid-ization (π-bond )0)to 120°for sp 2(π-bond )1)to 180°for sp (π-bond )2)based on the geometry of the central atom j and its neighbors.In addition to including the effects of π-bonds on the central atom j ,eq 8d also takes into account the effects of over-and under-coordination in central atom j (∆j )on the equilibrium valency angle,including the influence of a lone electron pair.The functional form of eq 8d is designed to avoid singularities when SBO )0and SBO )2.The angles in eqs 8a -d are in radians.To reproduce the stability of systems with two double bondssharing an atom in a valency angle,like allene,an additional energy penalty,as described in eqs 9a and 9b,is imposed forsuch systems.Equation 9b deals with the effects of over/undercoordination in central atom j on the penalty energy.2.5.Torsion Angles.Just as with angle terms we need to ensure that dependence of the energy of torsion angle ωijkl accounts properly for BO f 0and for BO greater than 1.This is done by eqs 10a -c.The V 2-cosine term in eq 10a depends on the bond order of the central bond BO jk .In torsion angles with a central double bond (BO jk )2)the V 2-term is at its maximum (about 30kcal/mol,see Table 6).If BO jk deviates from 2the magnitude of the V 2-term rapidly diminishes.The valence-angle-dependent term sin(Θijk )‚sin(Θjkl )in eq 10a ensures that the torsion energy contribution disappears when either of the two valence angles (Θijk or Θjkl )approaches π.To avoid excessive torsion contributions in systems containing two attached over-coordinated sp 3-carbon atoms,like an ethane molecule in which the central C -C bond length is reduced from its equilibrium value of about 1.52Åto 1.35Å,we include eq 10c,which reduces the influence of BO jk on the V 2-term in eq 10a when atoms j and k are over-coordinated [∆j >0,∆k >0].Equation 10b describes the smooth disappearance of the torsion energy contribution when one of the bonds in the torsion angle dissociates.2.6.Conjugated Systems.Equations 11a and 11b describe the contribution of conjugation effects to the molecular energy.A maximum contribution of conjugation energy is obtained when successive bonds have bond-order values of 1.5as in benzene and other aromatics.2.7.Nonbonded van der Waals Interactions.In addition to valence interactions which depend on overlap,there are repulsive interactions at short interatomic distances due to Pauli principle orthogonalization and attraction energies at long distances due to dispersion.These interactions,comprised ofTABLE 5:Valence Angle Parameters As Used in Equations 8a -dvalence angle units Θo,o degree k akcal/mol k b(1/radian)2p v,1p v,2C -C -C 71.31a 35.4 1.370.010.77C -C -H 71.5629.65 5.29H -C -H 69.9417.37 1.00C -H -C 028.5 6.00H -H -C 00 6.00H -H -H27.96.00aThis value leads to an equilibrium angle of 180-71.31)108.69°for the single-bond C -C -C valence angle (eq 8d).TABLE 6:Torsion and Conjugation Parameters (V 2and V 3in kcal/mol)As Used in Equations 10a -ctorsion angle a V 2V 3p t C -C -C -C 21.70.00-2.42C -C -C -H 30.50.58-2.84H -C -C -H26.50.37-2.33aTorsion angles not defined in this table (i.e.,C -H -C -C)are assigned torsion barrier energies of 0kcal/mol.E val )f 7(BO ij )‚f 7(BO jk )‚f 8(∆j )‚{k a -k a exp[-k b (Θo -Θijk )2]}(8a)f 7(BO ij ))1-exp(-λ11‚BO ij λ12)(8b)f 8(∆j ))2+exp(-λ13‚∆j )1+exp(-λ13‚∆j )+exp(p V ,1‚∆j )‚[λ14-(λ14-1)‚2+exp(λ15‚∆j )1+exp(λ15‚∆j )+exp(-p V ,2‚∆j )](8c)SBO )∆j -2‚{1-exp [-5‚(12∆j)λ16]}+∑n )1neighbors(j )BO jn ,π∆j ,2)∆j if ∆j <0∆j ,2)0if ∆j g 0SBO2)0if SBO e 0(8d)SBO2)SBO λ17if 0<SBO <1SBO2)2-(2-SBO)λ17if 1<SBO <2SBO2)2if SBO >2Θ0)π-Θ0,0‚{1-exp[-λ18‚(2-SBO2)]}E pen )λ19‚f 9(∆j )‚exp[-λ20‚(BO ij -2)2]‚exp[-λ20‚(BO jk -2)2](9a)f 9(∆j ))2+exp(-λ21‚∆j )1+exp(-λ21‚∆j )+exp(λ22‚∆j )(9b)E tors )f 10(BO ij ,BO jk ,BO kl )‚sin Θijk ‚sin Θjkl[12V 2‚exp {p l(BO jk-3+f 11(∆j ,∆k ))2}‚(1-cos 2ωijkl )+12V 3‚(1+cos 3ωijkl )](10a)f 10(BO ij ,BO jk ,BO kl ))[1-exp(-λ23‚BO ij )]‚[1-exp(-λ23‚BO jk )]‚[1-exp(-λ23‚BO kl )](10b)f 11(∆j ,∆k ))2+exp[-λ24‚(∆j +∆k )]1+exp[-λ24‚(∆j +∆k )]+exp[λ25‚(∆j +∆k )](10c)E conj )f 12(BO ij ,BO jk ,BO kl )‚λ26‚[1+(cos 2ωijkl -1)‚sin Θijk ‚sin Θjkl ](11a)f 12(BO ij ,BO jk ,BO kl ))exp [-λ27‚(BO ij -112)2]‚exp [-λ27‚(BO ij -112)2]‚exp [-λ27‚(BO kl -112)2](11b)ReaxFF:A Reactive Force Field for HydrocarbonsJ.Phys.Chem.A,Vol.105,No.41,20019399van der Waals and Coulomb forces,are included for all atom pairs,thus avoiding awkward alterations in the energy descrip-tion during bond dissociation.In this respect,ReaxFF is similar in spirit to the central valence force fields that were used earlier in vibrational spectoscropy.To account for the van der Waals interactions we use a distance-corrected Morse-potential (eqs 12a,b).By including a shielded interaction (eq 12b),excessively high repulsions between bonded atoms (1-2interactions)and atoms sharing a valence angle (1-3interactions)are avoided.Figure 3shows how the bond energies,derived from eq 5,combine with the van der Waals interactions for diatomic C -C,C -H,and H -H systems to give a dissociation energy curve.2.8.Coulomb Interactions.As with the van der Waalsinteractions,Coulomb interactions are taken into account between all atom pairs.To adjust for orbital overlap between atoms at close distances a shielded Coulomb potential is used.Atomic charges are calculated using the Electron EquilibrationMethod (EEM)approach.20-21The EEM charge derivation method is similar to the Q Eq scheme;6the only differences,apart from parameter definitions,are that EEM does not use an iterative scheme for hydrogen charges (as in Q Eq)and that Q Eq uses a more rigorous Slater orbital approach to account for charge overlap.However,the γij in eq 13can be optimized to reproduce the Q Eq orbital overlap correction.The initial values for the EEM-parameters ( ,η,and γ,Table 2)were taken fromNjo et al.,22but these parameters were allowed to change during the FF optimization procedure.Intraatomic contributions of atomic charges,to account for the energy required to polarize the atoms,are taken into account in the energy scheme,23thus allowing a straightforward expansion of the force field for ionic compounds.With the EEM-parameter values from Table 2,ReaxFF assigns a charge of -0.113to the carbon atoms in cyclohexane and charges of +0.050and +0.063to the equatorial and axial hydrogens,respectively.A Mulliken distribution,from a DFT calculation with the 6-31G**-basis set,results in charges of -0.174,+0.0859,and 0.0876to the cyclohexane carbon,equatorial and axial hydrogens while the Q Eq method 6gets a -0.24,+0.104,+0.137charge distribution.2.9.Force Field Optimization Procedure.The FF was optimized using a successive one-parameter search technique as described by van Duin et al.24In general,our aim was to reproduce heats of formation to within 4.0kcal/mol,bond lengths to within 0.01Å,and bond angles to within 2°of their literature values.To use the QC data in the FF optimization procedure,structures relating to these data were added to the FF training set.All molecules used in the heat of formation and geometry data comparisons were continuously energy minimized during the FF optimization while the structures relating to the QC data were kept fixed or were optimized with appropriate bond length or torsion angle restraints.3.Results3.1.Non-Conjugated Systems.3.1.1.Energy and Geometry Reproduction.Figure 4a and Table 7show how well the ReaxFF reproduces the heat of formation for nonconjugated closed shell molecules.The heat of formation predictions for ReaxFF are compared with those of the semiempiricalMOPAC-method,Figure 3.Interatomic distance dependency of the carbon -carbon,carbon -hydrogen,and hydrogen -hydrogen bond-and van der Waals-energy terms of diatomic C -C,C -H,and H -H systems.Energy contributions from Coulomb interactions and energy effects related to under and overcoordination are ignored in the total energy curve.E vdWaals )D ij ‚{exp [R ij ‚(1-f 13(r ij )r vdW )]-2‚exp[12‚R ij ‚(1-f 13(r ij )r vdW)]}(12a)f 13(r ij ))[r ij λ29+(1λw)λ28]1/λ28(12b)E Coulomb )C ‚q i ‚q j[r ij 3+(1/γij )3]1/3(13)Figure parison of calculated heats of formation with experi-mental data for nonconjugated (a),conjugated (b),and radical systems (c).9400J.Phys.Chem.A,Vol.105,No.41,2001van Duin et al.using the PM3-parameters39and with those of the nonreactive MM3force field.Heats of formation data were calculated from the total system energy,as determined from eq1,by adding the terms described in eq14.POP in eq14reflects the contribution of high-energy conformations,defined as the difference in heat of formation between the most stable conformation and the mixture of conformations.These high-energy conformation contributions,which are primarily signifi-cant for the larger(C6+)monocyclic saturated ring system in the training set,are also listed in Table7.I C and I H in eq14are the heat increments for the carbon and hydrogen atoms, respectively,as given in Table2.n C and n H are the number of carbon and hydrogen atoms in the molecule.4RT is added to account for translation,rotation,and pV-work in nonlinear polyatomic molecules.The values for the heat increments were determined by calculating the system energies of the compounds in the force field training set,addition of the POP and4RT-contribution and subsequent optimization of I C and I H to minimize the difference between calculated and literature heats of formation.As a result,the heat increments values bear no direct physical meaning,as,apart from the heats of formation of the elements,they also contain corrections for vibrational and zero-point energy.Table8shows the ReaxFF geometry data reproduction for nonconjugated molecules.3.1.2.Reproduction of Quantum Chemical Data.Figures5-7 show the carbon-carbon dissociation energies for ethane, ethylene,and ethyne as obtained from DFT calculations,the Brenner FF,PM3,and ReaxFF.The DFT calculations were performed at the B3LYP-level using the6-31G**basis set, which includes Generalized Gradient Corrections and exactTABLE7:Heat of Formation(kcal/mol)from ReaxFF for Non-conjugated Systemscompound H f(calc)H f(exp)a-POP diff cyclohexane-30.65-29.49-1.16 ethane-18.33-20.02 1.69 isobutane-30.62-32.07 1.45 neopentane-40.91-40.18-0.73 anti-n-butane-29.69-30.20b0.31 ethylene8.7512.55-3.80 propene 2.64 4.78-2.14 allene40.2346.40c-6.17 hydrogen8.850.008.85 cyclopentane-17.59-18.450.86 trans-decaline-45.14-43.52-1.62 cis-decaline-41.13-40.45-0.68 cyclobutene42.5437.50 5.04 cyclopentene 6.358.56d-2.21 cyclohexene-2.57-1.20-1.37 norbornane-8.00-12.42 4.42 norbornene20.6721.52-0.85 ethyne63.0654.508.56 propyne46.4344.20 2.23 cyclobutane 2.89 6.80-3.91 cyclopropane18.8212.70 6.12 protoadamantane-20.38-20.54e0.16 cis-hydrindane-30.11-30.410.30 perhydroquinacene-21.40-22.000.60 1,1-dimethylcyclopentane-31.80-33.33 1.53 2,2,3,3-tetramethylbutane-49.56-53.65 4.09 gauche-butene-2.31-0.50-1.81 cyclopropene88.3566.2322.12 cycloheptane-27.99-28.44f0.45 cyclooctane-28.51-30.12g 1.61 cyclononane-30.76-32.26h 1.50 cyclodecane-36.62-37.56I0.94 bicyclo[2.2.2]octane-23.80-23.68-0.12 cis-bicyclo[3.3.0]octane-21.77-22.200.43 bicyclo[3.3.1]nonane-31.32-30.50-0.82 trans-bicyclo[3.3.0]octane-14.06-15.91 1.85 di-tert-butylmethane-55.13-57.60 2.47 diamantane-37.30-34.87e-2.43 tst-perhydroanthracene-58.12-58.210.09 adiene19.8725.24-5.37 carbene93.8498.00-4.16 methane-14.25-17.80 3.55a Experimental heats of formation were taken from Pedley et al.25 unless noted otherwise.b POP)-0.20kcal/mol.c ref26.d ref27.e ref 28.f POP)-0.21kcal/mol.g POP)-0.36kcal/mol.h POP)-0.52 kcal/mol.i POP)-0.68kcal/mol.TABLE8:Geometry Predictions from ReaxFF(bond lengths inÅ,angles in degrees)for Non-Conjugated Systems compound bond/anglej calcd expt ethane a a 1.555 1.53b 1.1198 1.10ab110.6110bb′108.3107 ethylene b a 1.312 1.337b 1.117 1.08ab121.0121.8bb′118.0116.4 ethyne c a 1.241 1.202b 1.104 1.06 hydrogenc a0.780.7414 cyclohexaned a 1.551 1.54b 1.121 1.10aa′111.2111.0bb′103.7107.0 cyclopentene e a 1.324 1.348b 1.552 1.52c 1.564 1.55ab111.9112.8bc103.1103.3 cyclohexene f a 1.343 1.34b 1.546 1.50c 1.561 1.53 norbornane g ab90.993.9 norbornene h ab90.195.3bc101.196.12,2,3,3-tetramethylbutane i a 1.553 1.54b 1.569 1.58a ref20.b ref30.c ref31.d ref32.e refs33-34.f ref35.g ref36.h ref37.i ref38.j See Figure24for bond and angledefinitions.Figure5.Ethane C-C bond dissociation.Crosses indicate the data used in the force field parametrization.∆Hf)Esystem+4RT+POP+nCIC+nHIH(14)ReaxFF:A Reactive Force Field for Hydrocarbons J.Phys.Chem.A,Vol.105,No.41,20019401。
俄罗斯科学院西伯利亚分院

俄罗斯科学院西伯利亚分院
俄罗斯科学院西伯利亚分院(苏维埃社会主义共和国联盟科学院西伯利亚分院,今俄罗斯科学院西伯利亚分院)在院士米· 阿· 拉夫连季耶夫、谢· 利· 索伯列夫和谢· 阿· 赫里斯基安诺维奇的倡议下于1957 年成立。
俄罗斯科学院西伯利亚分院自建立之初就以科研的综合性、科学和教育的整体性及积极促进科研成果在经济创新领域的转化为主要活动原则。
西伯利亚分院—俄罗斯科学院最大的区域性分院。
坐落于西比利亚及邻近地区,总面积约为 11,000,000 km 2。
俄罗斯科学院西伯利亚分院的科研中心位于新西伯利亚、托木斯克、克拉斯诺亚尔斯克、伊尔库茨克、雅库茨克、乌兰乌德、克麦罗沃、秋明、鄂姆斯克市,独立的研究所位于巴尔瑙尔、比斯克、克孜勒、赤塔市。
俄罗斯科学院西伯利亚分院下设79 个科研所从事数学与信息学、动力学、力学和过程控制、纳米技术和信息技术领域的科研工作,及物理、化学、生物、地球科学、经济学和人文科学领域的科研工作,并开展交叉学科基础上的跨学科研究。
分院的半数科研力量集中于新西伯利亚科研中心。
西伯利亚的科学家以其众多卓越的成果享誉世界科研协会。
他们参与了大型强子对撞机里“ 希格斯玻色子” 的发现和新人种 - “ 德尼索瓦人” ( Homo altaiensis, Homo denisovans )的发现,就癌症抗争方法提出了建议。
俄罗斯科学院西伯利亚分院主席是亚历山大· 列奥尼多维奇· 阿谢耶夫院士,学术秘书长—俄罗斯科学院通讯院士瓦列里· 伊万诺维奇· 布赫季亚洛夫。
哈里顿(Haryton,U B,1904-1996)俄国物理学家(Physicist,Russia)

哈里顿(Haryton,U B,1904-1996)俄国物理学家
(Physicist,Russia)
佚名
【期刊名称】《《光谱实验室》》
【年(卷),期】2007(024)001
【摘要】哈里顿是前苏联发展原子弹的第二把手。
他于1925年毕业于列宁格勒多科工业大学。
1926-1928年,他被前苏联政府选派到卡文迪许实验室,在卢瑟福指导下工作,并获博士学位。
他是卢瑟福的学生中去世最晚的。
1931年他在化学物理研究所工作,研究金属蒸气的凝结和离心法分离气体的理论。
1939年他和泽尔多维奇对铀的链式裂变反应进行了计算,他们发表于1939-1941年的一系列论文为后来苏联发展原子武器打下了基础。
【总页数】1页(P71)
【正文语种】中文
【中图分类】O4-09
【相关文献】
1.阿基莫维奇(Artsimovich,Lev Andreevich,1909—1973)俄国物理学家(Physicist,Russia) [J],
2.邦奇-布鲁耶维奇(Bontch-Bruyervitch,M A,1888-1940)俄国物理学家(Physicist,Russia) [J],
3.阿耳费罗夫(Alferof,Zhores I,1930-)俄国物理学家(Physicist,Russia)[J],
4.亚历山大罗夫(Alexanderroy,A P,1903-1994)俄国物理学家(Physicist,Russia) [J],
5.切伦科夫(Cherenkov,Pavel Alekseyevich,1904-1990)俄国物理学家(Physicist,Russia) [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实力雄厚的俄罗斯晶体学研究所吴自勤 1 王进萍2( 1 中国科学技术大学物理学院合肥230026)( 2 物理编辑部北京100190)俄罗斯科学院晶体学研究所是俄罗斯一所名闻遐迩的研究机构。
该所历史悠久,研究成果丰硕,拥有多名著名学者,不仅发表了许多重要的论文,而且出版了多册经典的专著,并且还用英文出版晶体学和表面科学期刊。
是什么原因让这样一个研究所取得了如此的成功?在深入了解它的发展历程后,我们感触颇深。
他们的成功经验对当前处于快速发展中的中国物理学或许能有所启发和借鉴。
晶体学是一门发展了三个多世纪的悠久学科。
它的基本概念和规律早已确立,如对称性理论中的32种晶体点群分别在1830和1867年被独立发现;晶体的14种空间点阵1848年被布拉菲发现;晶体的230种空间群在1890年由俄国科学家费多洛夫(稍后是熊夫利)发现;费多洛夫的晶体对称性著作1901年以俄文发表。
由此可见,俄罗斯的晶体学研究早已具备了深厚的基础和积累。
俄罗斯科学院晶体学研究所的前身包括了1925年在列宁格勒成立的苏联科学院矿物博物院晶体学实验室,1934年在莫斯科成立的苏联科学院罗蒙诺苏夫地球化学、矿物学和石油工学研究所晶体学分部,以及1937年成立的苏联科学院地球科学部晶体学实验室。
1943年晶体学实验室被调整到苏联科学院数学物理部,并正式更名为晶体学研究所。
1944年2月在学界享有盛誉的A V舒勃尼科夫(Shubnikov)教授被任命为该研究所第一任所长。
1961年晶体学研究所迁往莫斯科列宁大街59号新址,后来在Butlerova街17a和莫斯科市郊Kaluga 还建有两处分所。
图 1 俄罗斯科学院晶体学研究所(莫斯科列宁大街59号)群英荟萃、硕果累累俄罗斯晶体学研究所悠久的历史上曾涌现了一大批优秀的晶体物理学家,其中包括苏联科学院、俄罗斯科学院院士、通信院士多名,列宁奖金等国家奖获得者20名,俄罗斯荣誉科学家18名。
限于篇幅,本文仅列举其中几位杰出的代表。
图2 A V舒勃尼科夫(Alexey Vasilyevich Shubnikov 1887-1970 -)教授是晶体学研究所第一任所长舒勃尼科夫院士和别洛夫(N V Belov)院士一起系统建立了色对称性,即舒勃尼科夫对称性理论。
他们利用反对称性,即除了原子的位置之外,采用黑、白色表示原子带有方向相反的磁矩;利用色对称性,即采用几种彩色表示原子带有若干不同方向的磁矩,表示磁性晶体原子结构的对称性,并在此基础上发展了费多洛夫、熊夫利等的晶体原子结构的对称性理论。
1964年,舒勃尼科夫和别洛夫发表了专著“Colored Symmetry”1),系统论述了晶体的反对称性理论和色对称性理论。
由于他们的卓越贡献,舒勃尼科夫曾两次获得社会主义劳动英雄称号,而在晶体学研究所工作了40余年的别洛夫则获得过社会主义劳动英雄称号和列宁奖金等国家奖。
1)Shubnikov A V, Belov N V. 1964. Colored Symmetry, Oxford :Pergamon Press, Oxford图3 从1962年到1996年间B K 伐因斯坦(Boris Konstantinovich Vainshtein)一直担任晶体学研究所的所长1962年,年仅41岁的的伐因斯坦(Vainshtein)教授被任命为晶体学研究所所长。
他在1945年和1947年本别获得了莫斯科大学物理系硕士学位和前苏联钢铁学院博士学位后,来到了晶体学研究所电子衍射实验室,从事博士后研究工作。
早在1956年,年仅35岁时,他便出版了《电子衍射结构分析》俄文著作(Structure Analysis by Electron Diffraction),并于1964年出版了英文版。
这本俄文著作早在1950年代末期就在我国流传。
当时国内的高校教师、研究生大多可以阅读俄文文献。
在这一专著中,他把电子衍射发展成为一种独立的结构分析方法。
由于原子对电子的射散比原子对X射线的射散显著得多,从而有利于测定晶体中超轻原子(如氢)的位置。
他还和合作者给出了用于结构分析的原子对电子衍射的振幅的表格。
这种电子衍射结构分析方法特别适用于研究带织构的粘土矿物晶体,适用便于电子显微镜下研究的小晶体样品。
可达60度的倾斜织构电子衍射花样指标化后所得到的电子衍射振幅,可以提供这些晶体结构的相当完整的信息,从而测定了许多粘土矿物的晶体结构. 2)别洛夫对伐因斯坦等人的工作给予了高度的评价:“他们把简单晶体中得到的漂亮的对称电子衍射照片,发展成为了一种用于测定轻原子位置的精确的结构分析方法”。
伐因斯坦和合作者Zvyagin的经典性工作后来发表在权威的晶体结构分析的国际表3)和纪念电子衍射发现50周年的纪念文集上。
伐因斯坦在这方面的工作持续了多年。
在上世纪五十年代末期到六十年代初期,他还特别关注聚合物、液晶和其他比较无序材料的结构分析。
1963年他出版了专著《链分子的X射线衍射》。
他和合作者在1996年6月的瑞典国际学术会议上还发表了有机LB膜的电子衍射结果.4)1959年伐因斯坦受到英国学者Perutz和Andrew的影响,并且在舒勃尼科夫的支持下,白手起家,在研究所内建立并领导了蛋白质结构实验室,把工作的重点转向了生物大分子结构的分析。
他没有把工作局限于蛋白质晶体X射线的结构分析,而是从研究方法和研究对象这两个方面入手,把实验研究确定为用X射线小角衍射、电子衍射研究溶液中生物聚合物的结构。
1974年他和合作者测定的第一个蛋白质是分子量达17000Da的豆血红蛋白(leghaemoglobin),结构的分辨率达到0.5nm。
他们一共测定了30个蛋白质,40多个蛋白质变态的结构。
2) Zvyagin B B. Electron Diffraction Analysis of Clay Mineral Structures,1967, Plenum3) Vainshtein B K, Zvyagin B B. in International Tables for Crystallography, Ed. by U Shmueli,Kluwer, Dordrecht, 1993, Vol. B, p3104)Vainshtein B K,Klechkovskaya V V. Electron Diffraction by Langmuir–Blodgett Films, Proc. Roy. Soc. Lond. 1993, A 442, 73–84在上世纪八十年代和九十年代,他们测定的实竹属植物病毒结构的分辨率达到了0.35nm。
而随后测定的含有1200个非氢原子的蛋白质的结构的分辨率甚至达到了0.2nm。
由此他们揭示了蛋白质和氧结合后的结构变化。
伐因斯坦是前苏联和俄罗斯应用X射线、电子、中子衍射进行结构分析和蛋白质晶体学研究的奠基人之一。
从他一生的论文和著作来看,他擅长利用短波长的波进行材料结构分析。
无论是在散射理论研究还是仪器设计制备等方面,他均有卓越贡献。
他对晶体学的贡献无疑是十分广博和全方位的,堪称是一位百科全书式的科学家。
自1962年由伐因斯坦担任所长后,晶体学研究所得到了迅速的发展。
1971年该所被改名为舒勃尼科夫晶体学研究所。
1996年研究所为纪念伐因斯坦75岁寿辰,出版了《晶体结构研究》文集。
不久他不幸突然去世。
从1962年到1996年间他一直但任该研究所的所长,长达34年。
1998年,俄罗斯科学院通信院士M V Koval'chuk被选为新的晶体学研究所所长。
晶体学研究所在伐因斯坦的领导下,建立了不少实验和理论研究室,如液晶、电子显微镜、小角散射、激光晶体、X射线光学、同步辐射、高温结晶实验室等。
研究所的科学家还参加了大科学实验装置及实验方法(如同步辐射和中子衍射)的发展工作。
所内还建立了基地,能够批量生产激光晶体。
1997年以晶体学研究所部分实验室为基础,在莫斯科市郊组建了空间材料科学研究中心(Space Materials Science Research Center)。
研究所的规模接近1000人,其中一半分布在生产晶体(如美国空间项目所需的大尺寸蓝宝石晶体)和商品实验仪器的车间中。
几十年的发展使得晶体学研究所逐步成为了一个以研究晶体生长、晶体结构(特别是生物大分子结构)分析和晶体性质为主要特色的研究所,并且成为前苏联和俄罗斯科学院先进的重点研究所之一。
伐因斯坦院士多年来还长期担任前苏联科学院和俄罗斯科学院晶体物理科学委员会主席,俄罗斯晶体学全国委员会主席(1984年起)。
他曾是科学院晶体学和天文学学部副秘书长(1990-1996)。
他从1957年起积极参加了所有的国际晶体学会议。
他曾分别担任了1966年在莫斯科举行的第VII届国际晶体学会议组委会主席,1969-1975年国际晶体学联合会(IUCr)执行委员会委员,以及1975-1978年IUCr的副主席。
1990年他获得IUCr的最高奖-Ewald 奖。
他1996年10月28日突然去世后,晶体学的国际权威期刊迅速发表了悼念他的文章5),2001年第46卷的“Crystallography Reports”发表了多篇纪念他80岁冥寿的文章。
例如B. B. Zvyagin发表的“从粘土的电子衍射到组件晶体学”一文,介绍了利用电子衍射得到的一系列粘土的晶体结构,并因此形成了一个新的分支学科—组件晶体学。
5)Simonov V I, Feigin L A. Acta Crystallography, 1997, A53, 531-534图4 A A 契尔诺夫(Aleksander Aleksandrovich Chernov 1931-)在晶体学研究所长期任晶体生长基本过程研究室主任晶体学研究所的另一个主要研究方向是晶体生长。
这一领域,最著名学者是契尔诺夫(A A Chernov)。
他毕业于莫斯科大学物理系。
他在晶体学研究所曾长期任晶体生长基本过程研究室的主任。
他于1966年获得苏联科学院合成金刚石特别奖, 1983年获得苏联科学院费多洛夫奖, 1987年当选苏联科学院院士,1986年和1994年两次获得晶体学研究所舒勃尼科夫奖,1989年获得国际晶体生长学会(IOCG)F.C. Frank奖。
1977–1983, 1989–1991年,他曾担任国际晶体生长学会副会长,2007-2010年担任国际晶体生长学会会长。
1992-1994年,契尔诺夫到美国国家标准和技术研究所(NIST,National Institute of Standards and Technology)半时工作。
1995年到美国NASA/ Marshall 空间飞行中心(Space Flight Center)工作。