药理学中的受体激动剂和拮抗剂
药理学中的药物分类

药理学中的药物分类
在药理学中,药物可以根据其作用机理、来源及化学结构等方面进行分类。
根据作用机理分类
按作用部位分类
药物可以按照其作用部位进行分类,例如:
- 中枢神经系统作用药物:如苯二氮卓类药、镇痛剂等,作用于中枢神经系统。
- 心血管系统作用药物:如降压药、心脏药等。
- 消化系统作用药物:如胃肠病用药、泻药、胆石症用药等。
按作用方式分类
药物可以按照其作用方式进行分类,例如:
- 拮抗剂:阻止生物体内某个物质的作用,如受体拮抗剂。
- 激动剂:能够增强生物体内某个物质的作用,如受体激动剂。
- 酶抑制剂:阻止某些重要酶的活性,如靶向药物。
根据来源分类
药物可以按照其来源进行分类,例如:
- 人工合成的药物:如青霉素类、头孢类、西咪替丁等。
- 天然药物:如马钱子碱、数字is等,这些化合物在植物或动
物体内自然存在。
- 半合成药物:如阿司匹林、氯霉素等,这些药物在天然物质
基础上,进行人工化学合成得到。
根据化学结构分类
药物可以按照其化学结构进行分类,例如:
- 萘类药物:包括磺胺类、氯霉素等。
- 嘌呤类药物:如咖啡因、茶碱等。
- 对氨基苯酰胺类药物:如吩噻嗪、利尿药等。
总的来说,分类有助于我们更好地了解药物,选择适当的药物治疗疾病。
当然,药物也存在着副作用,我们需要根据药物的不同分类,在应用药物时注意潜在的风险。
拮抗剂和激动剂的名词解释

拮抗剂和激动剂的名词解释在生物学和医学领域中,拮抗剂和激动剂是常见的术语,用于描述某些物质对生物系统的影响。
拮抗剂和激动剂的作用相反,但它们在药物研究、临床治疗以及生理学研究中都起到了重要的作用。
拮抗剂是指能够与生物体内的结构或信号系统相互作用,通过干扰特定的生物过程来阻断或减弱某种效应的物质。
拮抗剂通常与生物体内的受体结合,阻碍病理过程中的信号传递,从而产生治疗作用。
举个例子,许多抗生素通过阻断细菌细胞壁合成的过程,达到抗菌的效果。
这些抗生素作为细胞壁合成酶的拮抗剂,能够阻止细菌生长和复制。
拮抗剂也被广泛应用于调节神经递质的信号传递,在治疗神经系统疾病方面发挥重要作用。
与拮抗剂相反,激动剂是指能够增强或模拟生物系统中特定效应的物质。
激动剂通过与受体结合激活生物过程,促进特定的生理反应。
举个例子,肌肉收缩需要神经冲动来触发,而某些药物具有作为肌肉激动剂的作用,可以增强神经冲动的传导,从而促进肌肉收缩。
在临床医学中,激动剂被广泛应用于治疗多种疾病,如心脏病、哮喘等。
拮抗剂和激动剂的研究与开发在药物领域中至关重要。
根据疾病的不同,科学家们进行了大量的研究,以寻找新型的拮抗剂和激动剂来治疗各种疾病。
药物的拮抗剂和激动剂特性可以通过多种方法进行研究,如体内和体外实验、分子对接模拟等。
研究者们希望找到具有高效性和选择性的药物,以实现减轻疼痛、减缓疾病进展、延长生命等目标。
除了药物研究外,拮抗剂和激动剂的概念也在生理和行为科学的研究中起着关键作用。
在生理学研究中,科学家们使用拮抗剂研究生物体内不同化学物质的作用机制。
这些研究有助于我们更好地理解生物体内各种生理过程。
在行为学研究中,激动剂被广泛用于研究动物和人类的行为反应,以便更好地理解行为变化的原因和机制。
总之,拮抗剂和激动剂是描述物质对生物系统的影响的重要术语。
拮抗剂通过阻断或减弱特定生物过程的效应发挥治疗作用,而激动剂则通过增强或模拟生物过程的效应来促进特定生理反应。
药理学名词解释

1.药理学:研究药物与生物体之间相互作用规律及机制的科学。
2.药效学:研究药物对机体作用,包括药物作用,作用机制,临床应用,不良反应。
3.药动学:研究机体对药物作用,包括药物在机体的吸收,分布,代谢及排泄过程。
4.半衰期:指血浆药物浓度下降到一半所需要的时间。
5.不良反应:指不适合用药目的而给病人带来不适或痛苦的反应。
5肝肠循环:是指某些药物经肝脏转化为极性较大的代谢产物并自胆汁排出后,又在小肠中被相应的水解酶转化成原型药物,再被小肠重新吸收进入体循环的过程。
6.副作用:在治疗剂量下,与治疗目的无关的作用(选择性低所致)7.毒性反应:用药剂量过大或时间过长,药物蓄积过多引起。
8.三致:致癌、致畸胎、致突变9.后遗效应:停药后血药浓度已降至阈浓度以下时残存的药物效应。
10.停药反跳:突然停药后原有疾病加剧(可能是受体向上调节所致)。
11.变态反应:又称为过敏反应(具有敏化的过程,与药物原有效应无关,用药理性拮抗药无效)12.特异质反应:无敏化过程,作用与药物原有作用有关,药理性拮抗剂无用。
13.生物利用度:指血管外给药时,药物吸收进入血液循环的相对数量。
14.耐药性:病原体及肿瘤细胞等对化学治疗药物敏感性降低。
15.耐受性:连续用药后机体对药物的反应强度递减,增加剂量才可以保持药效不减。
16.依赖性长期使用某种药物后,机体对这种药物产生生理或精神的敏感性。
17.安慰剂:本身没有药理活性的中性物质制成的外形似药的制剂。
18.肝药酶诱导剂:能诱导肝药酶的活性,加速自身或其它药物的代谢,便药物效应减弱。
19.肝药酶抑制剂:能抑制肝药酶的活性,降低其它药物的代谢,使药物的效应增强,甚至引起毒性反应。
20. 首关消除:指口服给药后,部分药物在胃肠道,肠粘膜和肝脏被代谢灭活,使进入体循环的药量减少的现象。
21.治疗作用:对症治疗,对因治疗,补充治疗。
22.二重感染:长期大剂量应用广谱抗生素,敏感菌被抑制,破坏了体内正常菌群生态平衡,致使一些抗药菌和真菌乘机繁殖,造成的再次感染,又称菌群交替症。
2023年初级药师考试复习笔记药理学胆碱能受体激动剂和拮抗剂肾上腺素受体激动剂和拮抗剂
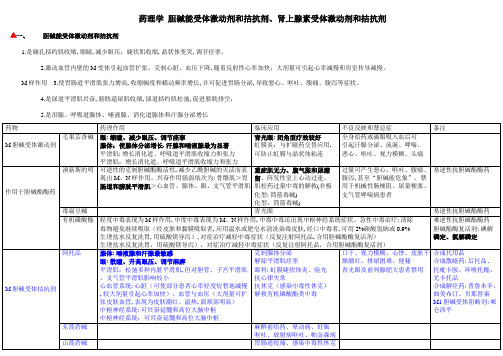
药理学 胆碱能受体激动剂和拮抗剂、肾上腺素受体激动剂和拮抗剂
胆碱能受体激动剂和拮抗剂
1.是瞳孔括约肌收缩, 缩瞳, 减少眼压;睫状肌收缩, 晶状体变突, 调节痉挛。
2.激动血管内壁的M 受体引起血管扩张;克制心脏、血压下降, 随着反射性心率加快;大剂量可引起心率减慢和房室传导减慢。
M 样作用
3.使胃肠道平滑肌张力增高, 收缩幅度和蠕动频率增长, 并可促进胃肠分泌, 导致恶心、呕吐、腹痛、腹泻等症状。
4.是尿道平滑肌兴奋, 膀胱逼尿肌收缩, 尿道括约肌松弛, 促进膀胱排空;
5.是泪腺、呼吸道腺体、唾液腺、消化道腺体和汗腺分泌增长
N胆碱受体激动剂: 尼古丁。
药理学重点笔记

1稳态血药浓度:药物在连续恒速给药或分次恒量给药的过程中,血药浓度会逐渐增高,经4~5个半衰期可达稳定而有效的血药浓度,此时药物吸收速度与消除速度达到平衡,血药浓度相对稳定在一定水平,这时的血药浓度称为稳态血药浓度,也称坪值。
P412副作用:也称副反应,是在治疗剂量下发生的不符合用药目的的反应。
P73.首关效应:指口服药物时某些药物在通过胃肠道,肠粘膜及肝脏时,部分可被代谢灭活而使进入体循环的药量减少,从而使药效降低,又称首关消除。
P304.半衰期:指血药浓度降低一半所需要的时间。
P415.拮抗剂:只有亲和力而无内在活性的药物。
P156.肝药酶抑制剂:药物等外源性物质使肝药酶的合成减少或活性降低,从而使药酶代谢能力减弱的现象,称为药酶的抑制,具有药酶抑制作用的化学物质称药酶抑制剂。
P347.受体激动剂:具有亲和力和内在活性的药物为受体激动剂。
P158.抗菌后效应:停用抗菌药后仍然持续存在的抗微生物效应。
P3239.耐药性:耐药性又称抗药性,是指细菌与抗菌药反复接触后对药物的敏感性降低甚至消失。
P325影响简单扩散因素的应用P26①分子量的大小,转运速率与分子量成反比②脂溶性:脂溶性越大跨膜转运速度越快、③膜面积和膜两侧的浓度差:膜面积越大,转运速度越快,浓度差越大,扩散速度越快④药物的解离度:解离度越大,越不容易扩散。
拟胆碱药分类、代表药P61或者P73小结(1)直接拟胆碱药①M,N胆碱受体激动药:乙酰胆碱,卡巴胆碱。
②M胆碱受体激动药:毛果芸香碱③N胆碱受体激动药;烟碱,络贝林。
(2)间接拟胆碱药:①易逆性抗胆碱脂酶药:新斯的明,毒扁豆碱。
②难逆性抗胆碱脂酶药:有机磷酸酯类。
其他:促乙酰胆碱释放药。
阿托品药理作用P75-P76(1)阻断M胆碱受体①抑制腺体分泌②扩瞳,升高眼内压和调节麻痹③解除内脏平滑肌痉挛④解除迷走神经对心脏的抑制(2)扩血管(3)兴奋中枢神经系统:阿托品可以通过血脑屏障。
临床应用P76-P77①抑制腺体分泌:)用为麻醉前给药,可减少麻醉过程中支气管粘液分泌,预防术后引起肺炎,并可消除吗啡对呼吸的抑制。
药理学名词解释

21、药理效应:药物作用的结果,机体反应的表现,对不同脏器有选择性。
22、不良反应:不符合用药目的并给患者带来不适或痛苦的反应。
14、受体拮抗剂:与受体有较强的亲和力而无内在活性的药物。
15、受体激动剂:与受体有较强的亲和力和有内在活性的药物。
16、受体部分激动剂:与受体有较强的亲和力和有较弱内在活性的药物。
17、兴奋作用:机体器官原有的功能水平提高。
18、竞争性拮抗药:与激动药互相竞争相同受体,与受体可逆性结合,从而阻断激动剂作用的药物。
60、内在拟交感活性:有些受体阻断药如吲哚洛尔和醋丁洛尔除了有阻断β
受体的作用外,其本身尚具有微弱的β受体激动作用。
61、竞争性α受体阻断药:是一类与α受体可逆性结合,竞争性阻断α受体,
从而拮抗肾上腺素的α型作用的药物。
62、反跳现象:长期使用受体阻断药后突然停药,可引起疾病复发或恶化,
可能是受体向上调节所致。
34.生物转化第二相反应:为结合反应,与体内物质如葡萄糖醛酸结合,使药物活性降低或灭活并使极性增加。
35.肝肠循环: 是指某些药物经肝脏转化为极性较大的代谢产物并自胆汁排出后,又在小肠中被相应的水解酶转化成原型药物,再被小肠重新吸收进入体循环的过程。
36.被动转运:药物依赖于膜两侧浓度差,从高浓度的一侧向低浓度的一侧扩散转运的过程。
73、反跳现象: 指患者症状基本控制后,突然停药或减量过快,引起原病复发或恶化的现象。
74、停药症状:停药出现原有疾病所没有的一些症状如肌痛、关节病、乏力、情绪消沉等。
药学院药理学名词解释

药学院药理学名词解释安全指数(SI)最小中毒量与最大治疗量之比半衰期(t1/2)血浆药物浓度降低一半所需的时间。
表观分布容积(vd)体内药物总量按血浆药物浓度推算时所需的体液总体积。
不良反应:在治疗剂量下,药物在发挥治疗作用的同时,可能产生一些其他作用,大都是人们不愿发生的。
变态反应(过敏反应allergic drug reaction):药物作为抗原或者半抗原刺激机体产生免疫反应引起生理功能障碍和组织损伤。
被动转运(passive transport):指药物借助细胞膜两侧存在的药物浓度梯度或电位差,以电化学势能差为驱动力,从高浓度侧向低浓度侧扩散。
迟后除极(DAD):细胞内钙超载下,发生在动作电位完全接近完全复极时的一种短暂的震荡性除极。
毒性反应(toxic reaction):在用药剂量较大和用药时间地过长情况下发生的机体组织、器官以器质性损伤为主的严重不良反应。
二重感染:广谱抗生素长期应用,使敏感菌受到抑制,而不敏感菌在体内繁殖生长,造成二重感染。
副反应:应用治疗量药物后出现的与治疗无关的反应。
肝肠循环:由胆汁排入十二指肠的药物,经肠粘膜上皮细胞吸收,再经门静脉进入肝脏干扰素(IFN):机体受到病毒或者其他微生物感染时,体内产生的一类抗毒糖蛋白物质,具有抗肿瘤和免疫调节作用。
后遗效应(residual effect):停药后血药浓度虽已降至有效浓度以下,但仍存留的生物效应。
化疗指数(CI)抗菌药物具有抑制或杀灭病原菌的能力激动剂(agonist):与受既有高亲和力,也有高内在活性,能产生最大效应。
拮抗剂(antagonist):与受体结合后本身不引起生物学效应,,但阻断激动剂介导的效应。
解离常数竞争性拮抗剂药物与受体有亲和力,但不产生受体激动效应,可以阻断激动剂与该受体的结合。
金鸡纳反应:应用奎尼丁的不良反应,耳鸣,听力减退,精神失常等。
继发性反应:由于药物治疗作用引起的不良后果。
简单扩散:抗生素:来自真菌或细菌的具有干扰细菌繁殖和生长的药物。
药理学中的受体激动剂和拮抗剂

药理学中一系列受体(肾上腺素受体α1、α2,β1、β2、β3,胆碱受体M1、M2、M3……;N1(NN)、N2(NM)),被激动时,什么时候什么地方哪些收缩哪些舒张,一直没有没搞清楚,也一直没贯通的去总结过,困惑了我五年,问过同学问过度娘,没有一个满意的答案。
现在纵览各受体,突然发现了一点大体的规律,有少数特殊的不符合这个规律,有些地方有点另类或牵强,能方便记忆才是王道!把兴奋性质的,如收缩、收缩增强、自律性增高、心率加快、传导加快、瞳孔开大肌收缩所致的散瞳,瞳孔括约肌收缩所致的缩瞳,统一归为收缩把其它相反性质的,如舒张、松弛、收缩减弱、自律性降低、心率减慢、传导减慢,统一归为舒张那么有如下规律:激动β(β1、β2)、M2的效应为舒张但激动β(β1、β2)对心脏、括约肌(胃)为收缩激动其它受体:α(α1、α2)、M(M、M1、M3)、N2的效应均为收缩但激动α对胃肠运动和张力为减弱,激动M3对除瞳孔括约肌外的胃肠、膀胱括约肌为舒张α1、β、M、N1均为增加分泌但α1对体内腺体(支气管、肠)的作用为抑制分泌α1、β2、β3对肝脏各项代谢均为增加代谢肾上腺素受体、胆碱受体M 在心脏和胃肠处的效应相反更精简的话就一句话了:激动β、M2 舒张,其它的为收缩,激动各受体均为增加分泌与代谢。
(但有红色的那些例外,要注意)PS:α受体主要分布于血管平滑肌、瞳孔开大肌、心脏等β 1受体主要分布于心脏、肾小球旁系细胞β 2受体主要分布于平滑肌、骨骼肌、肝脏M受体主要分布于胆碱能神经节后纤维支配的效应器:心脏、胃肠平滑肌、膀胱逼尿肌、瞳孔括约肌、各种腺体N1(N N)受体分布于神经节、肾上腺髓质N2(N M)受体主要分布于神经肌肉接头(骨骼肌)多巴胺受体主要分布于肾、肠血管平滑肌肾上腺受体、M胆碱受体均为G蛋白偶联型受体N受体为配体门控离子通道型受体典型药物:M激动-毛果芸香碱N激动-烟碱M、N激动-卡巴胆碱抗胆碱酯酶-溴新斯的明、有机磷酸酯类M 拮抗-阿托品N1 拮抗-美卡拉明N2 拮抗-筒箭毒碱、琥珀胆碱胆碱酯酶复活-氯解磷定α、β激动-肾上腺素α激动-去甲肾上腺素β激动-异丙肾上腺素α1 激动-去氧肾上腺素α2 激动-可乐定β1 激动-多巴酚丁胺β2 激动-沙丁胺醇α、β拮抗-拉贝洛尔α拮抗-酚妥拉明(短效)、酚苄明(长效)β拮抗-普萘洛尔α1 拮抗-哌唑嗪α2 拮抗-育享宾β1 拮抗-阿替洛尔β2 拮抗-布他沙明间接激动-麻黄碱其他机制-利舍平(利血平)(耗竭周围交感神经末梢的肾上腺素,心、脑及其他组织中的儿茶酚胺和5-羟色胺达到抗高血压、减慢心率和抑制中枢神经系统的作用)融会发散:关于肾上腺素的细节在皮肤、肾脏、胃肠道的血管平滑肌(大多数血管)上α受体占优势,骨骼肌、肝的血管上β2受体占优势,小剂量肾上腺素以兴奋β2为主,引起血骨骼肌、肝的血管舒张(降压),大剂量时对α受体作用明显,引起大多数血管收缩,总外周阻力增大(升压),由此可以得出,如果同时使用α受体阻断药,因为α受体阻断药选择性地阻断了与血管收缩有关的α受体,留下与血管舒张有关的β受体;所以能激动α、β受体的肾上腺素的血管收缩作用被取消,而血管舒张作用得以充分地表现出来,由升压作用翻转为降压作用,此乃肾上腺素作用的翻转,氯丙嗪,酚妥拉明有此作用,使用时应注意。
激动剂和拮抗剂的名词解释

激动剂和拮抗剂的名词解释在生物医学领域中,人们常常会听到激动剂和拮抗剂这两个词,它们是药物研究和治疗中非常重要的概念。
激动剂和拮抗剂在药物的作用方式和效果上有着截然不同的作用,对于理解药物的机制和效果具有重要意义。
本文将对激动剂和拮抗剂进行详细的解释和探讨。
激动剂(Agonist)激动剂是指能够与生物体内的受体结合,并激活受体引发效应的物质。
在生物学和药理学中,激动剂可以是天然产物或合成化合物。
当激动剂与受体结合时,它们会引起一系列的生理和药理效应。
在神经系统中,激动剂可以刺激神经递质的释放,或增加神经递质与受体之间的相互作用。
以神经递质多巴胺为例,多巴胺激动剂能够结合多巴胺受体并模拟多巴胺的作用,因此可用于治疗帕金森病等多巴胺缺乏疾病。
除了神经递质,激动剂还可以作用于其他类型的受体,如激素受体、细胞表面受体等。
例如,肾上腺素能受体激动剂可以用于治疗心衰、支气管痉挛等疾病。
拮抗剂(Antagonist)相对于激动剂,拮抗剂是一类能与受体结合,但不能引发效应的化合物。
拮抗剂在生物体内能与激动剂竞争受体结合位点,从而阻断激动剂与受体的结合,减弱或抑制激动剂的效应。
拮抗剂中的一类被称为竞争性拮抗剂。
它们与激动剂竞争同一受体位点,由于结合力较强,使激动剂无法结合受体。
例如,贝他受体阻滞剂是一种广泛应用的药物,能够与β受体结合并抑制它的激动作用,用于治疗心脏病和高血压等疾病。
除了竞争性拮抗剂外,还有一类被称为非竞争性拮抗剂。
它们与受体结合,但作用不同于激动剂。
非竞争性拮抗剂通过改变受体的构象,使激动剂难以结合或无法产生效应。
这类拮抗剂的作用通常是不可逆的。
例如,氟虫腈是一种γ-氨基丁酸(GABA)受体的非竞争性拮抗剂,用于治疗癫痫和焦虑症等疾病。
激动剂与拮抗剂的临床应用激动剂和拮抗剂在药物研究和临床应用中发挥着不可替代的作用。
通过设计和合成具有特定效应的激动剂和拮抗剂,科学家和医生可以更好地了解生物体内的药物作用机制,并开发出更有效的药物。
药理学重点总结

一、名词解释:耐受性:指机体对药物反应性降低的一种状态。
半衰期:资血浆药物浓度下降一半所需要的时间。
毒性反应:指药物在用要药剂量过大,用药时间过长或机体对药物敏感性过高时产生的危害性反应。
半数致死量(LD50):反应药物毒性大小的重要数据。
副作用:药物在治疗剂量时出现的与用药目的无关的作用。
受体激动剂:与受体有较强亲和力,又有较强内在活性的药物。
交叉耐药性:机体对某药产生耐受性后,对另一种药物也的敏感性也降低。
后遗效应:停药后血药浓度已降至阀浓度以下时残存的药理效应。
首关消除:口服药物在胃肠黏膜吸收后,首先经门静脉进入肝脏,当通过肠黏膜及肝脏时部分药物发生转化,使进入体循环的有效药量减少的现象。
疫苗;激活一种或多种免疫活性细胞,增强机体免疫功能的药物。
抗菌药物:是指对病原菌具有抑制或杀灭作用,主要用于防治细菌性感染疾病的一类药物;属于抗微生物药物的范畴。
抗微生物药物:对病原微生物有抑制或杀灭作用,用于防治病原微生物感染性疾病的药物。
化学治疗:化学药物抑制或杀灭机体内的病原微生物、寄生虫及恶性肿瘤细胞,消除或缓解由它们所引起的疾病。
抗菌谱:是指药物的抗菌范围窄谱:仅对单一菌种或单一菌属有抗菌作用。
广谱:对多数革兰阳性、革兰阴性细菌有抗菌作用,还对某些衣原体、支原体、立克次体、螺旋体及原虫等也有抑制作用。
抗生素后效应:抗生素在撤药后其浓度低于最低抑菌浓度时,细菌仍受到持久抑制的效应。
如青霉素类和头孢菌素类抗菌药的抗生素后效应十分明显。
PAE的确切机制尚不清楚。
固有耐药性:是由细菌染色体基因决定而代代相传的耐药,如肠道杆菌对青霉素的耐药。
获得耐药性:指细菌与药物多次接触后,对药物的敏感性下降甚至消失,致使药物疗效低或无效。
1、*有机磷酸酯类中毒的原理及解救措施原则。
中毒的原理:有机磷酸酯类+AChE——磷酰化AChE ---中毒时间过长——酶老化——递质Ach被AChE水解的量减少——突触间隙ACh 堆积——中毒症状。
(完整版)药理学重点汇总笔记全

药理学一、名词解释:1不良反应:对机体带来不适,痛苦或损害的反应。
2血浆半衰期:是指体内血药浓度下降一半所需要的时间,是表示药物消除速度的一种参数。
3选择性作用:在一定剂量范围内,多数药物吸收后,只对某一.两种器官或组织产生明显的药理作用,而对其它组织作用很小甚至无作用,药物的这种特性称为选择性。
4激动剂:药物与受体有较强的亲和力,也有较强的内在活性。
它兴奋受体产生明显效应。
5拮抗剂:药物与受体亲和力较强,但无内在活性,故不产生效应,但能阻断激动药与受体结合,因而对抗或取消激动药的作用。
6部分激动剂:本类药物与受体的亲和力较强,但只有弱的内在活性,能引起较弱的生理效应,较大剂量时,如与激动药同时存在,能拮抗激动药的部分效应。
7半数致死量(LD50):如以死亡为指标,则称为半数惊厥量或半数致死量。
8安全范围:有人用1%致死量与99%有效量的比值来衡量药物的安全性,5%致死量与95%有效量之间的距离称为药物的安全范围。
9生物利用度:指药物吸收进入血液循环的速度和程度,生物利用度高,说明药物吸收良好,反之,则药物吸收差。
10首关消除:口服某些药物时,在胃肠道吸收后,经肝门静脉进入肝脏,在进入体循环前被肠粘膜及肝脏酶代谢灭活或结合贮存,使进入体循环的药量明显减少。
称首关消除。
12.首过效应:口服经门静脉进人肝脏的药物,在进人体循环前被代谢灭活或结合储存,使进人体循环的药量明显减少。
11肝肠循环:药物自胆汁排泄到十二指肠后,在肠道被再吸收又回到肝脏的过程12量效关系:在一定的范围内,药物的效应与靶部位的浓度成正相关,而后者决定于用药剂量或血中药物浓度,定量地分析与阐明两者间的变化规律称为量效关系。
药物剂量与效应之间的规律性变化为量效关系。
13有效量:出现疗效的剂量。
14肝药酶诱导剂:是指有些药物长期使用后能加速肝药酶的合成并增强其活性,这类药物就称为肝药酶诱导剂。
15最小有效量:在一定剂量范围内,随剂量的增加药物效应逐渐增强,出现疗效的最小剂量称为最小有效量。
拮抗剂名词解释药理学

拮抗剂名词解释药理学
拮抗剂是指一类药物,通过与生物体内的特定受体结合,阻止或减弱受体的激活能力,从而达到调节生理或病理状态的药理作用。
拮抗剂的作用机制可以分为竞争性和非竞争性两种。
竞争性拮抗剂与受体结合的亲和力较强,与激动剂争夺受体结合位点,从而阻止激动剂与受体结合,减少受体的激活。
非竞争性拮抗剂则通过与受体的其他位点结合,改变受体的构象或功能,使其无法被激动剂结合或激活。
拮抗剂在药理学研究和药物治疗中具有重要的应用价值。
它们可以用于研究受体的结构和功能,揭示受体与激动剂之间的相互作用机制。
此外,拮抗剂还可以用于治疗疾病,例如高血压、哮喘、抑郁症等。
通过选择合适的拮抗剂,可以阻断异常的生理反应或病理过程,恢复机体的平衡状态。
不同类型的药物可以作为拮抗剂,例如β受体阻断剂、抗组胺药物、抗精神病药物等。
这些药物可以选择性地作用于不同的受体亚型或亚单位,从而实现特定的药理效应。
例如,β受体阻断剂可以选择性地结合β1受体或β2受体,用于治疗心脏病或哮喘。
总之,拮抗剂是一类能够与特定受体结合,阻止或减弱受体的激活能力的药物。
它们在药理学研究和药物治疗中具有重要的作用,可以揭示受体的结构和功能,治疗疾病并调节机体的生理状态。
5-HT受体激动剂和拮抗剂

5-HT受体激动剂和拮抗剂在胃肠病中的应用之宇文皓月创作作者:佚名科研信息来源:本站原创点击数:157 更新时间:2005-12-16[关键词]:功能性胃肠病中,司琼类,必利类健康网讯:5-羟色胺(5-hydroxytryptamine,5-HT)又称血清素(serotonin),是重要的神经递质,人体内95%的5-HT在胃肠道的肠嗜铬细胞(enterochromaffin cells,EC)及肠神经元中合成,5-HT通过与其受体相互作用,在胃肠道动力、感觉和分泌中发挥重要作用。
5-HT受体超家族可分为7种亚型(5-HT1~7受体)和更多的亚亚型。
胃肠道内至少有5种受体,其中5-HT3受体和5-HT4受体与胃肠运动和分泌功能最为密切。
本文将讨论5-HT受体激动剂和拮抗剂在功能性胃肠病中的应用(见表1)。
5-HT1受体激动剂舒马曲坦(sumatriptan)是选择性5-HT1B/D受体激动剂。
在健康人静注舒马曲坦后不单可以使胃液体排空延缓,还可以延缓胃固体餐的排空。
舒马曲坦激活中枢和周围5-HT1B/D受体,释放非肾上腺素能非胆碱能(non-adrenergic non- cholinergic, NANC)神经递质,松弛胃底、胃窦和幽门平滑肌,改善餐后胃的容受性;并增加食管的敏感性,降低食管顺应性。
Tack等报导,舒马曲坦治疗功能性消化不良(functional dyspepsia, FD)患者,可降低胃壁张力,增加胃平均容量,并改善早饱不适感。
但也有分歧结果的陈述,因而需更多的临床研究的证实。
5-HT3受体激动剂动物中的研究显示,5-HT3受体激动剂YM- 31636可促进动物的排便、增加排便量,这可能与增加结肠的动力有关,这一效应可被5-HT3受体拮抗剂雷莫司琼所阻断。
YM-31636不增加内脏疼痛阈值,有望用于治疗慢性便秘、以便秘为主的肠易激综合征(irritable bowel syndrome, 1BS)等,但目前尚无临床报导。
药理学

1、从药物与受体作用角度简述激动剂与拮抗剂的特点。
激动剂:有很大的亲活力和内在活性,能与受体结合并产生效应拮抗剂:很强的亲和力,能与受体结合,但缺乏内在活性,结合后非但不能产生效应,同时由于占据受体而拮抗激动剂的效应。
与吗啡相比哌替啶的药理作用及临床用途有那些不同答:药理作用:(1)中枢神经系统:与吗啡相似,作用于中枢的阿片受体而发挥作用。
有镇静作用、镇痛作用,但持续时间比吗啡短。
镇痛作用弱于吗啡(2)对平滑肌作用:作用时间短,不引起便秘,也无止泻作用,对胃肠道平滑肌作用比吗啡弱临床应用:不能用于止泻,可用于麻醉前给药及人工冬眠试述硝酸酯类与β受体阻断药联合应用治疗心绞痛的药理基础答:β受体阻断药和硝酸酯类合用,能协同降低耗氧量,同时β受体阻断药可取消硝酸酯类所引起的反射性心率加快和心肌收缩力加强;硝酸酯类可缩小β受体阻断药所致的心室容积增大和射血时间延长。
两药合用可互相取长补短,合用时用量减少,副作用也减少。
比较非去极化肌松药和去极化肌松药的药理作用特点非去极化肌松药:(1)不引起终版电位,骨骼肌松弛前无肌肉兴奋现象;(2)肌肉松弛作用可被抗胆碱酯酶所拮抗,过量时可用新斯的明解救;(3)吸入性全麻药和氨基酸糖苷类抗生素能增强和延长本类药物的作用;(4)肌肉松弛作用可被同类药物所增强;(5)可有程度不等的神经节阻断作用和组释放作用。
去极化肌松药:(1)肌松前常先出现短时间的肌束颤动,不同部位的骨骼肌去极化出现的时间先后不同,故出现不协调的肌束颤动,而后处于麻痹状态;(2)连续用药可产生快速耐受性;(3)抗胆碱酯酶药不能拮抗其肌松作用,甚至使之加剧;(4)治疗量时,无神经节阻断作用,相反有兴奋作用。
5、抗高血压药是怎样分类的?列举各类的代表药。
答:根据药物在血压调节系统中的主要影响及部位,可将抗高血压药分成以下几类:(1)主要影响血容量的抗高血压药,如利尿药。
(2)b受体阻断药,如普萘洛尔。
(3)钙拮抗药,如硝苯地平。
神经递质、受体、激动剂和拮抗剂的类型

2、神经递质、受体、激动剂和拮抗剂的类型神经递质受体激动剂拮抗剂胆碱类: 乙酰胆碱M-受体:M1-M5N-受体:N1、N2M:毒菌碱毛果芸香碱槟榔碱氧化震颤素N:烟碱M、N:杀虫剂促使Ach释放:蝎毒黑寡妇蜘蛛毒液α-银环蛇毒Ca2+、Mg2+胆碱酯酶抑制剂:新斯的明毒扁豆碱腾喜龙有机磷脂类M:阿托品N1:六烃季胺十烃季胺美加明N2:箭毒抑制Ach合成:密胆碱-3三乙基胆碱4-吡啶抑制Ach释放:肉毒毒素河豚毒单胺类:(1)儿茶酚胺 :a 去甲肾上腺素b 多巴胺c 肾上腺素(2) 吲哚胺:5-羟色胺血清紧张素去甲肾上腺素受体:α1、α2β1、β2多巴胺受体:D1—D5 受体5-羟色胺受体:5-HT1—5-HT7受体多巴胺激动剂:左旋多巴苯丙胺(安非他明)可卡因哌甲酯(利他灵)司来吉米肾上腺素激动剂:咪唑克生5-羟色胺激动剂:氟西汀芬氟拉明MDMALSD去甲肾上腺素激动剂:α:异丙肾上腺素、NEβ:NE、E多巴胺拮抗剂:AMPT氯内嗪氯氮平利血平肾上腺素拮抗剂:镰刀菌酸5-羟色胺拮抗剂:PCPA去甲肾上腺素拮抗剂:α:酚妥拉明β:心得安心得平心得静氨基酸类:(1)抑制性氨基酸类:甘氨酸(2) 兴奋性氨基酸类:谷氨酸天冬氨酸谷氨酸门控离子通道受体:NMDA受体非NMDA受体(AMPA受体、KA受体)G蛋白耦联谷氨受体:ACPD受体L-AP4 NMDA受体谷氨酸激动剂:NMDAAMPA红藻氨酸γ-氨基丁酸激动剂:毒蝇蕈醇巴氯芬苯二氮卓类巴比妥酸盐类固醇谷氨酸拮抗剂:AP5酒精PCPγ-氨基丁酸拮抗剂:荷牡丹碱CGP335348印防己毒素烯丙基甘氨酸甘氨酸拮抗剂:士的宁多肽类:神经肽类阿片肽类胃肠肽类激肽类阿片肽类受体:κ、δ、μ阿片肽类激动剂:吗啡海洛因杜冷丁芬太尼美沙酮阿片肽类拮抗剂:纳洛芬纳洛酮纳曲酮其他:前列腺素组胺内皮源性舒张因子(NO、CO)核苷类核苷类的阻断剂:咖啡因NO的拮抗剂:L-NAME。
药理学中的受体激动剂和拮抗剂

药理学中的受体激动剂和拮抗剂This manuscript was revised by the office on December 22, 2012药理学中一系列受体(肾上腺素受体α1、α2,β1、β2、β3,胆碱受体M1、M2、M3……;N1(NN)、N2(NM)),被激动时,什么时候什么地方哪些收缩哪些舒张,一直没有没搞清楚,也一直没贯通的去总结过,困惑了我五年,问过同学问过度娘,没有一个满意的答案。
现在纵览各受体,突然发现了一点大体的规律,有少数特殊的不符合这个规律,有些地方有点另类或牵强,能方便记忆才是王道!把兴奋性质的,如收缩、收缩增强、自律性增高、心率加快、传导加快、瞳孔开大肌收缩所致的散瞳,瞳孔括约肌收缩所致的缩瞳,统一归为收缩把其它相反性质的,如舒张、松弛、收缩减弱、自律性降低、心率减慢、传导减慢,统一归为舒张那么有如下规律:激动?β(β1、β2)、M2的效应为舒张但激动?β(β1、β2)对心脏、括约肌(胃)为收缩激动其它受体:α(α1、α2)、M(M、M1、M3)、N2的效应均为收缩但激动α对胃肠运动和张力为减弱,激动M3对除瞳孔括约肌外的胃肠、膀胱括约肌为舒张α1、β、M、N1均为增加分泌但α1对体内腺体(支气管、肠)的作用为抑制分泌α1、β2、β3对肝脏各项代谢均为增加代谢肾上腺素受体、胆碱受体M在心脏和胃肠处的效应相反更精简的话就一句话了:激动?β、M2?舒张,其它的为收缩,激动各受体均为增加分泌与代谢。
(但有红色的那些例外,要注意)PS:α受体主要分布于血管平滑肌、瞳孔开大肌、心脏等β1受体主要分布于心脏、肾小球旁系细胞β2受体主要分布于平滑肌、骨骼肌、肝脏M受体主要分布于胆碱能神经节后纤维支配的效应器:心脏、胃肠平滑肌、膀胱逼尿肌、瞳孔括约肌、各种腺体N1(N N)受体分布于神经节、肾上腺髓质N2(N M)受体主要分布于神经肌肉接头(骨骼肌)多巴胺受体主要分布于肾、肠血管平滑肌肾上腺受体、M胆碱受体均为G蛋白偶联型受体N受体为配体门控离子通道型受体典型药物:M激动-毛果芸香碱N激动-烟碱M、N激动-卡巴胆碱抗胆碱酯酶-溴新斯的明、有机磷酸酯类M拮抗-阿托品N1拮抗-美卡拉明N2拮抗-筒箭毒碱、琥珀胆碱胆碱酯酶复活-氯解磷定α、β激动-肾上腺素α激动-去甲肾上腺素β激动-异丙肾上腺素α1?激动-去氧肾上腺素α2?激动-可乐定β1?激动-多巴酚丁胺β2?激动-沙丁胺醇α、β拮抗-拉贝洛尔α拮抗-酚妥拉明(短效)、酚苄明(长效)β拮抗-普萘洛尔α1?拮抗-哌唑嗪α2?拮抗-育享宾β1?拮抗-阿替洛尔β2?拮抗-布他沙明间接激动-麻黄碱其他机制-利舍平(利血平)(耗竭周围交感神经末梢的肾上腺素,心、脑及其他组织中的儿茶酚胺和5-羟色胺达到抗高血压、减慢心率和抑制中枢神经系统的作用)融会发散:关于肾上腺素的细节在皮肤、肾脏、胃肠道的血管平滑肌(大多数血管)上α受体占优势,骨骼肌、肝的血管上β2受体占优势,小剂量肾上腺素以兴奋β2为主,引起血骨骼肌、肝的血管舒张(降压),大剂量时对α受体作用明显,引起大多数血管收缩,总外周阻力增大(升压),由此可以得出,如果同时使用α受体阻断药,因为α受体阻断药选择性地阻断了与血管收缩有关的α受体,留下与血管舒张有关的β受体;所以能激动α、β受体的肾上腺素的血管收缩作用被取消,而血管舒张作用得以充分地表现出来,由升压作用翻转为降压作用,此乃肾上腺素作用的翻转,氯丙嗪,酚妥拉明有此作用,使用时应注意。
药理学名词解释汇总

药理学名词解释汇总1.药理学:研究药物与机体相互作用规律及作用机制的科学。
2.药效:研究药物对机体的作用规律和机制。
3.药代动力学:是阐明机体对药物的作用,即药物在机体内吸收、分布、代谢和排泄过程的药效和血药浓度消长的规律。
4.不良反应:用药后出现与治疗目的无关的作用。
副反应:也称副作用,指在治疗剂量下出现的与治疗目的无关的反应。
5.治疗量(有效量):能对机体产生明显药效而又不引起毒性反应的剂量。
6.极量:是由国家药典规定允许使用的最大剂量,也是医生用药选择剂量的最大限度。
7.安全范围:最小有效量和极量之间的范围。
8.受体激动剂:药物与受体有较强的亲和力,并有较强的内在活性,能激动受体,产生明显效应。
9.受体拮抗剂:药物与受体亲和力强,但无内在活性,能阻断激动剂一受体的结合,拮抗激动剂作用。
10.首过效应:指某些口服用药后经肠粘膜及肝脏被代谢灭活,进入体循环的药量明显减少的现象。
11.生物利用度:指药物被机体吸收进入体循环的相对分量和速度。
12.血浆蛋白结合率:指治疗剂量下药物与血浆蛋白结合的百分率。
13.肝肠循环:某些药物或代谢物经胆汁排泄进入肠道朋解后,再吸收入血,这种胆汁排泄又重吸收的现象称肝肠循环。
14.血浆半衰期:指血浆中药物浓度下降一半所需时间。
15.稳态血药浓度:恒速恒量或按半衰期连续多次给药后经5个t1/2,药物吸收与消除速度达平衡,血药浓度相对稳定在一定水平,称稳态血浓度。
16.药物的机体消除:包括代谢及排泄两个过程。
17:血浆蛋白结合率:指血中与血浆蛋白结合的药物占总药量的百分数。
18.血脑屏障(blood—brain barrier):脑组织内的毛细血管内皮细胞紧密相连,内皮细胞之间无间隙,且毛细血管外表面几乎均为星形胶质细胞包围,这种特殊结构形成了血浆与脑脊液之间的屏障。
血浆与脑脊液之间的屏障。
19.胎盘屏障(placental barrier):胎盘绒毛与子宫血窦之间的屏障称为胎盘屏障。
5-HT受体激动剂和拮抗剂

5-HT受体激动剂和拮抗剂在胃肠病中的应用作者:佚名科研信息来源:本站原创点击数: 157 更新时间:2005-12-16 [关键词]:功能性胃肠病中,司琼类,必利类健康网讯:5-羟色胺(5-hydroxytryptamine,5-HT)又称血清素 (serotonin),是重要的神经递质,人体内95%的5-HT在胃肠道的肠嗜铬细胞(enterochromaffin cells,EC)及肠神经元中合成,5-HT通过与其受体相互作用,在胃肠道动力、感觉和分泌中发挥重要作用。
5-HT受体超家族可分为7种亚型(5-HT1~7受体)和更多的亚亚型。
胃肠道内至少有5种受体,其中5-HT3受体和5-HT4受体与胃肠运动和分泌功能最为密切。
本文将讨论5-HT受体激动剂和拮抗剂在功能性胃肠病中的应用(见表1)。
5-HT1受体激动剂舒马曲坦(sumatriptan)是选择性5-HT1B/D受体激动剂。
在健康人静注舒马曲坦后不但可以使胃液体排空延缓,还可以延缓胃固体餐的排空。
舒马曲坦激活中枢和周围5-HT1 B/D受体,释放非肾上腺素能非胆碱能(non-adrenergic non- cholinergic, NANC)神经递质,松弛胃底、胃窦和幽门平滑肌,改善餐后胃的容受性;并增加食管的敏感性,降低食管顺应性。
Tack等报道,舒马曲坦治疗功能性消化不良(functional dyspepsia, FD)患者,可降低胃壁张力,增加胃平均容量,并改善早饱不适感。
但也有不同结果的报告,因而需更多的临床研究的证实。
5-HT3受体激动剂动物中的研究显示,5-HT3受体激动剂YM- 31636可促进动物的排便、增加排便量,这可能与增加结肠的动力有关,这一效应可被5-HT3受体拮抗剂雷莫司琼所阻断。
YM-31636不增加内脏疼痛阈值,有望用于治疗慢性便秘、以便秘为主的肠易激综合征(irritable b owel syndrome, 1BS)等,但目前尚无临床报道。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
?药理学中一系列受体(肾上腺素受体α1、α2,β1、β2?、β3?,胆碱受体M1、M2、M3……;N1(NN)、N2(NM)),被激动时,什么时候什么地方哪些收缩哪些舒张,一直没有没搞清楚,也一直没贯通的去总结过,困惑了我五年,问过同学问过度娘,没有一个满意的答案。
? ? ??现在纵览各受体,突然发现了一点大体的规律,有少数特殊的不符合这个规律,有些地方有点另类或牵强,能方便记忆才是王道!
把兴奋性质的,如收缩、收缩增强、自律性增高、心率加快、传导加快、
瞳孔开大肌收缩所致的散瞳,瞳孔括约肌收缩所致的缩瞳,统一归为收缩
把其它相反性质的,如舒张、松弛、收缩减弱、自律性降低、心率减慢、传导减慢?,统一归为舒张
那么有如下规律:
激动??β(β1、β2)、M2?的效应为舒张
但激动??β(β1、β2)对心脏、括约肌(胃)为收缩
激动其它受体:?α(α1、α2)、M(M、M1、M3)、N2的效应均为收缩??
但激动α对胃肠运动和张力为减弱,激动M3对除瞳孔括约肌外的胃肠、膀胱括约肌为舒张?
α1、β、M、N1均为增加分泌
但α1对体内腺体(支气管、肠)的作用为抑制分泌
α1、β2、β3对肝脏各项代谢均为增加代谢
肾上腺素受体?、胆碱受体M 在心脏和胃肠处的效应相反
更精简的话就一句话了:激动??β、M2?舒张,其它的为收缩,激动各受体均为增加分泌与代谢。
(但有红色的那些例外,要注意)
PS:
α?受体主要分布于血管平滑肌、瞳孔开大肌、心脏等
β?1受体主要分布于心脏、肾小球旁系细胞
β?2受体主要分布于平滑肌、骨骼肌、肝脏
M受体主要分布于胆碱能神经节后纤维支配的效应器:心脏、胃肠平滑肌、膀胱逼尿肌、瞳孔括约肌、各种腺体N1(N N)受体分布于神经节、肾上腺髓质
N2(N M)受体主要分布于神经肌肉接头(骨骼肌)
多巴胺受体主要分布于肾、肠血管平滑肌
肾上腺受体、M胆碱受体均为G蛋白偶联型受体
N受体为配体门控离子通道型受体
典型药物:
M激动-毛果芸香碱
N激动-烟碱
M、N激动-卡巴胆碱
抗胆碱酯酶-溴新斯的明、有机磷酸酯类
M 拮抗-阿托品
N1 拮抗-美卡拉明
N2 拮抗-筒箭毒碱、琥珀胆碱
胆碱酯酶复活-氯解磷定
α、β?激动-肾上腺素?
α?激动-去甲肾上腺素
β?激动-异丙肾上腺素
α1?激动-去氧肾上腺素
α2?激动-可乐定
β1?激动-多巴酚丁胺
β2?激动-沙丁胺醇
α、β?拮抗-拉贝洛尔
α?拮抗-酚妥拉明(短效)、酚苄明(长效)
β?拮抗-普萘洛尔?
α1?拮抗-哌唑嗪
α2?拮抗-育享宾
β1?拮抗-阿替洛尔
β2?拮抗-布他沙明
间接激动-麻黄碱
其他机制-利舍平(利血平)(耗竭周围交感神经末梢的肾上腺素,心、脑及其他组织中的儿茶酚胺和5-羟色胺达到抗高血压、减慢心率和抑制中枢神经系统的作用)
融会发散:
关于肾上腺素的细节
在皮肤、肾脏、胃肠道的血管平滑肌(大多数血管)上α受体占优势,骨骼肌、肝的血管上β2受体占优势,小剂量肾上腺素以兴奋β2为主,引起血骨骼肌、肝的血管舒张(降压),大剂量时对α受体作用明显,引起大多数血管收缩,总外周阻力增大(升压),由此可以得出,如果同时使用α受体阻断药,因为α受体阻断药选择性地阻断了与血管收缩有关的α受体,留下与血管舒张有关的β受体;所以能激动α、β?受体的肾上腺素的血管收缩作用被取消,而血管舒张作用得以充分地表现出来,由升压作用翻转为降压作用,此乃肾上腺素作用的翻转,氯丙嗪,酚妥拉明有此作用,使用时应注意。
对于主要作用于血管α受体的去甲肾上腺素,它们只能取消或减弱其升压效应而无“翻转作用”。
再反观药理学口诀中相应片段,已经比较好理解
肾上腺素
α、β受体兴奋药,肾上腺素是代表;
血管收缩血压升,局麻用它延时间,
局部止血效明显,过敏休克当首选,
心脏兴奋气管扩,哮喘持续它能缓,
心跳骤停用“三联”,应用注意心血管,α受体被阻断,升压作用能翻转。
去甲肾上腺素
去甲强烈缩血管,升压作用不翻转,
只能静滴要缓慢,引起肾衰很常见,
用药期间看尿量,休克早用间羟胺。
异丙肾上腺素
异丙扩张支气管,哮喘急发它能缓,
扩张血管治“感染”,血容补足效才显。
兴奋心脏复心跳,加速传导律不乱,
哮喘耐受防猝死,甲亢冠心切莫选。
α受体阻断药
α受体阻断药,酚妥拉明酚苄明,
扩张血管治栓塞,血压下降诊治瘤,NA释放心力增,治疗休克及心衰。
β受体阻断药
β受体阻断药,普萘洛尔是代表,
临床治疗高血压,心律失常心绞痛。
三条禁忌记心间,哮喘、心衰、心动缓。
传出神经药在休克治疗中的应用(一)药物的种类
抗休克药分二类,舒缩血管有区分;
正肾副肾间羟胺,收缩血管为一类;
莨菪碱类异丙肾,加上α受体阻断剂;还有一类多巴胺,扩张血管促循环。
(二)常见休克的药物选用:
过敏休克选副肾,配合激素疗效增;
感染用药分阶段,扩容纠酸抗感染,
早期需要扩血管,山莨菪碱为首选;
后期治疗缩血管,间羟胺替代正肾。
心源休克须慎重,选用“二胺”方能行。
说明:“二胺”指多巴胺和间羟胺。