原料药和制剂质量标准
原料药和制剂质量标准

原料药质量研究的主要容一、性状(一)外观聚集状态、色泽、臭、味、结晶性是对药物感官的一般性描述,一般无法定方法检查,但仍应准确描述。
(二)溶解性1. 溶剂的选择应根据药物的结构和性质,考查药物在水与常用溶剂中的溶解度。
应选择与药物溶解特性密切相关的溶剂,配制制剂、制备溶液和精制操作有关的溶剂,避免使用昂贵的、毒性大的、不常使用的溶剂。
2. 表述方式用极易溶解”易溶”溶解”等术语来表示(见《中国药典》凡例)。
3•试验方法按《中国药典》凡例的规定操作。
25±2C,每隔5分钟强力振摇30秒钟,观察30分钟的溶解情况。
(三)理化常数理化常数包括:熔点,馏程,凝点,比旋度,折光率,黏度,相对密度,酸值、碘值、羟值、皂化值,吸收系数等。
测定物理常数可以鉴别药物,也可以反映药物的纯杂程度。
注意事项:应按药典附录规定的方法进行测定。
测定前应按规定对仪器进行校正。
测定理化常数时应用精制品进行测定;质量标准中规定的理化常数围,用试制的药物来测定。
固体药物:熔点、吸收系数。
液体药物:馏程、相对密度、黏度、折光率。
油脂类还需测定:酸值、碘值、羟值、皂化值。
有手性中心的药物:比旋度。
1、熔点照《中国药典》”熔点测定法”测定。
(1)中国药典使用毛细管法测定熔点,以供试品局部液化的温度作为初熔的温度,以全部液化的温度作为全熔的温度。
Word 文档(2)所用的传温液、毛细管、升温速度等对测定结果有影响,应按药典要求进行测定。
Word 文档Word 文档(3) 应使用分浸型的温度计,温度计应用熔点标准品进行校正。
(4) 应用DSC 予以佐证。
2、吸收系数(1)测定方法配制高、低两种不同浓度的溶液(吸光度在0.3~0.4和0.6~0.8之间)各2份,或配制吸收度在0.1~0.8的系列浓度的溶液,在 5台不同型号的紫外分光光度计上测 定,按下式计算吸收系数。
要求:应使用对照品进行测定。
测定前应对容量仪器进行校正。
测定前应对紫外分光光度计进行校正。
复方制剂质量标准

复方制剂质量标准
复方制剂质量标准是指对复方制剂的质量进行规范化、标准化的要求,以确保其药效和安全性。
在建立复方制剂质量标准时,需要从多个方面进行考虑和评估,以下是一些主要方面:
1. 原料药:原料药是复方制剂的主要成分,其质量对复方制剂的质量有着至关重要的影响。
因此,需要对原料药的来源、生产工艺、质量标准等进行严格的控制和评估。
2. 生产工艺:生产工艺是影响复方制剂质量的重要因素之一。
在制定复方制剂质量标准时,需要明确生产工艺的流程和参数,并制定相应的控制指标,以确保生产过程中的稳定性和一致性。
3. 包装材料:包装材料对复方制剂的质量也有一定的影响。
在选择包装材料时,需要考虑其对复方制剂的保护作用、阻隔性能、安全性等方面的因素,并制定相应的质量标准和控制指标。
4. 稳定性:复方制剂的稳定性是其质量的重要指标之一。
在制定复方制剂质量标准时,需要对复方制剂的稳定性进行严格的测试和评估,以确保其在规定的时间内保持稳定。
5. 有效性:有效性是复方制剂质量标准的另一个重要方面。
在制定复方制剂质量标准时,需要对其有效性进行充分的测试和评估,以确保其能够达到预期的治疗效果。
6. 安全性:安全性是复方制剂质量标准的首要考虑因素。
在制定复方制剂质量标准时,需要对可能存在的安全隐患进行充分的评估和控制,以确保其安全性。
总之,建立复方制剂质量标准需要考虑多个方面,包括原料药、生产工艺、包装材料、稳定性、有效性和安全性等。
只有制定科学、合理、全面的质量标准,才能确保复方制剂的质量和安全性。
药品质量控制

药品质量控制药品质量控制是指通过一系列的质量管理措施和标准,确保药品的质量符合规定的要求,以保障药品的安全性、有效性和一致性。
药品质量控制是药品生产和销售过程中的重要环节,对于保护公众的健康和利益具有重要意义。
下面将详细介绍药品质量控制的内容和相关标准。
一、药品质量控制的内容1. 原料药质量控制:原料药是制药过程中的重要组成部分,其质量直接影响到最终药品的质量。
原料药质量控制主要包括对原料药的来源、纯度、杂质、稳定性等方面进行检验和控制。
2. 制剂质量控制:制剂是指将原料药按一定比例和工艺制备成的成品药品。
制剂质量控制包括对制剂的外观、理化性质、含量、溶解度、稳定性等进行检验和控制。
3. 包装质量控制:包装是保护药品的重要手段,包装质量直接关系到药品的安全性和有效性。
包装质量控制主要包括对包装材料的选择、包装工艺的控制以及包装后的药品质量检验等方面。
4. 生产过程质量控制:药品生产过程中的各个环节都需要进行质量控制,包括原料的采购、仓储、生产工艺、设备设施、人员操作等。
生产过程质量控制主要通过建立标准操作规程、设备验证、人员培训等方式来保证药品生产过程的质量。
5. 质量管理体系:质量管理体系是指建立一套完整的质量管理体系,包括质量方针、质量目标、质量手册、程序文件、记录等。
质量管理体系可以帮助企业建立科学、规范的质量管理体系,保证药品质量的稳定性和可靠性。
二、药品质量控制的相关标准1. 国家药品质量标准:国家药品质量标准是指国家卫生健康委员会或国家药监局制定和发布的关于药品质量的标准。
国家药品质量标准包括药典标准、药品注册标准、药品GMP标准等,对药品的质量要求进行了详细规定。
2. 国际药典标准:国际药典是指国际药典委员会制定和发布的关于药品质量的标准。
国际药典标准是全球公认的药品质量标准,包括欧洲药典、美国药典、英国药典等,药品生产和质量控制过程中通常会参考国际药典标准。
3. GMP要求:GMP(Good Manufacturing Practice)是药品生产质量管理的基本要求。
原料药和制剂测试方法及标准

新分析技术在不断出现,现有技术在不断改进,当认为这些新技术能提供更进一步的质 量保证或经过论证,就应予采用。
2.10 原料药对制剂规范的影响
通常,原料药中已控制的质检项目在药物制剂中不必重复。例如,制剂中就不必检查已 在原料药中控制过的并且不属于降解产物的杂质。在 ICH“新药制剂杂质”指导原则中有详 细说明。
是逐批检验,前提是认为未测定的批次仍必须符合这一产品的所有认可标准。这表示试验数 少于整个试验计划,因而在执行前需经论证并报有关当局批准。这一概念可应用于固体口服 制剂残留溶剂和微生物试验等。申报时可能只有有限的数据(见 2.5)。因此,本概念通常批 准后才能实施。定期试验结果如不符合认可标准,应通报有关管理机构,如果这些试验结果 证明需恢复常规试验,则需逐批进行出厂试验。
指导原则中末提及的一些其他方法也可以使用和接受。申报者应论证其替代的方法。论 证应以新原料药和制剂工艺中的数据为基础,论证可以考虑给定方法或认可标准的理论偏 差,但所得的实际结果决定了使用哪种方法。
在制订和论证规范时,应考虑稳定性和生产规模或论证批次的试验结果,特别是初步稳 定性试验批次的结果。如果计划有多个生产工厂,在建立最初的检验项目和认可标准应考虑 这些工厂的数据。如果根据一个有代表性的场所的数据来制定检验项目和认可标准,那么各 个其他生产场所生产出来的产品应符合该标准。
规范由一系列的试验、有关分析方法和认可标准组成,这些标准以限度值、范围或其他 描述来表示。它建立了一套新原料药和制剂都必须遵循的、与其用途相适应的标准。“符合 规范”是指原料药和制剂按照给定的方法试验,符合认可标准规范是重要的质量标准,它由 生产商提出和验证、管理机构批准,并作为批准产品的依据。
除了出厂试验外,规范中可能还列出了生产过程中的试验(见 2.3)、定期试验和其他不 必每批必检的试验。在这种情况下,申报者应弄清哪些试验是每批必检的,哪些试验不必每 批必检,并对实际的实验次数进行论证和说明。在没有进行每批必检的情况下,原料药或制 剂检验结果应符合认可标准的规定。
原料药及制剂的质量标准制定(2011年)

制剂 Description
• Description: 制剂的定性描述 A qualitative description of the dosage (e.g., size, shape, shape and color) color).
– 例子:Light green oval tablet printed using black ink with “JJ” on one side and 100 on the JJ other side, no obvious physical defects.
杂质顺序 Impurity Listing
• 指定结构确定杂质 Each specified identified degradation product • 指定结构不确定杂质 Each spec ed u de ac specified unidentified ed degradation product y p • 任何单一非指定杂质 Any unspecified degradation product with an acceptance criterion of not more than (≤) the figure in the identification h h ld in Attachment 1 Q3B(R) id ifi i threshold i A h 1, • 总杂质 Total degradation products
ICH 限度 Threshold
Qualification and Specification
• Qualification level ≠ acceptance criterion in specification • Production capability and stability data are also factors
原料药和制剂测试方法及标准

原料药和制剂测试方法及标准规范:新原料药和新药制剂的测试方法和认可标准:化学物质1.介绍1.1 指导原则的目的本指导原则旨在尽可能建立起一套全球性的新原料药和新制剂的规范,它提供了化学合成的新原料药及其制剂认可的标准的制定、论证和试验方法的选择,这些新药尚未在美国、欧盟、日本注册。
1.2背景规范由一系列的试验、有关分析方法和认可标准组成,这些标准以限度值、范围或其他描述来表示。
它建立了一套新原料药和制剂都必须遵循的、与其用途相适应的标准。
“符合规范”是指原料药和制剂按照给定的方法试验,符合认可标准。
规范是重要的质量标准,它由生产商提出和验证,管理机构批准,并作为批准产品的依据。
规范是确保原料药与制剂质量和一致性的总方案的一部分。
方案的其他部分包括制订规范所依据的开发期间的产品全部性质、遵循 GMP,如合适的设施、已论证的生产工艺、已论证的试验方法、原材料的检验、生产过程中的检验、稳定性试验等。
规范用于确定原料药和制剂的质量而不是确定全部性质,应着重考虑能确保药物安全、有效的性质。
1.3指导原则的范围新原料药和制剂的质量是由设计、开发、生产过程的控制、GMP 控制和生产工艺的论证,以及贯穿于开发和生产始终的规范的应用所决定的。
本指导原则阐述规范,即用于确保新原料药及制剂在投放市场和货架寿命期间质量的检验项目、方法及认可标准。
规范是质量保证的重要内容,但不是唯一的内容,上述各项对确保生产高质量的原料药及制剂是必不可少的。
本指导原则只涉及批准上市的新药及制剂(包括复方制剂),不涵盖临床研究阶段的药物。
本指导原则可适用于合成与半合成抗生素和低分子合成肽;然而它尚不适合应用于高分子肽、多肽、生物/生物技术制品。
ICH 指导规范:“生物/生物技术制品的试验方法和认可标准”阐述了对生物/生物技术制品的规范、试验项目和方法。
ICH 不包括放射药物、发酵制品、寡聚核苷酸、草药和来源于动植物的粗制品。
指导原则提供了新原料药和制剂认可标准,即常规的认可标准,也提供了对某些原料药和(或)剂型的专门标准,但它不包罗万象。
中药制剂的生产工艺与质量标准

中药制剂的生产工艺与质量标准中药制剂是指通过加工、提取、配制中药材或中药复方药物,制成符合药品管理法规要求的剂型和规格的药品。
中药制剂作为一种特殊药物形式,其生产过程需要严格遵循工艺要求和质量标准,以确保药品的安全、有效和稳定。
一、中药制剂的生产工艺中药制剂的生产工艺包括原料药提取、制剂配制和包装等环节,每个环节都需要细致精确的操作,以确保药品的质量和稳定性。
1. 原料药提取原料药提取是中药制剂生产的第一步,通过适当的溶剂提取中药材中的有效成分,得到包含中药有效成分的浸膏或浸膏干燥粉。
提取工艺中需要控制温度、时间和溶剂比例等参数,以确保提取效果和成分稳定。
2. 制剂配制制剂配制是将提取得到的中药浸膏与其他辅料按一定比例混合,制成符合规定剂型与规格的制剂。
制剂配制需要确保配方准确、操作规范,避免添加过量或不足的辅料,以保证药品的质量和安全性。
3. 包装中药制剂的包装需要符合相关药品管理规定,采用适当的包装材料和包装工艺,确保药品在运输和储存过程中不受外界环境的影响。
包装过程中需要注意密封性、防潮防光等要求,以保持药品的稳定性和使用寿命。
二、中药制剂的质量标准中药制剂的质量标准是制定和评价药品质量的重要依据,主要包括药材质量、制剂质量和药品规格。
1. 药材质量中药制剂的药材应符合中药材质量标准,包括外观、性状、含量测定和微生物限度等指标。
药材质量的好坏直接影响制剂的质量,因此在采购和使用药材时,应选择符合质量标准的优质药材。
2. 制剂质量中药制剂的质量标准主要包括外观、含量测定、溶出度、微生物限度、纯度和稳定性等指标。
这些指标通过合适的检测方法来评定,制剂质量的好坏直接影响药品的疗效和安全性。
3. 药品规格中药制剂的规格是指药品的剂型、规格和使用方法。
药品规格需要符合国家药品管理规定,包括药品命名、剂型定义和适应症等内容。
药品规格的准确性和明确性对医生、患者和药师具有指导意义,避免误用和滥用药品。
三、中药制剂质量控制的重要性中药制剂质量控制是确保药品安全、有效和合规的重要环节,对于维护患者用药权益和保障用药安全具有重要意义。
中药质量标准

②测定方法选择:目前常用的有: • 容量法 • 重量法 • 分光光度法 • 薄层层析-分光光度法 • 薄层扫描法 • 气相色谱法 • 高效液相色谱法 • 高效毛细管电泳
③方法学考察:
• 提取、分离、纯化条件的选定:
• 测定条件的选定:检测波长、色谱条件等
• 线性关系: • 稳定性试验: • 精密度试验: • 重现性试验: • 回收率试验: • 代表性样品的测定:临床三批、生产10批
(3)制法:按资料四的制备工艺进行简 要描述。 (4)性状:制剂的性状指成品的颜色、 形状、形态、气味等。片剂、丸剂如有包 衣的还应描述除去包衣后片芯的颜色及气 味,硬胶囊剂应写明除去胶囊后内容物的 性状。
(5)鉴别: ①鉴别对象的选择: 成药一般药味多,不可能逐一鉴别,应选择君药或 臣药进行鉴别,对药味多,如20~30味,可分别选出君、 臣、佐、使各类代表之一,设计鉴别试验。毒剧药必须 进行鉴别和限量检查。贵重药:必须进行鉴别。 ②鉴别方法: 显微鉴别:以原粉入药的药味可采用显微鉴别。 一般理化鉴别:包括呈色反应、沉淀反应、荧光反 应等。 色谱鉴别:主要包括薄层色谱、气相色谱、液相色 谱等方法,以薄层色谱鉴别使用较多。注意:要求有阴 性和阳性对照(标准品或标准药材)。 光谱法:包括紫外、红外、核磁共振、质谱等光谱 方法。
接与药品接触的包装材料对药品稳定性的影响。
• 于常温下进行考察,一般可在初步稳定性考察后
继续考察,即在初步稳定性考察三个月后,放置
三个月再考察一次,然后每半年一次。
• 样品考察批数与报送的稳定性试验报告 要求均与初步稳定性试验要求相同。 • 药品的稳定性试验在到达规定的考核时 间后还可在正常室温下继续考核,每半 年一次,不超过5年,为该药品在审定正 式使用期限提出依据。
原料药质量标准

药品质量研究与质量标准的制定,是新药研究的主要内容之一,研究开发新药,必须对产品质量进行详细研究,并制订合理的质量标准,以保证药品安全有效。
一、原料药质量研究原料药在确证化学结构或组份的基础上,应对该药品进行质量研究,并参照现行版《国家药品标准工作手册》制订质量标准,一些中国药典附录已有详细规定的常规测定方法,对方法本身可不作验证,但用于申报原料药测定的特殊注意事项应明确标明。
1.性状应分别记述药品的外观、嗅、味和一般稳定情况、溶解度以及有关物理常数。
1.1外观、色泽、形状、嗅、味。
在贮存期内发生的变化应予以研究记述,如遇光变色、易吸湿、风化等。
1.2溶解度溶解度是药品的一种物理性质,在一定程度上反映药品的纯度。
表示溶解度的术语应按照药典规定分极易溶解、易溶解、溶解、略溶、微溶、极微溶解、几乎不溶或不溶。
试验法可参照中国药典2000年版二部附录。
一般用与该药品溶解特性密切相关、配制制剂、制备溶液或精制操作所需用的常用溶剂作试验,不必罗列过多。
1.3熔点已知结构的化学原料药,熔点是重要的物理常数之一,利用熔点或熔矩数据,可以鉴别和检查该药品的纯杂程度。
测定原料药的熔点常用中国药典附录第一法。
适于测定熔点的药品应是在熔点以下遇热时晶型不转化,其初熔与全熔易于判断的品种。
应详细记录初熔与全熔时的温度,并应在规定范围内。
化学药品的熔点范围一般为3℃-4℃,熔矩一般不超过2℃,不宜过宽。
对熔融时同时分解的药品,要记录熔融时的现象,如变色,产生气泡等。
通常当供试品开始局部液化,毛细管中出现液滴或开始产生气泡时的温度作为初熔温度,至供试品固相消失全部液化时作为全熔温度。
有时固相消失不明显,则以供试品分解并开始膨胀时的温度作为全熔温度。
对某些药品无法分辨初熔与全熔现象时,可以记录其发生突变(如气泡很快上升,颜色明显变深)时的温度,作为熔融分解温度。
对熔点难以判断或熔融时同时分解的品种以及一、二类新药的熔点应用DSC热分析法予以说明。
药物制剂的质量控制与质量评价

药物制剂的质量控制与质量评价药物制剂是指将药物原料通过一系列工艺加工、配制而形成的可供使用的药品。
为了确保药物制剂的质量、安全和疗效,质量控制和质量评价是不可或缺的环节。
本文将从药物制剂的质量控制和质量评价两个方面进行探讨。
一、药物制剂的质量控制1. 原料药的质量控制原料药是药物制剂的基础,其质量对制剂的质量至关重要。
原料药的质量控制包括外观、理化性质、纯度、溶解度等指标的检测。
合格的原料药能够确保制剂的稳定性和一致性。
2. 辅料的质量控制辅料在药物制剂中起着辅助作用,如填充剂、分散剂等。
辅料的质量控制包括外观、纯度、杂质和溶解度等方面的检测。
合格的辅料能够保证制剂的稳定性和安全性。
3. 制剂工艺的质量控制制剂工艺是指药物原料加工和配制的过程,对制剂的质量具有关键影响。
制剂工艺的质量控制包括工艺参数、操作规程、设备清洁等方面的管理和监控。
合理的工艺能够保证制剂的一致性和稳定性。
4. 成品药的质量控制成品药是指最终制得的药物制剂,其质量控制是保证药物安全和有效性的重要环节。
成品药的质量控制包括外观、规格、标签、包装等方面的检测。
合格的成品药能够确保病患使用的药物质量可靠。
二、药物制剂的质量评价1. 质量评价指标药物制剂的质量评价旨在确定制剂是否符合相关标准和规范要求。
其中包括外观、理化性质、溶解度、释放度、稳定性和生物等效性等指标的评价。
这些指标能够客观地反映制剂的质量情况。
2. 质量评价方法质量评价方法包括定性分析和定量分析两种。
定性分析是通过观察和比较,判断制剂质量是否符合要求。
定量分析则是使用一系列分析方法,对制剂中的活性成分或特定指标进行测定。
常用的评价方法包括色谱法、光谱法、电导法等。
3. 质量评价标准药物制剂的质量评价标准是根据相关法规和规范制定的,能够判断制剂是否符合质量要求。
常见的评价标准包括中国药典、美国药典和欧洲药典等。
这些标准对制剂的质量要求进行了明确规定。
三、质量控制与质量评价的重要性质量控制和质量评价是确保药物制剂质量的重要保障措施。
原料药质量标准
药品质量研究与质量标准的制定,是新药研究的主要内容之一,研究开发新药,必须对产品质量进行详细研究,并制订合理的质量标准,以保证药品安全有效。
一、原料药质量研究原料药在确证化学结构或组份的基础上,应对该药品进行质量研究,并参照现行版《国家药品标准工作手册》制订质量标准,一些中国药典附录已有详细规定的常规测定方法,对方法本身可不作验证,但用于申报原料药测定的特殊注意事项应明确标明。
1.性状应分别记述药品的外观、嗅、味和一般稳定情况、溶解度以及有关物理常数。
1.1外观、色泽、形状、嗅、味。
在贮存期内发生的变化应予以研究记述,如遇光变色、易吸湿、风化等。
1.2溶解度溶解度是药品的一种物理性质,在一定程度上反映药品的纯度。
表示溶解度的术语应按照药典规定分极易溶解、易溶解、溶解、略溶、微溶、极微溶解、几乎不溶或不溶。
试验法可参照中国药典2000年版二部附录。
一般用与该药品溶解特性密切相关、配制制剂、制备溶液或精制操作所需用的常用溶剂作试验,不必罗列过多。
1.3熔点已知结构的化学原料药,熔点是重要的物理常数之一,利用熔点或熔矩数据,可以鉴别和检查该药品的纯杂程度。
测定原料药的熔点常用中国药典附录第一法。
适于测定熔点的药品应是在熔点以下遇热时晶型不转化,其初熔与全熔易于判断的品种。
应详细记录初熔与全熔时的温度,并应在规定范围内。
化学药品的熔点范围一般为3℃-4℃,熔矩一般不超过2℃,不宜过宽。
对熔融时同时分解的药品,要记录熔融时的现象,如变色,产生气泡等。
通常当供试品开始局部液化,毛细管中出现液滴或开始产生气泡时的温度作为初熔温度,至供试品固相消失全部液化时作为全熔温度。
有时固相消失不明显,则以供试品分解并开始膨胀时的温度作为全熔温度。
对某些药品无法分辨初熔与全熔现象时,可以记录其发生突变(如气泡很快上升,颜色明显变深)时的温度,作为熔融分解温度。
对熔点难以判断或熔融时同时分解的品种以及一、二类新药的熔点应用DSC热分析法予以说明。
药品生产质量控制标准

药品生产质量控制标准引言:药品是人类医疗保健的重要组成部分,药品质量直接关系到患者的生命健康。
为了确保药品的质量安全和有效性,各国纷纷制定了药品生产质量控制标准。
本文将从药品质量控制的角度出发,分别从原料药的生产、药品制剂的生产、药品包装的规范以及药品贮存和运输的标准等方面,对药品生产质量控制标准进行探讨。
一、原料药的生产原料药是药品制剂的核心组成部分,对药品的质量直接影响重大。
为了确保原料药的质量,药品生产必须按照以下标准进行控制:1. 优质原料药的选择:选择高纯度、高活性、无杂质的原料药,保证其符合药典规定的质量要求。
2. 合理的生产工艺:建立稳定可靠的原料药生产工艺,确保原料药的纯度和活性。
3. 严格的质量控制参数:制定原料药的质量控制参数,包括理化性质、纯度、含量等,确保各项参数符合要求。
4. 严格的质量检验:建立健全的原料药质量检验制度,对进货的原料药进行全面检验,确保原料药的质量符合标准要求。
二、药品制剂的生产药品制剂是指将原料药按照一定比例和工艺合成为制剂的过程。
为确保药品制剂的质量,药品制剂的生产必须按照以下标准进行控制:1. 质量稳定性研究:对药品制剂进行质量稳定性研究,确定其保存期限和适宜的贮存条件。
2. 生产工艺标准化:制定科学合理的生产工艺标准,确保生产过程可控。
3. 质量控制检查点:设定药品制剂生产中的关键控制点,并建立相应的质量控制措施,确保每个生产环节符合质量要求。
4. 药品制剂质量控制:制定药品制剂的质量控制参数,包括外观、理化性质、有效成分含量等。
三、药品包装的规范药品包装是保护药品质量的重要环节,合理的药品包装可以防止药品在贮存和运输过程中受到光、湿度、氧化等不良环境的影响。
为保证药品包装的质量,需要制定以下规范:1. 包装材料选择:选择符合药品包装要求的包装材料,如塑料瓶、铝塑泡腾板等,确保其符合相关标准。
2. 包装工艺标准化:制定合理的药品包装工艺标准,确保每个环节的包装操作规范、一致。
原料药及制剂成品质量标准的制定

Met-R003P
NO
NMT 350 µm
Polymorphism
XR-PDP: Form II by absence of a peak at 20.6°C 2 theta for Form I
Met-R003J
NO / YES
制剂成品的质量标准 Specification
性状
• 外观形态、色泽、片面有无印字或刻痕或有商标记号等的描述
溶出限度一般与原研对照药一致并用已通过的BE批溶出度行为来确证
如与原研对照药有显著差异,但体内数据可接受的情况下,允许设置 不同的限度
一旦溶出限度已设定,产品在有效期内必须符合此限度
制剂成品的质量标准 :溶出度或释放度
速释药的常规质控 • 单点溶出限度(对于高溶速释药,i.e., BCS class I/III drug: 85% dissolution in 0.1N HCl in 15 min) • 双点溶出限度或溶出曲线(对于某些慢释或低溶药)
• 手性含量测定方法 chiral assay method • 或 手性杂质检测方法 enantiomeric impurity method
含量测定
• 手性含量测定方法 chiral assay method • 或 非手性含量测定方法结合手性杂质检测方法
(总含量 – 对映体杂质含量 = 手性含量)
• Meet USP<232> for US (2015, 12) • USP<233> method or in-house method
制剂成品的质量标准 :溶出度或释放度
对于口服固体制剂以及混悬剂
生物等效性试验 Bioequivalence (BE): 指用生物利用度研究的方 法,以药代动力学参数为指标,比较同一种药物的相同或者不同 剂型的制剂,在相同的试验条件下,其活性成份吸收程度和速度 有无统计学差异的人体试验。
药品质量评价标准

药品质量评价标准随着药品的广泛应用和不断增长的需求,药品质量评价标准对于保障患者用药安全和推动药品行业的健康发展至关重要。
本文将从药品质量评价的背景和重要性出发,分别对药品质量评价的各个环节进行详细阐述,包括原料药评价、制剂评价、包装材料评价以及药品现场质量评价。
一、原料药评价原料药是药品的核心组成部分,对其质量进行评价非常重要。
首先,原料药的外观和理化性质需要满足相应的标准,确保其清晰、无异物、溶解性好等特性。
其次,药品的化学成分必须符合规定的纯度标准,而且应当进行相关性能和稳定性测试,以保证其在药品制备过程中的稳定性。
二、制剂评价制剂是指将原料药和辅料按照一定比例混合加工制成的药品。
制剂评价主要针对制剂的安全性、有效性和稳定性进行,其中安全性包括毒理学评价以及接触安全性的评估;有效性则需要通过药效学评价和药代动力学评价来确定;稳定性评价则主要关注药品在不同存储条件下是否能够保持稳定,不发生分解等情况。
三、包装材料评价药品的包装材料的质量直接影响到药品的保质期和使用安全性。
在包装材料评价中,包装材料的选择应当符合药典中制定的要求,例如瓶盖应当符合密封性能要求,包装袋应当耐热、耐潮湿等。
同时,在使用过程中需要对包装材料进行不同环境条件下的透气性、防漏性等性能的评价。
四、药品现场质量评价药品现场质量评价主要关注药品制备过程中各个环节的合规性和规范性。
包括与生产车间环境相关的评价,如车间温度、湿度、洁净度等要求;与人员操作相关的评价,如生产工艺、设备操作要求等;以及与质量管理相关的评价,如记录的规范和完整性要求等。
综上所述,药品质量评价标准在保障患者用药安全和推动药品行业的健康发展中起着至关重要的作用。
而药品质量评价的过程包括原料药评价、制剂评价、包装材料评价以及药品现场质量评价等环节,每个环节都需要按照相应的标准进行评价。
只有通过严格的质量评价体系,我们才能确保药品的质量安全,为患者提供高效、安全、可靠的治疗药物。
原料药与制剂认证的差别

一、首先看看原料药的生产工艺与制剂生产存在以下差别:(1).原料药的生产通常为化学过程,而制剂的生产通常为物理过程; (2).原料药通常涉及纯化过程,并以纯度为标准,微量的杂质可能会引起严重的毒性反应,因而强调工艺中对杂质的控制,而制剂的生产则通常为混合和分散的过程,原料本身通常不会产生新的杂质,因而强调混合或分散的均一性和对污染的控制;(3).原料药生产的起始物质是化工产品,通常没有药用标准,而制剂生产所用的原料药、赋形剂和溶剂都具有药用标准;(4).原料药的质量未达到标准时,可以通过返工和再加工工艺补救,而制剂通常不能;(5).原料药生产中的溶剂可以回收再利用,而制剂生产通常不会有也不允许有这样的工艺。
再说二者GMP认证的不同以上差别使得以药品剂型生产为参照起草的GMP无法适应原料药的生产,因此国外有专门针对原料药的GMP,如美国FDA在1996年9月公布了新的原料药GMP指南讨论稿,1998年3月开始实施,药品检查条约(PIC)于1987年6月发布了原料药GMP指南,ICH制定了原料药GMP指南,作为质量部分的一个主要的专题(ICH Q7A,以下简称Q7A ), 2001年7月,欧盟将其作为GMP的附录18正式发布,目前得到国际社会普遍的执行。
综合世界卫生组织药品GMP和ICH药品GMP中有关原料药的规定,原料药GMP认证起始步骤划分表达为:“原料药生产应从对原料药质量有关键性影响的步骤开始按照GMP生产。
原则上说,化学合成的原料药的生产质量控制应从形成原料药的主要结构单元的工艺过程开始,其它工艺方法(如发酵、提取、纯化等)制备的原料药,起始步骤应根据每个原料药的具体特性制定,并获得国家食品药品监督管理部门的批准。
企业实际执行的生产规程至少从“粗品”前一步反应开始按药品GMP要求生产。
(关于原料药起始步骤划分问题(药品认证交流第一期))前面已经有战友说了原料药按品种进行认证,制剂按剂型认证.至于楼主提出的为什么很多公司拿到原料药的批件却不生产的问题,我认为可能的原因有:1.按申报的原料药工艺根本就无法生产,如合成路线根本就不通;2.按申报的原料药工艺生产成本很高,比直接买其他已经有批准文号厂家的原料药成本还要高,或者就直接买没有批准文号厂家的原料药(现在的一些精细化工生产厂家合成原料药或中间体的很多)。
比较原料药及其制剂的质量标准
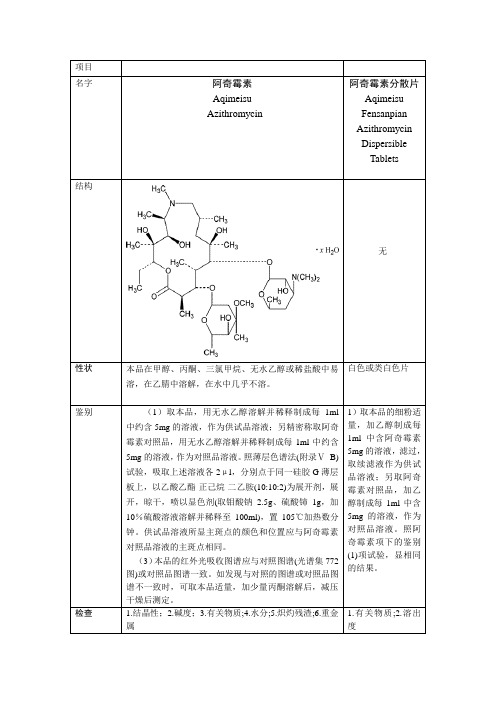
名字
阿奇霉素
Aqimeisu
Azithromycin
阿奇霉素分散片
Aqimeisu Fensanpian
Azithromycin Dispersible
Tablets
结构
无
性状
本品在甲醇、丙酮、三氯甲烷、无水乙醇பைடு நூலகம்稀盐酸中易溶,在乙腈中溶解,在水中几乎不溶。
白色或类白色片
鉴别
(1)取本品,用无水乙醇溶解并稀释制成每1ml中约含5mg的溶液,作为供试品溶液;另精密称取阿奇霉素对照品,用无水乙醇溶解并稀释制成每1ml中约含5mg的溶液,作为对照品溶液。照薄层色谱法(附录ⅤB)试验,吸取上述溶液各2μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-正己烷-二乙胺(10:10:2)为展开剂,展开,晾干,喷以显色剂(取钼酸钠2.5g、硫酸铈1g,加10%硫酸溶液溶解并稀释至100ml),置105℃加热数分钟。供试品溶液所显主斑点的颜色和位置应与阿奇霉素对照品溶液的主斑点相同。
(3)本品的红外光吸收图谱应与对照图谱(光谱集772图)或对照品图谱一致。如发现与对照的图谱或对照品图谱不一致时,可取本品适量,加少量丙酮溶解后,减压干燥后测定。
1)取本品的细粉适量,加乙醇制成每1ml中含阿奇霉素5mg的溶液,滤过,取续滤液作为供试品溶液;另取阿奇霉素对照品,加乙醇制成每1ml中含5mg的溶液,作为对照品溶液。照阿奇霉素项下的鉴别(1)项试验,显相同的结果。
检查
1.结晶性;2.碱度;3.有关物质;4.水分;5.炽灼残渣;6.重金属
1.有关物质;2.溶出度
含量鉴定
照高效液相色谱法(附录V D)测定
照高效液相色谱法(附录V D)测定
类型
大环内酯类抗生素
原料药和制剂的质量标准制定的基本流程

原料药和制剂的质量标准制定的基本流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor.I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!原料药与制剂质量标准制定的基本流程解析在医药行业中,原料药和制剂的质量直接关系到药品的安全性和有效性。
原料药和制剂质量标准

原料药质量研究的主要内容一、性状一外观聚集状态、色泽、臭、味、结晶性是对药物感官的一般性描述;一般无法定方法检查;但仍应准确描述..二溶解性1. 溶剂的选择应根据药物的结构和性质;考查药物在水与常用溶剂中的溶解度..应选择与药物溶解特性密切相关的溶剂;配制制剂、制备溶液和精制操作有关的溶剂;避免使用昂贵的、毒性大的、不常使用的溶剂..2. 表述方式用“极易溶解”、“易溶”、“溶解”等术语来表示见中国药典凡例..3. 试验方法按中国药典凡例的规定操作..25±2℃;每隔5分钟强力振摇30秒钟;观察30分钟内的溶解情况..三理化常数理化常数包括:熔点; 馏程; 凝点; 比旋度; 折光率; 黏度; 相对密度; 酸值、碘值、羟值、皂化值;吸收系数等..测定物理常数可以鉴别药物;也可以反映药物的纯杂程度..注意事项:应按药典附录规定的方法进行测定..测定前应按规定对仪器进行校正..测定理化常数时应用精制品进行测定;质量标准中规定的理化常数范围;用试制的药物来测定..固体药物:熔点、吸收系数..液体药物:馏程、相对密度、黏度、折光率..油脂类还需测定:酸值、碘值、羟值、皂化值..有手性中心的药物:比旋度..1、熔点照中国药典”熔点测定法”测定..1中国药典使用毛细管法测定熔点;以供试品局部液化的温度作为初熔的温度;以全部液化的温度作为全熔的温度..2所用的传温液、毛细管、升温速度等对测定结果有影响;应按药典要求进行测定..3应使用分浸型的温度计;温度计应用熔点标准品进行校正..4应用D S C予以佐证..2、吸收系数1测定方法配制高、低两种不同浓度的溶液吸光度在0.3~0.4和0.6~0.8之间各2份;或配制吸收度在0.1~0.8的系列浓度的溶液;在5台不同型号的紫外分光光度计上测定;按下式计算吸收系数..要求:应使用对照品进行测定..测定前应对容量仪器进行校正..测定前应对紫外分光光度计进行校正..应按干燥品计算..以平均值作为吸收系数;RSD应小于1.5%..3、晶型1 方法 X射线粉末衍射法中国药典附录Ⅸ F..X射线衍射法:用一束准直的单色X射线照射单晶或粉末晶体时;便发生衍射现象;发生衍射的条件符合布拉格方程:X射线衍射图谱的横坐标一般为2θ;纵坐标为衍射强度..X射线粉末衍射法可以用来鉴别药物;测定含量;也可以用来对晶型进行考察..要求:固体药物除水溶性好的外;均应作..若不同晶型药物的生物利用度不同;应将晶型的检查列入质量标准..二、鉴别鉴别试验:是使用化学的、物理化学的或生物学的方法;来辨别药物的真伪..是对取自有标签容器中样品的确认..要求:专属性强、重现性好、灵敏、简便..通常采用 2 种以上不同类型的方法进行研究;从不同角度验证目标化合物..常用的方法有:1、制备衍生物测定熔点本法较繁琐、费时;但专属性强;必要时可使用..1成酯的反应例炔雌醇和苯甲酰氯反应成酯;炔雌醇苯甲酸酯的熔点为201℃..2成腙的反应例异烟肼和香草醛反应生成异烟腙;熔点为226℃..3酯的水解例丙酸睾酮水解为睾酮和丙酸;睾酮的熔点为150~156℃..4与生物碱沉淀剂的反应2.呈色反应或沉淀反应要求:反应现象明显;专属性强..一般鉴别试验:为一类药物共有的反应;收载在中国药典附录Ⅲ“一般鉴别试验”项下..例水杨酸盐中国药典附录第20页1取供试品的稀溶液;加三氯化铁试液1滴;即显紫色..2取供试品;加稀盐酸;即显水杨酸的白色沉淀;分离;沉淀在醋酸铵溶液中溶解..又如:“丙二酰脲类”、“有机氟化物”、“钠盐”、“钾盐”的鉴别反应等..2个别药物的鉴别试验例维生素C的鉴别取本品0.2g;加水10ml溶解后;照下述方法试验..取溶液5ml;加硝酸银试液0.5ml;即生成银的黑色沉淀..取溶液5ml;加二氯靛酚钠试液1~2滴;试液的颜色即消失..3.紫外特征吸收方法:1核对光谱的数据a.核对吸收光谱峰、谷的位置..b.核对吸收光谱峰、谷处的吸收度、吸收系数或吸收度比值.. C.核对经反应处理后反应产物的吸收特征..2核对紫外吸收光谱例1 叶酸的鉴别p.137取本品;加0.4%的氢氧化钠制成每1ml中含10μg的溶液;照分光光度法测定;在256、283±2和365±4nm的波长处有最大吸收;在256与365nm波长处的吸收度比值应为2.8~3.0..例2 苯妥英钠的鉴别p.440取本品约0.3g;加高锰酸钾10mg;氢氧化钠0.25g与水10ml;小火加热5分钟;放冷;取上清液5ml;加正庚烷20ml振摇提取;静置分层后;取正庚烷提取液;照分光光度法测定;在248nm的波长处有最大吸收..4.红外分光光度法中国药典附录Ⅳ C主要用于原料药的鉴别;特别适合于同类药物用其它理化方法不易区别者..方法:按要求录制红外吸收光谱;与药品红外光谱集中的标准图谱比较;应一致..注意事项:1测定前应对仪器进行校正用聚苯乙烯薄膜对仪器的波数和分辨率进行校正..2确定制样的方法..3如果是多晶型的样品;应收载有效的晶型;并应提出晶型转化的条件..4对图谱中的主要吸收峰进行解释..5.色谱法主要用于制剂的鉴别;对原料药仅在没有其它更好的方法时; 或检查和含量测定中已使用了色谱法时使用..方法:取供试品和对照品;在相同的色谱条件下进行色谱分离;其R f 或t R 应一致..例1 盐酸苯海索片的鉴别p.726取本品的细粉适量;加氯仿制成每1ml中含盐酸苯海索2mg的溶液;滤过;作为供试品溶液;另取盐酸苯海索对照品;加氯仿制成每1ml中含盐酸苯海索2mg的溶液;作为对照品溶液;照薄层色谱法试验;吸取上述溶液各10μl;分别点于同一硅胶G薄层板上;以氯仿-甲醇9∶1为展开剂;展开后晾干;喷以稀碘化铋钾试液使显色..供试品溶液主斑点的位置应与对照品溶液主斑点相同..例2 盐酸林可霉素的鉴别p.727取本品与盐酸林可霉素对照品;分别加流动相制成每1ml中含2mg的溶液;照含量测定项下的色谱条件进行试验;供试品与对照品主峰的保留时间应一致..6、生物化学或生物学方法四. 检查检查项下包括有效性、均一性、纯度要求和安全性四个方面..一有效性指与疗效有关;但在鉴别、纯度检查和含量测定中不能控制的特殊性质的检查..如:结晶性利福平;制酸力氢氧化铝;吸着力药用炭;凝冻浓度明胶;乙炔基炔诺孕酮;含氟量诺氟沙星例氢氧化铝制酸力的检查p.553取本品约0.12g;精密称定;置250ml具塞锥形瓶中;精密加盐酸滴定液0.1mol/L50ml;密塞;在37℃不断振摇1小时;放冷后;加溴酚蓝指示液6~8滴;用氢氧化钠滴定液0.1mol/L滴定;每1g消耗盐酸滴定液0.1mol/L不得少于230ml..二纯度的检查中国药典2005年版附录新增了“药品杂质分析指导原则”..指导原则是参照ICH人用药品注册技术要求国际协调会的文本Q3A和Q3B制订的..适用于化学合成的或半合成的有机原料药及其制剂..1、杂质的分类及其在药品质量标准中项目的名称1 药物中的杂质杂质:影响药品纯度的物质均称为杂质..杂质的来源:生产过程引入:按规定工艺、规定原辅料生产;引入成品的合成的起始物、中间体、副产物、试剂等;贮存过程引入:经稳定性试验确认在贮存过程中产生的降解产物..药品质量标准中的杂质不包括变更工艺或原辅料时产生的新的杂质;也不包括掺入或污染的外来物质..2 杂质的分类:按化学类别和性质分类:有机杂质、无机杂质、有机挥发性杂质..有机杂质:合成的起始物、中间体、副产物、降解产物、试剂、催化剂、配位体等..无机杂质:试剂、催化剂、配位体、重金属或其他残留金属、无机盐、其他物质..有机挥发性杂质:残留的有机溶剂..按毒性分类:毒性杂质和普通杂质..3. 杂质项目的名称:明确为某一物质时;以该物质的化学名称作为项目名称..如磷酸可待因中的“吗啡”;阿司匹林中的“游离水杨酸”等..仅知为某一类物质时;用该类物质的名称..如“其他甾体”、“还原糖”、“有关物质”等..对未知杂质;可根据检测方法命名..如“杂质吸收度”、“易炭化物”、“不挥发物”等..4、质量标准中杂质检查项目的确定根据生产和贮存过程可能引入的杂质来确定;包括存在的杂质和潜在的杂质..考察产品中是否引入了合成的起始物、中间体、副产物、试剂..通过影响因素试验、加速试验和长期试验来确定降解产物..对表观含量>0.1%或虽<0.1%;但具有强烈生物作用或毒性的杂质应确定结构..对可能存在的无机杂质进行考察并制定检查项目..有机挥发性杂质:应根据工艺所使用的有机溶剂及残留情况;确定检查的项目..除降解产物和毒性杂质外;在原料中已控制的杂质在制剂中一般不再控制..共存的异构体和抗生素多组分一般情况不作为杂质检查项目;作为共存物质..5、要求1在保证用药安全、有效的前提下;允许有一定量的杂质存在..2一般为限量检查..3检查项目的确定既要保证药物的安全性、有效性;又要根据药物在生产和贮存中可能产生的杂质来确定..4应重点检查影响药物疗效、危害人体健康或能真实反应药品内在质量的项目..5限量的确定;既要根据生产的实际水平;又要根据药典的惯例..杂质检查方法1. 酸碱度分为酸度、碱度或酸碱度..方法:酸碱滴定法、pH值测定法、指示剂法..2. 溶液的澄清度与颜色中国药典附录Ⅸ A; B1溶液的澄清度中国药典附录Ⅸ B 主要用于注射用原料药的检查..方法:与溶剂或浊度标准液比较..“澄清”:供试品溶液的澄清度应相同于所用的溶剂或不超过0.5号浊度标准液..2溶液的颜色中国药典附录Ⅸ A 检查样品中的有色杂质..检查方法:第一法:与标准比色液比较第二法:测定吸收度第三法:色差计法3.无机杂质主要有氯化物Cl-、硫酸盐SO42-、铁盐Fe3+、重金属Pb2+、砷盐AsO43-; AsO33-、氰化物CN-等..方法:收载于中国药典附录Ⅷ..如“氯化物检查法”、“砷盐检查法”等..供试品与对照品在相同条件下比色、比浊;以确定杂质的量是否超过限量..注意:应完全按照药典附录的方法进行检查..注意遵循平行原则..研究时;可取系列浓度的杂质对照品溶液与供试品溶液进行比较;以确定杂质含量的范围..应考察样品本身对检查是否有影响..4.有机杂质有机杂质是指残存在产品中的原料、中间体、副产物和降解产物等..杂质能够确定的;检查的项目一般以杂质的名称命名;也可以杂质的类别命名;不能确定的原料、中间体、副产物一般称为“有关物质”..检查方法:1色谱法纸色谱法主要用于极性较大的杂质的检查..如地高辛中洋地黄毒苷的检查中国药典2000年版..薄层色谱法薄层色谱法分离效率高、简便、不需要特殊的设备;有多种检测的方法;是有关物质检查中应用最多的方法..常用的方法:杂质对照品法;供试品自身稀释对照法..HPLC;GC法中国药典附录Ⅴ D; E1. 内标法加校正因子测定某个杂质的含量..2. 外标法测定主成分或某个杂质的含量..3. 加校正因子的主成分自身对照法..4. 不加校正因子的主成分自身对照法5. 面积归一化法方法的考察:1. 色谱系统适用性试验..2. 测定杂质含量: 专属性、准确度、精密度、定量限、线性、范围、耐用性..3. 限量检查:专属性、检测限、线性、耐用性..2分光光度法控制特定波长处的吸收度..或控制两波长处吸光度的比值..3化学法容量法、比色或比浊的方法..5.残留溶剂的检查1溶剂的分类 ICH将溶剂分为三类:第一类溶剂:致癌物、危害环境的物质;应避免使用的溶剂..第二类溶剂:具有非基因毒性;不可逆或可逆毒性;应限制使用的溶剂..第三类溶剂:毒性低;对人体危害较小的溶剂..2残留溶剂测定法: 中国药典2010年版附录Ⅷ P色谱柱:载气: N或He ;2检测器:FID; 对于氯代的溶剂也可以使用MS或ECD..系统适用性试验:供试品溶液的制备:测定方法:附注:6. 干燥失重或水分干燥失重测定法中国药典附录Ⅴ L:水分测定法:中国药典附录Ⅴ M:第一法:费休氏法Karl Ficher法;第二法:甲苯法..7. 炽灼残渣中国药典附录Ⅷ N三安全性的检查热原或细菌内毒素;异常毒性;降压物质;无菌..五.含量测定1.重量法用适当的方法将被测组分与样品中的其他组分分离;转化为一定的称量形式;用称量来测定该组分含量的方法..特点:精确;但操作繁琐;费事..必要时采用..例如:苯妥因钠的含量测定中国药典 2005年版顺铂的含量测定中国药典 2005年版2. 容量法容量法是将一种已知、准确浓度的试剂溶液─滴定液滴加到供试品溶液中;至化学反应按剂量关系反应完全;根据滴定反应的化学剂量关系以及消耗滴定液的体积和浓度来计算测定组分含量的方法..特点:精确;简便;一般不需要对照品;是原料药和制剂含量测定的首选方法..常用的方法:中和滴定法;沉淀滴定法;配位滴定法;碘量法;溴酸钾法;溴量法;高锰酸钾法;铈量法;亚硝酸纳法;非水溶液滴定法等..方法的建立:应根据药物的结构和性质选择适当的方法;作滴定曲线;以考察突跃的范围;并确定终点时指示剂颜色的变化..方法的考察:准确性作对照实验、精密度重复性..3.紫外分光光度法中国药典 附录 ⅣA特点:灵敏;简便、较准确..测定方法:1吸收系数法 配制一定浓度的供试品溶液;测定吸收度;按吸收系数计算含量.. g/100mllE A C %cm 11特点:不需要对照品;但测定条件应符合要求..2 对照品比较法配制一定浓度的供试品溶液和对照品溶液;测定吸收度;按下式计算含量..特点:需要对照品;但可以克服测定条件对结果的影响;使提倡的方法..例如:注射用硫喷妥钠的含量测定p367;甲硝唑片的含量测定p124..3 计算分光光度法方法的建立:选择测定波长:应选择最大吸收波长;选择溶剂:易于溶解样品;吸收度值稳定;无毒、价廉.. 确定供试品溶液的制备方法和浓度吸光度应在0.3~0.7之间..方法的考察:线性和范围;专属性;准确度;精密度..4.色谱法 GC 和HPLC1 加校正因子的内标法2 外标法方法的建立1色谱条件的选择根据药物的结构和性质选择色谱条件HPLC 首选C18柱、硅胶柱;检测器首先选择紫外-可见检测器..GC 首先选择通用性的固定相甲基聚硅氧烷SE-30; OV-1、5%苯基甲基聚硅氧烷SE-54、聚乙二醇2万PEG-20M ;检测器首先选择FID..2 色谱系统适用性试验柱效n;分离度R;拖尾因子T;精密度等..3 方法的考察专属性;线性和范围;精密度;准确度;耐用性等..制剂质量研究的主要内容制剂质量研究的内容;应在原料药质量研究的基础上;结合剂型的特点、处方工艺以及临床使用方法来确定..一、性状制剂的外观、色泽;或内容物的色泽..二、鉴别参考原料药的方法..注意辅料的干扰..三、检查一杂质检查重点:有关物质降解产物原料药已经控制的杂质;原则上不再检查..二制剂的检查1.片剂常规检查项目:重量差异、崩解时限..特殊检查项目:溶出度、释放度、含量均匀度1溶出度中国药典附录ⅩC溶出度系指药物从片剂或胶囊剂等固体制剂中溶出的速度和程度..凡检查溶出度的制剂;不再进行崩解时限的检查..重点检查溶解度低;体内吸收不良或治疗剂量和中毒剂量接近的制剂..主要的研究内容:测定方法的选择转篮法;桨法;小杯法;介质的选择;转速的选择;含量测定方法的选择;溶出曲线的测定;取样的时间和限度的确定2释放度中国药典附录ⅩD系指口服药物从缓释制剂、控释制剂、肠溶制剂及透皮贴剂等在规定溶剂中释放的速度和程度..检查释放度的制剂;不再检查崩解时限..第一法用于缓释制剂或控释制剂第二法用于肠溶制剂第三法用于透皮贴剂2含量均匀度中国药典附录Ⅹ E系指小剂量的片剂、膜剂、或注射用无菌粉末单剂含量偏离标示量的程度..重点检查主药含量低;不易混匀的制剂..2.注射剂常规检查项目:装量装量差异;可见异物附录Ⅸ H 可见异物检查法;无菌;不溶性微粒静脉注射用注射液;注射用无菌粉末;注射用浓溶液;细菌内毒素或热原静脉注射用注射液..特殊检查项目:溶剂油的检查;容器玻璃的检查..3. 胶囊剂同片剂..4. 颗粒剂常规检查项目:粒度;干燥失重≤ 2.0%;溶化性;装量差异单剂量包装;装量多剂量包装..四、含量测定结合原料药的方法和剂型的特点来选择..着重考虑方法的专属性和灵敏度..。
原料药与制剂之间究竟有哪些差别?

原料药与制剂之间究竟有哪些差别?展开全文很多养殖场使用原料药,是认为原料药便宜;以为国内大部分兽药厂的制剂产品,都是在原料药中加入辅料做一下简单混合,这跟直接使用原料药没有大的区别。
但是,制剂的价格远远高于原料药,所以认为用原料药可避免花“冤枉钱”。
也有用户认为用药只要成分一样, 用原料药多用点效果会更好,这是不对的。
因为不同的原料药,由于其自身分子结构的物理和化学性质不同,会具有一定的毒性、不稳定性、难溶性、吸收差等各种缺陷,用的量大不代表利用的多,可能反而对机体有害。
只有把原料药经过科学的制剂生产工艺加工,制成好的兽药制剂产品,这样才能充分发挥药物的作用,降低或避免其原药的毒副作用。
好兽药制剂的3大要素由高品质的合格原料与辅料,以及科学的制剂技术与生产工艺好兽药制剂的这三大要素“缺一不可”。
原料药质量是影响的第1要素首先,原料药质量是影响药品质量的第一要素。
原料药除了鉴别、含量等指标外,其中的杂质控制是非常重要的,过量杂质进入动物体内不但起不到药物的治疗作用,反而会给机体带来不必要的伤害。
其次,辅料的作用是很重要的。
它能够帮助增加原料药的溶解度、控制原料药的溶出度等,从而提高药品的生物利用度、增强药效。
在某些情况下,合适的辅料可以增加原料药的稳定性,提高药物的安全性。
因此,每一种辅料在产品制剂的配方中,它们具体都起到什么作用,为什么要加这个剂量,是非常严谨和科学的。
不合适的辅料,就无法保证药物的安全性、吸收和生物等效性等问题。
科学的工艺很关键先进的制剂都需要科学的工艺才能实现兽药的高品质为什么都是一样的含量,国外原研兽药产品的效果更确实?好的兽药制剂,其生产工艺是决定药物在体内吸收、分布、代谢和排泄好坏的重要因素。
生产工艺对药物品质和效果起到了关键作用。
如果生产工艺没做好,一种情况是药物进入动物体内,没有有效利用,或只利用了一部分;则另一部分就排泄出来,或者被破坏掉,在体内没有起到应有的治疗作用。
第4节原料药及制剂检查

篮法
桨法
小杯法-缩小版桨法
溶出度应用的指导原则
1. 重点用于难溶性的药品品种一般指在水中微溶或不溶的;
2. 用于因制剂处方与上产工艺造成临床疗效不稳定的品种以及 治疗量与中毒量相接近的口服固体制剂(包括易溶性药品), 对后一种情况应控制两点溶出量(第一点不应溶出过多);
3. 检测方法的选择:转篮法,以100 r/min为主,桨法,以50 转/分钟为主。溶出量一般为 45 分钟达 70 %,第三法用于 规格小的品种; 4. 溶出介质应以水、0.1 mol/L盐酸、缓冲液(pH值3~8为主); 若介质中加适量有机溶剂如异丙醇、乙醇等或加分散助溶剂 如十二烷基硫酸钠(0.5%以下),应有文献数据,并尽量选用 低浓度,必要时应与生物利用度比对。
色谱条件 考察色谱法 的有效性
(三)确定杂质检查及其限量的基本原则
•
1. 针对性
如果研究的药物属新药,应按照新药报批的要求逐 项进行研究并将试验结果整理成报批资料。 对一般杂质的检查 ,针对剂型及生产工艺,应尽可能考察 有关项目。
对特殊杂质或有关物质的研究 ,也应针对工艺及贮藏过程 确定待检查杂质的数量及其限度。
(七)注射液中不溶性微粒检查
装量在100 ml以上的供静脉滴注用注射液,在澄 明度检查符合规定后,再检查不溶性微粒方法与 限度规定均按《中国药典》附录。
(八)异常毒性检查
有的注射液必要时要检查异常毒性,过敏试验、降压 物质、热原,细菌内毒素等项:方法均按《中国药典 》附录,但热原剂量(即家兔体重每1 kg注射多少)要经 实验探索,或参考国外药典及有关文献。
作为缓释、控释口服固体制剂,释放度是必订的主要 项目
(五)有关物质
制剂加工工艺 原料药 稳定 有关物质增加 制剂质量标准√
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
原料药质量研究的主要容一、性状(一)外观聚集状态、色泽、臭、味、结晶性是对药物感官的一般性描述,一般无法定方法检查,但仍应准确描述。
(二)溶解性1. 溶剂的选择应根据药物的结构和性质,考查药物在水与常用溶剂中的溶解度。
应选择与药物溶解特性密切相关的溶剂,配制制剂、制备溶液和精制操作有关的溶剂,避免使用昂贵的、毒性大的、不常使用的溶剂。
2. 表述方式用“极易溶解”、“易溶”、“溶解”等术语来表示(见《中国药典》凡例)。
3. 试验方法按《中国药典》凡例的规定操作。
25±2℃,每隔5分钟强力振摇30秒钟,观察30分钟的溶解情况。
(三)理化常数理化常数包括:熔点, 馏程, 凝点, 比旋度, 折光率, 黏度, 相对密度, 酸值、碘值、羟值、皂化值,吸收系数等。
测定物理常数可以鉴别药物,也可以反映药物的纯杂程度。
注意事项:应按药典附录规定的方法进行测定。
测定前应按规定对仪器进行校正。
测定理化常数时应用精制品进行测定;质量标准中规定的理化常数围,用试制的药物来测定。
固体药物:熔点、吸收系数。
液体药物:馏程、相对密度、黏度、折光率。
油脂类还需测定:酸值、碘值、羟值、皂化值。
有手性中心的药物:比旋度。
1、熔点照《中国药典》”熔点测定法”测定。
(1)中国药典使用毛细管法测定熔点,以供试品局部液化的温度作为初熔的温度,以全部液化的温度作为全熔的温度。
(2)所用的传温液、毛细管、升温速度等对测定结果有影响,应按药典要求进行测定。
(3)应使用分浸型的温度计,温度计应用熔点标准品进行校正。
(4) 应用D S C 予以佐证。
2、吸收系数(1)测定方法 配制高、低两种不同浓度的溶液(吸光度在0.3~0.4和0.6~0.8之间)各2份,或配制吸收度在0.1~0.8的系列浓度的溶液,在5台不同型号的紫外分光光度计上测定,按下式计算吸收系数。
要求:应使用对照品进行测定。
测定前应对容量仪器进行校正。
测定前应对紫外分光光度计进行校正。
应按干燥品计算。
以平均值作为吸收系数,RSD 应小于1.5%。
3、晶型(1) 方法 X 射线粉末衍射法(中国药典 附录 Ⅸ F )。
X 射线衍射法:用一束准直的单色X 射线照射单晶或粉末晶体时,便发生衍射现象,发生衍射的条件符合布拉格方程:X 射线衍射图谱的横坐标一般为2θ,纵坐标为衍射强度。
X 射线粉末衍射法可以用来鉴别药物,测定含量,也可以用来对晶型进行考察。
要求:固体药物除水溶性好的外,均应作。
若不同晶型药物的生物利用度不同,应将晶型的检查列入质量标准。
二、鉴别鉴别试验:是使用化学的、物理化学的或生物学的方法,来辨别药物的真伪。
是对取自有标签容器中样品的确认。
要求:专属性强、重现性好、灵敏、简便。
通常采用 2 种以上不同类型的方法进行研究,从不同角度验证目标化合物。
常用的方法有:1、制备衍生物测定熔点θλsin n d hkl 2=本法较繁琐、费时,但专属性强,必要时可使用。
(1)成酯的反应例炔雌醇和苯甲酰氯反应成酯,炔雌醇苯甲酸酯的熔点为201℃。
(2)成腙的反应例异烟肼和香草醛反应生成异烟腙,熔点为226℃。
(3)酯的水解例丙酸睾酮水解为睾酮和丙酸,睾酮的熔点为150~156℃。
(4)与生物碱沉淀剂的反应2.呈色反应或沉淀反应要求:反应现象明显,专属性强。
一般鉴别试验:为一类药物共有的反应,收载在中国药典附录Ⅲ“一般鉴别试验”项下。
例水酸盐(中国药典附录第20页)(1)取供试品的稀溶液,加三氯化铁试液1滴,即显紫色。
(2)取供试品,加稀盐酸,即显水酸的白色沉淀;分离,沉淀在醋酸铵溶液中溶解。
又如:“丙二酰脲类”、“有机氟化物”、“钠盐”、“钾盐”的鉴别反应等。
(2)个别药物的鉴别试验例维生素C的鉴别取本品0.2g,加水10ml溶解后,照下述方法试验。
取溶液5ml,加硝酸银试液0.5ml,即生成银的黑色沉淀。
取溶液5ml,加二氯靛酚钠试液1~2滴,试液的颜色即消失。
3.紫外特征吸收方法:(1)核对光谱的数据a.核对吸收光谱峰、谷的位置。
b.核对吸收光谱峰、谷处的吸收度、吸收系数或吸收度比值。
C.核对经反应处理后反应产物的吸收特征。
(2)核对紫外吸收光谱例1 叶酸的鉴别(p.137)取本品,加0.4%的氢氧化钠制成每1ml中含10μg的溶液,照分光光度法测定,在256、283±2和365±4nm的波长处有最大吸收;在256与365nm波长处的吸收度比值应为2.8~3.0。
例2 苯妥英钠的鉴别(p.440)取本品约0.3g,加高锰酸钾10mg,氢氧化钠0.25g与水10ml,小火加热5分钟,放冷,取上清液5ml,加正庚烷20ml振摇提取,静置分层后,取正庚烷提取液,照分光光度法测定,在248nm的波长处有最大吸收。
4.红外分光光度法(中国药典附录ⅣC)主要用于原料药的鉴别,特别适合于同类药物用其它理化方法不易区别者。
方法:按要求录制红外吸收光谱,与《药品红外光谱集》中的标准图谱比较,应一致。
注意事项:(1)测定前应对仪器进行校正(用聚苯乙烯薄膜对仪器的波数和分辨率进行校正)。
(2)确定制样的方法。
(3)如果是多晶型的样品,应收载有效的晶型,并应提出晶型转化的条件。
(4)对图谱中的主要吸收峰进行解释。
5.色谱法主要用于制剂的鉴别,对原料药仅在没有其它更好的方法时,或检查和含量测定中已使用了色谱法时使用。
方法:取供试品和对照品,在相同的色谱条件下进行色谱分离,其R f 或t R 应一致。
例1 盐酸苯海索片的鉴别(p.726)取本品的细粉适量,加氯仿制成每1ml中含盐酸苯海索2mg的溶液,滤过,作为供试品溶液,另取盐酸苯海索对照品,加氯仿制成每1ml中含盐酸苯海索2mg的溶液,作为对照品溶液,照薄层色谱法试验,吸取上述溶液各10μl,分别点于同一硅胶G薄层板上,以氯仿-甲醇(9∶1)为展开剂,展开后晾干,喷以稀碘化铋钾试液使显色。
供试品溶液主斑点的位置应与对照品溶液主斑点相同。
例2 盐酸林可霉素的鉴别(p.727)取本品与盐酸林可霉素对照品,分别加流动相制成每1ml中含2mg的溶液,照含量测定项下的色谱条件进行试验,供试品与对照品主峰的保留时间应一致。
6、生物化学或生物学方法四. 检查检查项下包括有效性、均一性、纯度要求和安全性四个方面。
(一)有效性指与疗效有关,但在鉴别、纯度检查和含量测定中不能控制的特殊性质的检查。
如:结晶性(利福平);制酸力(氢氧化铝);吸着力(药用炭);凝冻浓度(明胶);乙炔基(炔诺孕酮);含氟量(诺氟沙星)例氢氧化铝制酸力的检查(p.553)取本品约0.12g,精密称定,置250ml具塞锥形瓶中,精密加盐酸滴定液(0.1mol/L)50ml,密塞,在37℃不断振摇1小时,放冷后,加溴酚蓝指示液6~8滴,用氢氧化钠滴定液(0.1mol/L)滴定,每1g消耗盐酸滴定液(0.1mol/L)不得少于230ml。
(二)纯度的检查中国药典(2005年版)附录新增了“药品杂质分析指导原则”。
指导原则是参照ICH(人用药品注册技术要求国际协调会)的文本Q3A和Q3B制订的。
适用于化学合成的或半合成的有机原料药及其制剂。
1、杂质的分类及其在药品质量标准中项目的名称(1)药物中的杂质杂质:影响药品纯度的物质均称为杂质。
杂质的来源:生产过程引入:按规定工艺、规定原辅料生产,引入成品的合成的起始物、中间体、副产物、试剂等;贮存过程引入:经稳定性试验确认在贮存过程中产生的降解产物。
药品质量标准中的杂质不包括变更工艺或原辅料时产生的新的杂质,也不包括掺入或污染的外来物质。
(2)杂质的分类:按化学类别和性质分类:有机杂质、无机杂质、有机挥发性杂质。
有机杂质:合成的起始物、中间体、副产物、降解产物、试剂、催化剂、配位体等。
无机杂质:试剂、催化剂、配位体、重金属或其他残留金属、无机盐、其他物质。
有机挥发性杂质:残留的有机溶剂。
按毒性分类:毒性杂质和普通杂质。
3. 杂质项目的名称:明确为某一物质时,以该物质的化学名称作为项目名称。
如磷酸可待因中的“吗啡”,阿司匹林中的“游离水酸”等。
仅知为某一类物质时,用该类物质的名称。
如“其他甾体”、“还原糖”、“有关物质”等。
对未知杂质,可根据检测方法命名。
如“杂质吸收度”、“易炭化物”、“不挥发物”等。
4、质量标准中杂质检查项目的确定根据生产和贮存过程可能引入的杂质来确定,包括存在的杂质和潜在的杂质。
考察产品中是否引入了合成的起始物、中间体、副产物、试剂。
通过影响因素试验、加速试验和长期试验来确定降解产物。
对表观含量>0.1%或虽<0.1%,但具有强烈生物作用或毒性的杂质应确定结构。
对可能存在的无机杂质进行考察并制定检查项目。
有机挥发性杂质:应根据工艺所使用的有机溶剂及残留情况,确定检查的项目。
除降解产物和毒性杂质外,在原料中已控制的杂质在制剂中一般不再控制。
共存的异构体和抗生素多组分一般情况不作为杂质检查项目,作为共存物质。
5、要求(1)在保证用药安全、有效的前提下,允许有一定量的杂质存在。
(2)一般为限量检查。
(3)检查项目的确定既要保证药物的安全性、有效性,又要根据药物在生产和贮存中可能产生的杂质来确定。
(4)应重点检查影响药物疗效、危害人体健康或能真实反应药品在质量的项目。
(5)限量的确定,既要根据生产的实际水平,又要根据药典的惯例。
杂质检查方法1. 酸碱度分为酸度、碱度或酸碱度。
方法:酸碱滴定法、pH值测定法、指示剂法。
2. 溶液的澄清度与颜色(中国药典附录ⅨA, B)(1)溶液的澄清度(中国药典附录ⅨB)主要用于注射用原料药的检查。
方法:与溶剂或浊度标准液比较。
“澄清”:供试品溶液的澄清度应相同于所用的溶剂或不超过0.5号浊度标准液。
(2)溶液的颜色(中国药典附录ⅨA)检查样品中的有色杂质。
检查方法:第一法:与标准比色液比较第二法:测定吸收度第三法:色差计法3.无机杂质主要有氯化物(Cl-)、硫酸盐(SO42-)、铁盐(Fe3+)、重金属(Pb2+)、砷盐(AsO43-, AsO33-)、氰化物(CN-)等。
方法:收载于中国药典附录Ⅷ。
如“氯化物检查法”、“砷盐检查法”等。
供试品与对照品在相同条件下比色、比浊,以确定杂质的量是否超过限量。
注意:应完全按照药典附录的方法进行检查。
注意遵循平行原则。
研究时,可取系列浓度的杂质对照品溶液与供试品溶液进行比较,以确定杂质含量的围。
应考察样品本身对检查是否有影响。
4.有机杂质有机杂质是指残存在产品中的原料、中间体、副产物和降解产物等。
杂质能够确定的,检查的项目一般以杂质的名称命名;也可以杂质的类别命名;不能确定的原料、中间体、副产物一般称为“有关物质”。
检查方法:(1)色谱法纸色谱法主要用于极性较大的杂质的检查。
如地高辛中洋地黄毒苷的检查(中国药典(2000年版))。