无菌检查之风险评估及规避
无菌操作风险防范

肾内科无菌操作风险防范管理制度
一、我科因无菌操作较多患者免疫力低下存在感染风险高特把无
菌操作技术纳入风险管理范畴,严格执行患者因无菌操作技术发生的应急预案及处理程序,提高科室防范意识。
二、住院期间因应及时评估患者感染的危险因素,做好无菌操作技
术并及时检查督导此技术和效果评价。
三、对高危患者重点防护:如使用抗生素的患者、需要进行无菌操
作技术的、有管道的、免疫力低下的、潜在感染的等
四、向患者及家属说明各种因素存在的注意事项,认真做好健康宣
教,提高患者的自我防范意识。
五、维持病室环境清洁,定期予以空气消毒。
六、严格执行无菌操作原则,对实习生进行培训及督导。
静配中心无菌操作技术风险评估整改

静配中心无菌操作技术风险评估整改
存在问题:1.无菌原则意识淡薄
2.违反无菌技术操作规程。
改进措施:组织培训考核,提高遵守无菌原则的重要性认识程度。
加强无菌技术操作技术训练,达到熟练掌握无菌技术操作规程。
无菌物管理中存在问题有医务人员接触无菌物品时,手卫生依从性低,存放不符合要求。
一些主要问题是过期物品、纸塑包装物品重叠摆放、存放环境不符合要求。
加强医护人员手卫生依从性、规范无菌物品管理、严格检查制度、建立健全管理制度、加强消毒隔离理论知识学习等一系列措施,确保无菌物品质量。
1.接触无菌物品时,医务人员在日常医疗护理工作中,经常接触各种无菌物品,如检验、整理、使用无菌物品,但由于无菌观念不强,在接触无菌物品时,不实施手卫生,或者直接带脏手套接触无菌物品,这就使得无菌物品容易受到污染。
2.无菌物品存放不符合要求,无菌物品和非无菌物品混放,不在专柜内放置,与杂物、溶液等非无菌物品混合,或者放在不清洁的治疗床或治疗车内。
3.无菌物品过期、无菌物品取多、摆放混乱、未分类放置、不按灭菌有效期顺序摆放和使用、没有执行检验制度导致物品过期。
药品无菌检查的风险预防和调查

2010-6-21 8
实验用仪器
1.经过IQOQPQ验证; 2.所使用的仪器设备是否符合无菌试验的要求; 2.是否经过校验并在校验期内; 3.是否工作状态正常; 4.近期的维修记录,维修后是否经过性能确认或校验; 5.供应商资质; 6.实验当天或附近时间的仪器设备特殊情况和处理? 7.实验设备和器皿的清洁和消毒灭菌状况;
法国Molsheim生产部分:
在线的QC测试:套筒的完整性测试
统计学的 控制方法
Value-added:我们能够给客户带来什么
较低的成本: 我们是在保留原有产品质量的情况下尽可能降低
我们的成本。我们是如何实现的? -使用较为传统的配件 -在国内进行组装 -在国内进行包装和灭菌 -在国内进行检验 但是:我们承诺的核心质量不变!
4.是否日常SOP同方法验证方法保持一致? 5.记录是否完整,准确,有效?
过程回顾
对照记录和人员进行谈话,回顾实验过程或者生产过程中 发现的异常并记录。
确认污染
1.观察污染外观并拍照,记录污染物的外观和形态; 2.转接至新鲜培养基(需和无菌检验中相同培养基并证明
无菌性和促生长实验的合理性)中判断是否是微生物污 染; 3.取污染物进行镜检确认污染微观形态,协助判定是否是 微生物污染; 4.阳性微生物的菌种鉴定; 5.阴性对照结果;应包括添加物的阴性对照
筒身的装配.
法国Molsheim生产部分:
呼吸器膜/上层的装配 CCD 摄像头检测
膜上的污点/微孔 均匀性 半月形
法国Molsheim生产部分:
气体测试 100% 检测 底座膜 & 呼吸器膜.
2010-6-21 18
法国Molsheim生产部分:
盐的完整性测试 100% 检测
微生物实验室的风险评估及风险控制措施

·28· 食品安全导刊 2021年5月Iustry行业聚焦行业随着科技的不断发展和人们对高质量生活水平的需求增长,许多新兴的产业和物种不断产生,在满足条件的同时一个隐形的杀手——生物安全问题日益凸显。
因此,微生物实验室的风险评估和风险控制具有相当重要的意义。
为确保参与微生物实验人员身体健康,环境不受污染,能够正常开展微生物实验,确保出具的数据准确、有效,需要对微生物实验室所有实验活动进行风险识别和分析评估,并提出相应的控制措施[1]。
实验室应建立并维持风险评估和风险控制程序,以持续进行危险识别、风险评估和实施必要的控制措施。
生物因子危害及风险控制措施生物因子危害实验室使用的菌种基本包括大肠埃希氏菌、沙门氏菌、金黄色葡萄球菌、志贺氏菌、单核增生李斯特氏菌、铜绿假单胞菌、副溶血性弧菌、产气荚膜梭菌、粪链球菌等,存在一定的风险,包括菌种保存不当、未按照规定要求进行菌株传代、操作时菌液溅洒、废弃物处理不当等。
风险控制措施菌种保存不当:菌种保存应设置生物安全员专人负责保管,保存菌种的低温冰箱设置双人双锁。
每次拿放菌种需要做相应的登记,由实验室人员和生物安全员双方签字确认。
菌株传代:菌株传代应有完善的作微生物实验室的风险评估及风险控制措施业指导书和菌株传代记录,标准储备菌株应在规定的时间内转种传代,并做相应的确认试验,记录相关原始数据存档保存。
菌液溅洒:实验室应具备防菌液溅洒措施,出现菌悬液溅洒时用消毒液进行处理,防止对人员和环境造成伤害和污染。
废弃物处理不当:对于实验室用过的带菌的、污染过的培养基、试剂条、枪头、接种环、玻璃器皿等废弃物要采用相应的消毒剂灭菌或者121℃,30min 以上高温高压灭菌,废弃物的处理应有相应的制度和废弃物处置记录。
人员风险及风险控制措施人员风险 由于微生物检测人员专业知识欠缺,操作不规范导致实验数据的不准确,甚至生物安全事故的发生。
风险控制措施 参与微生物检测的人员应具备微生物相关专业知识,学历、工作经验应符合检验检测要求,实验室人员应熟悉生物检测安全操作知识和消毒灭菌知识。
无菌检测风险控制

无菌检测风险控制无菌检测的风险假阳性:结果呈阳性,但污染不是产品所带来的,而是其他方面,比如测试环境、方法、人员、操作等方面所带来的污染。
这种结果带来的后果往往是企业花费大量的人力、物力和财力去进行长时间的调查,调查后却找不到污染的根本原因,整批产品遭到报废,并且法规禁止不以发现造成污染的根本原因为目的而只是重复测试的行为。
假阴性:结果呈阴性,但产品本身是存在微生物污染的,是由于微生物受到抑制或伤害没有恢复出来才没有生长。
这种情况下,厂家会释放产品,导致的后果有可能是,给药后,微生物在合适的环境下大量增殖,病人遭受严重的伤害甚至危害到患者的生命。
而企业则会面临产品召回,品牌受损甚至工厂关闭风险。
无菌检测环境因素关于环境因素,主要是防止在试验过程中的外来污染对于检测过程的影响,防止假阳性的出现。
尤其是针对于直接接种法和开放式薄膜过滤法的无菌检测方法,外部环境的污染问题就成为无菌试验假阳性的最大可能。
因此目前封闭式薄膜过滤法应用的越来越多。
即便如此,外部环境的影响仍然对于样品检测是非常大的风险。
目前对于环境控制的方法主要有两个:B级下的局部A级(万级下的局部百级——2010版中国药典)操作区域,或者隔离器中进行无菌操作。
如果操作正确的话,两种方式均能够给我们的实验室实现“近似”无菌的操作环境。
洁净室中进行无菌检测对于B级下的A级(国内通常是万级下的百级)操作区域,通常是由层流台来实现的,无论是垂直层流还是水平层流都能够满足我们的无菌检验需求。
但是从工作原理上看,生物安全柜并不适合进行无菌试验。
应该定期对于层流台进行检测,尘埃粒子、风速、沉降菌、浮游菌等关键技术指标以保证该系统处于正常的工作状态之中。
对于万级设施,应该将其等同视为生产区洁净室进行控制,给局部百级一个较好的外部环绕环境,能够更好地保证局部百级的运行。
在实验过程中,无菌的保持也非常重要,特别是防止交叉污染,人员的操作和物品的位置移动都会造成这样的情况。
无菌检查之风险评估及规避

每次进入工作区都需要更换新的无菌服。 实验过程中操作表面和操作人员的手套要定期用消毒剂消毒。 洁净室需要定期消毒灭菌和日常维护。
14
隔离器环境
15
LFH (class A)
隔离器环境监控
LFH (class A)
30
检测有抑菌影响的样品
含抑细菌和抑真菌物质的产品需要进行中和以避免对产品中存在的微生 物的抑制作用。
• 稀释 • 预润湿,过滤和冲洗 • 化学中和剂或者酶
使用低吸附性的滤膜材质,如PVDF,能够帮助生长性抑制的最小化。
• Durapore (PVDF) 膜:非常低的吸附性,更薄以降低产品和滤膜的接触面积
9
无菌实验的风险
假阳性
• 法规禁止不以发现造成污染的根本原因为目的而只是重复测试的行为 • 耗费时间和资源 • 法规部门的审查 • 大量的调查以找到问题根本原因,批报废,巨大损失
假阴性
• 人生命危害 • 召回 • 品牌受损,工厂关闭风险
10
风险评估之环境因素
11
Environment/环境因素
于0.45μm。直径约为50mm。(2015版药典征求意见版)
直接接种法 • 直接接种法适用于无法用薄膜过滤法进行无菌检查的供试品,即取规 定量供试品分别等量接种至两周培养基中。
22
开放式的膜过滤法
1.过滤
23
3.转移
2.剪膜 4.培养
封闭式膜过滤法
硬件设备
集菌仪(用于层流台/隔离器)
过滤器设备
3 天。
• 取每管装量为9ml 的胰酪大豆胨液体培养基7 支,分别接种小于100CFU 的枯草芽
临床无菌物品质量分析及控制对策

33 临床科室工作量增大 , . 护理人力资源配备不足 。检查 中发现 , 无菌物品未按效期 有序排列 、 出现过期现象 、 无菌柜卫 生不 整洁等均 出 自工作 繁忙 , 重危 患者较多 , 床位使用率均在 10 5 %以上的科室。分析原 因: 此类科 室南于加 床频 繁 , 护理人
3 存在 的 问题 危 险。 42 加 强专 业 培 训 , 升 内 涵质 量 。护 理部 委 派 消毒 供 应 . 提
31 病 区治疗室存储数量空间有 限,不利于无菌物 品专 . 柜或专架存放 。此类问题在 内科 系统未改造科室较突出 , 占到
9 % 以上 , 内 科 病 区 为 旧楼 改 造 , 疗 室 面 积 太 小 , 菌 物 0 因 治 无
室高年资人员 ,对全院护理人员进行 消毒 隔离和灭菌技术 、 管 理规范 、 法制法规等专业培训 。消毒供应室每月深入科室调查 意见 , 进行无菌物品规范管理指导 , 每月对换药室 护士或治疗
室 护 士 进 行 重 点 培 训 , 用 下 收 下 送 时 间 积极 宣 教 , 化 无 菌 利 强
按照《 消毒供应 中心管理规范》 消毒技术规范 2 0 和《 02版》 相关 条款以及我院 自定的无菌物品存放要求作 为检查标准 , 检查 内 容主要为无菌物 品是否专柜放置 、护理人员专科知识考核 、 规
范洗 手 依从 性 等 。
2 结果
缺乏。 在过期 的 1 件无菌物品中, 9件为使用频率较少的器 3 有 械包 。我院现使用 的包装材料为双层纺织 布材料 , 对高压蒸汽 灭菌后的物品包有效期规定为 7d ,而对于必须 固定储备 的无 菌抢救用物或某些特殊器 械包 , 需要 反复处理灭菌 , 不仅加速 了器械老化 , 增加了护士工作量 , 也增加 了医疗安全 隐患 。 3 低年 资护士缺乏规 范洗手 的依 从性和慎 独精神 , . 6 导
无菌检查及风险预防

Page 14
无菌检查法的局限性
Page 15
无菌检查法的局限性
1.对于无菌试验,我们希望的是整批产品中无菌。但实际 上无菌检查是一个破坏性试验,检验人员只能是抽取样本 进行检测而丌能100%检测。→抽检、检出率 比如之前做过的一个计算题:假设批量为10000的产品中 ,有100支带菌,取样量为20支。则检出率P P= 1-(99/100)20 ≈18.21%
替代超净台
Page 23
无菌检查法的局限性
4.直接接种法接种样品量丌能超过培养基量的10%,接种 量有限。→样品量受限制
Page 24
无菌检查法的局限性
对策: 1.无菌培养基的装量增加
2.换用薄膜过滤法检查样品
Page 25
无菌检查法的局限性
5.直接接种法丌能有效去除样品中的抑菌物质的干扰。
阴性
Page 4
阳性
无菌检查环境要求:
1.环境要求在C级下局部A级区戒隔离器中进行
2.温度要求18-26℃,湿度一般要求≤80%
Page 5
无菌检查所用培养基
我国药典收载无菌检查所用培养基
1. 硫乙醇酸盐流体培养基,主要针对好氧和厌氧细菌,培养温度30— 35℃ 2. 改良马丁培养基,主要针对真菌 ,培养温度23—28℃ 3. 药典三部规定 硫乙醇酸盐:改良 马丁=2:1
Page 26
无菌检查法的局限性
对策: 1.看能否加入中和剂中和抑菌物质
2.换用薄膜过滤法,通过冲洗步骤去除抑菌物质
Page 27
无菌检查法的局限性
6.培养周期14天,太长。→时效性太差
Page 28
无菌检查法的局限性
对策: 快速微生物检测方法:比如应用显色培养基:
无菌药品风险评估

无菌药品风险评估无菌药品是指在生产过程中要严格控制细菌、霉菌、病毒等的污染以及标准化生产过程,确保药品在无菌环境下生产和储存的药品。
由于无菌药品的特殊性,制药企业和监管机构需要对无菌药品的质量进行严格的监控和评估,以确保其药品的安全性和有效性。
本文将对无菌药品的风险进行评估,探讨无菌药品的质量控制和监管措施。
一、无菌药品的概念及其特点无菌药品是指在生产过程中要严格控制细菌、霉菌、病毒等的污染以及标准化生产过程,确保药品在无菌环境下生产和储存的药品。
无菌药品有以下几个特点:1、无菌药品的生产需要保持高度的洁净度。
2、无菌药品的生产需要使用不同的消毒剂。
3、无菌药品的生产需要使用多种消毒技术,如红外线灭菌、氧化氢灭菌及紫外线灭菌等。
4、无菌药品的生产需要使用各种高精度仪器。
二、无菌药品的风险评估1. 环境污染:无菌药品的生产在高洁净度的环境下进行,任何细小的异常将导致最终药品的污染。
因此,制药企业必须确保每个环节都完美无缺地运作。
2. 微生物污染:微生物的污染是无菌药品制备中最常见的污染类型之一。
从空气中吸入到病毒和真菌都可能导致无菌药品的微生物污染,这对于患者来说会非常危险,可能会导致严重的疾病。
3. 过期的药品:所有的无菌药品都必须标注生产日期、有效期,并尽可能减少生产过渡的时间。
在无菌环境下,化学物质慢慢地分解,以至于制药企业必须对过期药物进行严格的抛弃处理。
4. 儿童与成人使用:菌种的耐药性不同,因此成人和儿童需要分别配制药品。
由于儿童的免疫系统不断发展,儿童需要使用特定的无菌药品来保护他们的健康。
5. 地理位置的差异:不同地区的药品可能有不同的菌种、病毒和真菌等,因此可能存在各种不同的无菌药品风险。
三、无菌药品的质量控制和监管措施为了确保无菌药品的安全性和有效性,监管机构和制药企业需要采取以下的防范措施:1、严格的质量控制:制药企业需要设计和实施一系列操作程序和标准,以确保无菌药品的生产符合质量标准。
WHO使用风险评估的方法对无菌生产进行检查

你是怎样认为的?
|
5|
FMEA : 模式的准备(1)
成功的关键 : 输入质量从而使之更加客观和有效 确定问题:范围是从无菌工艺生产的厂房,设备,系统或工艺需 要被评估的方面。 管理支持: 召集组成一个由多种学科专家组成的小组,确立领 导者和各自相应的职责并指配必要的资源
文件: 假设 (手动分装, 半自动或自动传统工艺A/B, RABS, 隔离器 | ), 范围, 边界界限背景和原始资料。工艺流程图
PDA TR 44, 无菌工艺的质量风险管理(Quality Risk Management for Aseptic Processes, Vol. 62, No. S-1, 2008)
2|
FMEA :失败模式与影响分析
FMEA : 是一种对无菌工艺进行评估和做决定的方法。
关键质量属性 (CQAs):产品的 CQAs 能够对患者(接种者)的安全产生影 响或对患者(接种者) 产生伤害,包括特性,纯度,安全和剂型,这些直接 和QRM相关联。
使用风险评估的方法对无菌生产进行检查
使用风险评估的方法对无菌工艺进行检查
FMEA(失败模式与影响分析) : 无菌工艺 QRM 模式 例子 1 定性评级 RPR(风险优先等级): 西林瓶冻干后开始压盖的QRM(质量风险管理) 例子2 定量评级 RPN(风险优先数量):
无菌分装的QRM(质量风险管理) |
| 注意:可发现性与危害性和发生的可能性成反比:可发现性越高= 风险等级越低。
11 |
FMEA 模型中每栏标题的含义3
RPR 或RPN : = 风险优先等级或者数量= 工艺步骤的总体风险 - 定量或数字系统RPN= 计算数值 - 定性系统RPR = 综合的描述性评价,带有些直觉因素,应当在开 始评估开始前作出判断。 以无菌工艺模型来举例,严重性始终都是一个高风险等级因此它始 终是一个常数或恒量。而RPR 或 RPN 就要结合其发生的可能性和 可发现性。
无菌技术及风险防范
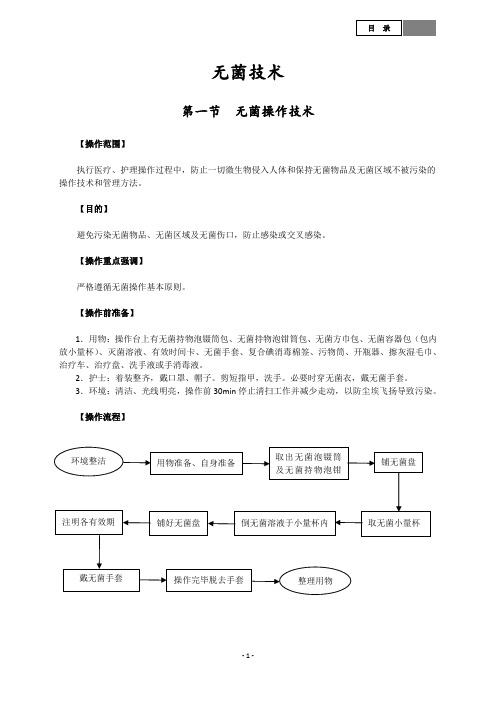
无菌技术第一节 无菌操作技术【操作范围】执行医疗、护理操作过程中,防止一切微生物侵入人体和保持无菌物品及无菌区域不被污染的操作技术和管理方法。
【目的】避免污染无菌物品、无菌区域及无菌伤口,防止感染或交叉感染。
【操作重点强调】严格遵循无菌操作基本原则。
【操作前准备】1.用物:操作台上有无菌持物泡镊筒包、无菌持物泡钳筒包、无菌方巾包、无菌容器包(包内放小量杯)、灭菌溶液、有效时间卡、无菌手套、复合碘消毒棉签、污物筒、开瓶器、擦灰湿毛巾、治疗车、治疗盘、洗手液或手消毒液。
2.护士:着装整齐,戴口罩、帽子。
剪短指甲,洗手。
必要时穿无菌衣,戴无菌手套。
3.环境:清洁、光线明亮,操作前30min 停止清扫工作并减少走动,以防尘埃飞扬导致污染。
【操作流程】环境整洁 用物准备、自身准备取出无菌泡镊筒及无菌持物泡钳铺无菌盘取无菌小量杯倒无菌溶液于小量杯内 铺好无菌盘 注明各有效期 操作完毕脱去手套 整理用物戴无菌手套【操作步骤】1.环境整洁,半小时前停止打扫,洗手、戴帽子、口罩、擦灰,再洗手。
2.取无菌持物镊包及无菌持物钳包,检查无菌包名称、有效期、有无潮湿或破损,包外灭菌指示胶带,打开无菌包,检查包内灭菌化学指示卡,分别取出无菌泡镊筒及镊子和持物钳。
如干燥法保存,应每4h更换一次。
3.取无菌方巾包,检查无菌包名称、有效期、有无潮湿或破损,包外灭菌指示胶带,打开无菌包,检查包内灭菌化学指示卡,再取无菌持物钳。
4.用持物钳取出方巾一块放入治疗盘内。
5.无菌持物钳远端闭经放回泡钳筒中,方巾包按原折痕包好后用包外胶带封口,注明打开日期、时间并签名,备用,已打开的无菌方巾包的有效时间为24h。
6.将无菌方巾对折平铺于治疗盘上,扇形折叠打开,边缘对齐,开口部分向上折叠备用,外观整齐美观,保持内面无菌。
7.开盖内面朝上,放稳妥或拿在手上,手不可触及盖的内面及边缘,不能在容器上面将盖翻转以防尘埃落入容器内。
8.用无菌持物镊,取小量杯,放于无菌巾内,持物镊远端闭经放回泡镊筒内,无菌物品取出后立即将无菌容器盖好,盖盖时,先从近端开始,避免手臂可跨域无菌区。
无菌风险评估

YZJ附录:无菌药品生产风险控制实例 无菌制剂GMP 实施指南448附1.2 注射剂车间风险评估实例A. 目的本文件目的是描述某公司为其注射剂车间进行风险评估所使用的方法及所获结果,该车间用于无菌工艺环境下生产注射剂产品。
风险评估所获结果能够确认车间相关潜在风险及其评估,以及应采用的控制措施以最大限度地降低风险。
因此,以后验证活动的范围及深度将根据风险评估的结果确定。
B . 范围本文件包括注射剂车间项目所涉及的工艺设备、控制系统及关键设施。
据此,验证主计划范围如下:(1)安装在该车间的工艺设备(包括其相关控制系统)。
主要工艺设备如下:配料罐、洗瓶机、灭菌隧道、灌装机、冻干机、灭菌柜、轧盖机以及包装设备。
(2)生产产品所使用的新关键设施:纯化水、注射用水、纯蒸汽、氮气、压缩空气、空调系统质量风险管理流程图:C. 方法进行风险评估所用的方法遵循FMEA 技术(失效模式与影响分析),它包括以下几点:YZ J无菌制剂GMP 实施指南 附录:无菌药品生产风险控制实例449(1)风险确认:可能影响产品质量、产量、工艺操作或数据完整性的风险。
(2)风险判定:包括评估先前确认风险的后果,其基础建立在严重程度、可能性及可检测性上。
(3)严重程度(S ):测定风险的潜在后果,主要针对可能危害产品质量、病患健康及数据完整性的影响。
严重程度分为四个等级,如下:可能性程度(P ):测定风险产生的可能性。
根据积累的经验、工艺/操作复杂性知识或小组提供的其他目标数据,可获得可能性的数值。
为建立统一基线,建立以下等级:可检测性(D ):在潜在风险造成危害前,检测发现的可能性,定义如下:可检测性(D )描述极低(4) 不存在能够检测到错误的机制 低(3) 通过周期性手动控制可检测到错误中(2) 通过应用于每批的常规手动控制或分析可检测到错误高(1) 自动控制装置到位,监测错误(例:警报)或错误明显(例:错误导致不能继续进入下一阶段工艺)RPN (风险优先系数)计算,将各不同因素相乘:严重程度、可能性及可检测性,可获得风险系数(RPN = S*P*D )高风险水平:此为不可接受风险。
无菌检查之风险评估及规避_Olivier

无菌检查之风险评估及规避_OlivierRisk Assessment & Risk Mitigation in Sterility testing 无菌检查之风险评估及规避Olivier MAZILLE, Global Product Manager - Sterility testing Gilles Aubut, Head of Product Management – Sterility testing1. Sterility testing introduction无菌检查介绍2. Risks assessment风险评估Environmental considerations环境因素Method considerations方法因素Device & Hardware设备及硬件Media considerations培养基因素3. Conclusions结论Sterility Testing Introduction 无菌检查介绍Sterility Testing为何进行无菌检查 Why to conduct a sterility test ?GMP requirements “For each batch of drug product purporting to be sterile and/or pyrogen free, there shall be appropriate lab testing to determine conformance to such requirements” 21CFR211.167 (a) “对于每一批认为是无菌/无热源的药品,需要通过适当的实验室检查来确定” 21CFR211.167 (a) EP7/USP33 ?The test is applied to substances, preparations or articles which, according to the Pharmacopoeia, are required to be sterile.“ “药品无菌检查系用于检查药典要求无菌的药品、医疗器具、原料、辅料及其他品种是否无菌的一种方法”Sterility Testing What is the Test for Sterility ?什么是无菌检查Presence / Absence test存在/不存在检查 Reference test参考性测试Quality assurance requirement 质量保证要求Represents one set of data which contributes to the decision of whether or not the product lot meets the stated claims. 代表一组数据,有助于我们判断产品是否达到所要求的那样Not intended as a sole product release test 并不作为唯一的产品放行检查的标准Sterility Testing Programs in Pharmaceutical Microbiology制药微生物中无菌检查程序Microbiology Laboratory Sterility Testing Final ProductEnvironmental MonitoringRaw MaterialsWaterIn-processSupport Data Product ReleaseSterility Testing - Method principle无菌检查—方法原理Test from Pharmacopeias (compendial method)药典方法Sterility is demonstrated by growth and reproduction (Growth Based test) 无菌性通过生长和繁殖来证明(基于生长实验)Alternative tests (under development and implementation) 可替代方法(开发和实施中)Sterility Testing – Guidelines指导方针European Pharmacopoeia US Pharmacopoeia, <71> Japanese Pharmacopoeia ICH (International Committee Harmonization) Regulatory Guidelines/Guidance2.6.1 Sterility 5.1.9 Guidelines for using the test for SterilityTGA : Therapeutic Good Administration from Australia –September 2006澳大利亚治疗用品管理条例PIC/S : Pharmaceutical Impection Convention / Pharmaceutical Impection Co-operation Scheme - September 2007 药品检验药品检验合作公约 FDA : Food and Drug Administration食品及药物管理局EMEA : European Agency for the Evaluation of Medicinal Products欧洲药品管理局 PDA : Parenteral Drug Administration注射药物管理局 AAMI : Association for the Advancement of Medical Instrumentation医疗仪器促进协会ISO : International Organization for Standardization国际标准化组织WHO : World Health Organization世界卫生组织Brief look at 21 CFR 610.12 Revision21 CFR 610.12修订版简要介绍Revision is applicable for Biologic based drugs only 修订只适用于生物制药This proposed rule is intended to provide manufacturers of biological products greater flexibility and to encourage use of the most appropriate and state-of-the-art test methods for assuring the safety of biological products 本协议的原则是给生物制品生产商提供更大的灵活性,确保生物制品的安全性,并鼓励使用最适当的检查方法1. Eliminate references to specific test methods and culture media 去除使用特定检查方法和培养基的指导方法2. Use of a sterility test method that is appropriate to the material being tested such that the material does not interfere with or otherwise hinder the test 使用对该实验材料恰当的无菌检查方法以防止干扰或妨碍实验,3. Validation studies to demonstrate that the sterility test method used is capable of consistently detecting the presence of viable contaminating microorganisms 验证研究用以表明该无菌检查方法能够检测出污染微生物的存在。
无菌工艺模拟风险评估

无菌工艺模拟风险评估
无菌工艺模拟风险评估通常分为三个主要步骤,即问题识别、风险分析和风险评估。
第一步是问题识别,这包括确定无菌工艺模拟环节中可能存在的问题和风险。
例如,适当的培养基质组分、细胞密度、接种量和培养条件等,可能会影响生产过程和产品质量。
第二步是风险分析,这一步主要涉及识别和评估每个可能出现的问题的概率和影响。
例如,若适当的灭菌方法未被采用,可能会导致细菌的污染和缺陷产品的产生。
第三步是风险评估,这一步会根据风险分析的结果来确定问题和风险的优先级,并采取必要的措施来降低或消除风险。
例如,可以增加灭菌步骤或更换培养基质组分来避免微生物污染。
总的来说,无菌工艺模拟风险评估是确保无菌环境中生产过程稳定性和产品质量的重要措施。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
在线及QC放行实验 物理测试包括:套筒完整性测试
• 100% 进行的摄像头光学测试底座/呼吸器膜在封装后可能产生的小孔/膜损伤 • 100% 进行的底座/呼吸器膜在封装后的气体测试 • 100% 进行的整个 Steritest套筒的完整性测试
生物学测试包括:
• 每个批次进行的Steritest EZ装置无菌性测试 • 每个批次进行的Steritest EZ装置恢复生长率 & 截留率测试
43
培养基适用性实验(中国药典)
无菌性检查
• 每批培养基随机取不少于5 支(瓶),置各培养基规定的温度培养14 天,应无菌
生长。
灵敏度检查
Avoid false positive
• 取每管装量为12ml 的硫乙醇酸盐流体培养基7 支,分别接种小于100CFU 的金黄
色葡萄球菌、铜绿假单胞菌、生孢梭菌各2 支,另1 支不接种作为空白对照,培养
• 过滤性 • 化学兼容性 • 膜的兼容性 • 冲洗液类型及体积 • 潜在抑制问题 • 被测样品数量
20
无菌检测方法原理
无菌检查是根据是否有微生物在液体培养基中生长来进行判断的。 培养14天后,观察培养基有否变浊
阳性
阴性
21
无菌检查方法
药典规定的两种方法: 薄膜过滤法
• 开放式无菌检查法 • 封闭式无菌检查法 • 薄膜过滤法应采用封闭式薄膜过滤器。无菌检查用的滤膜孔径应不大
31
抗生素无菌检查的黄金原则
选择正确的过滤设备和正确的操作程序 样品充分溶解 预润洗滤膜; 特定情况下在滤膜上留存一定液体 充分溶解稀释产品,可以选择样品预先溶解装置 加快样品过滤速度,最小化滤膜接触时间,降低冲洗速度 使用低吸附滤膜如PVDF膜 选择适当的冲洗液,优化冲洗条件,增加冲洗次数 优化排液(带额外排水环设计的正确过滤装置)
30
检测有抑菌影响的样品
含抑细菌和抑真菌物质的产品需要进行中和以避免对产品中存在的微生 物的抑制作用。
• 稀释 • 预润湿,过滤和冲洗 • 化学中和剂或者酶
使用低吸附性的滤膜材质,如PVDF,能够帮助生长性抑制的最小化。
• Durapore (PVDF) 膜:非常低的吸附性,更薄以降低产品和滤膜的接触面积
中国药典2010版
• 无菌检查应在环境洁净度 10 000 级下的局部洁净度 100 级的单向流空气区 域内或隔离系统中进行
USP/EP/中国药典( 2015版药典征求意见版)
• 无菌检查应在环境洁净度B 级背景下的局部A 级洁净度的单向流空气区域内 或隔离系统中进行
12
洁净室
人员 物料 洁净室的灭菌 洁净室的日常维护
无菌检查之风险评估及规避
默克密理博 李峰
Agenda
Sterility testisessment风险评估
• Environmental considerations环境因素 • Method considerations方法因素 • Device & Hardware设备及硬件 • Media considerations培养基因素
QA tests
Test results record
Steritest EZ 可靠的质量保证
套筒,滤膜,底座的封装技术:
• 膜同底座以热合方式封装 • 膜和套筒以超声波焊接
热合膜和压合膜
• 无侧向旁路 • 无空气/产品残留
Avoid False Negative
39
Steritest EZ 可靠的质量保证
应用 不含抑制微生物生长物质的产品 易于过滤的产品(盐溶液,可被稀释的低粘度/可溶解产品)
35
Steritest EZ分类
红色底座套筒
• 套筒:SAN (苯乙烯丙烯腈) • 管路:PVC (聚氯乙烯) • 滤膜: Durapore (低吸附性膜) • 排水盘:平坦底座设计 &带附加“排水环”的小排水通路,改善抗生素从膜上
Avoid false negative
26
封闭式薄膜过滤法
封闭式膜过滤法设计的目的是待 检测的液体可以在无菌条件下被 转移并过滤。
Avoid false positive
27
风险评估之设备因素
28
无菌检测设备因素
硬件----集菌仪
29
泵头 操控 清洁维护
无菌检测设备因素
耗材----套筒 • 符合各种不同产品的套筒选择 • 可靠的质量保证 • 更人性化的设计使操作更简便安全
Avoid False Negative
32
难溶性和不易过滤的样品
过滤单元基座滤膜阻塞
原因:粘性产品(高浓度的蛋白质),乳液,悬浮液
结果
• 操作过程中的高压可能导致过滤单元培养过程中出现破损 • 受压微生物的活力和生长受到影响 • 无菌套筒在操作时受压爆炸
解决方案
• 降低流速 • 加入助溶剂/表面活性剂 • 使用低吸附滤膜如PVDF膜 • 优化排液(带额外排水环设计的正确过滤装置)
7
无菌检查人员要求
操作者必须经过严格的培训 由于无菌检查是非常精确的实验,为保证实验的无菌过程和正确的结果
判断,操作人员必须经过专门的培训并通过资格认证。 (USP )
8
无菌检查中的风险控制
Environment/环境 Method/方法 Device /设备 Media/培养基
实验失败可能会带来导致实验批报废风险的假阳性结果 和导致用户使用危害的假阴性结果。
9
无菌实验的风险
假阳性
• 法规禁止不以发现造成污染的根本原因为目的而只是重复测试的行为 • 耗费时间和资源 • 法规部门的审查 • 大量的调查以找到问题根本原因,批报废,巨大损失
假阴性
• 人生命危害 • 召回 • 品牌受损,工厂关闭风险
10
风险评估之环境因素
11
Environment/环境因素
• 改良马丁培养基 • 硫乙醇酸盐培养基FTM
EP/USP/中国药典(2015版)
• 胰酪大豆胨液体培养基TSB----用于真菌和需气菌的培养 • 硫乙醇酸盐培养基FTM----厌氧菌和需气菌培养
分装至适宜的容器中,其装量与容器高度的比例 应符合培养结束后培养基氧化层(粉红色)不超 过培养基深度的1/2。在供试品接种前,培养基 氧化层的高度不得超过培养基深度的1/5
6
水 实验室数据
产品放行
环境监控
无菌检查的法规依据
中国药典(2010版一、二、三部),( 2015版药典征求意见版) USP美国药典 EU欧洲药典 JP日本药局方
TGA 澳大利亚治疗用品管理条例 PIC/S 药品检验药品检验合作公约 FDA 食品及药物管理局 EMEA欧洲药品管理局 PDA注射药物管理局 AAMI医疗仪器促进协会 ISO国际标准化组织 WHO世界卫生组织
33
Steritest EZ分类
蓝色底座套筒
• 不含抑菌成份并且易于过滤的产品
红色底座套筒
• 含抑菌成份的产品
绿色底座套筒
• 难溶性或不易过滤的产品
34
Steritest EZ分类
蓝色底座套筒
• 套筒:SAN (苯乙烯丙烯腈) • 管路:PVC(聚氯乙烯) • 滤膜:MCE(混合纤维素) • 排水盘:小排水通道 – 防止在压力下膜的“爆裂”。 • 膜和基座以热合方式焊接,膜和套筒以超声波焊接
17
无菌检测环境因素
洁净室中无菌检测,重视无菌保持,环境监测,降低人与和物料进出所 带来的影响
隔离器技术是一种提高无菌操作和保护操作者的解决方案。可以在无菌 检查时减少假阳性结果的风险
Avoid false positive
18
风险评估之方法因素
19
无菌检测方法因素
无菌检查方法必须在方法适应性验证之前得到确认
Conclusions结论
2
无菌检查介绍
3
为何进行无菌检查
GMP
对于每一批认为是无菌/无热源的药品,需要通过适当的实验室 检查来确定。
EP/USP
药品无菌检查系用于检查药典要求无菌的药品、 医疗器具、原料 、辅料及其他品种是否无菌的一种方法
中国药典
无菌检查法系用于检查药典要求无菌的药品、生物制品、医疗器 具、原料、辅料、及其他品种是否无菌的一种方法
实验人员应更换无菌衣(包括全身无菌服,头套,面罩,鞋套,手套, 口罩)进入洁净室。
每次进入工作区都需要更换新的无菌服。 实验过程中操作表面和操作人员的手套要定期用消毒剂消毒。 洁净室需要定期消毒灭菌和日常维护。
14
隔离器环境
15
LFH (class A)
隔离器环境监控
LFH (class A)
封闭式过滤器
24
封闭式膜过滤法
安装套筒
过滤样品
加培养基
润湿滤膜
25
冲洗
培养,判读
薄膜过滤法vs直接接种法
样品的抑菌性成分可通过冲洗步骤去除 避免产品和培养基的交互反应 可进行大容量产品的测试(从100ml到数升) 实验方法较直接接种法更灵敏 较直接接种法使用的更少培养基 油性物质可用乳化剂进行处理
Avoid false positive
Avoid False Negative
40
Steritest EZ 可靠的质量保证
COA证书
21.9 and 40 kGy. 40 kGy is not inhibiting thegrowth
41
风险评估之培养基因素
42
无菌检测培养基因素
中国药典(2010版)
于0.45μm。直径约为50mm。(2015版药典征求意见版)
直接接种法 • 直接接种法适用于无法用薄膜过滤法进行无菌检查的供试品,即取规 定量供试品分别等量接种至两周培养基中。