British Pharmacopoeia 2013 Levonorgestrel左炔诺孕酮英国药典2013
[宝典]欧洲药典附录3.1.3.
![[宝典]欧洲药典附录3.1.3.](https://img.taocdn.com/s3/m/f3357cec760bf78a6529647d27284b73f2423679.png)
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3%–1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3%–二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
英国国家处方集和其使用培训课件

文档仅供参考,不能作为科学依据,请勿模仿;如有不当之处,请联系网站或本人删除。
章节和单个药品举例
3 Respiratory system
3.1 Bronchodilators 3.1.1 Adrenoceptor agonists 3.1.1.1 Selective beta2 agonists 3.1.1.2 Other adrenoceptor agonists 3.1.2 Antimuscarinic bronchodilators 3.1.3 Theophylline 3.1.4 Compound bronchodilator preparations 3.1.5 Peak flow meters, inhaler devices and nebulisers
文档仅供参考,不能作为科学依据,请勿模仿;如有不当之处,请联系网站或本人删除。
How to use the BNF
Selecting the dose Selecting a suitable preparation Writing prescriptions Administering drugs Advising patients Monitoring drug treatment Identifying and reporting adverse drug reactions Finding significant changes in a new edition Nutrition Wound dressing ……
药学相关英文缩写

药学相关英文缩写API Active Pharmaceutical Ingredient 原料药(活性药物组分)BP British Pharmacopoeia 《英国药典》CAC Codex Alimentarius Committee (国际)食品法典委员会CBER FDA Center for Biologics Evaluation and Research (美国)FDA生物制品评价与研究中心CCD Certification Committee for Drugs(国家食品药品监督管理局)药品认证管理中心CDC Centers for Disease Control (美国)疾病控制中心CDE Center for Drug Evaluation (国家食品药品监督管理局)药品审评中心CDER FDA Center for Drug Evaluation and Research (美国)FDA药品评价与研究中心CDR Center for Drug Reevaluation (国家食品药品监督管理局)药品评价中心CDRH FDA Center for Devices and Radiological Health (美国)FDA医疗器械和辐射健康中心CFSAN FDA Center for Food Safety and Applied Nutrition (美国)FDA食品安全和应用营养中心CGMPs Current good manufacturing practices 现行良好制造规范;现行药品生产质量管理规范CMA Chinese Medical Association 中华医学会CNAO National Audit Office of the People's Republic of China 中华人民共和国审计署CNMA China Nonprescription Medicines Association 中国非处方药协会COS Certificate of Suitability (《欧洲药典》)适用性证书CP The Pharmacopoeia of the PRC 《中国药典》CPA China Pharmaceutical Association 中国药学会CPMA China Preventive Mediceine Association 中华预防医学会CVM FDA Center for Veterinary Medicine (美国)FDA兽药中心DE Drug evaluation 药品评价DEA Drug Enforcement Administration (美国)毒品强制执行管理局DIC Drug information center 药品信息中心DME Drug-metabolizing enzyme 药物代谢酶DMF Durg master file 药品主文件DUR Drug utilization review 药物利用评价DUR Drug use review 药物使用评论DHHS Department of Health and Human Services (美国)健康和人类服务部EFSA European Food Safety Authority 欧洲食品安全局ELA Establishment license application 机构许可申请EP European pharmacopoeia 《欧洲药典》EMEA European Agency for the Evaluation of Medicinal Products欧洲药品评价署EPA Environmental Protection Agency (美国)环境保护署EU European Union 欧盟FAO United Nations Food and Agricultural Organization 联合国粮农组织FD&C Act Federal Food, Drug and Cosmetic Act 《联邦食品、药品和化妆品法》FDA Food and Drug Administration (美国)食品药品管理局FSIS Food Safety Inspection Service (USDA) (美国农业部)食品安全检查服务局FTC Federal Trade Commission (美国)联邦贸易委员会GLP Good laboratory practice 良好实验室规范;药品非临床研究质量管理规范GMP Good manufacturing practices 药品生产和管理规范GPP Good pharmacy practice 良好药房规范;医疗机构制剂配制管理规范GPSP Good Post-marketing Surveillance Practice 良好上市后监督规范;上市药品监督规范GSP Good supplying practice 良好供应规范;药品经营质量管理规范HVAC Heating Ventilation Air Conditioning 空调净化系统ICH International Conference on Harmonization 国际(药品注册)协调会议IMIC International Medical Information Center 国际医学信息中心INCB International Narcotics Control Board 国际麻醉药品管制局IOM Institute of Medicine (美国国家科学院)医学研究所IPF International Pharmaceutical Federation 国际制药联合会IRC International Red Cross 国际红十字会IRCC International Red Cross Conference 国际红十字大会ISO International Standards Organization 国际标准化组织IVDC China Institute of Veterinary Drug Control 中国兽医药品监察所JIFSAN Joint Institute for Food Safety and Applied Nutrition (美国FDA)食品安全和应用营养联合研究所KFDA Korea Food and Drug Administration (韩国)食品药品管理局MHLW Ministry of Health, Labour and Welfare (日本)厚生劳动省MII China Ministry of Information Industry 中华人民共和国信息产业部MOF Ministry of Finance People's Republic of China 中华人民共和国财政部MoH Ministry of Health P.R.China 中华人民共和国卫生部MOST Ministry of Science and Technology of the People's Republic of China 中华人民共和国科学技术部NBS National Bureau of Statistics of China 国家统计局NCI National Cancer Institute (美国)国家癌症研究所NDA New drug application 新药申请NCTR FDA National Center for Toxicological Research (美国)FDA国家毒理学研究中心NIAID National Institute of Allergy and Infectious Diseases (美国)国家过敏症和传染病研究所NIDA National Institute on Drug Abuse (美国)国家药物滥用研究所NIH National Institute of Health (美国)国家健康研究所ORA FDA Office of Regulatory Affairs (美国)FDA监管事务办公室OTC Over the counter drug (Nonprescription drugs) 放在柜台上的药品(非处方药)PHS Public Health Service (美国)公众健康服务局Ph Gal Pharmacopoeia galisa 《法国药典》[拉]Ph J Pharmacopoeia japonica 《日本药典》PhI Pharmacopoeia internationalis 《国际药典》[拉]PLA Product license application 产品许可申请PRC People's Republic of China 中华人民共和国QA Quality assurance 质量保证QC Quality control 质量控制SAIC State Administration For Industry & Commerce 国家工商行政管理总局SAMHSA Substance Abuse and Mental Health Services Administration (美国)物质滥用和精神健康服务管理局SATCM State Administration of Traditional Chinese Medicine (中国)国家中医药管理局SETC State Economic and Trade Commission,PRC 中华人民共和国国家经济贸易委员会SFDA State Food and Drug Administration (中国)国家食品药品监督管理局SIPO State Intellectual Property Office of the People's Republic of China 国家知识产权局SOP Standard operating procedure 标准操作规程TFDA Thailand Food and Drug Administration (泰国)食品药品管理局TGA Therapeutic Goods Administration (澳大利亚)治疗产品管理局TQC Total quality control 全面质量控制UK United Kingdom (大不列颠)联合王国USP The united states pharmacopoeia 《美国药典》WHO United Nations World Health Organization (联合国)世界卫生组织WTO World Trade Organization 世界贸易组织ZDA Zhejiang Drug Administration 浙江省药品监督管理局。
1塞欧斯简介

塞欧斯简介
产品名:塞欧斯
成分:美洛昔康
规格:7.5mgХ6粒/板
适应症:
风湿性关节炎,类风湿性关节炎,骨关节炎,头痛,肌肉痛,痛经,牙痛,五官科疼痛
用法用量:
风湿、类风湿——15mg/天,根据治疗后反应,剂量可减至7.5mg/天,12周为一个疗程。
骨关节炎——7.5mg/天,假如需要,剂量可增至15mg/天
其他疼痛——7.5mg/天
禁忌症:对本品过敏者禁止使用
对于具有上消化道疾病史的病人和正在应用抗凝剂治疗的病人使用本药应该注意。
若出现消化道性溃疡或肠胃道出血应该停止使用本药。
产品特点:
1、新的高选择性环氧化酶—2(COX—2)抑制剂。
90年代初Needleman等人发现人体有两种不同的环氧化酶异构体,环氧化酶—1(COX—1)和环氧化酶—2(COX—2)。
COX—1是人体正常成分,它调节组织内生理性前列腺素合成,调节外周血管阻力,维持肾血流量,保护胃粘膜,调节血小板聚集等。
COX—2存在于炎症部位,如滑膜细胞,内皮细胞内,在外界刺激因子(IL —1)的作用下,促使炎症介质的释放,诱发炎症反应。
原有的抗炎镇痛药是通过抑制环氧化酶(COX)阻断花生四稀酸形成前列腺素,无选择的抑制了COX—1和COX—2,所以既有抗炎镇痛作用,也不可避免的出现胃肠道不良反应。
2、起效快——1小时即达到血浆药物峰值浓度,并且峰值浓度更高。
3、剂量小——7.5mg一粒(比较与其他镇痛药)
1 / 1。
各国药典比较

国际药典(Ph.Int) 国际药典(Ph.Int)
• 基本介绍
由联合国世界卫生组织主持编订。第一版于1951和 由联合国世界卫生组织主持编订。第一版于1951和 1955年分两卷用英、法、西班牙文出版,于1959出版增 1955年分两卷用英、法、西班牙文出版,于1959出版增 补本。第二版于1967年用英、法、俄、西班牙文出版。现 补本。第二版于1967年用英、法、俄、西班牙文出版。现 行版为第三版,于1979、1981、1988年、1994、2003分 行版为第三版,于1979、1981、1988年、1994、2003分 5卷出版,第1卷收载42项分析测试方法。第2、3两卷共 卷出版,第1卷收载42项分析测试方法。第2 收载药品383种。第4 收载药品383种。第4卷收载有关试验、方法的信息,以及 药品原料、赋形剂的一般要求和质量说明,以及剂型。第 5卷收载制剂通则以及药品原料和片剂的质量标准,这实 际上将涵盖目录上的有机合成药物以及一些抗疟疾药物及 其最广泛应用剂型的所有各论。
内容简介
• 美国药典正文药品名录分别按法定药名字
母顺序排列,各药品条目大都列有药名、 结构式、分子式、CAS登记号、成分和含量 结构式、分子式、CAS登记号、成分和含量 说明、包装和贮藏规格、鉴定方法、干燥 说明、包装和贮藏规格、鉴定方法、干燥 失重、炽灼残渣、检测方法等常规项目, 失重、炽灼残渣、检测方法等常规项目, 正文之后还有对各种药品进行测试的方法 和要求的通用章节及对各种药物的一般要 求的通则。可根据书后所附的USP和NF的 求的通则。可根据书后所附的USP和NF的 联合索引查阅本书。
图书版本
• • • • • •
最新版本: 最新版本: USP 33-NF 28重新发行版: 3328重新发行版 重新发行版: 2010年10月 日生效。 2010年10月1日生效。 增补版1 2010年 月出版,2010年10月 增补版1于2010年4月出版,2010年10月1日生效。 增补版2 2010年 月出版,2011年 增补版2于2010年6月出版,2011年1月1日生效。 U.S. Pharmacopeia / National Formulary《美国 Formulary《 药典/国家处方集》(简称USP/NF)。由美国政 药典/国家处方集》(简称USP/NF)。由美国政 府所属的美国药典委员会(The 府所属的美国药典委员会(The United States Pharmacopeial Convention)编辑出版。 Convention)编辑出版。
培美曲赛二钠英国药典

Pemetrexed Disodium HeptahydrateGeneral Notices(Ph. Eur. monograph 2637)C20H19N5Na2O6,7H2O 597.5 357166-29-1Action and useThymidylate synthetase inhibitor; cytostatic.Ph EurDEFINITIONDisodium(2S)-2-[[4-[2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedio ate heptahydrate.Content97.5 per cent to 102.0 per cent (anhydrous substance).CHARACTERSAppearanceWhite or almost white powder.SolubilityFreely soluble in water, very slightly soluble in anhydrous ethanol, practically insoluble in methylene chloride.IDENTIFICATIONCarry out either tests A, C, D, E or tests B, C, D, E.Results The 1H NMR spectrum obtained is qualitatively similar to the 1H NMR spectrum obtainedwith pemetrexed disodium heptahydrate CRS; disregard the peak located at approximately 5.0 ppm for the comparison.C. It gives reaction (a) of sodium (2.3.1).D. Enantiomeric purity (see Tests).E. Water (see Tests).TESTSSolution SDissolve 0.56 g in carbon dioxide-free water R and dilute to 10.0 mL with the same solvent.Appearance of solutionSolution S is not more opalescent than reference suspension II (2.2.1) and not more intensely coloured than reference solution GY4 or Y4(2.2.2, Method II).pH (2.2.3)7.5 to 8.4 for solution S.Enantiomeric purityLiquid chromatography (2.2.29). Prepare the solutions immediately before use or store them at 2-8 °C for not more than 24 h.Solution A Dissolve 8 g of β-cyclodextrin R in 900 mL of water for chromatography R. Add 15 mLof triethylamine R then 6 mL of phosphoric acid R and adjust to pH 6.0 with phosphoric acid R. Dilute to 1000 mL with water for chromatography R.Test solution Dissolve 12 mg of the substance to be examined in water for chromatography R and dilute to 50.0 mL with the same solvent.Reference solution (a) Dissolve 6 mg of pemetrexed for system suitability CRS (containing impurity E) in water for chromatography R and dilute to 25.0 mL with the same solvent.Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with water for chromatography R.Dilute 3.0 mL of this solution to 10.0 mL with water for chromatography R.Column:∙—size: l = 0.25 m, Ø = 4.6 mm;∙—stationary phase: octadecylsilyl silica gel for chromatography R (5 µm) with a pore size of 12 nm; ∙—temperature: 40 °C.Mobile phase acetonitrile R, solution A (5:95 V/V).Flow rate 1.0 mL/min.Detection Spectrophotometer at 230 nm.Injection 50 µL.Run time 1.5 times the retention time of pemetrexed.Relative retention With reference to pemetrexed (retention time = about 30 min):impurity E = about 0.94.System suitability:∙—symmetry factor: maximum 2.0 for the principal peak in the chromatogram obtained with reference solution (b);∙—peak-to-valley ratio: minimum 5.0, where H p = height above the baseline of the peak due to impurity E and H v = height above the baseline of the lowest point of the curve separating this peak from the peak due to pemetrexed in the chromatogram obtained with reference solution (a).Calculation of percentage contents:∙— for impurity E, use the concentration of pemetrexed disodium heptahydrate in reference solution (b).Limit:∙—impurity E: maximum 0.3 per cent.Column rinse The following program is given for information only.Use a gradient column rinse before column storage or after 30 sample injections to avoid build-up on the column. If a drifting baseline is observed, allow additional time for equilibration with the mobile phase. If a blank chromatogram exhibits broad humps, perform a gradient column rinse.Rinsing solution A water for chromatography R.Rinsing solution B acetonitrile R1.Related substancesLiquid chromatography (2.2.29). Prepare the solutions immediately before use or store them at 2-8 °C for not more than 24 h.Solution A 1.45 g/L solution of ammonium formate R in water for chromatography R, adjusted to pH 3.5 with anhydrous formic acid R.Test solution Dissolve 20 mg of the substance to be examined in water for chromatography R and dilute to 100.0 mL with the same solvent.Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with water for chromatography R.Dilute 1.0 mL of this solution to 10.0 mL with water for chromatography R.Reference solution (b) In order to prepare impurities B and C in situ, dissolve 30 mg of the substance to be examined in 10.0 mL of a 4.0 g/L solution of sodium hydroxide R, heat at 70 °C for 40 minutes and allow to cool. Dilute 1.0 mL of the solution to 10.0 mL with water for chromatography R.Reference solution (c) Dissolve the contents of a vial of pemetrexed impurity mixture CRS (impurities A and D) in 1.0 mL of water for chromatography R.Column:∙—size: l = 0.15 m, Ø = 4.6 mm;∙—stationary phase: base-deactivated octylsilyl silica gel for chromatography R (3.5 µm).Mobile phase:∙—mobile phase A: acetonitrile R, solution A (5:95 V/V);∙—mobile phase B: acetonitrile R, solution A (30:70 V/V);Flow rate 1.0 mL/min.Detection Spectrophotometer at 250 nm.Injection 20 µL.Identification of impurities Use the chromatogram supplied with pemetrexed impurity mixture CRS and the chromatogram obtained with reference solution (c) to identify the peaks due to impurities A and D;use the chromatogram obtained with reference solution (b) to identify the peaks due to impurities B andC.Relative retention With reference to pemetrexed (retention time = about 26 min): impurity A = about 0.82;impurity B = about 0.87; impurity C = about 0.88; impurity D = about 0.90.System suitability Reference solution (b):∙—peak-to-valley ratio: minimum 1.5, where H p = height above the baseline of the peak due to impurity B and H v = height above the baseline of the lowest point of the curve separating this peak from the peak due to impurity C.Calculation of percentage contents:∙— for each impurity, use the concentration of pemetrexed disodium heptahydrate in reference solution(a).Limits:∙—impurities A, D: for each impurity, maximum 0.15 per cent;∙—unspecified impurities: for each impurity, maximum 0.10 per cent;∙—total: maximum 0.6 per cent;∙—reporting threshold: 0.05 per cent.Heavy metals (2.4.8)Maximum 20 ppm.Solvent mixture acetone R, water R (40:60 V/V).0.250 g complies with test H. Prepare the reference solution using 0.5 mL of lead standard solution (10ppm Pb) R.Water (2.5.12)19.5 per cent to 22.1 per cent, determined on 0.050 g.Bacterial endotoxins (2.6.14)Less than 0.17 IU/mg.ASSAYLiquid chromatography (2.2.29). Prepare the solutions immediately before use or store them at 2-8 °C for not more than 24 h.Acetate buffer Mix 1.7 mL of glacial acetic acid R and 900 mL of water for chromatography R, adjust to pH 5.3 with a 760 g/L solution of sodium hydroxide R in water for chromatography R and dilute to 1000 mL with water for chromatography R.Test solution Dissolve 30.0 mg of the substance to be examined in water for chromatography R and dilute to 200.0 mL with the same solvent.Reference solution Dissolve 30.0 mg of pemetrexed disodium heptahydrate CRS in water forchromatography R and dilute to 200.0 mL with the same solvent.Column:∙—size: l = 0.15 m, Ø = 4.6 mm;∙—stationary phase: base-deactivated octylsilyl silica gel for chromatography R (3.5 µm); ∙—temperature: 30 °C.Mobile phase acetonitrile R, acetate buffer (11:89 V/V).Flow rate 2.0 mL/min.Detection Spectrophotometer at 285 nm.Injection 20 µL.Run time Twice the retention time of pemetrexed (retention time = about 3 min).Calculate the percentage content of C20H19N5Na2O6 taking into account the assigned contentof pemetrexed disodium heptahydrate CRS.IMPURITIESSpecified impurities A, D, E.Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034).It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10.Control of impurities in substances for pharmaceutical use): B, C.A.(2S)-2-[[4-[2-(2-amino-1-methyl-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino ]-pentanedioic acid,B.(2S,2′S)-2,2′-[[(5R)-2,2′-diamino-4,4′,6-trioxo-1,4,4′,6,7,7′-hexahydro-1′H,5H-5,6′-bipyrrolo[2,3-d]pyrimi dine-5,5′-diyl]bis(ethylenebenzene-4,1-diylcarbonylimino)]dipentanedioic acid,C.(2S,2′S)-2,2′-[[(5S)-2,2′-diamino-4,4′,6-trioxo-1,4,4′,6,7,7′-hexahydro-1′H,5H-5,6′-bipyrrolo[2,3-d]pyrimi dine-5,5′-diyl]bis(ethylenebenzene-4,1-diylcarbonylimino)]dipentanedioic acid,D.(2S)-2-[[(4S)-4-[[4-[2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-carboxybutanoyl]amino]pentanedioic acid,E.(2R)-2-[[4-[2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentane dioic acid.Ph Eur。
2013年6月FDA批准新药

BRISDELLE (NDA #204516) HYDROXYZINE 8 PAMOATE (ANDA #201507) MORPHINE SULFATE 9 (ANDA #202104) 编 药物名称 号 7
甲磺酸帕罗西汀 扑姆酸羟嗪 硫酸吗啡 有效成分
抗抑郁药 镇静,抗焦虑 镇痛药 适应症
批准 批准 批准
仿制
口服液
草酸依地普仑
IMIPRAMINE PAMOATE 仿制 (ANDA #202338) NORETHINDRONE (ANDA #200980) NORETHINDRONE (ANDA #201483) TESTOSTERONE CYPIONATE (ANDA #201720) TRANEXAMIC ACID (ANDA #201580) WARFARIN SODIUM (ANDA #200104) LAMOTRIGINE (ANDA #200694) LAMOTRIGINE (ANDA #202887) 仿制 仿制 仿制 仿制 仿制 仿制 仿制 仿制 仿制 仿制 仿制 类型
06/18/201 3 06/18/201 3 06/26/201 3 06/27/201 3 06/27/201 3 06/28/201 3
FLUDEOXYGLUCOSE 仿制 F18 (ANDA #203811) AMMONIA N 13 (ANDA 仿制 #203812) SODIUM FLUORIDE F仿制 18 (ANDA #203605)
仿制 仿制 仿制
片剂 片剂 注射剂
利鲁唑 利鲁唑 盐酸拓扑替康
本品用于治疗影响肌肉力量的神经系统 疾病:肌萎缩侧索硬化症。 本品用于治疗影响肌肉力量的神经系统 疾病:肌萎缩侧索硬化症。 用于治疗小细胞肺癌。
2013年7月FDA批准新药
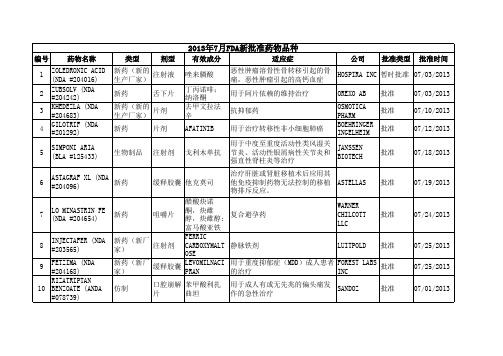
用于成人有或无先兆的偏头痛发 GLENMARK 作的急性治疗 GENERICS 用于成人有或无先兆的偏头痛发 APOTEX INC 作的急性治疗 用于成人有或无先兆的偏头痛发 AUROBINDO 作的急性治疗 PHARMA LTD 用于成人有或无先兆的偏头痛发 NATCO 作的急性治疗 PHARMA LTD 用于成人有或无先兆的偏头痛发 INVAGEN 作的急性治疗 PHARMS 用于偏头痛急性发作的治疗 APOTEX INC
42
仿制
胶囊
用于干燥综合征病人的口干症状 ROXANE 治疗。 用于治疗注意缺陷多动障碍 KUDCO IRELAND KUDCO IRELAND AUROLIFE PHARMA LLC CARACO
批准
07/08/2013
43
仿制
缓释片
批准
07/09/2013
44
仿制
缓释片
盐酸哌甲酯 硫酸右苯丙 胺 瑞格列奈 瑞格列奈 盐酸二甲双 胍
类型 仿制
剂型 片剂
有效成分 非那雄胺 去氧孕烯; 炔雌醇
适应症 用于治疗前列腺增生
公司 SUN PHARMA GLOBAL
批准类型 批准
批准时间 07/01/2013
31
仿制
片剂
避孕药
NOVAST LABS 批准 LTD WATSON LABS 批准 INC ALEMBIC PHARMS LTD ACTAVIS ELIZABETH TARO
6
ASTAGRAF XL (NDA 新药 #204096) LO MINASTRIN FE (NDA #204654) INJECTAFER (NDA #203565)
缓释胶囊 他克莫司
批准
07/19/2013
美国药典 阿司匹林 中英文对照

About 143 °C (instantaneous method).
IDENTIFICATION
First identification A, B.
Second identification B, C, D.
A.Infrared absorption spectrophotometry {2.2.24).
Reference solution (a) Dissolve 50.0 mg of salicylic acid R in the mobile phase and dilute to 50.0 ml with the mobile phase.Dilute 1.0 ml of this solution to 100.0 ml with the mobile phase.
Limits:
--any impurity: for each impurity, not more than the area of the principal peak in the chromatogramobtained with reference solution (a) (0.1 per cent);
ASSAY
In a flask with a ground-glass stopper, dissolve 1.000 g in 10 ml of ethanol (96 per cent) R. Add 50.0 ml of 0.5 M sodium hydroxide. Close the flask and allow to stand for 1 h.
Comparison acetylsalicyiic acid CRS.
B.To 0.2 g add 4 ml of dilute sodium hydroxide solution R and boil for 3 min. Cool and add 5 ml of dilute sulphuric add R
关于药业名词简称

关于药业名词简称美国药典Pharmacopoeia of the United States USP英国药典British Pharmacopoeia BP欧洲药典European Pharmacopoeia Eur Ph国际药典Pharmacopoeia Internationals Ph Int世界卫生组织WHO美国国家食品与药物管理局Foodand Drug Administration FDAcGMP是英文Current GMP,即动态药品生产管理规范,也为现行药品生产管理规范,它要求在产品生产和物流的全过程都必须验证药品生产质量管理规范GMP药物非临床研究质量管理规范GLP Good Laboratory Practice药物临床试验质量管理规范GCP中药材生产质量管理规范GAP品经营质量管理规范GSP药品使用质量管理规范GUP验证管理规范GVP标准管理规程SMP标准操作规程SOP:Standard Operating Procedure验证试验规程VTPCLP 表示设备在线清洁程序(clean line procedure)VP 表示验证方案(validation protocol)IQ 表示安装确认(installation qualification)OQ 表示运行确认(operational qualification)PQ 表示性能确认(performance qualification)PV 表示产品验证(product validation)或工艺验证(process validation) GVR 表示验证总报告(general validation report)P 表示计划或方案R 表示报告S 表示小结验证总计划(Validation Master Plan验证报告(Validation Report验证小结(Validation Summary 全面生产管理TPM全面成本管理TCM现代品质管理MQMQA质量保证quality assuranceOC质量控制qualitycontrolTQC total quality contr01全面质量控制TQM total quality management全面质量管理QMS质量管理体系环境管理体系EMS职业健康安全管理体系OHSMS质量管理Quality manatgement QM质量体系Quality system QS质量控制Quality control QC质量保证Quality assurance QA质量控制图Quality control charts世界卫生组织WHO国际标准化组织ISO质量管理体系标准ISO 9000ISO 9000 质量管理和质量保证标准的选择和使用指南ISO 9001 质量体系设计开发生产安装和服务的质量保证模式ISO 9002 质量体系生产和安装的质量保证模式ISO 9003 质量体系最终检验和试验的质量保证模式ISO 9004 质量管理和质量体系要素的指南环境管理体系标准ISO 14000英国标准协会BSI国际劳工组织ILO环境标准environmen-tal standard环境质量environmental quality环境评价environmental as-sessment非处方药Nonpre-scription Drug Over The Counter OTC药物不良反应adverse drugreaction ADR泡罩式包装PTP条带状热封合包装SP聚氯乙烯polyethylene echloride PVC而聚丙烯polypropylen PP聚乙烯polyethylene PETPM Total Product Maintenance全员生产维修RCM(Reliability Centered Maintenance)预防性维修①事后维修----BM(Breakdown Maintenance)②预防维修--PM(Preventive Maintanance)③改善维修--CM(Corrective Maintanance)④维修预防--MP(Maintenance Prevention)⑤生产维修--PM(Productive Maintenance)十分钟内完成开车准备和规格调换single-minuteexchangeofdie SMEDPQCDSM:效率productivity 质量quality 费用或成本cost 交货期delivery 安全safety环境卫生和劳动情绪morale5S:整理sort整顿setinorder清洁明亮shine标准化的清理standardized clean up保持sustain生产要素简称5M:man 人machine 机器materical 物料money 财method 方法准时生产JIT just in time成组技术GT group technology计算机集成制造系统CIMS computer integrated manufacturing system 计算机辅助设计CAD computer aided design计算机辅助制造CAM computer aided manufacturing计算机辅助工艺设计CAPP computer aided process planning企业资源计划ERP enterpise resource planning电子数据交换EDI主生产计划master production schedule MPS物料需求计划MRP车间作业管理PAC production activity control车间作业控制SFC shopfloorcontr01生产计划大纲production planning PP质量控制管理信息系统软件QCMAIN quality control management information system实验室信息管理系统软件LIMS laboratory information management system 制造资源计划Manufacturing Resource Planning MRP浓的Fortis e 稀的Dilums a nm 重的Pondero-SUS a am 轻的kevis e白的Albus a nih 黄的Flavus aum 红的Ruber bm brum 黑的Niger m rum液状的Liquidus aum 升华的Sublimatus a um片剂Tablets颗粒剂Granules胶囊剂Capsules丸剂Pills微丸Micropills滴丸Dropping Pills中药丸剂Traditional Chinese Medicinal Pill注射剂Injections滴眼剂Eye Drops软膏剂Ointments栓剂Suppositories液体药剂Liquid Preparations胶体溶液剂Colloidal Preparations混悬剂Suspensions乳剂Emulsions气雾剂Aerosprays控缓释制剂Controlled Release Preparations靶向制剂Target Praparations生物技术药品制剂Biotechnological Praparations固体分散技术Solid Dispersion Technic药物包合技术Drug Inclusion Technic药物微囊化技术Drug Microcapsulation Technic在线灭菌(SIP)前验证(Prospective Validation同步验证(Concurrent Validation回顾性验证(Retrospective Validatoin再验证(Revalidation空调净化系统即采暖通风和空气调节系统(Heating Ventilation and Air Conditioning HVAC)高效空气过滤器(High Efficiency Pariticle Airfilter HEPADOP 用粒子计数器监测泄漏。
国家药品监督管理局通告2020年第30号——国家药监局关于发布仿制药参比制剂目录(第二十七批)的通告

国家药品监督管理局通告2020年第30号——国家药监局关于发布仿制药参比制剂目录(第二十七批)的
通告
文章属性
•【制定机关】国家药品监督管理局
•【公布日期】2020.04.29
•【文号】国家药品监督管理局通告2020年第30号
•【施行日期】2020.04.29
•【效力等级】部门规范性文件
•【时效性】现行有效
•【主题分类】药政管理
正文
国家药品监督管理局通告
2020年第30号
国家药监局关于发布仿制药参比制剂目录(第二十七批)的
通告
经国家药品监督管理局仿制药质量与疗效一致性评价专家委员会审核确定,现发布仿制药参比制剂目录(第二十七批)。
特此通告。
附件:仿制药参比制剂目录(第二十七批)
国家药监局
2020年4月29日附件
仿制药参比制剂目录(第二十七批)。
国外药典介绍

USP–NF 的不断 修订
修订公告 IRA拟议的修订 说明
勘误表
2019/12/16
美国药典的修订
USP–NF 不断进行修订。 修订包括 USP–NF 年度修订和每年两 次增补,以及 USP 网站上的加速修订。 USP 使用加速修订过程 加快修订美国药典–国家处方集 (USP–NF)。加速修订包括修订公 告、临时修订声明 (IRA) 和勘误表。 是 USP 最快的修订途径,可取代在 USP–NF 及其增补(印刷版 和在线版)中发布的标准。 在 USP 网站上发布的修订公告指示 其正式日期和纳入正式出版物中的日期。 IRA 在 PF 中发布,征求公众意见期为 90 天。 在意见(如果有) 通过审查并且 IRA 得到相关专家委员会的批准后,最终 IRA 将发 布在 USP 网站上。 与修订公告一样,IRA 可取代在印刷版和在线 版的 USP–NF 及其增补中发布的标准。 IRA 被纳入下一个可用的 USP–NF 或增补中。 是指在 USP–NF 或其增补中发布的文字有误,不能准确地反映专 家委员会批准的预期要求。 勘误表发布在网站上,并立即成为正 式版本。 勘误表被纳入下一个可用的正式出版物中。
通则提供各论中所用术语的定义,以及解释各论要求所需 的信息。
2019/12/16
美国药典USP内容介绍
美国药典-国家处方集(USP-NF)是两个法定药品标准 USP 中提供关于原料药和制剂的质量标准。
NF 中提供关于辅料的质量标准。 各论中提到的测试和程序将在 USP-NF 附录中予以详细说明。
2019/12/16
2019/12/16
日本药典简介
简介
《日本药典》(The Jepanese Pharmacopoeia)的名称是《日本药局方 》,英文缩写JP 。由日本药局方编辑委 员会编制,厚生省颁布执行。1886年6月 25号颁布第一版,1887年7月1日开始实
世界卫生组织儿童标准处方集

WHO Model Formulary for ChildrenBased on the Second Model List of Essential Medicines for Children 2009世界卫生组织儿童标准处方集基于2009年儿童基本用药的第二个标准目录WHO Library Cataloguing-in-Publication Data:WHO model formulary for children 2010.Based on the second model list of essential medicines for children 2009.1.Essential drugs.2.Formularies.3.Pharmaceutical preparations.4.Child.5.Drug utilization. I.World Health Organization.ISBN 978 92 4 159932 0 (NLM classification: QV 55)世界卫生组织实验室出版数据目录:世界卫生组织儿童标准处方集基于2009年儿童基本用药的第二个标准处方集1.基本药物 2.处方一览表 3.药品制备 4儿童 5.药物ISBN 978 92 4 159932 0 (美国国立医学图书馆分类:QV55)World Health Organization 2010All rights reserved. Publications of the World Health Organization can be obtained fromWHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: ******************). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the aboveaddress(fax:+41227914806;e-mail:*******************).世界卫生组织2010版权所有。
国外药典介绍

包括欧盟在内共有37个成员。最新加入的成员:波兰(2006)。
包括世界卫生组织在内,加上7个欧洲国家(阿尔巴尼亚、亚美尼亚、 白俄罗斯、格鲁吉亚、哈萨克斯坦共和国、摩尔多瓦、俄罗斯联邦和乌 克兰)和16个非欧国家(阿尔及利亚、澳大利亚、巴西、加拿大、中国、 以色列、马达加斯加、马来西亚、摩洛哥、塞内加尔、叙利亚、突尼斯、 美国),共24个观察员。 最新加入的观察员:新加坡(HSA)(2012)、阿根廷、亚美尼亚和 摩尔多瓦(2008)、白俄罗斯共和国(2007)、俄罗斯联邦和乌克兰 (2006)。
第二卷
各论举例、各论、按字母的索引页 总索引 每卷均有侧面黑色索引标示
2014-11-3
欧洲各册内容介绍
增补举例:
目录、凡例、分析方法通论、试剂、通则、制剂各论、中草药及其 制剂各论、顺势疗法制剂各论、各论、增补内容简介(包括新增内 容、修订内容和更正内容)
索引:可以帮助用户索引到包含在最新增补本中的内容。
查看
在通则、附录及各论中,凡是和欧洲药典完全一致的, 均加入 标志,并在名称项下用斜体字标示出了
欧洲药典中的索引页码。
2014-11-3
欧洲药典简介
欧洲药典简介
《欧洲药典》由欧洲药品质量委员会(EDQM)编辑出版,有英文和法 文两种法定文本。其全称为European Pharmacopoeia,缩写为Ph.Eur.。
历史及版 本
USP于1820年出第一版,1950年以后每5年出一次修订版。年成立药典修订委员会, 并对第一版药典进行了修订,1906年,FDA将药典指定为官方标准,经过多次版 本的升级,到2014年已出版至第37版。从2002年(USP25-NF20)起每年一版。 NF1883年第一版,1980年15版起并入USP,但仍分两部分,前面为USP,后面为 NF。每一版本的《美国药典》包含4卷及2个增补版。美国药典最新版为USP 37NF 32;2013年12月出版;2014年5月1日生效。
医药行业专业英语词汇

医药行业专业英语词汇(非常有用)FDA和EDQM术语: CLINICAL?TRIAL:临床试验? ANIMAL?TRIAL:动物试验? ACCELERATED?APPROVAL:加速批准? STANDARD?DRUG:标准药物? INVESTIGATOR:研究人员;调研人员PREPARING?AND?SUBMITTING:起草和申报? SUBMISSION:申报;递交? BENIFIT(S):受益? RISK (S):受害? DRUG?PRODUCT:药物产品? DRUG?SUBSTANCE:原料药? ESTABLISHED?NAME:确定的名称? GENERIC?NAME:非专利名称? PROPRIETARY?NAME:专有名称;? INN (INTERNATIONAL?NONPROPRIETARY?NAME):国际非专有名称? ADVERSE?EFFECT:副作用? ADVERSE?REACTION:不良反应? PROTOCOL:方案? ARCHIVAL?COPY:存档用副本? REVIEW?COPY:审查用副本? OFFICIAL?COMPENDIUM:法定药典(主要指USP、?NF).? USP(THE?UNITED?STATES?PHARMACOPEIA):美国药典 NF(NATIONAL?FORMULARY):(美国)国家处方集? OFFICIAL=PHARMACOPEIAL=?COMPENDIAL:药典的;法定的;官方的? AGENCY:审理部门(指FDA)? IDENTITY:真伪;鉴别;特性? STRENGTH:规格;规格含量(每一剂量单位所含有效成分的量)? LABELED?AMOUNT:标示量? REGULATORY?SPECIFICATION:质量管理规格标准(NDA提供)? REGULATORY?METHODOLOGY:质量管理方法? REGULATORY?METHODS?VALIDATION:管理用分析方法的验证 COS/CEP?欧洲药典符合性认证 ICH(International?Conference?on?Harmonization?of?Technical?Requirements?for?Registrat ion?of?Pharmaceuticals?for?Human?Use)人用药物注册技术要求国际协调会议 ICH文件分为质量、安全性、有效性和综合学科4类。
药典基本知识

第二节 中国药典
中国药典(英文缩写:Ch.P)的沿革
我国建国后先后出版了八版药典: 1953、1963、1977、1985、1990、1995、2000、2005
年版 现行中国药典为2005年版,2005.7.1开始实施 1953年版:共一部,收载药品531种 1963-2000年版:分一、二两部
取样精密度与准确度 称定:指所称取重量的百分之一 精密称定:指所称取重量的千分之一 精密量取:指量体积移液管的精密度 “约”若干:指不超过规定量的±10%
精确度
恒重
除另有规定外,系指供试品连续两次干燥或炽灼后的重量差 异在0.3mg以下的重量;干燥至恒重的第二次及以后各次称重均在 规定条件下继续干燥1小时后进行;炽灼至恒重的第二次称重应在 继续炽灼30分钟后进行。
量 的比例,此外,根据需要可采用下列符号:
%(g/g) 表示溶液100g中含有溶质若干克; %(ml/ml) 表示溶液100ml中含有溶质若干毫升; %(ml/g) 表示溶液100g中含有溶质若干毫升; %(g/ml) 表示溶液100ml中含有溶质若干克; 液体的滴 系指在20℃时,以1.0ml水为20滴进行换算。
最新版本为USP(30)-NF(25)
《英国药典》 (BP)
始于1618年,近现代世界上最早的药典,是英 国制药标准的重要来源。英国药典不仅提供了药 用和成药配方标准以及公式配药标准,而且也展 示了许多明确分类并可参照的欧洲药典专著。
英国药典出版周期不定,最新版本BP(2007)
《日本药局方》(JP)
《国际药典》(Ph.Int)
由联合国世界卫生组织WHO主持编订。 第一版于1951和1955年分两卷出版。 第二版于1967年出版。 第三版目前有5卷 国际药典更多关注发展中国家的需要,并且只推
英国药典制剂通则

英国药典制剂通则摘要:一、英国药典制剂通则概述二、英国药典制剂通则的主要内容1.原料药的质量要求2.制剂的生产和质量控制3.药品的包装、储存和运输4.药品的标签和说明书5.药品的注册和审批三、英国药典制剂通则在我国的应用和建议正文:在英国,药品的研发、生产、销售和使用都需遵循严格的法规要求,以确保药品的安全、有效和质量可控。
英国药典制剂通则(British Pharmacopoeia Preparation General Monographs,简称BP GM)是英国药典中的一部分,旨在为药品制剂的生产和质量控制提供全面、详尽的技术指南。
一、英国药典制剂通则概述英国药典制剂通则是一部涵盖药品制剂生产全过程的指南,包括原料药的质量要求、制剂的生产和质量控制、药品的包装、储存和运输、药品的标签和说明书以及药品的注册和审批等方面。
这些通则旨在确保药品在研发、生产、使用和监管全过程中符合相关法规和标准。
二、英国药典制剂通则的主要内容1.原料药的质量要求:英国药典对原料药的质量要求非常严格,包括化学纯度、生物活性和杂质控制等方面。
这些要求旨在确保原料药的安全性和有效性。
2.制剂的生产和质量控制:英国药典制剂通则对制剂的生产工艺、质量控制环节和检验方法等方面进行了详细规定。
这些规定有助于企业规范生产过程,确保产品质量。
3.药品的包装、储存和运输:英国药典制剂通则对药品的包装材料、储存条件、运输方式和时间等方面提出了明确要求。
这些要求有助于保护药品的质量和稳定性,确保药品在储存和运输过程中的安全。
4.药品的标签和说明书:英国药典制剂通则规定,药品标签和说明书需包含药品的适应症、用法用量、禁忌、不良反应等信息。
这些信息有助于患者和医务人员正确使用药品,降低潜在风险。
5.药品的注册和审批:英国药典制剂通则对药品的注册和审批流程进行了详细规定。
这些规定旨在确保药品在市场上销售前符合相关法规和标准,保障患者用药安全。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
LevonorgestrelGeneral Notices(Ph. Eur. monograph 0926)C21H28O2 312.5 797-63-7Action and useProgestogen.PreparationsLevonorgestrel TabletsLevonorgestrel and Ethinylestradiol TabletsPh EurDEFINITION13-Ethyl-17-hydroxy-18,19-dinor-17α-pregn-4-en-20-yn-3-one.Content98.0 per cent to 102.0 per cent (dried substance).CHARACTERSAppearanceWhite or almost white, crystalline powder.SolubilityPractically insoluble in water, sparingly soluble in methylene chloride, slightly soluble in ethanol (96 per cent).IDENTIFICATIONA. Specific optical rotation (see Tests).B. Infrared absorption spectrophotometry (2.2.24).Comparison levonorgestrel CRS.TESTSSpecific optical rotation (2.2.7)-35 to -30.∙—mobile phase B: acetonitrile R1;Flow rate 0.7 mL/min.Detection Spectrophotometer at 215 nm and, for impurity O at 200 nm.Injection 50 µL.Identification of impurities Use the chromatograms supplied with levonorgestrel for system suitability CRS and the chromatograms obtained with reference solution (a) at 215 nm to identify the peaks due to impurities A, H, K, M and S, and at 200 nm to identify the peak due to impurity O; use the chromatogram obtained with reference solution (c) to identify the peak due to impurity B; use the chromatogramobtained with reference solution (d) to identify the peak due to impurity U.Relative retention With reference to levonorgestrel (retention time = about 20 min):impurity H = about 0.5; impurity U = about 0.8; impurity K = about 0.85; impurity A = about 0.91;impurity M = about 0.95; impurity O = about 1.16; impurity B = about 1.26; impurity S = about 1.9.System suitability:∙—signal-to-noise: minimum 60 for the principal peak in the chromatogram obtained with reference solution (b);∙—peak-to-valley ratio: minimum 3.0, where H p = height above the baseline of the peak due to impurity M and H v = height above the baseline of the lowest point of the curve separating this peak from the peak due to impurity A.Limits:∙—correction factors: for the calculation of content, multiply the peak areas of the following impurities by the corresponding correction factor: impurity A = 0.4; impurity M = 3.1; impurity O = 2.6; ∙—impurities A, K: for each impurity, not more than 3 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.3 per cent);∙—impurity B: not more than 3 times the area of the corresponding peak in the chromatogram obtained with reference solution (c) (0.3 per cent);∙—impurity O at 200 nm: not more than 3 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.3 per cent);∙—impurities M, S: for each impurity, not more than twice the area of the principal peak in the chromatogram obtained with reference solution (b) (0.2 per cent);∙—impurity U: not more than twice the area of the corresponding peak in the chromatogram obtained with reference solution (d) (0.2 per cent);∙—impurity H: not more than 1.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.15 per cent);∙—unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (0.10 per cent);∙—sum of impurities other than O: maximum 1.0 per cent;∙—disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.05 per cent).Loss on drying (2.2.32)Maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 105 °C.Sulfated ash (2.4.14)Maximum 0.1 per cent, determined on 1.0 g.ASSAYDissolve 0.200 g in 45 mL of tetrahydrofuran R. Add 10 mL of a 100 g/L solution of silver nitrate R. After1 min, titrate with 0.1 M sodium hydroxide, determining the end-point potentiometrically (2.2.20). Carryout a blank titration.1 mL of 0.1 M sodium hydroxide is equivalent to 31.25 mg of C21H28O2.STORAGEProtected from light.IMPURITIESSpecified impurities A, B, H, K, M, O, S, U.Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): C, D, G, I, J, L, N, P, Q, R, T.A. 13-ethyl-17-hydroxy-18,19-dinor-17α-pregna-4,8(14)-dien-20-yn-3-one,B. 13-ethyl-17-hydroxy-18,19-dinor-17α-pregn-5(10)-en-20-yn-3-one,C. 13-ethyl-3-ethynyl-18,19-dinor-17α-pregna-3,5-dien-20-yn-17-ol,D. 13-ethyl-18,19-dinor-17α-pregn-4-en-20-yn-17-ol (3-deoxolevonorgestrel),G. 13-ethyl-6α,17-dihydroxy-18,19-dinor-17α-pregn-4-en-20-yn-3-one (6α-hydroxylevonorgestrel),H. 13-ethyl-6β,17-dihydroxy-18,19-dinor-17α-pregn-4-en-20-yn-3-one (6β-hydroxylevonorgestrel),I. 13-ethyl-10,17-dihydroxy-18,19-dinor-17α-pregn-4-en-20-yn-3-one (10-hydroxylevonorgestrel),J. 13-ethyl-17-hydroxy-18,19-dinor-17α-pregn-4-en-20-yne-3,6-dione (6-oxolevonorgestrel),K. 13-ethyl-17β-hydroxygon-4-en-3-one (18-methylnandrolone),L. 13-ethylgon-4-ene-3,17-dione (levodione),M. 13-ethyl-17-hydroxy-18,19-dinor-17α-pregna-4,6-dien-20-yn-3-one (Δ6-levonorgestrel),N. 13-ethylgon-5(10)-ene-3,17-dione (Δ5(10)-levodione),O. 13-ethyl-17-hydroxy-5α-methoxy-18,19-dinor-17α-pregn-20-yn-3-one (4,5-dihydro-5α-methoxylevonorgestrel),P. 13-ethyl-17-hydroxy-18,19-dinor-17α-pregn-5-en-20-yn-3-one (Δ5-levonorgestrel),Q. 13-ethyl-3-methoxygona-2,5(10)-dien-17β-ol,R. 13-ethyl-3-methoxygona-2,5(10)-dien-17-one,S. 13-ethyl-3-methoxy-18,19-dinor-17α-pregna-3,5-dien-20-yn-17-ol,T. 13-ethyl-3-methoxy-18,19-dinor-17α-pregna-2,5(10)-dien-20-yn-17-ol,U. 17-hydroxy-19-nor-17α-pregn-4-en-20-yn-3-one (norethisterone).。