核磁共振13C谱操作步骤
第5章 13C 核磁共振

(3) 空间效应
化学位移对分子的几何形状非常敏感,相隔几个键的碳, 如果它们空间非常靠近,则互相发生强烈的影响,这种短程的 非成键的相互作用叫空间效应。
例:苯乙酮中若乙酰基邻近有甲基取代,则苯环和羰基的共 平面发生扭曲,羰基碳的化学位移与扭曲角有关。
CH3 O CH3 O CH3
决定化学位移的主要因素。使共振移向低场。
(3) 邻近各向异性屏蔽 在所观察的碳核周围,有许多其他核 B 存在,这 些核电子环流的各向异性影响,对碳原子可产生正 或负的效应。 NB 只取决于 B 的性质与几何位置,而
与核N性质无关。
(4) 溶剂和介质
除位移试剂对化学位移影响很大外,溶剂影响较
小。
第5章
133C
核磁共振原理
5.2
5.3
13C
13C
NMR测定方法
NMR参数
5.4 各类碳的化学位移 5.5
13C
NMR的解析及应用
5.1
13C核磁共振原理
C H —— 构成有机化合物的主要元素
在有机物中,有些官能团不含氢 例如: 的结构信息
12C 13C
-C=O,-C=C=C-,-N=C=O等
利用偏共振去偶技术可以在保留NOE使信号增
强的同时,仍然看到CH3四重峰,CH2三重峰和CH
二重峰,不与1H直接键合的季碳等单峰。
通过比较宽带去偶和不完全去偶的碳谱可以得
出各组峰的峰形,从而可以判断分辨出各种CH基 团,峰的分裂数与直接相连的氢有关一般遵守n+1 规律。
二氯乙酸13C NMR图谱 质 子 宽 带 去 偶 偏 共 振 去 偶
15N 等)灵敏度很低。后来发现,在具有两种核的
自旋体系中,可以把高灵敏核( 1H )的自旋极化
核磁共振实验室操作规程

核磁共振实验室操作规程1. 打开空压机电源(电源开关向上推);2。
打开空压机的排气口;3。
取下磁体样品腔上端的盖子4。
将样品管插入转子中,然后用定深量筒控制样品管的高度。
这个步骤不能缺少,如果样品管插入的太长,有可能会损坏探头。
二、常规样品的测试1。
双击桌面上的图标,进入topspin2.1主界面,调出最近做过的一张谱图。
2. 在命令行中输入“new" 回车,跳出一窗口,建立一个新的实验,输入name、Solvent、Experiment等实验参数。
其中1H选proton,13C选C13CPD,点击OK.3.“ej”回车,打开气流,放入样品管;”ij"回车,关闭气流,样品管落入磁体底部。
4.“lock solvent(选用的溶剂)”回车,进行锁场,待锁场完场后,进行探头匹配调谐和自动匀场。
7。
“ased”回车,调出采样参数,根据具体的样品设置NS、DS、D1等。
8。
“getprosol”回车,调脉冲参数。
所有参数不用改动,尤其PL1不能修改。
9.“rga”回车,自动增益,“zg"回车,开始采样。
带采样完毕后,按进行傅立叶变换和自动相位校正13. 实验完毕后,脱锁、停止旋转,输入“ej”命令,把样品管吹出,取出样品管;“ij”关掉气流。
14、对样品数据处理,然后打印图谱填写实验记录三、注意事项1。
实验前空压机必须打开,保证气流流畅;2. 打开气流前,查看样品腔的上盖是否取下;3。
不要带具有磁性的物质靠近磁体;4. 本台电脑数据转移使用光盘,不允许使用其它工具如U盘等常见问题1。
购买核磁管和氘代溶剂?答:核磁管和常用的溶剂可以在校试剂库购买.也可向试剂代理公司购买,例如:青岛腾龙、百灵威、创美、柏卡、腾达远等公司,联系方式网上搜索即可。
2。
核磁管如何清洗?答:a.用原先的溶剂(非氘代)清洗;b。
不知道原先的溶剂时,则用二氯甲烷、丙酮清洗;c.如果仍洗不掉,在适当的溶剂中用超声波振荡,或者用棉棒擦洗(可能会损坏核磁管);d.如果必要的话,用混酸(HNO3/H2SO4,不可用铬酸)溶解;e。
13CNMR实验操作步骤

1H NMR 实验操作步骤1.测试前的准备1.1 样品的配制①使用干净和干燥的样品管以免污染样品②取少量样品放入管中③加入0.5mL 氘代溶剂,摇动,使样品溶解④使用量高器来确定样品的放置高度,待测2.谱仪的调整1.1 仪器的启动①打开谱仪谱仪24h不间断运行,一般情况下不需要开启谱仪②打开空气压缩机和空气净化器1.2 样品放入磁体①打开匀场界面,点击lift on-off,磁体上有气流声,将装有样品管的转子放入探头上方,点击lift on-off核磁管平稳进入探头1.4 核的选择①在操作窗口打命令edsp (回车)②在F1下面选择1H③在F2下面选择off④点击SA VE1.3 建立新的实验数据目录名①命令edc (回车)②设置新实验的名称(NAME)、实验号(EXPNO)、处理号(PROCNO)③点击SA VE1.5 探头调谐①打命令wobb (回车)②在采样窗口观察,打命令A(回车)③调tuning和matching④点击stop⑤点击return1.6 锁场①打命令Lock (回车)②点击所用氘代溶剂1.7 匀场调节Z,Z2,Z3,X,Y3.参数的选择1.1 采样参数选择①打命令ased (回车)②点击SA VE1.2 处理参数选择①SI=32K,LB=04.1H谱的测量1.1 采样①打命令zg (回车)②在采样窗口看FID信号,打A(回车)1.2 付立叶变换①打命令FT (回车)1.3 调相位①点击phase ②点击biggest ③点击pH0和pH1来调整相位④return ⑤save & retcen1.4 校正零点①点击calibrate1.5 积分,①点击integrate1.6 定最大、最小值,点击utilities1.7 打标题①打命令setti (回车)②打入标题③点击File中的save④点击File中的Exit1.8 定打印图的谱宽点击dp11.9 观看打印图打命令V (回车)1.10 打印图打命令plot (回车)13C NMR实验操作步骤步骤1和2中除1.4核的选择,1.5探头调谐和1H谱不一样,其它步骤同1H谱。
布鲁克核磁共振培训-13C-nmr-chs

杂核的性质
T1 驰豫时间的范围很宽:
14 14
9
107
CH
132 9.2
需要较长的 D1 时间,总的实验时间也较长
化学位移的分布范围也很广:
~ 250ppm 59Co: ~ 5000ppm 需要较短的90度激发脉冲 激发的偏共振效应较大
13C:
杂核实验技术
天然丰度和灵敏度低:
累加FID (S/N)NS = NS *(S/N)1 (S/N)100 = 10*(S/N)1 (S/N)1000 = 30*(S/N)1
144 143 142 141 140 139 138 137 136 135 134 133 132 131 130 129 128 127 126 125
ppm
对反转门控去偶的 13C 谱进行去卷积分析
Data set: D:/data/dmo/nmr/Strychnin/45/pdata/1 Fit type: Gaussian Fit Frequency ppm Hz Width ppm Hz Intensity Area %Lor.
decoupling during relaxation delay ('gated decoupling')
no decoupling at all
ቤተ መጻሕፍቲ ባይዱ180
170
160
150
140
130
120
110
100
90
80
70
60
50
40
30
ppm
去偶模式
不要只分析峰强 度,因为由于去 偶不完全的原因 半峰宽可能各不 相同
Bruker用户培训班
13C-核磁共振光谱 -2

例2.有两个有机物A、B,分子式均为C5H10,由气相色谱分离, 其13C谱数据如下: A:13(q),17(q),26(q),118(d),132(s) B:13(q),22(q),31(t),108(t),147(s) 解: 1、两个碳氢化合物均含有一个Ω,可知其分子内含有一个 双键(在100~160范围内均有两个峰)。 2、它们都没有等价的碳原子,由各峰的多重性可指示出分 子中所需的的10个氢原子; A化合物中含有3个甲基及1个 sp2杂化的CH;而B中有两个甲基,1个sp3的CH2和一个sp2 的CH2
酮,醛>羧酸>羧酸衍生物(酰胺、酰氯、酸酐、酯)
苯环碳谱出峰数目:
无对称性: 单取代: 对位取代: 6个峰 4个峰 4个峰
X
X
Y X Y
R
Y
邻位相同取代基: 3个峰 间位三相同取代基:2个峰 单个苯环不可能只有5个碳峰!
X X
X X X
例1. C5H11Cl ,COM如图。
22.0q 25.7d 41.6t 43.1t
2
照射3.91,7.40有NOE增益
3 1 2
1
1
COOH
OCH3 C 6 H4
1Hale Waihona Puke 0503、 C5H10O
HOCH2-CH2-CH2CH=CH2
C4H6O2 1
CH3-CH=CH-COOH
3 1
注:1H-NMR中,δ12处的积分高度对应于1个H的吸收峰未画出来。
解: 1、不饱和度Ω=0,所以,该化合物为链状饱和氯代烷。 2、13C-NMR谱中共有四个峰,而分子式中有五个碳,故分子结 构中有对称因素。 δ22.0处的甲基峰强度很大,表明有两个等价 的甲基(CH3)。 3、根据偏共振去偶13C谱中的裂分情况可知,分子中有如下的结 构单元: CH3(22.0q),CH(25.7d),CH2(41.6t),CH2(43.1t)
300 兆核磁碳谱操作步骤

300兆核磁碳谱操作步骤1.装样。
将装有样品(13C:100~300 mg)及0.5 ml氘代溶剂的核磁管(溶液高度不低于3.5 cm)用绸布擦干净,插入转子中,用量规(高度定为1.8 cm)确定好高度。
2.放样。
打开磁体顶端的安全盖,在BSMS控制板上点击LIFT-ON/Off(灯亮),听到磁体中部有气流声时,放入核磁管(切记:未听到气流声绝对不可放入样品!!!)。
再点击LIFT-ON/OFF(灯灭),样品进入磁体。
3.调实验指南。
点击菜单栏的Spectrometer,选Data Acquisition Guide,出现界面(左下图):4.建新实验。
点击“New Eexperiment”图标,出现界面(右上图):①NAME 输入测试者姓名(英文字母缩写)②EXPNO 实验采样号1000(下一个样品就是1001,依此类推)③USER 输入所在课题组名,一般以导师名。
④其余部分不改动。
注: 如果已经建立了碳谱文件夹(USER),则在左侧的数据浏览器中(D盘)找出USER 及NAME并选中最后的实验号,点中鼠标左键直接拖入即可。
5.锁场。
点击Lock,回车,选择所用氘代试剂(如:CDCl3,Acetone,DMSO等);锁场需等待几分钟,待状态栏显示finished后,再进行下一步操作。
注:连续测相同溶剂的样品时,其它样品锁场可在BSMS面板直接点击LOCK-ON/OFF 完成锁场。
6.探头调谐。
点击Probe Match/Tune,选第三项,等待仪器自动调节探头的谐振调谐(tuning)与阻抗匹配(matching),待显示“finished”后再进行下一步。
注: 1.若出现问题,即长时间不能结束,在命令行键入stop,退出操作软件,重新登陆,再重复探头调谐。
2.连续测相同溶剂的样品时,其它样品可省略这一操作。
7.匀场。
与氢谱操作相同。
8.累加次数。
键入NS(累加次数),回车,对话框显示NS值,NS数值与样品浓度有关,测定13C-NMR谱时,样品浓度越大则NS越小。
13CNMR 核磁共振碳谱化学位移总览表==
INEPT谱中不出现季 碳的信号
CH3和CH为正峰,而 CH2为负峰
只出现CH的正峰
CH3、CH2、CH为正值
2)DEPT法
DEPT谱中也不 出现季碳的信号
DEP-45°谱,CH3、 CH2和CH的峰均为正峰 DEPT-90°谱,只出现 CH的正峰 DEPT-135°谱,CH3 和CH为正峰,而CH2的 峰为负 常规宽带质子去偶13C谱
3.炔烃的化学位移值
炔基碳为sp杂化,其化学位移介于sp3与sp2杂化碳之间,为 67-92ppm。
4.芳环碳和杂芳环碳的δC值
芳环碳的化学位移值一般在120-160ppm范围内,峰往往出现 在较低场,这点与脂肪族季碳峰在较低场是类似的。
稠环芳烃和杂环芳烃中芳环碳的化学位移值也在苯及衍生物的δC值 范围内。
3.能给出不连氢碳的吸收峰 在1H NMR中不能直接观察到C=O、C=C、C≡C、C=N、季 碳等不连氢基团的吸收信号,只能通过相应基团的化学位移值、 分子式不饱和度等来判断这些基团是否存在。而13C NMR谱可 直接给出这些基团的特征吸收峰。由于碳原子是构成有机化合 物的基本元素,因此从13C NMR谱可以得到有关分子骨架结构 的信息。 4.不能用积分高度来计算碳的数目 13C NMR的常规谱是质子全去偶谱。对于大多数碳,尤其是 质子化碳,它们的信号强度都会由于去偶的同时产生的NOE效 应而大大增强,如甲酸的去偶谱与偶合谱相比,信号强度净增 近2倍。季碳因不与质子相连,它不能得到完全的NOE效应, 故碳谱中季碳的信号强度都比较弱。由于碳核所处的环境和弛 豫机制不同,NOE效应对不同碳原子的信号强度影响差异很大, 因此不等价碳原子的数目不能通过常规共振谱的谱线强度来确 定。
4.13C金属原子的偶合常数
核磁共振波谱法-碳谱

1
2
图谱简单。虽然碳原子与氢原子之间的偶合常数较大,但由于它们的共振频率相差很大,所以-CH-、-CH2-、-CH3等都构成简单的 AX、AX2、AX3体系。因此可用一级谱解析,比氢谱简单的多。
三 13C NMR谱图
典型碳谱图谱 最常见的碳谱是宽带全去偶谱,每一种碳原子只有一条谱线。在去偶的同时,由于核的NOE效应,信号更为增强。但不同碳原子的T1不同,这对峰高影响不一样;不同核的NOE也不同; 峰高不能定量地反映碳原子数量。
*
5、缺电子效应
如果碳带正电荷,即缺少电子,屏蔽作用大大减弱,化学位移处于低场。
1
例如:叔丁基正碳离子(CH3)3C+的达到 327.8ppm。这 个效应也可用来解释羰基的13C 化学位移为什么处于较低 场,因为存在下述共振:
2
*
6、电场效应
在含氮化合物中,如含 -NH2的化合物,质子化作用后生成 – NH3+,此正离子的电场使化学键上电子移向 或碳,从而使它们的电子密度增加,屏蔽作用增大,与未质子化中性胺相比较,其 或碳原子的化学位移向高场偏移约 0.5-5ppm。这个效应对含氮化合物的碳谱指认很有用。
用于区分碳类型的一种技术。 INEPT称为低灵敏核的极化转移增强法。 DEPT称为不失真的极化转移增强法。 即去偶呈现单峰,又可以区分出碳的类型。 INEPT通过脉冲把灵敏度高的1H的自旋极化转移到13C核上去,13C信号强度增加4倍,进行测定,故灵敏度好。 DEPT谱法是INEPT法的一种改良方法。 DEPT的信号强度仅与脉冲的倾倒角有关。通过改变照射1H的的倾倒角(),使作45,90 ,135 变化并测定其13C NMR谱
*
磁各向异性的基团对核屏蔽的影响,可造成一定的差异。这种差异一般不大,而且很难与其它屏蔽的贡献分清(这一点与1H不同)。 但有时这种各向异性的影响是很明显的。如异丙基与手性碳原子相连时,异丙基上两个甲基由于受到较大的各向异性效应的影响,碳的化学位移差别较大,而当异丙基与非手性碳原子相连时,两个甲基碳受各向异性效应的影响较小,其化学位移的差别也较小。
第4章 13C核磁共振

N B
+ 介质
核外电子云密度大, 抗磁大,在高场共振,δ小。
σ σ σ
抗磁为核外局部电子环流产生的抗磁屏蔽,与外磁场方向相反。 顺磁为各项异性的非球形电子环流产生的顺磁屏蔽,与外场方向相同。 NB为邻近核B的各向异性对核的屏蔽作用,取决于B的性质和几何位置。
介质表示溶剂和介质的影响。
14092(高斯)
23500 (高斯)
25MHz(13C)
H splitting
弧度 H 26753
秒 高斯
1 I= 2
C splitting
C =6726
弧度 秒 高斯
E
1 I= 2
B0
13C核磁共振碳谱的特点
适合含有长碳链或含碳原子化合物的分析, 特别可以得 到1H谱不能直接测得的羰基、腈基和季碳的信息 . 由于13C核的化学位移范围(δc=0~240 ppm)远大 于H核的化学位移范围(δH=0~15 ppm),因此13C 谱分辨率高. 由于自然界中13C核的丰度太低,另外13C的旋磁比只 有1H核的1/4, 13C NMR的灵敏度比1H NMR要低得多.
第四章 核磁共振碳谱
核磁共振碳谱的特点 核磁共振碳谱的测定方法 13C的化学位移 13C的自旋偶合及偶合常数 核磁共振谱的解析及应用 二维核磁共振谱
4.1 13C核磁共振碳谱的特点
B0
60MHz(1H) 15MHz(13C) 100MHz (1H)
1 1 13 19 I 1H 7 C 9 F 2 15 31 29 N 15 P 14 Si 8
求仪器有多脉冲器,射频脉冲有45o、90o、135o等相位移控 制装置。 1)INEPT法(低灵敏核的极化转移增强法) 2)DEPT法(无畸变极化转移增强法)
核磁共振波谱检测的基本流程

核磁共振波谱检测的基本流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!核磁共振波谱检测(NMR,Nuclear Magnetic Resonance)是一种用于分析物质结构和性质的重要技术。
波谱分析-研究生-13C核磁共振谱概述

CW灵敏度低,做13C-NMR(1%)极其困难。 解决方法: (1)降低实验温度,使N1↑ (2)增强磁场强度,使N1↑ (3)增大射频功率,可提高灵敏度,但受饱和的限制。 (4)连续波(CW)仪器上加累加器(CaT),n次累加 后
可提高信信噪比 倍N ,对磁场的稳定度要求较高。 (5) 进行13C富集. (6) 增加样品浓度与样品体积.
T1 Processes
dM z M z M0
dt
T1
Mz = Mo ( 1 - e-t/T1 )
Time Domain NMR Signal ——> NMR Spectrum
§1.3 有关脉冲缚里叶变换核外共振的一些概念
(Pulse Fourier Transform NMR, PFTNMR)
S 限制
磁隙限制
在通常的连续波核磁共振(CW-NMR)中,射频频率 是固定的,且连续作用在样品上,采用扫频或扫场方式记录 信号,即在某一时刻谱仪只对一个很窄的频带起作用强共振。
一:关于PFT-NMR
脉冲序列
频谱分析
根据频谱分析,样品所受的不再是的单一的射频频率fc, 而是以fc为中心的一个频谱,频谱的频率间隔为1/T,宽度为 若干千Hz,这个频率范畴远远超过化学位移的范围。
二: 受激跃迁过程 在外磁场的作用下, 粒子发生受激跃迁后, 粒子差数是
按指数规律减小的,如无其它因素影响, 粒子受激跃迁将使 两个能级上的粒子是趋于相等, 这时看不到吸收信号。
第一节 基本原理
三: 驰豫过程
从上面开始的时候,自旋系统吸收能力较多,信 号较大,但很快上下能级粒子数相等,信号消失,这种现 象叫“饱和”。
类核矩之间的能量交换驰豫。其驰豫时间为:
C13核磁共振谱全解说课讲解

+aa
No Image
4.炔烃
烷基取代炔烃的化学位移在δ65~90范围 内。乙炔碳的化学位移为71.9ppm,炔碳值 的经验计算公式:Ci=71.9+ ∑Ai • 式中71.9为炔碳的化学位移值;Ai为取代参数。 炔烃的通式为 :
• 所谓宽带去耦( broad band decoupling)又叫质 子噪音去耦proton noise decoupling,是在扫描 时,同时用一个强的去耦射频在可使全部质子共 振的频率区进行照射,使得1H对13C的耦合全部去 掉。质子宽带去耦简化了图谱,每种碳原子都出 一个单峰。一般来说,在分子中没有对称因素和 不含F和P等元素时,每个C原子都出一个峰,互 不重叠。而且由于多重耦合峰合并成单峰,提高 了信噪比,使信号增强。
0.5~1000Hz的质子去偶频率,使与13C核直接相连 的1H和13C之间还留下一些自旋偶合作用,偶合常 数N+11J规C-H律比,C原H来3显的示偶四合重谱峰小,等,2J等,3J. 则表现不出来,按
• 偏共振去偶使远程偶合完全消失,而仅观 察到一个键13C-1H偶合的残余裂分,此时每 个13C信号的多重性与相连质子数有关,符 合(n+1)规则,这样甲基、亚甲基、次甲
h核共振的中心频率051000hz的质子去偶频率使与13c核直接相连13c之间还留下一些自旋偶合作用偶合常ch比原来的偶合谱小j则表现不出来按n1规律ch13c信号的多重性与相连质子数有关符合n1规则这样甲基亚甲基次甲基和季碳分别以四重峰三重峰二重峰和单峰出现
C13核磁共振谱全解
• C,H 为有机化合物的骨架元素。 • 在有机物中,有些官能团不含氢: • 例如:-C=O,-C=C=C-,-N=C=O,季碳
300兆核磁操作步骤

---------------------------------------------------------------最新资料推荐------------------------------------------------------300兆核磁操作步骤300 兆核磁操作步骤 BRUKER DPX 300 13C 谱: 100~300 mg)及适当氘代溶剂 (约1. 预备。
将装有样品(1H 谱: 1~40 mg;0.5 ml)的核磁管(溶液高度一般不低于 4cm)用绸布擦干净,插入转子中,用量规(1. 8cm) 量好高度。
2. 放样。
打开 BSMS 控制面板,点击 Lift On-Off(灯亮) ,听到磁体中部有气流声时,方可放入核磁管(切记:未听到气流声绝对不可放入样品!!! ) 。
再点击Lift On-Off(灯灭) 使样品进入磁体。
将 BSMS 面板最小化(点右上角小圆点) 。
3. 建立新的文件或查找已有文件。
键入 edc,回车,出现以下界面:Current data Parameter name of current data set Experiment number PROCNO 1 DU /u USER guest Owner of data TYPE nmr NAME EXPNO processed data number disk unit Data type ①Name 输入自己的姓名 (英文字母) ②Expno 实验采样号 1(下一个样品就是 2,依此类推) ③其余部分不需要改动。
④如果您已经建立了自己的文件名,则从 File 中点search 寻找自己的名字,点中最后的实验号,点 append 后再点1/ 5apply,将出现主界面 Xwin-nmr 将实验号改为新的号码,并保存。
4. 锁场。
键入 Lock 后回车,选择所用氘代试剂 (如:CDCl3, Acetone, DMSO 等) ,待主界面(Xwinnmr 下面) 显示finished 后,打开 Lock 和 BSMS 两个界面,点击 spin on-off,该灯亮而且不闪则表明样品旋转稳定。
核磁操作指南.

5. “ atma ”回车,进行探头匹配调谐。
6. “ edte ”回车,设置气流在温度不超过313K ;点击set max,调节max power为probe heater后的off ,使其变为on ,打开控温。实验温度超过313K时需通入氮气。一般一维谱不用控温,二维谱常用。
四、谱图的处理
1.手动相位校正:若自动相位校正效果不佳,可点击手工校正图标进行校正,通过0、1来相位校正,校正完毕,保存,退出。
分别表示近端和远端的相位校正
保存并退出
不保存退出
2.定标:如测试试剂中还有TMS ,选取TMS的峰,点击图标,把TMS峰值定为O。
3.标峰:点击图标
,进入标峰界面。
对选定的范围进行标峰
2.在命令行中输入“ new ”回车,跳出一窗口,建立一个新的实验, NAME、Solvent、Experiment等实验参数。其中1H选13C选13C定量谱选13Cdept
Байду номын сангаас谱选择
。点击OK。
3. “ ej ”回车,打开气流,放入样品管; ”ij”回车,
关闭气流,样品管落入磁体底部。
4. “ lock solvent(选用的溶剂”回车,进行锁
标定单个峰
删除所有标定的峰值
保存并退出
不保存并退出4.
积分:点击积分图标“
”
,进入积分界面
对某一特定峰进行积分
积分面积拆分
保存并退出
不保存退出
五、画图及谱图的输出
在命令行输入“
plot ”或点击图标
进入作图界面
超导核磁共振操作指南(Bruker AVANCEIII 400 MHz)选定区域对插入的谱图放大某一区域插入谱图具体步骤:插入标题1.点击图标,在谱图上点击鼠标右键,出一对话框可以看到有一系列的参数。点击Eidt,出现另一对话框,用于限定谱图的显示范围,峰值、积分值的显示以及修改坐标轴的位置、颜色等。点击1D/2D-Edit,出现一对话框,此对话框主要用来调整谱图,提高谱图的质量2.如果需要把谱图的局部放大,点击,在谱图比较空的区域按住鼠标左键拖拽。可以看到另一个谱图,然后点击,对所要放大的区域截取。结合5.1操作,调整放大的谱图。3.谱图的输出(1)点击主界面菜单栏的“file”下的子菜单“eport”,跳出一对话框,保存谱图(PDF或第6页共7页
核磁共振C谱(13C-NMR)13C-NMR

核磁共振C谱(13C-NMR)13C-NMR⼆、13C-NMR的去偶技术2、偏共振去偶三、13C的化学位移及影响因素3、影响δC的因素(2)诱导效应(3)共轭效应(4)空间效应四、13C-NMR的解析例1、推测C8H18的结构例2:未知物分⼦式为C7H9N,核磁共振碳谱如下,推测其结构。
不饱和度U=41号峰为饱和碳,为四重峰,故是CH3,按?C值可能为CH3Ph2~7号峰为sp2杂化碳,从多重峰的组成及?C值看是双取代苯上的碳除以上两个结构单元CH3和C6H4外,还剩⼀个NH2,故可能结构为CH3PhNH2结构C的取代苯上的碳只出4个峰,可排除。
A和B可⽤计算碳原⼦?C值,排除A。
化合物为B核磁共振碳谱(13CNMR)13CNMR核磁共振的特点13CNMR的去偶技术13CNMR的化学位移及影响因素13C-NMR谱图解析⼀、13CNMR核磁共振的特点化学位移范围宽,分辨能⼒⾼。
1H-NMR常⽤δ值范围为0-15ppm。
13C-NMR常⽤δ值范围为0-250ppm(正碳离⼦达300ppm),其分辨能⼒远⾼于1H-NMR。
13C-NMR给出各种类型碳(伯、仲、叔、季)的共振吸收峰。
不能⽤积分曲线获取碳的数⽬信息。
13C-1H偶合常数较⼤,1JCH=110~320Hz。
偶合谱的谱线交迭,谱图复杂。
常规13CNMR谱为全去偶谱,所有的碳均为单峰。
灵敏度低。
13C峰度仅1.11%,⽐1H信号弱得多,约1/6400。
为提⾼信号强度,采⽤:(1)增加样品浓度,以增⼤样品中13C核的数⽬。
(2)采⽤共振技术,利⽤NOE效应增强信号强度。
(3)多次扫描累加,是最常⽤的有效⽅法。
(4)改变仪器测量条件。
13C-NMR谱中,1JCH约100-200Hz,偶合谱的谱线交迭,谱图复杂。
常采⽤⼀些特殊的测定⽅法。
1、质⼦宽带去偶(噪⾳去偶)和NOE增强:双共振技术⽤射频场(B1)照射碳核,使其激发产⽣13C核磁共振吸收,同时附加另⼀个射频场(B2,去偶场)使其覆盖全部质⼦的共振频率范围,⽤强功率照射使所有质⼦达到饱和,从⽽使1H对13C的偶合全部去掉。
[整理版]核磁共振氢、碳普解析的步骤
![[整理版]核磁共振氢、碳普解析的步骤](https://img.taocdn.com/s3/m/76bbb61402d8ce2f0066f5335a8102d276a26118.png)
三. 核磁共振氢谱核磁共振技术发展较早,20世纪70年代以前,主要是核磁共振氢谱的研究和应用。
70年代以后,随着傅里叶变换波谱仪的诞生,13C—NMR的研究迅速开展。
由于1H—NMR的灵敏度高,而且积累的研究资料丰富,因此在结构解析方面1H—NMR的重要性仍强于13C—NMR。
解析图谱的步骤1.先观察图谱是否符合要求;①四甲基硅烷的信号是否正常;②杂音大不大;③基线是否平;④积分曲线中没有吸收信号的地方是否平整。
如果有问题,解析时要引起注意,最好重新测试图谱。
2.区分杂质峰、溶剂峰、旋转边峰(spinning side bands)、13C卫星峰(13C satellite peaks) (1)杂质峰:杂质含量相对样品比例很小,因此杂质峰的峰面积很小,且杂质峰与样品峰之间没有简单整数比的关系,容易区别。
(2)溶剂峰:氘代试剂不可能达到100%的同位素纯度(大部分试剂的氘代率为99-99.8%),因此谱图中往往呈现相应的溶剂峰,如CDCL3中的溶剂峰的δ值约为7.27 ppm处。
(3)旋转边峰:在测试样品时,样品管在1H-NMR仪中快速旋转,当仪器调节未达到良好工作状态时,会出现旋转边带,即以强谱线为中心,呈现出一对对称的弱峰,称为旋转边峰。
(4)13C卫星峰:13C具有磁距,可以与1H偶合产生裂分,称之为13C卫星峰,但由13C 的天然丰度只为1.1%,只有氢的强峰才能观察到,一般不会对氢的谱图造成干扰。
3.根据积分曲线,观察各信号的相对高度,计算样品化合物分子式中的氢原子数目。
可利用可靠的甲基信号或孤立的次甲基信号为标准计算各信号峰的质子数目。
4.先解析图中CH3O、CH3N、、CH3C=O、CH3C=C、CH3-C等孤立的甲基质子信号,然后再解析偶合的甲基质子信号。
5.解析羧基、醛基、分子内氢键等低磁场的质子信号。
6.解析芳香核上的质子信号。
7.比较滴加重水前后测定的图谱,观察有无信号峰消失的现象,了解分子结构中所连活泼氢官能团。
13C核磁课程

因NOE使谱线强度增大。
42
方法:
调节去偶频率v2恰好等于某质子的共振吸收频率,
且B2场功率又控制到足够小(低于宽带去偶采用功
率)时,与该质子直接相连的碳会发生全去偶变成
尖锐的单峰,并因NOE而使谱线强度增大。 选择某特定的质子作为去耦对象,用去耦频率照 射该特定的质子,使被照射的质子对13C的耦合去 掉,13C成为单峰,以确定信号归属。分子中其它 碳核,仅受到不同程度的偏移照射,产生偏共振 去偶。所测得的 13C NMR称为质子选择性去偶谱。
时间累加不至分辩率下降。
22
2)溶剂 大多数氘代试剂都含有碳,会出现溶剂
13C共振吸收峰,且由于D与13C间偶合,溶剂 13C共振吸收峰往往被裂分为多重峰。分析
时,要先识别出溶剂峰,常用溶剂的13C的溶
剂δ 值见表
重水(D2O)不含碳,在13C NMR谱中无 干扰,是理想的极性溶剂。
23
常用有机溶剂的13C核的化学位移和峰形
13C核磁共振波谱
Nuclear Magnetic Resonance Spectrascopy (13C NMR)
13C
1
5.1
13C
NMR的特点
1.灵敏度低 NMR的信号可用下式表示
S ∝ N
nB0γ3I(I+1)
T
式中:S/N---信噪比(信号与噪声的比值) B0---磁场强度 n---共振核数目 γ---磁旋比 I---自旋量子数 T---热力学温度,单位为K。
25
5.2.2
13C
NMR的去偶技术
由于自身连接的H和邻位C上H的偶合
作用,使13C谱非常复杂难以辨认,故常采
用去偶合的方法,除去H核对13C共振吸收
波谱分析之13C核磁共振与二维核磁共振篇

第5章 13C 核磁共振与二维核磁共振大多数有机化合物分子的骨架是由碳原子组成的,通过13C 核磁共振(13C -NMR)研究有机分子的结构是十分有用的。
但由于13C 的天然丰度只占1.108%,所以含碳化合物的13C -NMR 信号很弱,致使13C -NMR 的应用受到了极大的限制。
六十年代后期,脉冲付立叶变换(PFT)谱仪的出现,才使13C -NMR 成为可实用的测试手段。
近年来13C -NMR 技术及应用有了飞速的发展。
成为化学、生物、医学和化工等领域不可缺少的分析工具。
5.1 13C 核磁共振基本原理13C -NMR 的原理与1H -NMR 是一样的。
在一个频率为υ的射频场中,只要13C 核的实受磁场B 满足υ=γπ2B,13C 就发生核磁共振。
在此式中γ是13C 核的旋磁比。
γC ≅γH4。
核磁共振的信号强度∝[NB 02γ3I(I+1)]/T N 一共振核的数目 γ一旋磁化I 一自旋量子数 T 一绝对温度由上式可见,共振信号与旋磁比的立方成正比。
而γC ≅γH4,13C 的天然丰度也只有1.1%。
所以13C 核的测定灵敏度是很低的,大约是H 1核的1/6000,所以测定很困难。
为了提高信号强度,常采用下述方法: (a)提高仪器灵敏度。
(b)提高仪器外加磁场强度和射频场功率。
但是射频场过大容易发生饱和。
这两条都受到限制。
(c)增大样品浓度,增大样品体积,以增大样品中13C 核的数目。
(d)采用双共振技术,利用NOE 效应增强信号强度。
(e)多次扫描累加,这是最常用的有效方法。
在多次累加时,信号S 正比于扫描次数,而噪音N 正比于扫描次数,所以S/N(信噪比,即信号强度) 正比于扫描次数。
若扫描累加100次,S/N 增大10倍。
13C 的测定灵敏度很低,信号弱,必须累加多次。
为了解决这个问题,只有采用脉冲付立叶变换NMR 仪。
脉冲付立叶变换NMR 仪采用脉冲发射,可以同时使各种不同的13C 核发生跃迁,便它们同时被激发。
13C核磁课程
因NOE使谱线强度增大。
42
方法:
调节去偶频率v2恰好等于某质子的共振吸收频率,
且B2场功率又控制到足够小(低于宽带去偶采用功
率)时,与该质子直接相连的碳会发生全去偶变成
尖锐的单峰,并因NOE而使谱线强度增大。 选择某特定的质子作为去耦对象,用去耦频率照 射该特定的质子,使被照射的质子对13C的耦合去 掉,13C成为单峰,以确定信号归属。分子中其它 碳核,仅受到不同程度的偏移照射,产生偏共振 去偶。所测得的 13C NMR称为质子选择性去偶谱。
近年来又发展了几种区别碳原子级数
(伯、仲、叔、季)的方法。往往分子中
有几个不同类C,就有几组峰,能直接
提供有机物碳骨架信息。 较之氢谱信
息更丰富、结论清楚。
11
4. 不能用积分高度计算碳的数目
13C
NMR 常规谱是质子全去偶谱。对大多
数碳,尤其是质子化碳,信号强度由于去
偶同时产生的NOE效应而大大增强。
24
c (ppm) 96.0
峰重数 s
8.
13C
13CNMR化学位移参照标准
NMR基本原理与1H NMR相同。
在外磁场中13C核吸收电磁波,从低能级跃 迁到高能级。 13C受环境影响,也有顺磁屏 蔽效应和抗磁屏蔽效应,使不同类型碳在 不同的磁场强度处发生共振吸收。吸收峰 的位置用化学位移 表示,也用TMS作参 照物。
43
(a) 选择去偶谱 (b) 偏共振去偶谱
44
全去耦、偏共振去耦、选择性去耦谱比较
45
4.门控去偶(gated decoupling 交替脉冲
去偶或预脉冲去偶):
质子宽带去偶失去了所有偶合信息,偏共振去偶
保留了13C与1H之间部分偶合信息,但因分子中
核磁共振13C谱操作步骤
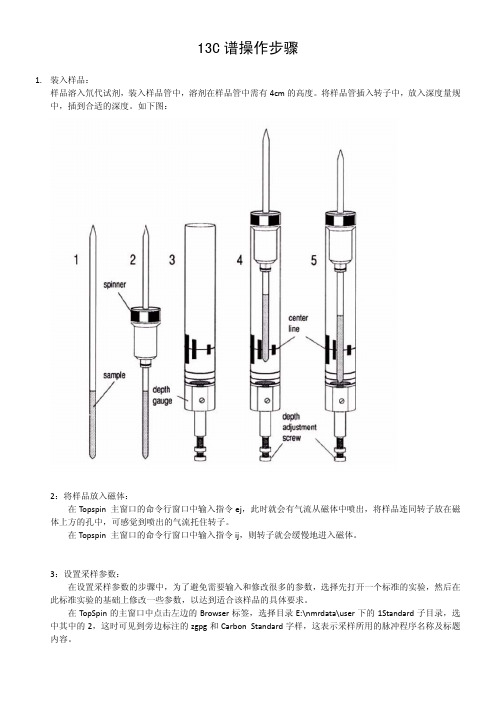
13C谱操作步骤1.装入样品:样品溶入氘代试剂,装入样品管中,溶剂在样品管中需有4cm的高度。
将样品管插入转子中,放入深度量规中,插到合适的深度。
如下图:2:将样品放入磁体:在Topspin 主窗口的命令行窗口中输入指令ej,此时就会有气流从磁体中喷出,将样品连同转子放在磁体上方的孔中,可感觉到喷出的气流托住转子。
在Topspin 主窗口的命令行窗口中输入指令ij,则转子就会缓慢地进入磁体。
3:设置采样参数:在设置采样参数的步骤中,为了避免需要输入和修改很多的参数,选择先打开一个标准的实验,然后在此标准实验的基础上修改一些参数,以达到适合该样品的具体要求。
在TopSpin的主窗口中点击左边的Browser标签,选择目录E:\nmrdata\user下的1Standard子目录,选中其中的2,这时可见到旁边标注的zgpg和Carbon Standard字样,这表示采样所用的脉冲程序名称及标题内容。
继续展开此子目录,双击下面的1,此时读入了一个标准的13C实验。
点击Start标签下的Create Dataset图标,则会出现以下的对话框:在MANE一行中输入你的姓名的拼音,在EXPNO一行中输入2(如在此目录中2#文件已存在,则输入3,以此类推),选择Use current Parameters, 选择Keep parameters,在DIR一行中输入E:\nmrdata\user。
然后点击OK。
4.锁场点击Topspin主窗口的Acquire标签下的Lock图标此时会出现一张溶剂表。
选择正确的溶剂,并点击OK5.自动调谐探头:点击Lock图标旁的Tune图标,则探头会进行自动调谐。
6.自动匀场:点击Shim图标,则会进行自动匀场(自动匀场需要一些时间,直到锁场线重新恢复到原来的高度或更高,并且在左下角的提示中显示:topshim completed表示自动匀场已进行完毕。
7.读入探头脉冲强度与宽度点击Prosol图标,则探头的脉冲参数读入。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
13C谱操作步骤
1.装入样品:
样品溶入氘代试剂,装入样品管中,溶剂在样品管中需有4cm的高度。
将样品管插入转子中,放入深度量规中,插到合适的深度。
如下图:
2:将样品放入磁体:
在Topspin 主窗口的命令行窗口中输入指令ej,此时就会有气流从磁体中喷出,将样品连同转子放在磁体上方的孔中,可感觉到喷出的气流托住转子。
在Topspin 主窗口的命令行窗口中输入指令ij,则转子就会缓慢地进入磁体。
3:设置采样参数:
在设置采样参数的步骤中,为了避免需要输入和修改很多的参数,选择先打开一个标准的实验,然后在此标准实验的基础上修改一些参数,以达到适合该样品的具体要求。
在TopSpin的主窗口中点击左边的Browser标签,选择目录E:\nmrdata\user下的1Standard子目录,选中其中的2,这时可见到旁边标注的zgpg和Carbon Standard字样,这表示采样所用的脉冲程序名称及标题内容。
继续展开此子目录,双击下面的1,此时读入了一个标准的13C实验。
点击Start标签下的Create Dataset图标,则会出现以下的对话框:
在MANE一行中输入你的姓名的拼音,在EXPNO一行中输入2(如在此目录中2#文件已存在,则输入3,以此类推),选择Use current Parameters, 选择Keep parameters,在DIR一行中输入E:\nmrdata\user。
然后点击OK。
4.锁场
点击Topspin主窗口的Acquire标签下的Lock图标
此时会出现一张溶剂表。
选择正确的溶剂,并点击OK
5.自动调谐探头:
点击Lock图标旁的Tune图标,则探头会进行自动调谐。
6.自动匀场:
点击Shim图标,则会进行自动匀场(自动匀场需要一些时间,直到锁场线重新恢复到原来的高度或更高,并且在左下角的提示中显示:topshim completed
表示自动匀场已进行完毕。
7.读入探头脉冲强度与宽度
点击Prosol图标,则探头的脉冲参数读入。
8.自动增益调节:
点击Gain图标,则进行自动增益调节,结束后左下角提示行中显示:rga finished.
9.进行采样:
点击Go图标,则采样开始进行。
此时会进入采样窗口,如未进入采样窗口,可点击Acqu标签。
此时在数据窗口的右上角有一信息窗口,其中有预定采样次数及已采样次数,总采样时间及剩余采样时间等信息。
在数据窗口上还有些图标,其功能如下:
将目前的FID数据存盘(存盘后可将其进行傅立叶变换,以查看数据的信噪比)。
停止采样,并将当前数据存盘。
停止采样(数据不保存)。
采样至某次后停止并保存数据。
采样至预定的次数后结束,并将数据存盘。
10.进行傅立叶变换:
点击Topspin主窗口中的Process标签,点击下面的Proc. Spectrum 图标,即进行傅立叶变换。
11. 相位校正:
点击Adjust Phase图标,进入相位校正子窗口
点住0级相位校正图标拖曳进行0级相位校正,点住1级相位校正图标拖曳进行1级相位校正,校正完成后点保存并退出图标。
12.校正化学位移:
先将溶剂的峰在屏幕上扩展,点击Calib. Axis图标,进入定标子窗口。
将光标移到溶剂峰中心,点击,在出现的对话框中输入化学位移值。
13. 标出各峰的化学位移值:
缩拢图谱,仅将要标出化学位移值的峰展现在屏幕上,点击Pick Peaks图标,
进入标峰子窗口
点击设定标峰范围图标,使其高亮后用鼠标左键在图谱区域你拖曳,可产生一个框,在此框中的峰的化学位移会被标注,如框的大小不满意可点击调整标峰范围图标,使其高亮后可用鼠标调节该款的大小,完成后点击保存并退出图标。
14.绘图
在Topspin主窗口的命令行窗口中输入plot指令,进入打印窗口
在谱图区域鼠标右击,在弹出菜单中选择1D/2D Edit,在新弹出的菜单中点击上面一排的粗黑体箭头拖曳,可对谱图进行调整,
完成后点击Close 退出调整。
完成后点击打印机图标打印图谱。
完成后点击右上角的红色关闭图标,退出打印程序,在弹出的对话框中须选择No。
15.取出样品:
在Topspin的命令行窗口中输入 ej, 则样品就会从磁体中被吹出。