国家药品监督管理局药品审评中心
执业药师资格考试-药事管理与法规考试重点(六)

执业药师资格考试-药事管理与法规考试重点第一节药品研制与注册管理(6)药物临床试验机构运行管理新药Ⅰ期临床试验或者临床风险较高需要临床密切监测的药物临床试验,应当由三级医疗机构实施。
药物临床试验机构应当于每年1月31日前在备案平台填报上一年度开展药物临床试验工作总结报告。
实战演练【例-B型题】A.Ⅰ期临床试验B.Ⅱ期临床试验C.Ⅲ期临床试验D.Ⅳ期临床试验根据《药品注册管理办法》1、新药上市后应用研究阶段的试验是()。
2、初步的临床药理学及人体安全性评价试验属于()。
3、治疗作用确证阶段;为药品注册提供充分依据()。
4、考查广泛使用条件下的药物疗效和不良反应,评价改进给药剂量()。
答案:D、A、C、D二、药品注册管理制度2020年3月30日,国家市场监管总局发布了新修订的《药品注册管理办法》(市场总局令第27号),于2020年7月1日起实施。
(一)药品注册与药品注册事项药品注册,是指药品注册申请人依照法定程序和相关要求提出药物临床试验、药品上市许可、再注册等申请以及补充申请,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,决定是否同意其申请的活动。
(二)药品注册类别药品注册,按照中药、化学药和生物制品等进行分类注册管理。
1、中药注册按照中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等进行分类。
2、化学药注册按照化学药创新药、化学药改良型新药、仿制药等进行分类。
3、生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药)等进行分类(三)药品注册管理机构和事权划分1、国家局事权(1)国家药品监督管理局主管全国药品注册管理工作。
(2)国家药品监督管理局药品审评中心,负责药物临床试验申请、药品上市许可申请、补充申请和境外生产药品再注册申请等的审评。
(3)中国食品药品检定研究院:承担依法实施药品注册管理所需的药品注册检验。
2、省级局事权省、自治区、直辖市药监部门负责本行政区域内以下药品注册相关管理工作:①境内生产药品再注册申请的受理、审查和审批;②药品上市后变更的备案、报告事项管理;③组织对药物非临床安全性评价研究机构、药物临床试验机构的日常监管及违法行为的查处;④参与国家药品监督管理局组织的药品注册核查、检验等工作;⑤国家药品监督管理局委托实施的药品注册相关事项。
药品审评中心的主要职责

药品审评中心的主要职责
国家药品审评中心是国家药品监督管理局的直属事业单位,主要负责药品的技术审评工作。
以下是药品审评中心的主要职责:
1. 负责对药品注册申请进行技术审评:对药品临床试验、生产工艺、质量控制等方面进行评估,确保药品的安全性、有效性和质量可控性。
2. 制定药品审评技术标准和规范:参与制定国家药品审评技术指导原则、审评规范和标准,为药品审评提供技术支持。
3. 开展药品审评科学研究:对药品审评中的关键技术问题进行研究,为审评决策提供科学依据。
4. 提供技术咨询和指导:为药品研发机构、生产企业提供技术咨询和指导,帮助其提高药品研发和生产水平。
5. 参与药品监管国际交流与合作:参与国际药品审评组织的交流与合作,跟踪国际药品审评技术发展动态,推动我国药品审评技术与国际接轨。
6. 承担药品审评相关培训工作:开展药品审评技术培训,提高审评人员和药品研发、生产人员的专业水平。
7. 协助国家药品监督管理局开展药品监管工作:参与药品注册现场核查、药品生产质量管理规范检查等工作,为药品监管提供技术支持。
总之,药品审评中心在保障公众用药安全、促进医药产业发展方面发挥着重要作用,通过严格的审评工作,确保上市药品的质量和安全性,维护公众的健康权益。
国家药品监督管理局药品审评中心的主要职责

国家药品监督管理局药品审评中心的主要职责国家药品监督管理局药品审评中心是中国药品监管体系中非常重要的机构之一,其主要职责是为保障人民群众用药安全、推动药品创新和提升药品质量提供支持和保障。
下面将从几个方面来介绍国家药品监督管理局药品审评中心的主要职责。
1. 药品审评和审批作为国家药品监督管理局直属机构,药品审评中心负责对国内外生产的新药、化学药品、生物制品和药材进行审评,以确定其安全性、有效性和质量符合国家和国际标准。
药品审评中心通过严格的评估和审查,提供给药品监督管理局合理的药品审批意见,确保药品的质量和安全。
2. 制定和修订药品审评准则药品审评中心负责制定和修订药品审评准则,确保评估和审查仅关注于药品的质量、安全性和有效性。
准则的制定和修订是基于国家和国际法规的变化、科学和技术的进步以及行业的需求。
药品审评中心将准则的制定和修订过程公开透明,接受专家和行业的意见和建议,并根据最新的科学证据进行更新。
3. 加强国际合作与交流为了提高国内药品审评领域的专业水平,药品审评中心积极开展国际合作与交流。
这包括与国际监管机构和组织的合作,如世界卫生组织、美国食品药品监督管理局等,以及与国外药品审评中心的合作。
通过与国际机构的交流,药品审评中心可以获取国际领先的科学理论和技术,提高药品审评的水平和质量。
4. 提供药品临床试验指导和监管药品审评中心还负责提供药品临床试验的指导和监管,确保临床试验的设计和执行符合法规要求和科学伦理原则。
为了保护病患的权益和安全,药品审评中心会对临床试验方案进行审查,包括试验设计、人员选择、伦理委员会审批和数据管理等。
通过提供指导和监管,药品审评中心确保临床试验的可靠性和有效性。
5. 监测和评估药品安全性药品审评中心负责监测和评估国内外研发和上市药品的安全性。
一旦发现药品的安全性问题,药品审评中心将积极采取措施,包括但不限于发出风险提示、通知药品生产企业进行改进和召回等。
通过对药品安全性的监测和评估,药品审评中心保障人民群众的用药安全。
国家药监局药品审评中心征求《生物利用度和生物等效性试验用药品的处理和保存要求技术指导原则》意见

国家药监局药品审评中心征求《生物利用度和生物等效性试验用药品的处理和保存要求技术指导原则》意见
佚名
【期刊名称】《中国医药生物技术》
【年(卷),期】2012(7)6
【摘要】为规范制剂生物利用度和生物等效性研究试验用药品的处理和保存,国
家药监局药品审评中心成立专题工作小组,在FDA相关指导原则的基础上,经过
前期的撰写和专家讨论,形成《生物利用度和生物等效性试验用药品的处理和保存要求技术指导原则》征求意见稿,现公开征求意见,时间至2012年12月31日。
【总页数】1页(P468-468)
【关键词】生物等效性;生物利用度;药品审评;国家药监局;保存;验用;专家讨论;FDA 【正文语种】中文
【中图分类】R969.1
【相关文献】
1.《化学药品注射剂与塑料包装材料相容性研究技术指导原则》公开征求意见 [J],
2.国家食品药品监督管理局药品审评中心对《灭菌/无菌工艺验证指导原则》征求
意见 [J],
3.关于《已上市化学药品生产工艺变更研究技术指导原则》征求意见的通知 [J], ;
4.关于征求化学仿制药生物等效性试验备案管理规定(征求意见稿)意见的公告
国家食品药品监督管理总局 2015年第221号 [J], ;
5.关于征求《关于解决药品注册申请积压实行优先审评审批的意见(征求意见稿)》意见的公告国家食品药品监督管理总局 2015年第227号 [J], ;
因版权原因,仅展示原文概要,查看原文内容请购买。
【免费下载】国家药品审评中心审评人员公示名单04

药品审评中心审评人员公示名单 (2015年4月)部门姓名 所负责工作分机中心主任许嘉齐611中心副主任尹红章602中心副主任侯仁萍603中心领导中心主任助理胡 军协助分管主任负责中药药品审评的技术管理工作604负责审评技术资料管理、审评任务分发调度及相关督查协调工作。
承担相关审评报告综合以及审评文件管理,组织开展与新药审评相关的核查、验证工作;承担主动撤回申请和超过规定时限未按要求提交补充资料等品种的相关处理工作;负责与国家食品药品监督管理局相关部门协调技术审评工作;承担业务咨询及业务信息管理工作。
负责中心文电、会务、档案等日常运转工作以及安全保密、印章管理、外部联络等工作。
承担中心交办的其他工作。
副部长温宝书303副部长王 敏172 何燕萍101张晓东102王 鹏104黄清竹105业务管理部协调员刘 璐106负责中药、民族药及天然药物临床试验申请和注册申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。
负责中药、民族药及天然药物7~8类临床试验申请、7~9类注册申请、各类注射剂注册申请、相关补充申请以及进口再注册申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。
承担中心交办的其他工作。
部长胡 军604 副部长周跃华486阳长明401张永文402曲建博406金 芳407李计萍408周 刚409韩 炜412中药民族药药学部成员马秀璟413负责化学药物1~3类临床试验申请和注册申请、国际多中心临床试验申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。
负责化学药物3类临床试验申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。
承担中心交办的其他工作。
副部长马玉楠577王 旸502任连杰505康建磊506王亚敏507于 红508王宏亮511马 磊512张 宁513霍秀敏514化药药学一部成员刘宗英515 负责化学药物4~5类临床试验申请、进口药注册申请、进口再注册申请、相关补充申请及其他申请的药学研究资料及生物等效性试验资料的技术审评工作,提出药学专业和相应生物等效性资料的审评意见并形成药学专业审评报告。
国家食品药品监督局药品审评中心化学药品药学研究的技术要求及常见问题分析

幻灯片9.3
性状:药物特性和质量的表征
1、外观:色泽、嗅、味、结晶形状,一般稳定性情况 2、溶解度:采用药典凡例中分等级方式,溶剂可采用
极性不同与工艺相关的(尤其精制溶剂). 3、物理常数:熔 点:范围3-4℃ ,熔矩<2℃
比旋度:光学活性化合物的固有特性及 纯度。注意:温度、浓度对测 定影响。 药典规定:200C,589um
幻灯片4.2.1
制剂辅料的要求(药监注函568号文)
1、国家标准 (中国药典、部颁)
2、进口辅料 附进口许可证、质量标准及口岸检验报告
3、习用辅料,提供依据并制订相应的质量标准 4、特殊需要、用量较小辅料:指国外药典上收载,
国外制剂上使用过的辅料,提供依据,质量标准 和检验结果。 5、食品添加剂:提供依据,质量标准。 6、国内外未使用过辅料,按新辅料与制剂同时申报。
1、按中国药典规定进行波长校正,并报告 测定数据。
2、供试品制备: ⑴ 尽量采用易溶中性溶剂; ⑵ 发色团上存在酸性或碱性基因,化合 物可增加0.1N HCL、0.1N NaOH的水溶 液以观察吸收带移动情况。
幻灯片7.3(续)
紫外—可见吸收光谱(UV-VIS)分析要求
3、制图要求: ⑴ 录制紫外可见区的全部吸收峰,不得遗 漏,不得截止,最强吸收度不得高于1.0。 ⑵ 必要时可分段以不同浓度试样溶液录制 图谱。
幻灯片1
注册分类:
1、未在国内外上市销售的药品。 2、改变给药途径尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药
品。 4、改变已上市销售盐类药物的酸根、碱基(或者
金属元素),但不改变其药理作用的原料药及其 制剂。 5、改变国内已上市销售药品的剂型,但不改变给 药途径的制剂。 6、已有国家药品标准的原料药或者制剂。
2011年药品审评中心CDE-审评人员完整名单及联系方式
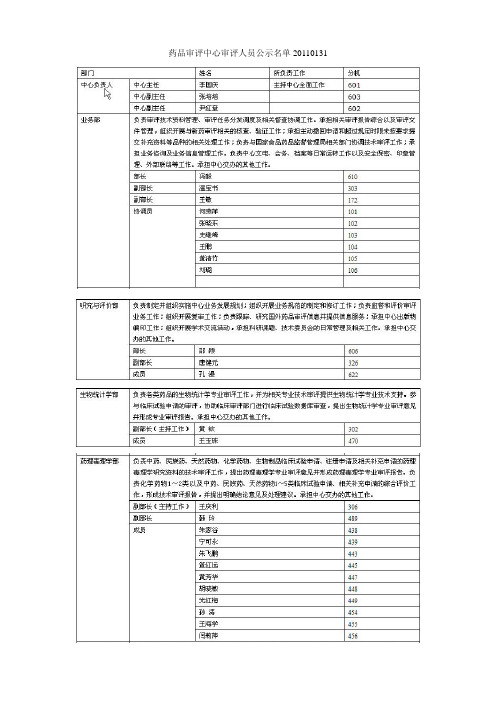
药品审评中心审评人员公示名单20110131关于国家食品药品监督管理局药品审评中心调整主要职责和内设机构的批复国食药监人函[2010]219号(一)业务管理部负责审评技术资料管理、审评任务分发调度及相关督查协调工作。
承担相关审评报告综合以及审评文件管理,组织开展与新药审评相关的核查、验证工作;承担主动撤回申请和超过规定时限未按要求提交补充资料等品种的相关处理工作;负责与国家食品药品监督管理局相关部门协调技术审评工作;承担业务咨询及业务信息管理工作。
负责中心文电、会务、档案等日常运转工作以及安全保密、印章管理、外部联络等工作。
承担中心交办的其他工作。
(二)中药民族药药学部负责中药、民族药及天然药物临床试验申请和注册申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。
负责中药、民族药及天然药物7~8类临床试验申请、7~9类注册申请、各类注射剂注册申请、相关补充申请以及进口再注册申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。
承担中心交办的其他工作。
(三)化药药学一部负责化学药物1~3类临床试验申请和注册申请、国际多中心临床试验申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。
负责化学药物3类临床试验申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。
承担中心交办的其他工作。
(四)化药药学二部负责化学药物4~5类临床试验申请、进口药注册申请、进口再注册申请、相关补充申请及其他申请的药学研究资料及生物等效性试验资料的技术审评工作,提出药学专业和相应生物等效性资料的审评意见并形成药学专业审评报告。
负责化学药物4~5类临床试验申请以及5~6类注册申请、进口药临床试验申请、进口再注册申请、相关补充申请及其他申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。
承担中心交办的其他工作。
(五)生物制品药学部负责生物制品临床试验申请、注册申请及相关补充申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。
国家药品审评中心审评人员公示名单2015.04.doc

(2015年4月)
部门
姓名
所负责工作
分机
中心领导
中心主任
许嘉齐
611
中心副主任
尹红章
602
中心副主任
侯仁萍
603
中心主任助理
胡 军
协助分管主任负责中药药品审评的技术管理工作
604
业务管理部
负责审评技术资料管理、审评任务分发调度及相关督查协调工作。承担相关审评报告综合以及审评文件管理,组织开展与新药审评相关的核查、验证工作;承担主动撤回申请和超过规定时限未按要求提交补充资料等品种的相关处理工作;负责与国家食品药品监督管理局相关部门协调技术审评工作;承担业务咨询及业务信息管理工作。负责中心文电、会务、档案等日常运转工作以及安全保密、印章管理、外部联络等工作。承担中心交办的其他工作。
副部长(主持工作)
杨建红
607
副部长
黄晓龙
576
9
李 丽
520
田 洁
524
宁黎丽
525
张玉琥
526
张 震
527
李晓峰
529
许真玉
530
李志万
531
何 伍
532
蒋 煜
533
张星一
536
成海平
537
李雪梅
539
生物制品药学部
负责生物制品临床试验申请、注册申请及相关补充申请的药学研究资料的技术审评工作,提出药学专业审评意见并形成药学专业审评报告。负责生物制品临床试验申请及注册申请、相关补充申请的综合评价工作,形成技术审评报告,并提出明确结论意见及处理建议。承担中心交办的其他工作。
部长
胡 军
药品审评中心补充资料工作程序(试行)

附件药品审评中心补充资料工作程序(试行)第一章总则第一条为规范药品注册审评补充资料管理工作,明确补充资料的依据和要求,提高申请人补充资料的质量和效率。
根据《药品注册管理办法》第八十七条的规定,制定本程序。
第二条国家药品监督管理局药品审评中心(以下简称药审中心)根据审评需要,通知药品注册申请人(以下简称申请人)在原申报资料基础上补充新的技术资料的(以下简称发补),或仅需要申请人对原申报资料进行解释说明的,适用本程序。
第三条药审中心通过发补前的专业审评问询和发补后的补充资料问询程序,请申请人进行解释说明或提供相关证明性材料,主动与申请人进行沟通交流,提高补充资料的质量和效率。
第四条补充资料过程中应当遵循依法、科学、公正、公平、及时、准确的原则。
第二章专业审评问询第五条药审中心在专业审评期间或综合审评期间,专业主审或主审报告人在充分审评基础上对申报资料有疑义或认为内容存在问题,经审评部门负责人审核后,通过药审中心网站向申请人发出“专业审评问询函”,告知申请人存在问题的具体内容、依据和要求等,并要求在5个工作日内进行解释说明或书面回复。
审评部门在审评过程中对需要发补的问题应发送“专业审评问询函”提前告知申请人。
但“专业审评问询函”并不是正式书面补充资料通知,也不代表最终审评决策意见,审评计时不暂停。
第六条药审中心通过“专业审评问询函”告知申请人以下信息:1)无需开展研究即可提供的证明性材料;2)不需要补充新的技术资料,仅需要对原申报资料进行解释说明;3)审评认为可能需要补充完善的缺陷问题。
第七条申请人应在“专业审评问询函”发出5个工作日内进行解释说明或书面回复。
对于需要书面回复的,申请人应在5个工作日内进行电子提交,同时在时限内寄出与电子版一致的纸质版资料,通过药审中心网站下载打印“专业审评问询函”作为接收补充资料及纳入档案的依据。
第三章正式发补、发补咨询和异议程序第八条在审评过程中需要申请人在原申报资料基础上补充新的技术资料的,结合“专业审评问询函”的答复情况,根据《药品注册管理办法》规定,药审中心原则上提出一次补充资料要求,列明全部问题后,以书面方式通知申请人在80个工作日内补充提交资料。
药审中心_精品文档

药审中心1. 简介药审中心(Center for Drug Evaluation,简称CDE)是中国国家药品监督管理局(National Medical Products Administration,简称NMPA)下属的机构,负责药品注册审核和审评工作。
其主要职责是对国内外药品进行审评、安全性评价和疗效评价,并根据相关法律法规,决定药品的注册、变更和注销等事项。
2. 药审中心的组成和职能药审中心由多个部门组成,包括临床评价部、儿童药物研究部、药物不良反应监测部等。
每个部门负责不同的药品审评工作,协同合作,确保药品的评价工作科学、准确、高效。
药审中心的主要职能包括:2.1 药品注册审评药审中心对申请注册的药品进行审评,包括药物的质量标准、生产工艺、临床试验数据等方面的审查。
通过评估药品的安全性、有效性和合理性,来确定是否批准该药品的上市。
2.2 药物研发支持药审中心为药物研发提供技术支持和政策指导,协助企业设计合理的研发方案,提供相关要求和标准,以确保药品能够符合国家的药品注册要求。
2.3 药品监管药审中心负责对上市药品的监管工作,包括药品生产、销售和使用过程中的质量监管、不良反应监测和风险评估等。
通过监管工作,保障药品的质量和安全性,减少药品相关风险。
2.4 国际交流合作药审中心积极开展与国际药品审评机构的交流合作,加强和吸收国际上的先进经验和技术,提高我国药品审评的水平和国际影响力。
同时,也积极参与国际药品审评准则的制定和修订工作。
3. 药审中心的工作流程药审中心的工作流程包括以下几个主要步骤:3.1 申请受理在药品注册申请提交后,药审中心将对申请材料进行初审,确认申请是否完整并符合要求。
同时,受理人员会对申请进行登记,并发放申请受理通知书。
3.2 审评准备受理通过后,药审中心会组织相关部门和专家对药品进行审评准备工作。
这包括对药品注册申请材料的再次审查和评估,准备审评意见和技术要求。
3.3 临床评价在临床评价阶段,药审中心会组织专家对药品的临床试验数据进行评价,包括疗效、安全性和用药指导等方面的考虑。
国家药品监督管理局关于国家药品监督管理局药品审评中心主要职责内设机构和人员编制规定的通知

国家药品监督管理局关于国家药品监督管理局药品审评中心主要职责内设机构和人员编制规定的通知文章属性•【制定机关】国家药品监督管理局•【公布日期】2000.06.24•【文号】国药管办[2000]255号•【施行日期】2000.06.24•【效力等级】部门规范性文件•【时效性】现行有效•【主题分类】正文国家药品监督管理局关于国家药品监督管理局药品审评中心主要职责内设机构和人员编制规定的通知(国药管办[2000]255号2000年6月24日)各省、自治区、直辖市药品监督管理局或卫生厅(局)、医药管理部门,局各直属单位,各有关协会:根据中央机构编制委员会办公室《关于卫生部药典委员会等5个单位更名并成建制划转国家药品监督管理局管理的批复》(中编办字[1998]32号),国家药品监督管理局药品审评中心为国家药品监督管理局直属事业单位。
经国家药品监督管理局党组研究批准,国家药品监督管理局药品审评中心主要职责、内设机构和人员编制规定如下:一、主要职责(一)负责按照《新药审批办法》、《新生物制品审批办法》及有关法规,对化学药品、生物制品、体外诊断试剂的新药申请进行技术审评。
(二)负责按照《新药审批办法》及有关法规,对中药新药申请进行技术审评。
(三)负责按照《进口药品管理办法》及有关法规,对进口药申请进行技术审评。
(四)负责按照《仿制药品审批办法》及有关法规,对仿制药申请进行技术审评。
(五)承办国家药品监督管理局交办的其他事项。
二、内设机构根据以上职责,国家药品监督管理局药品审评中心设置7个职能处(室):(一)行政处协调中心日常行政事务,负责文电处理、档案管理、资产、财务、后勤保障等工作。
(二)人事监察处负责人事、党务、纪检、监察等工作。
(三)业务综合处负责药品申请资料的接受、借阅、保管等管理工作,并通过对资料的管理实现对各业务室审评工作的规范化制约及进度平衡;负责中心计算机管理及信息化建设;负责对审评业务的动态管理,组织研讨审评工作中的普遍性问题;负责审评业务工作协调及其他相关事宜。
国家食品药品监督管理总局药品审评中心拟录用工作人员情况

9
张新房
男
汉族
1979年5月
本科
副主任药师
药学
山东大学
山东省食品药品监督管理局审评认证中心
审评员及审评员助理
10
郑保华
女
汉族
1986年10月
博士研究生
无
药物化学
沈阳药科大学
北京大学生命科学学院
审评员及审评员助理
11
贾增荣
女
汉族
1981年3月
博士研究生
无
药剂学
沈阳药科大学
昆泰企业管理上海有限公司北京分公司
审评员及审评员助理
12
高建超
男
汉族
1986年4月
博士研究生
主管药师
人体解剖与组织胚胎学
北京大学医学部
国家食品药品监督管理总局药品评价中心
审评员及审评员助理
13
黄云虹
女
汉族
1977年10月
博士研究生
无
微生物与生化药学
北京协和医学院
罗氏(中国)投资有限公司
审评员及审评员助理
14
戴学栋
男
汉族
1984年9月
国家食品药品监督管理总局药品审评中心拟录用工作人员情况
序号
姓 名
性别
民族
出生年月
学 历
职称
专业
毕业院校
原所在单位
拟录用岗位
1
王功富
男
汉族
1982年5月
硕士研究生
工程师
药物化学
烟台大学
山东省食品药品监督管理局审评认证中心
审评员及审评员助理
2
王 统
男
汉族
简述有哪些药品监督管理技术支撑机构及其职责

简述有哪些药品监督管理技术支撑机构及其职责(1)中国食品药品检定研究院:是国家食品药品监督管理总局的直属事业单位,是国家检验药品、生物制品质量的法定机构。
(2)国家药典委员会:是法定的国家药品标准工作专业管理机构,其主要职责:1)组织编制、修订和编译《中华人民共和国药典》及配套标准;2)组织制定修订国家药品标准。
参与拟定有关药品标准管制度和工作机制;3)组织《中国药典》収载品种的医学和药学遴选工作。
负责药品通用名称命名。
4)组织评估《中国药典》和国家药品标准执行情况;5)开展药品标准发展战略、管理政策和技术法规研究。
承担药品标准信息化建设工作。
(3)国家药品监督管理局药品审评中心:是国家药品注册技术审评机构。
其主要职责:1)负责药物临床试验、药品上市许可申请的受理和技术审评;2)负责仿制药质量和疗效异型性评价的技术评审。
(4)国家药品监督管理局食品药品审核查验中心:组织制定修订药品、医疗器械、化妆品检查制度规范和技术文件。
(5)国家药品监督管理局药品评价中心(国家药品不良反应检测中心),其主要职责:1)组织制定修订药品不良反应、医疗器械不良事件监测、化妆品不良反应监测与上市后安全性评价及药物滥用监测的技术标准和规范。
2)组织开展药品不良反应、医疗器械不良事件、药物滥用,化妆品不良反应监测工作。
(6)国家中药品种保护审评委员会:负责组织国家中药品种保护的技术审评工作。
(7)国家药品监督管理局行政事项受理服务和投诉举报中心:以12315一个号码对外,全国一个12315平台受理,为企业和社会公众提供便捷高效的市场监管投诉举报服务。
(8)国家药品监督管理总局执业药师资格认证中心。
国家药品审评中心

家
药
品
审
评
中
心
注射剂的无菌保证
与工艺研究
2008.10
国
家
药
品
审 评
药品管理法与药品生产
中
心
• 2001年2月28日修订的《药品管理法》第十条,
经修订后明确要求:
“药品必须按照〔原法规:工艺规程〕国家药品 标准和国务院药品监督管理部门批准的生产工 艺进行生产,…。改变影响药品质量的生产工 艺的,必须报原批准部门审核批准。〞
P=〔1-q〕n=〔1-0.05〕20 =0.359
即有35.9%的可能性,该批药品将会被判
定为无菌。
国
家
药
品 审
无菌检查特性数据表
评
中
心
中国药典现取样数为:20,如污染率为5%,从曲线 得,检出概率为64%。
国 家 药 品 审 评 中 心
如何保证注射剂
无菌合格呢?
国
家
药
品
审 评
质量控制的三种模式
• 首先要考虑被选剂型可采用的灭菌工艺的无菌 保证水平的上下。原则上首选剂型应能采用终 端灭菌工艺,并保证SAL不小于6。
• 对于有充分的依据证明不适宜采用终端灭菌工 艺且临床必需注射给药的品种,可考虑选择采
用无菌生产工艺的剂型。通常无菌生产工艺仅 限于粉针剂或局部小容量注射剂。
国
家
药
品
审 评
输液产品灭菌工艺的选择原则
以理化性质等硬指标来压无菌的软指标。
• 可最终灭菌产品一定要首选最终灭菌方式,以 保证产品的无菌特性。
• 无菌制造工艺的基本特征是:低安全+高成本。 应尽可能防止采用。
• 当灭菌可能会造成副产物等问题时,尽可能从 工艺上创造条件,使灭菌成为可行。
药品审评中心审评人员公示名单[1][1]
![药品审评中心审评人员公示名单[1][1]](https://img.taocdn.com/s3/m/b3b608f29e314332396893ce.png)
治疗用生物制品(呼吸)
常卫红/555
赵超/559
胡晓敏/463
李娅杰/461
治疗用生物制品(精神神经)
常卫红/555
赵超/559
王庆利/525
王水强/518⑤
治疗用生物制品(抗感染)
常卫红/555
赵超/559
孙涛/454
谢松梅/458
治疗用生物制品(抗肿瘤、放射)
罗建辉/549
罗建辉/549
吕佳康/427
呼吸B
黄芳华/425
李计萍/424
黄芳华/425
吕佳康/427
其他
朱飞鹏/426
李计萍/424
黄芳华/425
吕佳康/427
审评四室
消化
笪红远/437
张永文/439
笪红远/437
裴小静/438
妇科B
张永文/439
张永文/439
笪红远/437
裴小静/438
妇科A
裴小静/438
张永文/439
室主任
吕东
复核审评五室所承担品种的综合审评报告
455
审评六室
室主任
赵德恒
复核审评六室所承担品种的综合审评报告
464
审评四部
部长
左晓春
审核审评四部所承担品种的综合审评报告
607
副部长
黄晓龙
协助部长负责审评四部工作
576
审评七室
室主任
高晨燕
复核审评七室所承担品种的综合审评报告
501
审评八室
室主任
陈海峰
复核审评八室所承担品种的综合审评报告
③朱家谷同志负责的五官项目负责人工作暂由马秀璟同志负责。朱家谷同志负责的药理毒理第一专业审评员工作暂由宁可永同志负责(分机号码:419)。
国家食品药品监督管理局医疗器械技术审评中心

国家食品药品监督管理局医疗 器械技术审评中心
政府机构
目录
01 中心简介
03 办公室职能
02 主要职责 04 综合业务处职能
目录
05 审评一处职能
07 审评三处职能
06 审评二处职能 08 审评四处职能
国家食品药品监督管理局医疗器械技术审评中心是中华人民共和国国家食品药品监督管理局下设的直属机构。 内设办公室、综合业务处、审评一处、审评二处、审评三处、审评四处等。
办公室职能
负责研究和制定与中心事业发展相关的规划;负责中心日常行政管理和后勤保障工作;负责中心信息化建设; 负责中心宣传工作的组织与实施;负责起草与人事管理相关的规定并监督落实;负责中心日常人事管理工作;负 责中心党的基层工作、党风廉政建设、纪检监察、外事、统战以及离退休人员的管理等工作;负责中心事业发展 所需资金的保障与筹措;负责建立和完善保障中心日常工作正常运转的财务和资产管理制度并监督执行;负责中 心日常财务管理工作;负责中心国有资产的核算与监管;负责中心交办的其他工作。
审评三处职能
负责对申请注册的进口临床检验分析仪器及试剂、境内第三类临床检验分析仪器及试剂
审评四处职能
负责对申请注册的进口无源医疗器械产品及境内第三类无源医疗器械产品(体外循环管道、口腔科材料、注 射穿刺器械等,详见附录)进行技术审评;组织起草相关专业医疗器械产品技术审评指南;接受相关产品的临床 试验方案的备案;组织相关的业务培训及咨询;负责中心交办的其他工作。
药品审评中心名词解释

药品审评中心是指国家药品监管部门设立的负责药品注册申请审评的专门机构。
其主要职责是对药品注册申请进行审评,评估药品的安全性、有效性、质量可控性以及合规性等方面的科学性和技术性问题,为药品监管部门决策提供科学依据。
药品审评中心在药品注册过程中发挥着至关重要的作用。
在药品研发、生产和上市过程中,药品注册是必不可少的一环。
而药品注册申请的审评是决定药品能否上市的关键环节之一。
因此,药品审评中心的工作对于保障公众用药安全和促进医药产业健康发展具有重要意义。
药品审评中心在审评过程中需要综合考虑多方面的因素,包括药品的安全性、有效性、质量可控性以及合规性等方面。
对于安全性问题,药品审评中心需要对药品的原料药、辅料、生产工艺、质量控制等方面进行全面评估,确保药品的安全性不受影响。
对于有效性问题,药品审评中心需要对临床试验结果进行分析和评估,确保药品能够达到预期的治疗效果。
对于质量可控性问题,药品审评中心需要对药品的生产工艺、质量控制标准等方面进行审核,确保药品质量的稳定性和可控性。
对于合规性问题,药品审评中心需要对药品注册申请的资料进行审核,确保申请资料的完整性和合规性。
总之,药品审评中心是药品监管体系中的重要组成部分,负责对药品注册申请进行审评,为保障公众用药安全和促进医药产业健康发展发挥着重要作用。
药品审评中心药品技术审评工作程序(试行).doc

8
召开此类会议的情况。 (七)对于聘请外部专家参与药学专业审评的生物制
品,审评部应提出召开“专家咨询会议”的申请,经管协部 审核后,提交中心领导批准。 第二十六条 中心主任或被授权人签发后,由管协部负责呈 送国家食品药品监督管理局进行行政审批。 第二十七条 注册申请项目完成行政审批后,由管协部负责 对于该注册申请项目的所有资料及相关文件归档立卷,移交 国家食品药品监督管理局保管。
第二章 机构与职责
第四条 药品审评中心是国家食品药品监督管理局药品注 册管理的技术审评部门,为药品注册提供技术支持。负责按 照国家食品药品监督管理局颁布的药品注册等规章,对有关 药品注册申请进行技术审评。承办国家食品药品监督管理局 交办的其他工作。 第五条 中心主任领导中心的全面工作,对国家食品药品监
7
处理: (一)对于签发结论为非书面发补者,由项目负责人按
照《非书面补充通知管理规范》的规定,通知注册申请人。 (二)对于签发结论为书面发补者,由管协部负责制作
文件,向注册申请人发送通知。 (三)对于签发结论为会议讨论者,由管协部按照相关
规定组织会议。 (四)对于审评部长审核结论为批准者: 1、属于涉及药品质量标准、说明书、包装标签等文件
负责组织宣传工作和对外咨询。 负责中心对外的公共事务。 负责提出中心信息化建设纲要和年度发展计划的建议; 研究以信息化技术手段,提高中心业务及行政管理工作的质 量与效率。负责提出人力资源配置规划的建议。 负责中心交办的其他工作。 第九条 审评一部负责按照有关规范组织所辖审评室进行 中药和天然药物中心血管、五官、儿科、肿瘤、消化、风湿、 外科、骨科、内分泌适应症注册申请的技术审评,提出相应 处理建议。负责研究解决审评工作中发现的共性问题,负责 就重大技术问题提出处置建议。负责研究、起草与所承担技 术审评工作相关的技术规范和要求。负责组织开展相应的学 科建设工作。 负责中心交办的其他工作。 第十条 审评二部负责按照有关规范组织所辖审评室进行
国家食品药品监督管理总局药品审评中心新址办公用房

国家食品药品监督管理总局药品审评中心新址办公用房机房建设搬迁及系统集成项目采购需求一、项目介绍1、业务需求国家食品药品监督管理总局药品审评中心(以下简称药审中心)新址办公用房机房建设搬迁及系统集成项目,需完成药审中心新办公楼机房建设、原药审中心信息机房整体搬迁、信息系统数据备份与冗余保护建设的系统集成工作。
药审中心新址办公用房机房建设应按高起点、高水平、高质量,采用新设备、新材料、新工艺的原则建设一流信息化机房。
保证信息机房以安全、节能、易管理、环保的原则运行。
新机房需满足药审中心新址办公用房信息化办公整体要求,与新办公区网络无缝兼容、互联互通。
药审中心原信息机房的整体搬迁工作应统筹安排,详细规划搬迁工作计划、内容、步骤、时间、风险防范等具体事项。
原有信息机房范围内的应用系统、全部设备,在保证数据安全、设备安全的前提下,将现有业务系统在停机窗口内平稳迁移到新机房。
药品审评业务系统极其重要、影响广泛巨大,对业务系统的搬迁必须做到准时无误。
搬迁前须做好系统和数据的备份,尤其要防止系统故障,确保业务系统数据和设备在搬迁过程中不损坏、不崩溃、不丢失,满足业务系统外部运行条件,全部搬迁后在最短时间内恢复系统运行。
全部搬迁过程须考虑各种可能的风险,并提供相应的预防措施、补救措施,必须保证数据和各应用系统迁移后正常运行(业务中断时间不得超过24小时)。
同时,需建立信息系统数据备份与冗余保护机制,完成数据备份系统的系统集成工作,对中心现有数据存储保护机制进行资源整合及优化升级,并按照药审中心现有的安全等级保护要求进行加固。
形成运行稳定、高效、可靠、安全的数据备份与数据冗余保护机制。
2、技术需求投标方提供的项目方案需分别对机房建设部分(机房装修、机房网络综合布线、防雷接地系统、空调系统、配电系统、UPS系统、照明系统、安防视频系统、机房环境监控系统等)、机房搬迁部分(搬迁工作计划、内容、步骤、时间、搬迁设备的安全保障等)、数据备份与数据保护冗余系统优化升级部分、系统集成部分分项进行技术阐述。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
国家药品监督管理局药品审评中心2019年6月目录一、前言 (1)二、背景 (2)三、抗肿瘤药物临床试验的常用终点 (3)3.1 基于死亡事件的终点 (5)3.2 基于肿瘤测量的终点 (5)3.3 基于症状评估的终点 (8)四、晚期非小细胞肺癌新药注册临床试验的设计及终点考虑 (8)4.1 以肿瘤生物标志物富集人群的临床试验 (9)4.2 不以肿瘤生物标志物富集人群的临床试验 (13)五、晚期非小细胞肺癌新药注册临床试验主要终点获益考虑 (14)5.1 总生存期 (14)5.2 无进展生存期 (15)5.3 客观缓解率 (15)六、结语 (16)一、前言肺癌的发病率和病死率居全球和中国恶性肿瘤之首[1]。
2015年中国新发肿瘤患者392.9万例,死亡233.8万例,新发肺癌78.7万例,死亡63.1万例[2] 。
非小细胞肺癌(non-small cell lung cancer, NSCLC)占肺癌总体的85%,大部分初诊时已为晚期。
近年小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitors, TKI)、抗血管生成药物和免疫检查点抑制剂的应用已显著提高了患者生存。
晚期NSCLC为抗肿瘤新药研发的热点领域,创新药物众多,临床证据链日趋复杂,涌现出了复杂的终点指标和研究设计——包括替代终点、中间临床终点和其它创新终点;并出现共同终点平行检验、复合终点序贯检验等复杂设计。
现有指导原则尚不能涵盖。
因此,本文旨在阐述当前晚期NSCLC临床试验终点的一般性设计与审评考虑,期望为抗肿瘤药物研发人员在晚期肺癌的临床试验设计和终点选择上提供参考,科学、高效地确定药物疗效,提高临床研发效率,使患者更早获益。
本指导原则适用于支持晚期NSCLC适应症注册的临床试验设计及终点选择。
本指导原则所涉及的抗肿瘤药物试验设计同样应遵循临床试验设计的一般原则,包括但不限于人用药品注册技术要求国际协调会议(international conference for harmonization, ICH)所发布的E81、E92、E103和E174等指导原则,以及国家药品监督管理局(National Medical Products Administration, NMPA)已发布的《抗肿瘤药物临床试验终点技术指导原则》和《抗肿瘤药物临床试验技术指导原则》等相关内容。
本指导原则所涉及的观点代表当前NMPA对晚期NSCLC临床试验设计和终点选择的审评认识,不能涵盖在抗肿瘤新药研发中遇到的所有情况,鼓励研发从业人员探索科学创新的终点和试验设计,并及时与NMPA的审评部门沟通和交流。
二、背景当前晚期NSCLC的治疗目标为延长生命和提高生活质量,关键注册研究的试验终点应能有效反映临床获益的指标或事件。
上世纪九十年代前批准的抗肿瘤药物多以总生存期(overall survival, OS)为主要终点。
OS的定义明确、客观稳健,是反映患者生存获益的金标准,但试验耗时长且需较大样本量。
监管部门为加速药品上市、改善治疗可及性,对难治疾病背景下具有突出临床获益的药物实施加速审批,即允1E8 《General Considerations for Clinical Trials》;2E9 《Statistical Considerations in the Design of Clinical Trials》3E10《Choice of Control Group in Clinical Trials》4E17 《General Principles for Planning and Design of Muti-Regional Clinical Trials》许使用可合理预测临床获益的替代终点,如:客观缓解率(objective response rate, ORR)和无进展生存期(progression free survival, PFS)为主要终点支持新药批准上市[3-5]。
近年晚期NSCLC的治疗模式已从基于组织病理学的细胞毒化疗,转变为基于驱动基因突变等疗效预测生物标志物的分子靶向治疗,以及免疫、细胞毒和抗血管生成等药物联合治疗。
监管部门通常基于直接的临床获益批准新药,如OS 的延长和肿瘤相关症状的改善,也可接受可预测临床获益的替代终点批准新药,如较高的ORR和足够的缓解持续时间(duration of response, DOR),监管机构需要权衡替代终点和创新研究设计对临床获益评价的影响。
本指导原则将对疗效评估的一般性监管要求及支持批准的终点选择进行讨论。
三、抗肿瘤药物临床试验的常用终点抗肿瘤药物临床试验常用的终点依据来源可分为三类:基于死亡事件的终点,如OS及OS率;基于肿瘤测量的终点,如采用实体瘤疗效评价标准(response evaluation criteria in solid tumors, RECIST)评估的ORR,或基于RECIST和访视的至进展时间(time to progression, TTP)、PFS和至治疗失败时间(time to treatment failure, TTF)等;基于症状评估的终点如疼痛的减轻、生活质量(quality of life, QoL)和患者自评结果(patient report outcome, PRO)等。
终点的选择应结合肿瘤分期、既往治疗和起效特点等因素综合考量。
表1、晚期NSCLC药物常用临床试验终点比较3.1 基于死亡事件的终点OS定义为从随机化到任何因素导致患者死亡的时间。
OS的判定精确可靠,不易偏倚,常作为首选终点。
以OS为主要终点的临床试验需采用随机对照设计,常需较大样本量和更长的随访时间,易受到交叉和后续治疗影响。
OS率定义为自随机化至指定时间节点同一试验组内生存的受试者所占的比例,为OS的中间临床终点,在既往研究中可作为次要终点,如1年OS率。
虽然OS率的计算精确可靠并将更早达到,但OS率通常作为描述性终点,OS率的评估更多受到时间选择的影响,其临床意义和统计学意义尚不明确,提前分析OS率将导致破盲不利于生存获益的检验,现阶段OS率多作为次要终点和支持性证据。
3.2 基于肿瘤测量的终点肿瘤临床治疗常基于病灶的影像学评估结果决策,肿瘤测量的终点被视为具有临床获益相关性。
实体瘤疗效评价标准RECIST (目前为1.1版本)是目前广泛应用于NSCLC的疗效评价标准。
随着免疫检查点抑制剂的使用,免疫治疗疗效评价标准(immune response evaluation criteria in solid tumors, iRECIST)开始应用于临床试验,但对照组为标准化疗时不适用iRECIST。
目前在以新药注册为目的的肿瘤免疫治疗临床试验中,RECIST 1.1仍为最常用的肿瘤疗效判定标准,推荐在单纯免疫治疗及“头对头”设计的免疫治疗临床试验中增加iRECIST评估标准,并与传统RECIST结果进行对比。
ORR定义为肿瘤体积缩小达到预先规定值并能维持最低时限要求的患者比例。
缓解期指从开始出现客观缓解直至证实肿瘤进展的时间。
ORR为完全缓解(complete remission, CR)与部分缓解(partial remission, PR)的比例之和,ORR 不包括疾病稳定(stable disease, SD),排除了疾病自然病程的影响,相比疾病控制率(disease control rate, DCR),ORR 可更可靠反映药物的抗肿瘤活性,是单臂临床试验常用替代终点。
DOR定义为肿瘤第一次评估为客观缓解至第一次评估为PD或PD前任何原因死亡的时间,反映了ORR的持续时间。
PFS定义为从随机化至出现肿瘤客观进展或全因死亡的时间,是OS的替代终点。
与TTP相比,PFS包括了任何原因导致的死亡,与OS相关性更高,且不受后续治疗影响,是随机对照设计临床试验最常用的替代终点。
TTP定义为从随机化至出现肿瘤客观进展的时间,不包括死亡。
TTP能精确反映治疗带来的近期生存获益,由于排除了死亡,TTP对治疗临床获益的相关性差于PFS和TTF。
TTF定义为从随机化至治疗失败或退出试验的时间,退出试验的原因可为患者要求、疾病进展、死亡或不良事件等。
与PFS相比,TTF覆盖了非疾病进展导致的退出,并可包括疾病进展后的继续治疗,是综合的临床终点。
因TTF不能充分将药物的疗效和耐受性等因素区分,当前,不常用于抗肿瘤药物确证性研究的主要研究终点。
TTP、PFS和TTF结果的判定均受访视间隔设计和试验质量的影响,如由失访或在研究期间未观察到终点事件而产生的删失和截尾值过多,将影响以上终点结果的分析。
肺癌脑转移患者的纳入及终点指标考虑脑转移是导致晚期NSCLC疾病进展和治疗失败的重要原因,有症状患者需进行临床干预。
既往晚期NSCLC临床试验多排除脑转移患者,或仅入组经局部治疗病情稳定的无症状脑转移患者,因此临床试验结果不能反映药物对脑转移患者的疗效,将导致重要有效性信息的缺失。
考虑肺癌脑转移高发,且控制脑转移为转移性NSCLC重要治疗目标,鼓励基于药物前期的临床研究结果,在试验中纳入脑转移患者,注意在基线收集完整准确的脑转移信息,在疗效评估中增加局部转移获益的评价,如颅内缓解率、颅内缓解持续时间和至颅内疾病进展时间等。
3.3 基于症状评估的终点肿瘤患者症状和体征的改善被认为是直接的临床获益,而非替代终点。
监管当局可能基于显著的症状改善(如恶性积液的控制、癌性乏力的改善和骨相关事件的改善等)批准新药上市。
复合症状终点中的不同症状指标应具有相似的临床重要性,其临床获益不仅归因于单个指标的改善,对复合终点进行分析时应对具体单个指标进行分析。
当以症状和体征的改善作为支持抗肿瘤药物审批的主要终点时,应当能够区分是肿瘤相关症状的改善还是试验组药物毒性的减小或缺失。
如选择具体症状缓解作为终点,受试者需在基线时存在该症状。
数据缺失、评价不充分将增加症状终点的评价复杂性,应严格执行访视计划使访视完成率均衡和最大化,统计分析计划(statistical analysis plan, SAP)应说明如何处理缺失数据,当患者停止治疗时应该继续收集可供分析的信息。
应进行多种症状的前瞻性数据收集。
存在多重检验情况时,需要在SAP中说明必要的一类错误控制方法。
四、晚期非小细胞肺癌新药注册临床试验的设计及终点考虑晚期NSCLC的临床试验设计可根据有无疗效预测生物标志物分为富集人群试验和非富集人群试验。
4.1 以肿瘤生物标志物富集人群的临床试验以肿瘤生物标志物富集人群的试验设计,应考虑生物标志物和治疗分线等因素:(1)生物标志物生物标志物选择:需明确具体的生物标志物命名,例如明确某表皮生长因子受体酪氨酸激酶抑制剂(epidermal growth factor receptor-tyrosine kinase inhibitor,EGFR-TKI)适用于EGFR第19外显子缺失突变和第21外显子L858R突变。