αβ不饱和醛酮
第9章羰基化合物

第9章 羰基化合物大体要求:1. 把握醛和酮的命名(系统命名法,一般命名法)2. 把握醛和酮的结构及对化学性质的阻碍。
3. 了解醛和酮的物理性质和光谱特点。
4. 把握亲核加成的反映类型、机理、应用。
5. α—H 的酸性,α—H 的卤代反映及缩合反映(羟醛缩合)6. 氧化、还原反映及其在有机合成中的应用。
7. α,β—不饱和醛、酮的反映特点。
醛(aldehydes )和酮(ketones )都是分子中含有羰基(碳氧双键)的化合物,因此又统称为羰基化合物。
羰基与一个烃基相连的化合物称为醛,与两个烃基相连的称为酮。
CO R'C RO HC R(H)O羰基 醛 酮醛能够简写为RCHO ,基团—CHO 为醛的官能团,称为醛基,酮能够简写为RCOR ’, 基团—CO —为酮的官能团,称为酮基。
醛和酮是一类超级重要的化合物,这不仅是因为学多化学产品和药物含有醛、酮结构,更重要的是醛、酮能发生许多化学反映,是进行有机合成的重要原料和中间体。
醌(quinone )类是一类特殊的环状不饱和二酮类化合物。
第一节 醛和酮一、羰基的结构羰基是醛、酮的官能团,它与醛、酮的物理化学性质紧密相关。
依照醛、酮分子的结构参数(见表10-1),能够以为羰基碳原子以sp 2杂化状态参与成键,即碳原子以三个sp 2轨道与其它三个原子的轨道重叠形成三个σ键,碳原子上未参加杂化的p 轨道与氧原子上的p 轨道在侧面彼此重叠形成一个π键(见图10-1)。
(请在图左侧第一幅图中,下半个轨道中着淡灰色,如中间那幅图轨道的颜色)表10-1 醛、酮分子的结构参数醛、酮分子 键长(pm ) 键角(0) HCHO C — ∠ ∠ CH 3CHOC — C —∠ ∠ ∠ CH 3COCH 3 C =O 121.4 C —∠ ∠δ-+图10-1 羰基的结构由于氧原子的电负性比碳原子大,因此成键处的电子云就不均匀地散布在碳氧原子之间,氧原子处电子云密度较高,带有部份负电荷,而碳原子处的电子云密度较低,带有部份正电荷。
第六节 α,β- 不饱和醛、酮

C H 3 -C H = C H -C H = C H -C H = C H -C H O C H 2 -C H = C H -C H O
-
- H 2O
OH CH 3 -CHO + C H 3 -C H = C H -C H O
- H 2O /
CH 3 -CH=CH-CH=CH-CHO
插烯规律
插烯规律:在 H3C-CHO分子的 CH3与CHO
H
HB
O G C - C H 2 -C H 2 = C H
G
C - C H 2 -C H = C
H
O G C-H + C H 2 = C H -C H
B
-
O G C - C H 2 -C H 2 = C H
O CH 3 + C H 2 = C H -C O O B
-
O O CH 3 B
-
CH 3 O CH 2 -CH 2 -C O CH 3
间插入 [CH=CH] n,成为 H3C [CH=CH] n CHO,
反应仍可在共轭体系的两端进行,而共轭体系相
连的两个基团仍保持 [CH=CH] n插入前的关系,
即乙醛的CH3与CHO相互关系仍然存在,甲基上
的氢仍然活泼,称为“插烯规律”。
从结构上对比:
H H C H + CH O H H C H + CH CH + CH O
_ 稀 OH
-H 2 O
CH 3 CH=CH-CH=CHCHO
-H 2 O
CH 3 CH=CHCH=O
+ H-CH 2 CH=CHCHO
稀 OH
CH 3 CH=CHCH=CHCH=CHCHO
第六节_α_β-_不饱和醛、酮.

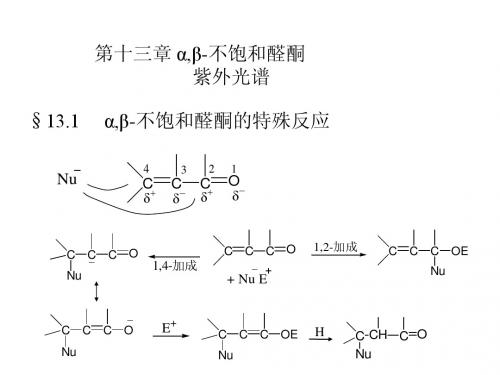
Nu -
+
C=C C=O
Nu C C=C O -
C=O Nu C C _
H+
Nu C C=C OH
互变异构
Nu C C C=O
H
在酸性条件下加成反应的机制
C C=C OH
C=C C=O
+ H+
+
C=C C OH
+
Z-
C C=C OH Z
互变异构
C C C=O Z
H
共轭加成的立体化学——反型加成
Ph
第六节 α,β- 不饱和醛、酮
一、亲电加成 二、亲核加成 三、插烯反应(羟醛缩合) 四、乙烯酮(自学)
C=C=O CH2=C=O 乙烯酮 不 烯酮 饱 C=C-C=O CH2=CH-C=O 和 α ,β 不饱和醛、酮 醛 C=C与 C=O 组成共轭体系 丙烯醛 H 、 C=C-(CH2)nC=O 酮 孤立不饱和醛、酮 n≥ 1
醌类化合物具有颜色,蒽醌类染料的重要组成部分。
O
OH
OH
O 茜素--1,2二羟基蒽醌(红色) (以糖苷的形式存在于茜草根中)
H+ OH甲基橙
NaO3S-
-N=N黄色
-N(CH3)2
NaO3S-
-NH-N= 红色
=N+(CH3)2
一、醌的化学性质
醌为非芳性的环烯酮,相当于α,β-不饱和酮。
O
◎与NH2OH加成
+
NH2O H
H+
N
OH
互变异构
H
N
பைடு நூலகம்
O
互变异构
NO
O
O 对苯醌单肟
O
有机化学第十一章 醛酮

NaCl + SO2↑ + H2O
Na2CO3 NaHCO3 + Na2SO3
(C)转化成α-羟基腈 α-羟基磺酸钠与NaCN作用,其磺酸基则被氰基取代生成α-
羟基腈。如:
CHO NaHSO3
OH C
Na C N
SO3Na
OH C
CN
HCl H2O
OH C
COOH
优点:可以避免使用易挥发、有毒的HCN,且产率较高。
O
O
CH3-C-CH2-C-CH3
2,4 戊二酮
H3C O
3 甲基环戊酮
练习
命名下列化合物。
O CH3 CH3CCH2C=CH2
4-甲基-4-戊烯-2-酮
CH3CHCH2CHO OH
3-羟基丁醛
O CH3CHCH2CCH3
Cl
4-氯-2-戊酮
O CCH3
CHO
CHO
苯乙酮
CH2CHO
苯甲醛
CH3
3-甲基苯甲醛
= = R
++ δ
δ
R δ+ δ
CO >
CO
H
R
羰基碳原子连有基团的体积↑,空间位阻↑,不利于亲核试剂
进攻,达到过渡状态所需活化能↑,故反应活性相对↓。
综上所述,下列醛、酮进行亲核加成的相对活性为:
Cl3C C=O
H
> C=O
> CH3 C=O
R
> C=O
Ar
> C=O
>
H
H
H
H
H
= CH3 C O >
α -二醛或酮:两个羰基直接相连。 β -二醛或酮:两个羰基间隔一个碳原子。
各类化合物的紫外吸收光谱

此外,由于引入含有n电子的N原子的,这类杂环化合物还可 能产生n*吸收带。
24
苯 、萘、蒽、并四苯的吸收光谱
25
直接分析烷烃和卤代烃的紫外吸收光谱,实用价值不大,
但是它们是测定紫外(或)可见吸收光谱的良好溶剂。
1
1.3.2 简单的不饱和化合物
1、烯烃、炔烃化合物
在不饱和烃类分子中,除含有键外,还含有键,它们 可以产生*和*两种跃迁。 例如,在乙烯分子中, *跃迁最大吸收波长为180nm。
22
在气态或非极性溶剂中,
苯及其许多同系物的B谱带有
许多的精细结构,这是由于
当苯环上有取代基时,
振动跃迁在基态电子上的跃 迁上的叠加而引起的。
在极性溶剂中,这些精 细结构消失。
苯的三个特征谱带都会发生 显著的变化,其中影响较大 的是E2带和B带。
23
稠环芳烃及杂环化合物
稠环芳烃,如萘、蒽、芘等,均显示苯的三个吸收带,但是 与苯本身相比较,这三个吸收带均发生红移,且强度增加。随 着苯环数目的增多,吸收波长红移越多,吸收强度也相应增加。
0nm +6nm
10
例3
1 2
3 4
胆甾-3,5-二烯
max=214nm(基数) +3×5nm(烷基取代) +5nm(环外双键)
=234nm 观察值max=235nm
11
例4
1
CH3COO 2
34
56
该化合物在1,4,6位上有三个烷基取代;3, 4位上的双键是环外双键;2位上是酰基取代。
max=254nm(基数)+30(延长一个共轭双键) +3×5nm(烷基取代或环的剩余部分) +5nm(环外双键)
第十三章 α,β-不饱和醛酮
N-OH
+ H2N-OH
O O
H2N-OH
N-OH
对苯醌单肟
对苯醌双肟
(2)烯键的加成(卤素、卤化氢等亲电试剂)
O
O Cl
O
+ Cl2
O
Cl2
Cl O
Cl
Cl
Cl O
Cl
(3) 1,4-加成(,-不饱和羰基化合物,亲核加成 )
O
பைடு நூலகம்OH
OH
+ HCl
Cl H
O
Cl OH
O
2、还原反应(易)
第二节 一、醌的结构特点
醌
O
环状不饱和二酮,两个羰基和两个 或两个以上碳碳双键共轭。 但不是芳环类的环闭共轭体系,不 具有芳香性。具有烯烃和羰基化 合物的性质。 二、醌的分类 苯醌、萘醌、蒽醌 三、命名 p319
O O
O
邻苯醌
对苯醌
四、化学性质
1、加成
(1)羰基的加成(羰基试剂、格氏试剂等亲核试剂)
(3) UV 的检测与表示
①检测----分光光度计 ②表示
A
λ
max
图谱: A~λ , ε ~λ , T~λ , log ε ~λ
数据:
λ / nm
λ
CH3OH max
252nm (12300)
二. 常见有机化合物的UV光谱:
1. 饱和有机化合物
σ σ* n σ *
CH3Cl 172 (弱)
CH3OH 183 (150) CH3OCH3 185 (2520)
CH3NH2 215 (600)
2. 不饱和有机化合物
(1) 孤立 C=C , C≡ C, C≡ N
醛酮反应机理二
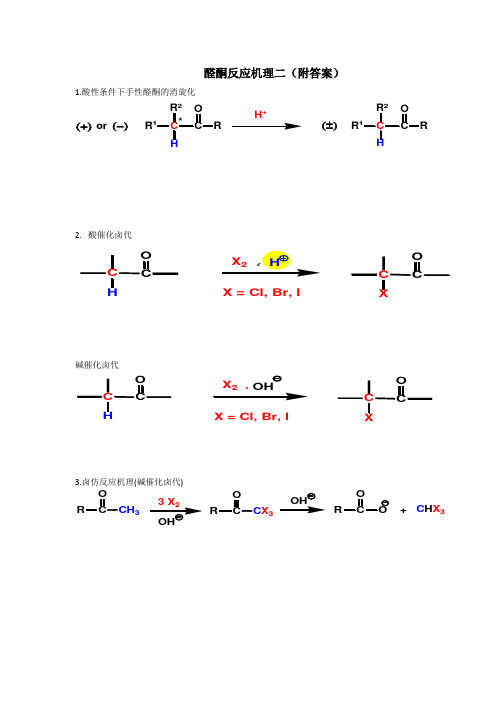
醛酮反应机理二(附答案)1.酸性条件下手性醛酮的消旋化RR 12CRR 1HR 2*H+(±)or (+)(-)2. 酸催化卤代碱催化卤代X,X = Cl, Br, I3.卤仿反应机理(碱催化卤代)3C C X 3CRC H X 3CR+OHX = Cl, Br, I4.羟醛缩合机理羟醛缩合产物的分解机理(羟醛缩合的逆反应)5.酸催化下的羟醛缩合机理:CH3CH3O2H3H3CH CH3+2Al[O(CH3)3]3OOOHOClHCl(Lewis 酸催化)6.Mannich反应(胺甲基化反应)CH 2R'+CH2O H+CHR'2NH1酸催化过氧酸氧化 —— 生成酯 ( Baeyer-Villiger 反应)(“O ”如何插入C -C 键)“O ”插入取代基多的基团一边(取代基多的基团易迁移)R 1R2R 1R 2R 122.Wolff-Kishner 还原酮羰基至亚甲基机理RR'O RCH 2R'NH -NH , Na N 2+3. Meerwein-Ponndorf 还原反应(i-PrO)AlCH 3CHOHCH 3(过量)O R'(H)ROH R'(H)4.醛酮被金属还原至醇或二醇Na or LiRR'(H)RC OH R'RC OH R'RR'O2Mg (Hg)5.Cannizzaro 反应(歧化反应)R浓 OHR RCH 2OH+H +R COOH+RCHPhHOO OHPhC O7. Benzoin 缩合反应(安息香缩合反应)8.Wittig 反应:Ph 32+R 2+Ph 32不饱和醛、酮,1. α, β-不饱和醛酮与亲核试剂的亲核加成反应。
1, 2 – 加成为主(Nu - : 强亲核试剂,如 RLi, 炔基钠, LiAlH4等)1β2. 与亲核试剂的1, 4 – 加成机理α, β-不饱和醛酮与亲电试剂的亲电加成反应 3.α, β-不饱和醛酮的羟醛缩合(插烯规则) 4.Michael加成COCC+碱4. Diels-Alder 反应+例 1:写出下列合成的路线1酸性条件下手性醛酮的消旋化RR 12CRR 1R 2*H+(±)or (+)(-)CO RR 1R R 2H H RR 1C RR 2R 1C R 1R2*2+H 3(±)H H2.酸催化卤代O+CXC烯醇化C HH 2O X+H X碱催化卤代X ,X = Cl, Br, IX = Cl, Br, ICHCXOH3.卤仿反应机理(碱催化卤代)3C C X3CR C H X3CR+OH23R3COHC H X3+2RXCHXX3HOR CHRXX4.羟醛缩合机理羟醛缩合产物的分解机理(羟醛缩合的逆反应)CCHH CHOHCOHO5.酸催化下的羟醛缩合机理:CH3O2H 3H 3CHCH 3+23)3]3O OHHCl(Lewis 酸催化)C HH+CHH +烯醇化H 2OCOH HCHHOH 2OH 2O++H +3O8. Mannich 反应(胺甲基化反应)CH 2+CH 2OR"NR"H+CH R'CH 2NR"HRCHR'H RRHC HHCR"NR"HH2CO HNH2NH2R"NR"OHR CHOHR"NR"CHR"NR"R'1酸催化过氧酸氧化——生成酯(Baeyer-Villiger反应)(“O”如何插入C-C键)“O”插入取代基多的基团一边(取代基多的基团易迁移)R1R2R1O R2R1O R2R1R2RCO O H+R1R2HH OR1C R2O HO+ OR1C R2O HOδ+~ R1OR C R2O HOH++OHR1O R2H+R1O R2(来自过氧酸)R迁移2.Wolff-Kishner 还原酮羰基至亚甲基机理R R'OR CH2R'NH-NH, Na~ 200CN2+R R'ONH2-NH2R C R'ONH-NH2R C R'OHN-NH2HOHRCR'HOH RCR'R CH2R' RCHR'N N HOHRCHR'225.Meerwein-Ponndorf 还原反应(i-PrO)AlCH 3CHOHCH3(过量)OR'(H)ROHR'(H)AlOOCHδRCOCH3C CH3+OH+AlO3)2 3333334.醛酮被金属还原至醇或二醇Na or LiR R'(H)R R'(H)e CO NaCORHR'eHO Na HRC OH RC OH RCR'O2Mg (Hg)自由基二聚RC O RC ORC OH RC OH PinacolH ORR'2eMg RC ORC O6. Cannizzaro 反应(歧化反应)RC OROHOHR +~ H ROO +HRC OH负H 迁移练习R浓 OHR RCH 2OH+H +R COOH+RCHPhHOO OHPhC O9. Benzoin 缩合反应(安息香缩合反应)8.Wittig 反应:Ph 32+R 2+Ph 3Ph 3P2R 2+Ph 3Ph 3C C O RPh 3C C R制备烯烃232OH或CH2Ph3P+3+或PPh3CH3I+PPh3+2CH3I(1) PPh32Ph323不饱和醛、酮共振式,OOE ENu Nu2.α, β-不饱和醛酮与亲核试剂的亲核加成反应。
第十二章醛酮不饱和-文档资料

2、共轭醛酮的特殊性质
1)特强的亲和试剂发生1,2-加成 亲核试剂进攻羰基,与饱和共轭醛酮类似。 典型试剂:烃基锂、炔化钠
H3C
O
H3C
Li
CH3
H3C
H3C
OH
CH3
H3C
O HC
CH3
C- Na+
H3C
OH
H3C
C
CH
勤读力耕,立己达人
思考:下列反应得到什么产物
O H3C Li
H3C O
C=C与C=O如果只相差一个CH2,酸、碱催化变成 共轭醛醛酮,E降低。
O
酸催化
H2C
CH3
H3C
O CH3
O
碱催化
H2C
CH3
H3C
O
CH3
勤读力耕,立己达人
碱催化异构机理:通过烯醇盐中间体进行
O
H2C
CH3
H HO-
O-
H2C
CH3
H2C
O
CH-
CH3
H2C-
O CH3
H2O
H3C
O CH3
O
H3C
CH2 O-
H3C
CH3
O
H3C O
H3C
CH3 O-
H2O
H3C O
CH3 Li CH3 H3C
CH3
H3C
H3C O- H3C
SOCl 2 H3C H3C Cl
CH3
Li CH3 H3C
H3C
CH3
H3C
CH3 H3C
勤读力耕,立己达人
第二节 醌 quinone
醌是环状共轭二酮—芳香化合物的衍生物。
第十二章 醛酮(不饱和)

4-nitrosophenol
勤力读耕,立己达人
3)1,4-加成反应
共轭酮与HCN、 HX、 MeOH/ZnCl2发生1,4-加成, 中间体重排取代二酚,一般反应式如下:
HO HX O O OH X HO X
O
O
X=卤素、CN、甲氧基(氯化锌催化)。
HO HCl O OH Cl HO Cl
-
H3C O H3C
CH3
H3C Mg Cl Ag
+
-
H3C Cl Mg O H3C
CH3 CH3
H5C2 O C2H5 CH3
+
H3O
+
H3C H3C CH3 O
勤力读耕,立己达人
合成化合物D
H3C CH3 O
H3C O H2O
CH3
KH
H3C O
-
CH2
H3C O H3C
CH3 O
-
H3C O H3C
C=C与C=O如果只相差一个CH2,酸、碱催化变成 共轭醛醛酮,E降低。
O H2C CH3
酸催化
H3C
O CH3
O H2C CH3
碱催化
H3C
O CH3
勤力读耕,立己达人
碱催化异构机理:通过烯醇盐中间体进行
O H2C CH3 CH
-
O H2C CH3
H HO
-
O H2C
O CH3 H2C
-
CH3
*Fries重排
苯酚在=与酰氯发生反应时先生成酚酯:
O HO
+
Cl
R
ACl 3
R
O
有机化学:第十章 醛 酮(1)
(2) O
O
C OC2H5
H+
O
HOCH2CH2OH
O
O C OC2H5
LiAlH4 (C2H5)2O
O
O
CH2OH H+, H2O
O
CH2OH + HOCH2CH2OH
上海交通大学化学化工学院
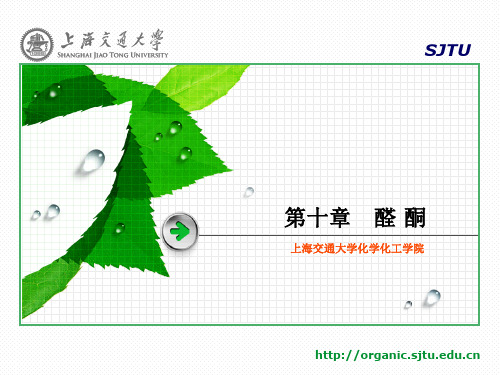
(3) 维尼纶:聚乙烯醇含有多个亲水基团,为了提高其耐水性能,可用 甲醛使其部分缩醛化,得到性能优良的合成纤维——维尼纶。
(Ph)3P
- R1 C
R2
上海交通大学化学化工学院
G.Wittg于1953年开始系统研究了它与醛酮的反应,并应用在合 成上。建立了独特的结构和合成方法。
R1 C O + Ph3P C
R2
Ph3P + C
O- C
(Ph)3PO +
R1 CC
R2
缩醛对氧化剂、还原剂、碱稳定,对酸不稳定。
上海交通大学化学化工学院
应用:保护羰基。
(1) CH2 CHCHO
CH2 CH CHO OH OH
2C2H5OH
CH2 CHCH
OC2H5
[O]
OC2H5 H2O
H+, H2O
CH2 CH OH OH
OC2H5 OC2H5
SJTU
第十章 醛 酮
上海交通大学化学化工学院
(一)结构和命名:
1. 结构:
通式:CnH2nO ,醛、酮互为同分异构体。
官能团:
O
O
C H 醛基
C
羰基
碳原子采用sp2 杂化,三个σ键共 平面,羰基碳原子和氧原子上的p轨道 在侧面重叠生成π键 ,氧原子上还有 两对未共用电子。
第13章 不饱和醛酮和取代醛酮

3-丁烯醛
OH
:
:
:
H2O +
:
CH2 CH CH CH O :
:
:
CH2 CH CH CH O :
在酸性溶液中3-丁烯醛通过烯醇转化为2-丁烯醛:
CH2 CH CH2 CH O : H+ H+
: : :
CH2 CH CH CH OH CH3 CH CH CH OH
+
: :
CH2 CH CH CH O:
CH3CH CHCH O:
2-丁烯醛
CH2 CH CH2 CH OH
+
CH2 CH CH2 CH OH
+
H+ H+
: :
: :
: :
CH3 CH CH CH OH
+
CH3 CH CH CH OH
+
:
:
CH3CH CHCH O:
13.1.1 α,β-不饱和醛酮的反应
H+
简单的烯烃与氢氰酸不起加成反应,因为生成的活性中间体——碳负离 子,非常不稳定。
R CH CH R' + CN R CH CH R' CN
:
α, β-不饱和醛酮与其他弱碱性的亲核试剂也容易起1, 4-加成反应:
O CH3 C CH CH C6H5 + HN
4-苯基-3-丁烯-2-酮 六氢吡啶
13.1.1.5 还原 α, β-不饱和醛酮用氢化铝锂还原生成α, β-不饱和醇:
CH3 CH CHCHO
2-丁烯醛
+
有机催化的不对称氧化反应

2008年第28卷有机化学V ol. 28 2008588~597第4期, 588~597 Chinese Journal of Organic Chemistry No. 4*E-mail:Received November 17, 2006; revised August 10, 2007; accepted October 9, 2007.No. 4宫斌等:有机催化的不对称氧化反应5891 α,β-不饱和醛酮有机催化不对称环氧化反应1.1 低聚肽为催化剂1980年, Julia 和Colonna [3]报道以低聚肽为催化剂, α,β-不饱和酮在氢氧化钠-过氧化氢水溶液、有机溶剂和低聚肽的三相体系中反应, 获得了具有光学活性的环氧化产物, 反应收率达96%, ee 值达96% (Eq. 1).然而, 苛刻的反应条件限制了该反应的应用, 如反应时间长(有时长达3 d), 反应中需要持续加入氧化剂和碱, 催化剂使用前需要活化处理, 反应底物有限, 凝胶状的催化剂回收困难等[4].Roberts 等[5]为解决这些问题, 从催化体系入手开发出两相催化体系. 用尿素-过氧化氢为氧化剂, 聚亮氨酸(PLL)为催化剂在四氢呋喃中反应, 不仅大大缩短了反应时间, 而且糊状的PLL 通过过滤回收, 循环使用6次, 反应转化率及对映选择性没有明显改变(>95% 转化率, 96%~98% ee 值). 该催化体系不仅对一些简单α,β-不饱和酮的不对称环氧化有较好的效果, 对一些复杂底物同样有很好的反应效果和选择性(Eq. 2), 在此条件下, 未观察到R 2酯基一侧双键的氧化.为进一步减少催化剂回收损失, Roberts[5,6]在稍后的工作中尝试将PLL 固载到载体硅胶上, 将硅胶负载的催化剂PLLSi 用于环氧化反应, 不仅减少了催化剂损失, 而且进一步提高了反应速率(<50 min); 另外, 催化剂用量即使降低至2.5 mol%, 反应依然保持较高的对映选择性(93% ee 值). 硅胶负载的聚丙氨酸(PLASi)催化活性不如PLLSi, 产物收率及ee 均有所降低(60%收率, 80% ee 值), 而硅胶负载的聚缬氨酸(PLVSi)和聚苯丙氨酸(PLPSi)的催化活性较差(30% ee 值). 聚氨基酸碳链的微小差异引起催化活性的巨大差异, 其原因至今仍不清楚. 硅胶负载的聚氨基酸催化剂在低活性底物的不对称环氧化过程中表现优秀. PLLSi 以及硅胶负载的聚非天然氨基酸——聚戊基氨基乙酸(PLNSi)是已知小范围低活性α,β-不饱和酮不对称环氧化反应最好的催化剂, 如催化不对称环氧化低活性化合物6~9生成相应的环氧化物10~13 (≥90%转化率, ≥95% ee 值).在研究两相催化体系的同时, 研究人员对三相催化体系的优化探索也在不断进行, 2004年研究有了突破性进展. Militzer, Geller 等[7]通过向三相体系中加入相转移催化剂——溴代四丁基铵(TBAB), 大大加快了Julia- Colonna 环氧化反应速率, 在室温下反应1.5 h, 转化率可达99%, ee 值达94%; 氧化剂和碱的用量也显著降低至底物用量的1.3倍, 而在此前反应所需氧化剂用量为底物用量的30倍. Geller 等[8]在制备催化剂PLL 时还意外地发现, 在相对高温下(甲苯中回流)制得的PLL 催化活性比市售品的催化活性显著提高, PLL 用量可降低至2~5 wt%, 即使降低至0.1 wt%仍能保持较好的催化效果(61%转化率, 80% ee 值)[9]. Gerlach [7b]利用此方法已将反应放大到100 g 级.其它有关低聚肽催化的α,β-不饱和醛酮的不对称环氧化反应的相关技术及研究报道可参考相关文献[10].由于肽本身在反应条件下可能会像酶一样发生各样的构象变化, 因此低聚肽催化的α,β-不饱和醛酮的不对称环氧化反应的确切机理仍不十分清楚[11], 不过大量的研究结果为反应机理的探讨提供了线索及支持.一般认为, 低聚肽的不对称催化活性来源于肽的二级结构及其氨基酸取代基之间相互作用. 无论是直接用低聚肽为催化剂还是用固载到载体上的低聚肽为催化剂, 反应结果提示: (1)低聚肽的N 末端区域是催化活性中心; (2)低聚肽的α-螺旋构象对催化活性有重要作用. 低聚肽的N 末端区域及α-螺旋构象通过氢键与底物相互作用, 形成具有手性环境的反应的过渡态, 从而完成手性催化氧化. Berkessel [12]研究了单体数为1~20的PLL 在三相催化体系中的催化效果, 结果表明5个L -Leu 单体聚合的低聚肽便可使反应ee 值达到最大(96%~98%), 而此时低聚肽恰好能形成一个α-螺旋构象; 随着肽链增长, 反应收率有所增加, 当单体数量达到14个时, 催化活性不再有明显改变. Berkessel [12]和Roberts [13]分别建立了相似的催化模型, 通过实验和计算的方法试图阐明作为催化活性中心的N 末端区域如何与底物相互作用, 但这两个模型对低聚肽中到底哪几个氨基与底物相互作用从而产生催化效果仍存在较大590有 机 化 学 V ol. 28, 2008分歧.当然, 高对映选择性的低聚肽催化剂的催化作用可能不仅局限于α-螺旋构象的存在, β-转角结构同样会使具有反应性的侧链官能团处于肽分子丰富的手性和作用中心, 使得这些官能团能在手性环境中产生催化作用.1.2 氨基酸及其衍生物为催化剂氨基酸及其衍生物属于胺类催化剂, 已成功地应用于许多类型的不对称反应中, 其中L -脯氨酸(L -Proline)及其衍生物是应用最成功的胺类催化剂[14].2005年, Lattanzi 等[15]报道了用商品化的手性胺14为催化剂, 叔丁基过氧化氢(TBHP)为氧化剂(Eq. 3), 反应获得较满意的结果(87% 收率, 80% ee 值), 但不足的是反应时间较长(105~190 h).对反应机理的研究发现(图1), 在不同的反应步骤中, 催化剂都是以单分子形式参与反应的, 催化剂14的双官能团OH 及NH 2分别与酮羰基氧原子及烷基过氧化氢作用, 形成稳定的过渡态, 底物酮的立体电子效应及分子构象对反应的反应性和对映选择性有重要的影响. 极性、质子、配位性溶剂都会影响过渡态中间体的稳定性, 降低反应的对映选择性, 因此反应在己烷、环己烷等非极性非配位的溶剂中效果较好.图1 Lattanzi 的氨基酸衍生物催化循环反应机理 Figure 1 Lattanzi’s catalytic cycle of amino acid derivatives同年, Jorgensen 小组[16]将手性胺催化剂17用于α,β-不饱和醛的不对称环氧化反应(Eq. 4). 用过氧化氢、尿素-过氧化氢为氧化剂, 反应转化率及对映选择性都很优秀(>90%转化率, 96% ee 值); 用叔丁基过氧化氢、枯烯基过氧化氢等有机氧化剂, 反应ee 值略有降低(93% ee 值), 但反应转化率有显著降低(30%~40%). 一般的, 反应溶剂对不对称有机催化反应影响较大, 但该反应在不同溶剂(CH 2Cl 2, 甲苯, 95% EtOH, 85% MeOH, 90% THF)中都有优秀的对映选择性(92%~96% ee 值). 值得注意的是, Jorgensen 等还发现该反应可使用便宜、安全及环境友好的水为溶剂, 反应18 h, 转化率28%, ee 值90%. 向水溶液中加入乙醇, 可以提高反应效率及对映选择性, 当V (乙醇)∶V (水)=1∶1时, 反应9 h, 转化率可高达97%, ee 值达92%.Jorgensen 以极性质子溶剂水或乙醇水溶液为反应溶剂得到了较好的反应结果, 这一结果与Lattanzi 等所得出的实验结论刚好矛盾, 尽管两者所用的反应底物类型略有不同, 但由此可以推测出, 反应底物、氧化剂、催化剂分子之间存在着更复杂的相互作用. Jorgensen 认为(图2)反应第一步是手性胺催化剂17与底物反应生成亚胺盐离子, 然后过氧化物作为亲核试剂进攻β-C 原子生成C —O 键得到烯胺中间体, 接着过氧化物氧原子进攻烯胺C 原子发生环氧化过程, 最后烯胺水解得到产图2 Jorgensen’s 氨基酸衍生物催化循环反应机理 Figure 2 Jorgensen’s catalytic cycle of amino acid derivativesNo. 4宫斌等:有机催化的不对称氧化反应591物.1.3 相转移催化剂为催化剂手性相转移催化在不对称合成中占有重要地位, 金鸡纳碱类相转移催化剂用于有机催化的不对称氧化反应的报道也屡见不鲜, 取得了较好的反应结果; 近年来, 研究人员又设计开发出许多新型的相转移催化剂用于有机催化的不对称氧化反应中, 也取得了较好的结果. 1.3.1 金鸡纳碱类相转移催化剂为催化剂自从Wynberg 用金鸡纳碱为相转移催化剂的先驱报道以来, 有许多小组参与了这一催化反应的研究[17]. Lygo, Corey, Adam, Arai 等[18]小组对金鸡纳碱催化剂20的母环结构进行改造, 提高了催化环氧化反应的效率和ee 值, 扩大了反应底物的范围, 使得三取代烯烃、顺式烯烃及缺电子烯烃也得到了很好的结果, 但相对而言, 反应ee 值仍然不高, 而且反应时间长, 反应温度低.2005年Jew [19]向反应体系中加入表面活性剂Span20, 用二聚的金鸡纳碱21为相转移催化剂, 反应只需0.5~12 h, 收率达94%~97%, ee 达97%~98%. 表面活性剂Span20的加入可显著加快金鸡纳碱相转移催化剂的催化反应速率, 并且提高环氧化反应的对映选择性,但可惜的是, 反应对脂肪烃基取代的底物效果较差.1.3.2 新型相转移催化剂为催化剂由于金鸡纳碱类催化剂分子结构的局限, 限制了人们对相转移催化剂更潜在本质的研究, 因此, 人们将注意力转向设计新型的相转移催化剂分子上. 目前, 设计新型相转移催化剂的基本理念是, 使催化剂分子具有分子识别力, 即能够识别具有潜手性的反应底物, 从而使反应获得足够的反应性及对映选择性; 另外, 希望该分子具有表面活性剂的作用.Maruoka 等[17]设计了新型相转移催化剂22, 分子中二芳基甲醇基通过氢键可以识别底物酮羰基, 二苯基甲基及手性萘具有空间识别能力, 将此催化剂用于化合物1的不对称环氧化中, 几乎能定量得到产物, ee 值89%~99%.Hori [20]设计了C 2对称的手性催化剂23, 与Maruoka 的不同, 该分子中的分子识别位点位于手性冠醚结构, 季铵盐N 原子上连有长度不同的碳链, 起到增加表面活性的作用. 此催化剂用于反应(1)中发现, 反应体系中的阳离子对反应效果有关键性的影响, 大的阳离子如K +, Cs +, N +(CH 3)4通常给出较好的ee 值, 分别为75%, 65%, 71%; 另外, 对于不同结构的底物, 通过调整催化剂碳链长度, 产物可获得最佳的ee 值.与Hori 设计的分子类似, Bako 等[21]设计的分子24也可通过调节分子中烃基的长度获得不同的反应结果. 当烃基碳数等于3时, 收率82%, ee 值92%, 反应时间可显著缩短到0.5~4 h.2 烯烃的有机催化不对称环氧化反应烯烃的有机催化不对称环氧化反应已有详细的综述[22], 本文简要概述其中的主要成果, 着重介绍最新的研究进展.2.1 手性胺为催化剂2000年, Aggarwal [23]首次报道了烯烃在手性胺催化剂27存在下, 用Oxone (2KHSO 5+KHSO 4+K 2SO 4)为氧化剂, 可获得中等ee 值46%的环氧化产物(Eq. 5); 大位阻及具有极性基团的手性胺催化剂(14, 28, 29)可提高产物的ee 值(54%~66%). 向反应体系中加入少量盐酸592有 机 化 学 V ol. 28, 2008溶液可以提高反应的对映选择性, 缩短反应时间[24].对催化机理的研究表明(图3), 质子化的手性胺30是反应的活性氧化剂, 30不仅作为反应的手性诱导试剂诱导不对称环氧化反应的进行, 同时也活化了氧化剂Oxone 氧化活性; 另外, 质子化的胺还可避免催化剂的氧化[25].图3 手性胺催化的烯烃环氧化反应机理Figure 3 Epoxidation mechanism of alkene catalyzed by chiral amines2.2 亚胺盐为催化剂1976年, Lusinchi 报道了氧杂氮杂环丙烷盐31可不对称催化环氧化烯烃为相应的环氧化物. 后来发现亚胺盐在Oxone 存在下也能不对称催化环氧化烯烃, 于是吸引了许多小组在这一领域探索设计选择性的不对称催化剂, 但即使用“最有效”的催化剂(32~35), 反应ee 值最多只达到70%[26].Page [27,28]在2004年打破了这种停滞的局面. 在催化剂36的存在下, 用Oxone 为氧化剂, 0 ℃下反应20~35 min, 产物(37, 38) ee 值最高达95%, 即使对末端烯, 产物39的ee 也达到29%, 这是已知用亚胺盐为催化剂所达到的最高值. 对反应催化剂用量研究发现, 催化剂用量降至0.5 mol%, 不影响反应ee 值, 只是反应时间延长至2 h. 对催化剂分子构效关系研究发现, 催化剂分子的乙缩醛结构是保持催化剂高催化活性的必要基团[29], 将O 原子用C 原子代替, 催化效果降低, 其可能原因是分子内邻近的氧原子对N 正电荷的稳定化作用, 增加了催化剂分子构象转动刚性, 从而增加了反应的对映选择性.带有砜基结构的催化剂40对苯并呋喃类烯烃有很高的催化活性[30], 反应ee 值可高达97% (Eq. 6). 用NMR 对反应中间体监测发现[31], 在-40 ℃下, 只检测到催化剂43的一个主要氧杂氮杂环丙烷盐中间体44, 此中间体与烯烃作用生成环氧化产物, 由此支持了此催化剂的高对映选择性催化机理(图4). 该反应已应用于抗高血压药Levcromvkalin 的合成[30](图5).图4 亚胺盐高对映选择性催化机理Figure 4 Catalytic mechanism of iminium salts with high enan-tioselectivityNo. 4宫斌等:有机催化的不对称氧化反应593图5 Levcromvkalin 的合成 Figure 5 Synthesis of levcromvkalin2.3 手性酮为催化剂非官能团化的反式、三取代及顺式烯烃的不对称环氧化反应长久以来一直存在对映选择性低的问题[32], 使用手性酮作催化剂为此问题提供了解决途径.手性酮催化剂通常由Oxone 与酮原位生成[32,33]. 目前, 高对映选择性的手性酮催化剂46, 47及48都是源于果糖分子结构设计制备的. 这三种催化剂对不同结构的烯烃有不同的适用性及催化效果, 见表1.表1 手性酮催化剂的应用及效果Table 1 The application and effect of chiral ketones催化剂 适用的烯烃ee/% 46 反式烯烃、三取代烯烃一般>9047 缺电子烯烃 87~97 48环状或非环状烯烃、末端烯烃 71~97这类催化剂在原位生成时受pH 值影响较大. 高pH 值会使Oxone 分解, 低pH 值会造成催化剂发生Baeyer-Villiger 副反应, 但实验结果表明, 通常高pH 值给出的反应结果较好. 虽然有研究称, 手性酮催化剂的催化活性足以与Oxone 的分解相竞争, 但这将使反应消耗大量的催化剂(20 mol%).Armstrong [34,35]使用外消旋的催化剂49, 对E -1,2-二苯乙烯反应给出较好的结果(71% 转化率, 98% ee 值); 可贵的是, 这一催化剂对其他类型的反式及三取代芳烯烃, ee 值一般也能达到70%~98%; 在反应条件下不发生Baeyer-Villiger 分解, 因此, 催化剂用量一般都<10 mol%. 但令人遗憾的是光学纯度的催化剂49制备比较困难. 若将催化剂49分子中的X 桥环去掉可得到容易制备的单环吡喃型催化剂50[36], 但反应的对映选择性会相应降低(ee 值最高83%).3 羰基化合物α-羟基化的反应光学活性的α-羟基羰基结构普遍存于天然产物及许多药物分子中; 另外, 这一结构也是合成其他重要结构, 如二醇化合物的合成子. 其有效的合成方法之一是用金属银配合物为催化剂, 亚硝基苯为氧化剂, 间接氧化锡烯醇化物[37,38]. 虽然这一催化氧化体系选择性较好, 但这一过程涉及多步反应, 步骤繁琐. 应用有机催化剂催化羰基化合物不对称α-羟基化步骤简单, 对映选择性高, 已经显示出巨大的应用潜力[39]. 3.1 氨基酸及其衍生物为催化剂 3.1.1 以有机过氧化物为氧化剂Zhong [40], MacMillan [41], Hayashi [42]几乎同时报道了以L -Proline 为催化剂, 亚硝基苯为氧化剂的醛的不对称α-氧化反应(Scheme 1). 反应首先生成O —N 化物, 之后经Adams 催化还原或用硫酸铜溶液处理, 使O —N 键断裂, 得到α-羟基化产物.Scheme 1极性溶剂, 如DMF, DMSO, CHCl 3, CH 3CN 等均适于此反应. 3个小组的反应条件及结果如表2.MacMillan [41]用5 mol%(最低可降为0.5 mol%) L -Proline 为催化剂, 在4 ℃反应2~4 h, 收率及ee 值都较高. 值得注意的是, Zhong [40]使用DMSO 为反应溶剂, 在室温下反应, 反应时间可大大缩短至10~20 min, 而反应收率及ee 值仍然较高. Hayashi 等[43]认为, 反应温度较高(>4 ℃)易使醛发生自身Aldol 反应, 因此他们将反应温度降低至-20 ℃, 产物ee 值略有提高, 但反应时间却因此延长至24 h, 而且催化剂用量也较多.与醛相比, 酮在此条件下的α-氧化并不顺利, 存在反应速率慢、收率低、ee 值低的问题, 并且α-氧化的非594有 机 化 学V ol. 28, 2008表2 α-羟基化反应条件及结果Table 2 Reaction conditions and results for α-hydroxylation作者 L -Proline 用量/mol% 溶剂 反应温度/℃ 反应时间/h 收率/% ee /%Zhong 20 DMSO 室温 0.12~0.3 60~86 97~99MacMillan 5(可降至0.5) CHCl 3 42~4 60~95 97~99Hayashi 30CH 3CN-20 2462~87 98~99对映选择性低, 酮两侧α位同时氧化的副反应较多. Hayashi [44]和Cordova [45,46]等尝试将相对大大过量的酮缓慢滴加到反应体系中, 不仅提高了反应收率(44%~91%), 而且还保证了较高的ee 值(96%~99%) (Eq. 7). Cordova [38]后来又发现, 用PhIO 及58为氧化剂, L -Proline 为催化剂, 在DMF 中反应可直接得到α-OH 化产物, ee 值达77%, 但收率较低, 只有29%.Barbas [47]将羰基化合物的α-羟基化反应用于对称螺酮去对称化反应(ADS), 通过一前一后的胺氧化/O —N 键断裂反应, 得到了含多个手性中心的产物(Eq. 8). 不仅反应的对映选择性十分优秀, 反应的非对映选择性也十分出众(ee >99%, de 最高>99%). 有趣的是, 底物分子a 位取代基对反应的选择性影响不显著, 而b 位羰基取代基却是反应表现出优秀的非对映选择性及对映选择性的关键所在.氨基酸催化不对称α-氧化过程类似与肽催化的α,β-不饱和酮的不对称环氧化过程. 催化剂α位的官能团都具有酸性质子, 酸性质子与催化剂分子中碱性的N 原子共同控制反应的区域选择性. Cordova [48]及Houk [49]通过量子力学计算表明, 反应过程中可能存在三种过渡态: O -anti , O -syn 及N -anti , 其中O -anti 过渡态能量E rel 相对最低, 因此反应的对映应选择性主要来自于O -anti 过渡态的贡献. 然而, Ramachary 和Jemmis [50]认为反应过渡态中还存在着静电力/双偶极-双偶极相互作用], 这种作用也是氨基酸具有选择性催化另一个因素. 但少量的反应动力学研究结果表明, 实际的催化反应过程比以上模型更复杂, 在每个催化循环过程中还可能存在自诱导或自加速作用, 从而使反应速率及ee 值获得提高[51].图 6 反应中可能存在的3种过渡态Figure 6 Three possible transitions existed in the reaction3.1.2 以分子氧为氧化剂分子氧作为安全、廉价、易得的氧化剂对经济和环境两方面都有益处[52]. 利用光或化学方法产生的活泼单线态氧1O 2[53]作为氧源已用于许多合成转化中, 但在不对称氧化反应中只有很少的报道[54].Cordova [54a,55]首次用分子氧实现了醛酮的直接有机催化不对称α-羟基化反应(Scheme 2). 溶剂对反应影响物. 对底物结构研究发现, 链状酮的氧化区域选择性很高, 分子氧通常在取代基最多的一侧氧化.Scheme 2多种氨基酸可用于醛的分子氧α-羟基化反应, 但L -Proline 最有效(45%~95%收率, 16%~48% ee 值). 对No. 4宫斌等:有机催化的不对称氧化反应595L -Proline 结构修饰发现, α-位修饰L -Proline 催化活性可大幅提高. α-甲基化的脯氨酸能显著提高产物的ee 值(54%~66%). 对位吸电基取代的苄基醛在三甲基硅取代的催化剂17, 63, 64催化下, 反应收率可提高到71%, ee 值可达98%, 催化活性63>64>17.用L -Proline 催化酮的分子氧α-羟基化反应所得ee 值较低(18% ee ), α-甲基脯氨酸可使反应ee 值提高到 48%; 使用直碳链的L -丙氨酸和L -缬氨酸为催化剂, ee 值可分别达到56%, 49%, 但是得到构型相反的产物. 反应虽然只得到中等ee 值的产物, 这一结果却丰富了氨基酸催化剂的种类, 并且将推动人们对催化机理的进步研究, 因为在此之前, 一般认为只有五元环的氨基酸才能保证反应的高效性及高对映选择性[55a]. 3.2 金鸡纳碱为催化剂Dupont 公司在开发一种新型农药的过程中非常幸运的发现金鸡纳碱(+)-辛可宁可催化不对称氧化β-二羰基化合物, 得到中等ee 值(50%)的α-羟基化产物(Eq. 9). 而在此之前所尝试的手性胺或氨基酸、手性季铵盐等有机催化剂对此反应无效, 过渡金属配合物催化剂催化所得产物也没有明显ee 值[56].对催化剂辛可宁(cinchonine)的结构进广泛的修饰研究发现, 手性碳原子上的羟基修饰后会显著降低反应的ee 值[57]; 另外, 喹啉母环6位上引入OCH 3基会降低反应ee 值; 而引入OH 后, ee 值有所提高(69% ee 值), 同时, 烯键单溴化后, ee 值进步提高(70%), 但二溴化ee 值略有降低(66% ee )[58]. Jorgensen [59]改用枯烯基过氧化氢为氧化剂, 二氢奎宁(dihydroquine)为催化剂, 使反应的ee 值达66%, 收率达88%, 只不过所得产物为R 型. 无水及非亲核性溶剂对反应速率及对映选择性无明显影响, 均可用于此反应, 但实验中发现, 此反应具有相转移催化反应特征[60], 当使用甲苯为溶剂时, 反应产物会随反应的进行不断析出, 这样简化了分离过程, 并且有利于催化剂的回收利用.4 小结与展望有机催化的不对称氧化反应在短短几年已经出现十分可喜的发展, 其具体表现在: (1)催化剂种类不断增加, 为各种类型的不对称氧化反应催化剂的选择提供了广泛的空间; (2)反应底物的范围不断扩大, 对一些金属有机催化效果较差的反应底物, 利用有机催化剂可以取得很好的反应收率和对映选择性; (3)催化剂回收方便, 多次循环利用不降低反应选择性; (4)保持有机催化优点的同时, 向更绿色的方向发展, 例如出现了一些使用水为溶剂、用氧气或空气为氧化剂的反应.当然, 有较好应用前景的催化反应除了具备以上几个要求, 催化剂用量需要降低到工业可接受的水平, 同时能得到高对映选择性的产物, 这点正是有机催化不对称氧化反应需要不断努力之处. 为了实现这一目标, 除了可采用传统的催化剂筛选修饰的方法, 利用一些新兴理念和技术(例如基于分子识别概念、纳米技术、分子自组装设计制备催化或离子液体为反应溶剂等)也可能获得可喜的结果; 另外, 对反应机理的深入研究是关键. 利用先进分析仪器跟踪反应进程、捕捉反应中间体推断反应机理是常用手段, 但这些推断目前常常缺少反应动力学数据的支持, 这是将来需要不断努力的. 利用计算机对反应过程进行量子化学计算, 从理论上提出更深层次或更新的反应机理模型或影响因素, 也是不应忽视的方向.有机催化的不对称氧化反应已经步入黄金时期, 相信今后的发展一定会秉承目前的发展趋势继续前进, 并且出现新的突破. 有机催化的不对称氧化反应将与酶催化、金属有机催化一起成为不对称催化氧化反应的三项有力工具.References1 Peter, I. D.; Lionel, M. Angew. Chem., Int. Ed. 2004, 43,5138.2 (a) List, B.; Yang, J. W. Science 2006, 313, 1584.(b) Armstrong, A. Angew. Chem., Int. Ed. 2004, 43, 1460. (c) Fu, B.; Xiao, Y.-M.; Tan, Z.-H.; Dong, Y.-H.; Li, N. Chin. J. Org. Chem. 2006, 26, 899 (in Chinese).596有机化学V ol. 28, 2008(傅滨, 肖玉梅, 覃兆海, 董燕红, 李楠, 有机化学, 2006, 26, 899.)(d) Jiang, H.-F.; Wang, Y.-G..; Liu, H.-L.; Liu, P. Chin. J.Org. Chem. 2004, 24, 1513 (in Chinese).(江焕峰, 王玉刚, 刘海灵, 刘鹏, 有机化学, 2004, 24, 1513.)3 (a) Julia, S.; Masana, J.; Vega, J. C. Angew. Chem., Int. Ed.Engl. 1980, 19, 929.(b) Julia, S.; Colonna, S.; Guixer, J.; Masana, J.; Rocas, J.;Annuziate, R.; Molinari, H. J. Chem. Soc., Perkin. Trans. 11982, 1317.4 Bentley, P. A.; Roberts, S. M. Chem. Commun. 1997, 739.5 Allen, J. V.; Roberts, S. M. J. Chem. Soc., Perkin. Trans. 11998, 3171.6 Baars, S.; Drauz, K.-H.; Krimmer, H. P.; Roberts, S. M.;Sander, J.; Skidmore, J.; Zanardi, G. Org. Process Res. Dev.2003, 7, 509.7 (a) Geller, T.; Gerlach, A.; Kruger, C. M.; Militzer, H. C.Tetrahedron. Lett. 2004, 45, 5065.(b) Gerlach, A.; Geller, T. Adv. Synth. Catal. 2004, 346,1247.8 Geller, T.; Gerlach, A.; Kruger, C. M.; Militzer, H. C. J.Mol. Catal. A: Chem.2006, 251, 71.9 Geller, T.; Kruger, C. M.; Militzer, H. C. Tetrahedron Lett.2004, 45, 5069.10 (a) Savizky, R. M.; Suzuki, N.; Bove, J. L. Tetrahedron:Asymmetry 1998, 9, 374.(b) Bentley, P. A.; Bickley, J. F.; Roberts, S. M.; Steiner, A.Tetrahedron. Lett. 2001, 42, 3741.(c) Flood, R. W.; Geller, T. P.; Petty, S. A.; Roberts, S. M.;Skidmore, J.; Volk, M. Org. Lett.2001, 3, 683.(d) Allen, J. V.; Drauz, K. H.; Roberts, S. M. TetrahedronLett. 1999, 40, 5417.(e) Pedrosa, L. J. M.; Pitts, M. R.; Roberts, S. M. Tetrahe-dron Lett. 2004, 45, 5073.11 Blank, J. T.; Miller, S. J. Biopolymers2006, 84, 38.12 Berkessel, A.; Gasch, N.; Glautibz, K.; Koch, G. Org. Lett.2001, 3, 3839.13 Kelly, D. R.; Roberts, S. M. Biopolymers 2006, 84, 74.14 List, B. Tetrahedron2002, 58, 5573.15 Lattanzi, A. Org. Lett. 2005, 7, 2579.16 Zhuang, W.; Marigo, M.; Jorgensen, K. A. Org. Biomol.Chem. 2005, 3, 3883.17 Ooi, T.; Maruoka, K. J. Am. Chem. Soc. 2004, 126, 6844.18 (a) Lygo, B.; To, Daniel, C. M. Chem. Commun. 2002,2360.(b) Corey, E. J.; Zhang, F. Y. Org. Lett. 1999, 1, 1287.(c) Adam, W.; Rao, P. B.; Degen, H. G.; Levai, A.; Patonay,T.; Saha-Moller, C. R. J. Org. Chem. 2002, 67, 259.(d) Arai, S.; Tsuge, H.; Oku, M.; Miura, M.; Shioiri, T.Tetrahedron 2002, 58, 1623.(e) Adam, W.; Rao, P. B.; Degen, H. G.; Saha-Moller, C. R.Tetrahedron: Asymmetry 2001, 12, 121.19 Jew, S. S.; Lee, J. H.; Jeong, B. S.; Yoo, M. S.; Kim, M. J.;Lee, Y. J.; Lee, J.; Choi, S. H.; Lee, K.; Lah, M. S.; Park, H.G. Angew. Chem., Int. Ed. 2005, 44, 1383.20 Hori, K.; Tamura, M.; Tani, K.; Nishiwaki, N.; Ariga, M.;Tohda, Y. Tetrahedron Lett. 2006, 47, 3115.21 Bako, T.; Bako, P.; Keglevich, G.; Bombicz, P.; Kubinyi,M.; Pal, K.; Bodor, S.; Mako, A.; Toke, L. Tetrahedron:Asymmetry 2004, 15, 1589.22 Zhang, Z.-G.; Wang, X.-Y.; Sun, C.; Shi, H.-C. Chin. J.Org. Chem. 2004, 24, 7 (in Chinese).(张治国, 王歆燕, 孙川, 石鸿昌, 有机化学, 2004, 24, 7.)23 (a) Adamo, M. F. A.; Aggarwal, V. K.; Sage, M. A. J. Am.Chem. Soc. 2000, 122, 8317.(b) Adamo, M. F. A.; Aggarwal, V. K.; Sage, M. A. J. Am.Chem. Soc. 2002, 124, 11223.24 Ho, C. Y.; Chen, Y. C.; Wong, M. K.; Yang, D. J. Org.Chem. 2005, 70, 898.25 Aggarwal, V. K.; Lopin, C.; Sandrinel, F. J. Am. Chem. Soc.2003, 125, 7596.26 Wong, M. K.; Ho, L. M.; Zheng, Y. S.; Ho, C. Y.; Yang, D.Org. Lett. 2001, 3, 2587.27 Page, P. C. B.; Buckley, B.; Blaker, J. Org. Lett. 2004, 6,1543.28 Page, P. C. B.; Barros, D.; Buckley, B. R.; Ardakani, A.;Marples, B. A. J. Org. Chem. 2004, 69, 3595.29 Page, P. C. B.; Rassias, G.. A.; Barros, D.; Ardakani, A.;Buckley, B. R.; Bethell, D.; Smith, T. A. D.; Slawin, Alexandra, M. Z. J. Org. Chem. 2001, 66, 6926.30 Page, P. C. B.; Buckley, B. R.; Heaney, H.; Blacker, A. J.Org. Lett. 2005, 7, 375.31 Page, P. C. B.; Barros, D.; Buckley, B. R.; Marples, B. A.Tetrahedron: Asymmetry 2005, 16, 3488.32 Shi, Y. Acc. Chem. Res. 2004, 37, 488.33 Shi, Y. Acc. Chem. Res. 2004, 37, 497.34 Armstrong, A.; Moss, W. O.; Reeves, J. R. Tetrahedron:Asymmetry 2001, 12, 2779.35 Armstrong, A.; Ahmed, G.; Fernandez, B. D.; Hayter, B. R.;Wailes, J. S. J. Org. Chem. 2002, 67, 8610.36 Armstrong, A.; Tsuchiya, T. Tetrahedron 2006, 62, 257.37 Marigo, M.; Jorgenson, K. A. Chem. Commun. 2006, 2001.38 Engqvist, M.; Casas, J.; Sunden, H.; Ibrahem, I.; Cordova,A. Tetrahedron. Lett. 2005, 46, 2053.39 Plietker, B. Tetrahedron: Asymmetry 2005, 16, 3453.40 Zhong, G. F. Angew. Chem., Int. Ed.2003, 42, 4247.41 Brown, S. P.; Brochu, M. P.; Sinz, C. J.; MacMillan, D. W.C. J. Am. Chem. Soc. 2003, 125, 10808.42 Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M.Tetrahedron Lett. 2003, 44, 8293.43 Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Hibino, K.; Shoji,M. J. Org. Chem. 2004, 69, 5966.44 Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Shoji, M. Angew.Chem., Int. Ed. 2004, 43, 1112.45 Bogevig, A.; Sunden, H.; Cordova, A. Angew. Chem., Int.Ed. 2004, 43, 1109.46 Sunden, H.; Ibrahem, I.; Adolfsson, H.; Cordova, A. Tetra-。
【有机化学】α, β-不饱和醛酮 、 醌、羟基醛酮 、酚醛和酚酮、紫外光谱

+ HCN(CH3)2 2) H2O
DMF
CHO 主要产物
反应机理:
POCl3 +
O
O H C N(CH3)2 Cl
: :
Cl2PH-O-CH-N(CH3)2
O
Cl2P O C=N+(CH3)2Cl-
H
H Cl
C=N+ (CH3)2Cl2PHO2-
OH
+
H
+
Cl C=N(CH3)2
OH
H2O
Cl C N(CH3)2 H
一 反应
1. 1-羟基酮的反应
(1) 银镜反应
(2) 与苯肼作用生成脎(osazones)(成脎反应).
CH2OH
OH-
R-C=O
1-羟基酮
CHOH OH-
R-C-OH 烯二醇
CHO
R-CH-OH a-羟基醛
C6H5COCH2OH + 3 C6H5NHNH2
N
H3C C
N
HC H N
C6H5
+
C6H5NH2
ArCHO
CN-
O-
Ar-C-H
H2O
OH-
OH
Ar-C-H
CN
CN
OH-
OH
Ar-C-
H2O
CN
ArCHO OHO-
O- OH
Ar-C-C-Ar
H2O
OH-
H CN
OHOH OH-
Ar-C-C-Ar
H2O H CN
-CN-
OHO
Ar-C-C-Ar +CN+ Ar-C-C-Ar
H CN
H
α、β-不饱和醛酮 羰基还原成亚甲基双键不含原

α、β-不饱和醛酮的羰基还原成亚甲基双键不含原是有机化学中的一种重要反应。
本文将从该反应的基本原理、反应机理、影响因素等方面进行介绍和分析。
一、基本原理α、β-不饱和醛酮的羰基还原成亚甲基双键不含原是一种亚甲基化反应,通常通过氢化试剂进行。
在该反应中,羰基醛酮分子中的CC双键被加成,生成羟基化合物。
一般情况下,这种反应是选择性还原羰基化合物中较弱的双键,而不影响较强的双键。
二、反应机理α、β-不饱和醛酮的羰基还原成亚甲基双键不含原的反应机理主要有三种方式:催化氢化、硼氢化和金属锂还原。
其中,催化氢化是最常见的反应方式,通常使用钯、铑、铂等过渡金属催化剂。
硼氢化方式则是通过硼氢化试剂将CC双键加成成为几何异构体,最后得到相应的醇。
金属锂还原则是利用金属锂将羰基化合物中的羰基还原成亚甲基双键。
三、影响因素α、β-不饱和醛酮的羰基还原成亚甲基双键不含原的反应受到多种因素的影响,包括反应条件、催化剂种类、底物结构等。
一般来说,合适的温度、压力和溶剂选择对于反应的进行起到重要作用。
而催化剂的选择则会影响反应的速率和产物的选择性,不同的底物结构也会对反应的进行产生一定的影响。
四、应用α、β-不饱和醛酮的羰基还原成亚甲基双键不含原反应在有机合成中应用广泛,特别是在制备医药、香料、农药等方面。
通过该反应,可以有效地合成具有特定生物活性的化合物,为医药、农药等领域的研究与开发提供了重要的化学手段。
α、β-不饱和醛酮的羰基还原成亚甲基双键不含原反应是一种重要的有机合成反应,具有广泛的应用前景。
对于该反应的基本原理、反应机理和影响因素的深入了解,将有助于提高反应的效率和选择性,进一步推动有机合成领域的发展和应用。
α、β-不饱和醛酮的羰基还原成亚甲基双键不含原反应在许多领域都有重要的应用。
其中,医药领域是该反应应用最为广泛的领域之一。
由于α、β-不饱和醛酮的羰基还原为亚甲基双键反应可以合成具有生物活性的化合物,因此在药物研发和合成中具有重要的地位。
α,β—不饱和醛酮的反应

α,β—不饱和醛酮的反应
不饱和醛酮是一类含有碳碳双键和醛基或酮基的有机化合物。
它们可以参与多种反应,下面列举了其中一些常见的反应。
1. 加成反应:不饱和醛酮可以与亲核试剂发生加成反应。
例如,它们可以与碱性溶液中的亲核试剂如水、胺或醇反应,生成相应的加成产物。
2. Michael加成:不饱和醛酮可以参与Michael加成反应,与
含有可负电荷的亲核试剂(如醇、胺、硫醇等)反应,形成
1,4-加成产物。
3. 氧化反应:不饱和醛酮可以被氧化剂如氧气、过氧化氢等氧化,形成相应的醛酸或酮酸。
4. 还原反应:不饱和醛酮可以通过还原反应还原为相应的醇。
还原剂包括金属铝、钠、锂等还原剂,还有氢气与催化剂(如铂或钯)反应。
5. 缩合反应:不饱和醛酮可以与胺反应形成亲缘结构的胺缩合产物。
6. 羟基化反应:不饱和醛酮可以与水或醇反应,羰基碳上的氢可以被羟基取代。
需要注意的是,不同的不饱和醛酮结构对应的反应也会有所不
同。
因此,具体的反应条件和产物取决于具体的不饱和醛酮结构和试剂条件。
四大谱图详解

/nm 177 178 204 214 186 339,665 280 300,665 270
max
13000 10000 41 60 1000 150000 22 100 12
跃迁类型
* * n* n*
n*,n*
n*, n* n* n*
常用术语
红移与蓝移 吸收峰向长波方向移动的现象叫红 移。 吸收峰向短波方向移动的现象叫蓝 移,也叫紫移。
基团基团对吸收带波长的贡献对吸收带波长的贡献共轭双烯共轭双烯基本值基本值217217环内双键环内双键3636每增加一个共轭双键每增加一个共轭双键3030每一个烷基或环烷取代每一个烷基或环烷取代55环外双键环外双键55oacoac0066srsr3030clclbrbr55nr2nr260601六元环不饱和酮基本值215取代1221个环外双键计算值244nm251nm不饱和酮基本值2152个烷基取代1221个烷基取代102个环外双键计算值259nm258nm不饱和酮基准值215延长1个共轭双键301个烷基取代181个烷基取代18计算值281nm281nm紫外吸收与分子结构关系紫外吸收与分子结构关系溶剂校正溶剂甲醇氯仿二氧乙醚己烷15711118紫外吸收与分子结构关系紫外吸收与分子结构关系不饱和羧酸酯酰胺紫外吸收与分子结构关系紫外吸收与分子结构关系苯的紫外吸收光谱溶剂
例:CH4 max= 125nm ②. n* 跃迁
CH3CH3 max= 135nm
分子中含有杂原子 S、N、O、X 等饱和化合物。
吸收波长:< 200nm(在远紫外区)
例:CH3OH max= 183nm(150) CH3CH2OCH2CH3 max= 188nm 某些含孤对电子的饱和化合物,如:硫醚、二硫化合物、硫醇、 胺、溴化物、碘化物在近紫外区有弱吸收。
13不饱和醛酮及取代醛酮

13--不饱和醛酮及取代醛酮§1.α,β-不饱和醛酮不饱和醛酮分子中,C=C 位于α-和β-碳原子间的称谓α,β-不饱和醛酮;位于β-和γ-碳原子间的,则称为β,γ-不饱和醛酮,由于前者和羰基组成共轭体系,所以比后者更稳定。
例如:H 2C CHCH 2CH OH 3CHC CHCH O△H =-25kJ/mol 放热 反 应在酸或碱催化下,3-丁烯醛容易转变成2-丁烯醛: ①在碱催化下:H 2C CHCH 2CH O H 3CHC CHCH O+OH H 2O +CH 2=CHCHCH=OCH 2=CHCH=CH OCH 2CH=CHCH=O②在酸催化下:H 2C CHCH 2CH O2C CH CH 2CH OHH 2C C HCH 2HC OHH +H 2C C H C H C H OH3CH C H CHOHCH 3CH=CHCH OHH 3CHC CH CH=OH- H +CH 3CH=CHCH=O1.α,β-不饱和醛酮 的反应 (1) 亲核加成:①与HCN 加成α,β不饱和酮与HCN 反应,主要生成1,4加成产物:α,β不饱和醛与HCN反应,主要生成1,2加成产物。
②与格氏试剂加成羰基上的取代基大小对1,2 和 1,4 加成有一定影响。
下列反应中的数据也说明了羰基上取代基大小对1,2和1,4 加成的影响:C6H5HC CHCOR(1)C H MgBr3C6H5HC CH COHRC2H5+C6H5CHCH2COR2H5 1,2-加成产物1,4-加成产物R=H CH3 C2H5 CH(CH3)2 C(CH3)3 C6H5 1,4-加成产物% 0 60 71 100 100 99③与烃基锂加成主要发生在1,2加成:④与二烃基酮锂加成以1,4 加成为主H OO(2)、亲电加成αβ不饱和醛酮与亲电试剂,一般都发生1,4加成OHBr(g)OBr (3)还原①使羰基还原②使双键还原③使羰基,双键同时被还原(催化加氢)H 3CH 2CH 2CHC CCHOCH 2CH 3H 2CH 3CH 2CH 2CH 2CHCH 2OH2CH 3(4)氧化αβ不饱和醛在温和条件下,可氧化为αβ不饱和羧酸(5)狄尔斯-阿尔德反应(Diels-Alder )反应2. α,β-不饱和醛酮的制备 有醛酮的缩合反应制备。
αβ不饱和醛酮
(2)只还原C=C 用 H2/Pd-C
O
O
H2/Pd-C
or 1)Li,NH3(l),-33℃
2) H3O+
(95%)
(3) C=C和C=O 同时还原
CH3CH2CH2CH=C(Et)CHO
O R1 R2
H2,Ni or Pt
CH3(CH2)3CH(Et)CH2OH
Zn/HCOONH4/C2H5OH/H2O
CH
C CH3
CH3
+
CH3CH CH2 C CH3
CH3
90% 1%
3% 95%
4.迈克尔反应
Michael reaction
O O
1.定义:烯醇负离子与α ,β -不饱和羰基化合物在碱性催化剂作用下1, 4-加成。 2.给体:能提供烯醇负离子的化合物:
CH3NO2
CH3C
N
3.受体:α ,β -不饱和羰基化合物共轭体系 O O
1, 4–加成 12%
5.如果的羰基和一个很大的基团如三级丁基相连, 无论哪一种格式试剂都得到1,4-加成产物
6.为得到1,4-加成产物,有一种常用方法是加入 0.05mol的卤化亚铜或者用二烃基铜锂进行反应
CH3MgBr CH C CH3 Et2O
OH
O
CH3CH
H3 O+
O
CH3CH
无Cu+ 存在微量Cu+
CH3C CH2 C OC2H5
C N
R
R CH CH C OR CH2 CH CH CH ቤተ መጻሕፍቲ ባይዱ R 4.碱性催化剂:三乙胺,六氢吡啶,氢氧化钾,乙醇钠,氨基钠 5.该反应可逆,升温对你反应有利
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
O
??
CCC HH
一,α, β-不饱和醛酮的结构与特 性
1. 定义:碳碳双键位于α,β-碳原子间的不饱和醛酮
CH3CH=CH CH=O
O
CH3CH=CHCCH3
2-丁烯醛
O
CH3CCH=CHC 6H5
3-戊烯-2-酮
O
4-苯基-3-丁烯-2-酮
甲基-2-环己烯-1-酮
2.特性:体系稳定 C=C与 C=O 组成共轭 体系
Unsaturated Aldehydes and Ketones
1. α, β-不饱和醛酮的结构与特性 2. 亲电加成 Nucleophilic addition 3. 亲核加成 Electrophilic addition 4. 迈克尔反应 Michael reaction 5. 还原反应 Reduction reaction 6. Diels-Alder 反应 7. 插烯作用
O
OH -
OH -
or
+
O
O
OO
O
O
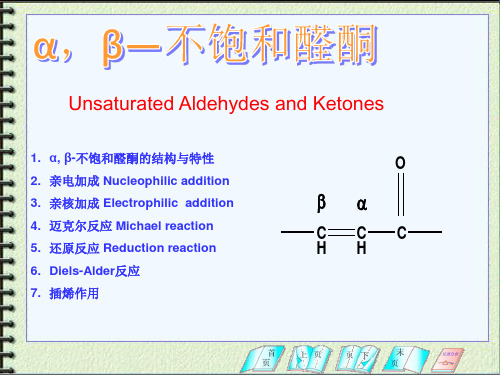
5.还原反应 Reduction reaction
(1)只 还原C=O 用LiAlH4 或 NaBH4
O LiAlH4 H2O
H OH
Et 2O
(97%)
(2)只还原C=C 用 H2/Pd-C
O H2/Pd-C
or 1)Li,NH3(l),-33℃
2) H3O+ (3) C=C和C=O 同时还原
如 CH2=CHCH2CH=O 3-丁烯醛
OH- CH3CH=CHCH=O
H+ CH3CH=CHCH=O 2-丁烯醛
3.制备:主要由羟醛缩合反应制备
C6H5CHO + CH3COC6H5 OH- C6H5CH=CHCOC6H5
HCHO + CH3COCH3 OH-
O
OH-
2CH3CHO
CHO
二,α, β-不饱和醛酮的化学性质
C2H5
1, 2–加成 40%
O
C6H5CH CH2 C CH3
C2H5
1, 4–加成 60%
O
C6H5CH CH C CH3
C6H5MgX
1. E2tO 2.H2O
C6H5CH
OH
O
CH C + CH3 C6H5CH CH2 C C6H5
C6H5
C6H5
1, 2–加成 88%
1, 4–加成 12%
(1)与HCN,氨和氨的衍生物,H2SO4,
RNH2等质子酸,H2O,ROH在酸催化下的1,
4–加成反应
O
O
R CH CH C H(R,) HCN RCH CH C H(R,)
HCN
CN H
O—H
RCH CH C H(R,)
CN
பைடு நூலகம்
O
例:R CH CH C H(R,) HX
O
RCH CH C H(R,)
CH3CH2CH2CH=C(Et)CHO
O
(95%)
H2,Ni or Pt
CH3(CH2)3CH(Et)CH2OH
O
Zn/HCOONH4/C2H5OH/H2O 饱和酮
R1
查耳酮类 R2
rt 30min
6.Diels-Alder 反应
? ,?? 不饱和醛酮是很好的亲二烯体
CHO
+
CHO
7.插烯作用
1. OH -
HCN
1,4- 加成产物为主
NH
1,4- 加成产物为主
C=C-C=O
1) R 2CuLi 2)H 2O
1,4- 加成产物为主
1) RMgX,CuCl 2)H 3O+ 1) RLi 2) H 2O
1) RMgX
2)H 2O
1,4- 加成产物为主
1,2- 加成产物为主 不饱和醛 (1,2- 加成产物为主 ) 不饱和酮 甲基酮 (1,2- 加成为主 ) 其他酮 (1,4- 加成为主 )
CH3
CH3
90%
3%
存在微量Cu +
1%
95%
4.迈克尔反应 Michael reaction
1.定义:烯醇负离子与α ,β -不饱和羰基化合物在碱性催化剂作用下1, 4-加成。
2.给体:能提供烯醇负离子的化合物: O
O
CH3NO2 CH3C N
CH3C CH2 C OC2H5
3.受体:α ,β -不饱和羰基化合物共轭体系
5.如果的羰基和一个很大的基团如三级丁基相连, 无论哪一种格式试剂都得到 1,4-加成产物
6.为得到1,4-加成产物,有一种常用方法是加入 0.05mol的卤化亚铜或者用二烃基铜锂进行反应
CH3CH
CH
O
C
CH3
CH3MgBr Et2O
H3O+
CH3CH 无Cu +
OH
O
+ CH C CH3
CH3CH CH2 C CH3
+ CH3CHO CH3CHO 2. △ CH3CH CHCHO
CH3CHO CH3CH CH CH CHCHO
在共轭体系中电子的流动性较大,羰基的电子效应可通过共轭体系系传递到碳上 插烯规则:在A-B化合物的A和B之间,插入一个或多个-CH=CH-,生成A(CH=CH)-nB型化合物后,原来A和B之间的相互影响仍然存在的规律
2.亲电加成 electrophilic addition 反应速率比单烯烃及共轭二烯烃慢 ,为什么?
?? ?? CH3CH=CHCOCH3
HCl(g)
CH3CH(Cl)CH2COCH3
Br2 CCl4
CH3CH(Br)CH(Br)COCH3
3.亲核加成 Nucleophilic addition
生1,4-共轭加成,形成加成物从溶剂中夺取一个质子形成烯醇,在互 变异构形成最终产物。
7.用途:用于合成环状化合物,通常用迈克尔反应和羟醛缩合一起合成环
状化合物
O
KOH
+ CH2=CHCOCH 3
O CH 2CH2COCH 3
O
O
O CH2CH 2COCH 3
NH C6H6
O
分子内缩合
O
+ H2O
O
O
O
R CH CH C R R CH CH C OR CH2 CH C N
4.碱性催化剂:三乙胺,六氢吡啶,氢氧化钾,乙醇钠,氨基钠
5.该反应可逆,升温对你反应有利
O
CH3
O
+CH2 CH C CH3
O
O
CH3
O
CH2 CH2 C CH3 KOH
KOH
O
O
O
6.反应机理: 碱夺取碳上的活泼氢,生成一个碳负离子,然后碳负离子与受体发
XH
R CH R CH R CH
O CH C
O CH C
O CH C
H(R,) H2NOH
O
RCH CH2 C H(R,)
H(R,) NaHSO3
NHOH
O
RCH CH2 C H(R,)
SO3Na
H(R,)
NH
O
RCH CH2 C H(R,)
N
+ (C2H5)3Al HCN
O
O
CN
85%
(2)和金属有机化合物反应
1.R2CuLi, RMgX,RLi
2.1,4-加成和1,2-加成均有,取决于羰基旁边的基团大 小,也与试剂的空间位阻有关
3.醛羰基旁边的空间位阻很小,因此与烃基锂,格 氏试剂时主要以 1,2-加成为主
4.与格氏试剂反应要做具体分析
1. C2H5MgBr 2. H3O+
C6H5CH
OH
+ CH C CH3