高分子化学第七章活性聚合
高分子化学第七章活性聚合

2019/1/17
高分子化学
CH3 O
H2C C
CH3 OLi
H2C C
CH3 O
H2C C
C OCH3 + R Li
C OCH3 R
C R + CH2O Li
CH3 CH2 C Li COOCH3
CH3O H3C C O CH 3 CH2 C COOCH3 H3C COOCH3
CH3 + H2C C COOCH3
休眠种752氮氧自由基tempo存在下自由基聚合氮氧自由基如2266四甲基1哌啶氮氧自由基tempo是一种稳定的自由基由于其空间位阻不能引发单体聚合但可快速地与增长链自由基发生偶合终止生成休眠种而这种休眠种在高温下100又可分解产生自由基复活成活性种即通过tempo的可逆链终止作用活性种与休眠种之间建立了一快速动态平衡从而实现活性可控自由基聚合
第七章 活性聚合
2019/1/17
高分子化学
7.1 概述
7.1.1活性聚合概念
不存在链转移和链终止的聚合称为活性聚合。为了保证所有的活 性中心同步进行链增长反应而获得窄分子量分布的聚合物,活性聚 合一般还要求链引发速率大于链增长速率。 典型的活性聚合具备以下特征: (1)聚合产物的数均分子量与 单体转化率呈线性增长关系; (2)当单体转化率达100%后, 向聚合体系中加入新单体,聚合 反应继续进行,数均分子量进一 步增加,并仍与单体转化率成 正比;
2019/1/17 高分子化学
相对于链增长反应,链终止反应速率对链自由基浓度的依赖性 更大,降低链自由基浓度,链增长速率和链终止速率均都下降,但 后者更为明显。假若能使链自由基浓度降低至某一程度,既可维持 可观的链增长速率,又可使链终止速率减少到相对于链增长速率而 言可以忽略不计,这样便消除了自由基可控聚合的主要症结 ─ 双 基终止,使自由基聚合反应从不可控变为可控。
高分子化学重点名词解释(2)

高分子化学重点名词解释(2)高分子化学重点名词解释p变动还不大,从而使kp/kt增加了近7-8倍,导致加速显著。
(自己总结,仅供参考)动力学链长ν,在聚合动力学研究中,常将一个活性种从引发开始到链终止所消耗的单体分子数定义为动力学链长ν,无链转移时,相当于每一链自由基所连接的单体单元数。
动力学链长应该考虑自初级自由基链引发开始,包括历次链转移以及最后双基终止所消耗的单体总数。
可控/“活性”聚合,传统自由基因为其慢引发、快增长、速终止的机理特性使得聚合物的微结构、聚合度和多分散性无法控制,如果能够降低自由基的浓度或活性,就可减弱双基终止,成为可控/“活性”聚合。
即令活性增长自由基与某化合物反应,经可逆的链终止或链转移,使之转化为低活性的共价休眠种,并使平衡倾向于休眠种一侧,以降低自由基浓度和链终止速率。
调聚反应,当聚合反应体系满足条件kp<<ktr,ka=kp时,聚合速率不变,但聚合物分子量较小,只能获得低聚体,这种聚合反应称为调聚反应。
第四章自由基共聚合无规共聚物是指大分子中两结构单元M1、M2按概率无规律排布,M1、M2连续的单元数不多(一至十几不等)的共聚物;交替共聚物是指M1、M2两单元严格交替相间的共聚物,是无规共聚物的特例;嵌段共聚物是指由较长的M1段链和另一较长的M2段链构成的大分子,每一链段可长达几百至几千结构单元;接枝共聚物是指主链由M1单元组成,支链则由另一种M2单元组成的共聚物。
竞聚率r,在研究共聚物的组成时,将均聚和共聚增长速率常数之比定义为竞聚率r,以表征两种单体的相对活性。
理想共聚合反应(理想恒比共聚):当一对单体满足r1=r2=1的条件时,就会发生理想共聚合反应。
前末端效应,带有位阻或极性较大基团的烯类单体在进行自由基共聚合时,前末端单元对末端自由基的活性将会产生影响,这种影响称作前末端效应。
第五章聚合方法本体聚合是指不加其他介质,单体在少量的引发剂、光或热等的作用下进行的聚合反应;溶液聚合是将单体和少量引发剂溶于适当的溶剂中,在溶液状态下进行的聚合反应;悬浮聚合是借助搅拌并在分散剂的作用下,将不溶于水的单体以小液滴状悬浮在水中进行的聚合反应;乳液聚合是指单体在乳化剂和机械搅拌的作用下,在水中分散成乳液状态进行的聚合反应。
第七章功能高分子的制备方法

第七章 功能高分子的制备方法
2. 环醚的开环聚合 环醚主要是指环氧乙烷、环氧丙烷、四氢呋喃
等。它们的聚合物都是制备聚氨酯的重要原料。 环氧乙烷和环氧丙烷都是三元环,可进行阴离
子聚合和阳离子聚合。四苯基卟啉/烷基氯化铝可引 发他们进行阴离子活性开环聚合。
17
第七章 功能高分子的制备方法
四氢呋喃为四元环,较稳定,阴离子聚合不能 进行,而只能进行阳离子聚合。碳阳离子与较大的 反离子组成的引发剂可引发四氢呋喃的阳离子活性 聚合。例如 Ph3C+SbF6- 可在-58℃下引发四氢呋 喃聚合,产物的相对分子质量分散指数为1.04。
第七章 功能高分子的制备方法
功能高分子材料的制备是通过化学或者物理的 方法按照材料的设计要求将功能基与高分子骨架相 结合,从而实现预定功能的。
从上一世纪50年代起,活性聚合等一大批高分 子合成新方法的出现,为高分子的分子结构设计提 供了强有力的手段,功能高分子的制备越来越 “随 心所欲”。
1
第七章 功能高分子的制备方法
7.2 高分子合成新技术
7.2.1 活性与可控聚合的概念 活性聚合是1956年美国科学家Szwarc等人在研
究萘钠在四氢呋喃中引发苯乙烯聚合时发现的一种 具有划时代意义的聚合反应。其中阴离子活性聚合 是最早被人们发现,而且是目前唯一一个得到工业 应用的活性聚合方法。目前这一领域已经成为高分 子科学中最受科学界和工业界关注的热点话题。
8
第七章 功能高分子的制备方法
7.2.3 阳离子活性聚合 阳离子聚合出现于20世纪40年代,典型工业产
品有聚异丁烯和丁基橡胶。 阳离子活性中心的稳定性极差,聚合过程不易
控制。多年来阳离子活性聚合的探索研究一直在艰 难地进行。
《高分子化学》教案第7章开环聚合

第六章 开环聚合开环聚合属于链式聚合,单体为环化合物,包括环醚、环缩醛、内酯、内酰胺、环硅氧烷等。
7.1 总论7.1.1 环单体的聚合活性环单体的聚合活性由热力学因素和动力学因素共同决定。
1. 热力学因素即环单体和相应的线形聚合物的相对稳定性,它与环大小、成环原子和环的取代基相关。
1) 环烷烃的稳定性:环烷烃进行开环聚合的热力学可行性顺序为:三元环、四元环>八元环>五元环,七元环>六元环。
2) 环的取代基:取代基的引入使聚合热增加、熵变增加,总体使开环聚合可能性降低。
3) 单环单体和多环单体:多环单体的环张力会有所增加,使开环聚合可能性增加。
如8-氧杂[4,3,0]环壬烷,反式的可开环聚合。
4) 成环原子:对于内酯而言,六元、七元环内酯可聚合,而五元环内酯则不可;环三硅氧烷的聚合活性高于环四硅氧烷。
2. 动力学因素环烷烃没有易受活性种攻击的键,因此动力学上仅环丙烷衍生物可进行开环聚合,并且仅能得到低聚物。
环醚、内酯、内酰胺等环单体,因有亲核或亲电子部位,易开环聚合。
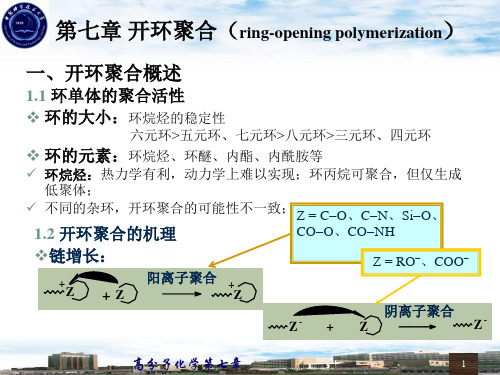
7.1.2 开环聚合机理和特征 1. 聚合机理开环聚合的引发剂为烯烃聚合进行离子型聚合所用的引发剂,引发反应包括初级活性种的形成和单体活性种的形成。
大多数阳离子开环聚合的链增长是通过单体对增长链末端的环状阳离子的亲核反应来进行的,其中的Z 基团为C-O (环醚)、C-N (环氮化合物)、Si-O (环硅氧烷)、酯键和酰胺键;而阴离子聚合的链增长则是增长链末端的阴离子对单体的亲核反应,Z 基团为RO -(环醚)、COO -(内酯)和Si-O -(环硅氧烷)。
;一般情况下,开环聚合的增长链末端带电荷,进行链增长的单体是中性的。
但是,开环聚合还有另一种链增长方式,即所谓的活化单体机理,增长链末端不带电荷,而单体是离子化的,如己内酰胺的阴离子聚合。
2. 开环聚合的基本特征Z-++Z单体加到增长链上进行高分子链的生长;聚合度随转化率增加缓慢,但是在许多场合下呈线性关系;溶剂对聚合反应影响同烯烃的离子聚合;动力学表达式通常类似于链式聚合,特别是活性聚合;许多开环聚合的单体平衡浓度较高,即临界聚合温度较低。
高分子化学(第五版)第7章

2)丙烯的配位聚合动力学 对于均相催化剂体系,可参照阴离子聚合增长速率
方程: Rp= kp[C*][M] α-TiCl3-AlEt3是微非均相体系,其聚合速率~时间(
Rp~t)曲线有2种类型:
A-衰减型 B-加速型
-TiCl3-AlEt3引发的丙烯
聚合动力学曲线
28
曲线A由研磨或活化后引发 体系产生:第Ⅰ段增长期,在 短时间内速率增至最大;第II 段衰减期;第III段稳定期,速 率几乎不变。
CH CH2
空位
R
环状过渡态
CH CH2 R
TiCl3和AlR3络合物在Ti上形成活性 点(或空位),丙烯在空位上配 位,形成σ-π络合物,配位活化后 的单体在金属-烷基链中插入增长。
配位和增长反复进行,形成大分子。
δ-
δ+
CH CH2 CH CH2 Mt
R
R
5
配位聚合的特点:
单体首先在过渡金属上配位形成σ-络合物;
Cl
Ⅳ B族过渡金属,如锆(Zr)、钛(Ti)、铪(Hf) +
茂型配体:至少一个环戊二烯基(Cp)、茚基(Ind)、芴基 (Flu)或它们的衍生物 +
非茂配体,如氯、甲基、苯基等。
18
空间几何构型
Ti
Cl
Cl
Cl
非桥链型 普通结构
X
R'
M
X
桥链型
R
R
R
R2
R
L
MCl2 2
R3
限定几何构型
19
茂金属引发剂的特点: 高活性,几乎100%金属原子都形成活性中心; 单一活性中心,可获得分子量分布很窄、共聚物组成
R代表速率,t为时间,下标0为起始最大值, 为后 期稳定值,k为常数,与丙烯压力有关,与三乙基铝浓度 无关。
高分子物理化学 第七章

2)2-2功能度体系 每个单体都有两个相同的功能 基或反应点,可得到线形聚合物, 如:
n HOOC(CH2)4COOH + n HOCH2CH2OH
HO CO(CH 2)4COOCH 2CH 2O
n
H + (2n-1) H2O
缩聚反应是缩合反应多次重复 结果形成聚合物的过程。
3)2功能度体系
同一单体带有两个不同 (或相同)且能相互反应的官 能团,得到线形聚合物,如:
按聚合产物分子链形态的不 同分类 线形逐步聚合反应 其单体为双功能基单体, 聚合产物分子链只会向两个方 向增长,生成线形高分子。
非线形逐步聚合反应 非线形逐步聚合反应的聚 合产物分子链不是线形的,而 是支化或交联的,即聚合物分 子中含有支化点,要引入支化 点,必须在聚合体系中加入含 三个以上功能基的单体。
n HO n HO
R COOH H O R CO n OH + (n-1) H2O R OH H O R n OH + (n-1) H2O
4) 2-3、2-4功能度体系
当功能度大于2时,分子链将向 多个方向增长,这样的话将得到支化 甚至是交联的聚合物。 例如: 通过苯酚和甲醛制备酚醛树脂时, 当反应程度较高时,可以得到支化甚 至交联的聚合物。
H (OROCOR`CO )m HO (COR`COORO)q ( OROCOR`CO)n OH ( COR`COORO)p H
+
既不增加又不减少功能基数目,不影响反应程度 特 不影响体系中分子链的数目,使分子量分布更均一 点 不同聚合物进行链交换反应,可形成嵌段缩聚物
线形缩聚动力学
1. 功能基等活性理论 缩聚反应在形成大分子的过程中 是逐步进行的,若每一步都有不同的 速率常数,研究将无法进行。Flory提 出了功能基等活性理论: 不同链长的端基功能基,具有相 同的反应能力和参加反应的机会,即 功能基的活性与分子的大小无关。
第七章开环聚合(ring-openingpolymerization)
高分子化学第七章
12
四、其它杂环单体
合成多肽阴离子开环聚合
单体:N-羧基-a-氨基酸酐(N-carboxy-a-amino-anhydride, NCA); 引发剂:胺类(伯胺、仲胺和叔胺)
CO HN O
CH CO R
-CO 2
R NHCHCO m
CO RNH2 + HN O
CH CO R
R COOH RNHCOCHNH
A CH2CH2O nCH2CH2O- M+ + ROH
A CH2CH2O nCH2CH2OH + RO- M+
环氧丙烷聚合的单体链转移反应:甲醇钠引发环氧丙烷聚合,
CM为0.013(70 °C)、0.027(93 °C)。
高分子化学第七章
3
2.2 (环氧化物的)阴离子聚合
CH3
O
CH2CHO- Na+ + CH3CH CH2 ktr,M
开环聚合应该 归类于
何种聚合方式 ???
O
O
R
CH3CH CH2 CH2 CH2 O
R
O
O
O
O
OO
O
高分子化学第七章
2
二、环醚(杂氧原为子什)么环醚中只有
环氧化物才能进行
2.1 环醚单体
阴离子聚合?
3, 4, 5元环醚(); 6元环如四氢吡喃、1,4-二氧六环();1,3,5-三
氧六环( )
聚合过程(活化单体链增长机理)
引发剂和单体反应生成活化单体; 活化单体与内酰胺反应形成N-酰基化内酰胺,进一步还原出活化单体; 活化单体不断与增长链末端的内酰胺单元反应; 聚合存在诱导期:以使N-酰基化内酰胺浓度达到足够大;
活性聚合 (ATRP)简介

ATRP法可以最大程度根据设计合成功 能聚合物刷,聚合过程可控
2.以ATRP技术合成的共嵌段聚合物
采用ATRP 技术合成多嵌段共聚物主要有以下两种方法:
一 采用单官能团引发剂,依次加入不同单体的活性聚合。
即先引发单体A 聚合,再与单体B 聚合,然后与单体A 或C 聚合,可形成ABA 型非 对称三嵌段共聚物或ABC 型三嵌段共聚物。
(3)ABC 型三嵌段共聚物
以单官能团小分子引发剂,通过ATRP 反应合成单体A 的均聚物,然后作为大分 子引发剂,引发单体B 的ATRP 反应,然后再引发单体C 的ATRP 反应,得到ABC 型三嵌 段共聚物。ABC 嵌段共聚物具有形成纳米形态的潜力,具有有趣的化学和物理性质。 利用不同分子量的PEO 大分子引发剂,通过DMA 和DEA 单体的连续ATRP 反应,合成了 聚[环氧乙烷-2-(二甲氨基) 乙基甲基丙烯酸酯-2-(二乙氨基) 甲基丙烯酸酯](PEODMA-DEA) 三嵌段共聚物(见图6) ,并研究了pH 诱发胶体自组装和胶束的尺寸与胶体 的稳定性核交联的影响。该聚合物在低pH 下溶解于水溶液中;pH = 7.1 时,出现胶束 化现象,形成三层“洋葱状”胶束,含DEA 核、DMA 内核与PEO 外晕。最近他们又采用 ATRP 技术,PEO 大分子引发剂首先与2-(二乙氨基) 乙基甲基丙烯酸酯(DEA) 聚合,然 后与2-羟乙基丙烯酸酯(HEMA) 的“一锅法”合成了三嵌段共聚物PEO-PDEA-PHEMA , 通过HEMA 嵌段上羟基的酯化形成相应的PEO-PDEA-PSEMA 两性离子三嵌段共聚物。在 室温下,通过调整溶液的pH 值,两性离子的PEO-PDEA-PSEMA 三嵌段共聚物形成三种胶 束聚集态。
近年来ATRP法在聚合物刷的制备中得到了广泛应用, 首先,在不同的基体表面如固体、球形分子以及大分子 表面引入烷基卤代烃引发剂,然后进一步在其表面引发 聚合,可以得到具有不同组成、聚合度和形状的聚合物 刷。 (1) 例如用ATRP法在硅片表面制备了低表面能的2,3, 4,5,6一五氟苯乙烯聚合物刷,利用椭圆偏正光测厚 仪、接触角测定仪和x射线光电子能谱仪对薄膜结构进 行了表征,结果表明,随着聚合时间的延长,聚合物刷 的厚度不断增加,反应16h后薄膜厚度增长变慢,接触角 数据证明引发剂已组装在硅片上制备了聚合物刷。又如 对聚偏氟乙烯(PVDF)进行化学处理使其表面羟基化,然 后与2一溴异丁酰溴反应在其表面接上ATRP引发剂,引 发三甲基硅保护的甲基丙烯酸羟乙酯(HEMAnTMS)聚合, 在PVDF表面形成PHEMA聚合物刷,动力学研究揭示出 PHEMA 的接枝浓度随反应时问的延长呈线性关系。
《材化高分子化学》第7章 配位聚合

(1)α-烯烃
H C* R
由于连接C*两端的分子链不等长,或端基不同, C*应当是手性碳原子,但这种手性碳原子并不 显示旋光性,原因是紧邻C*的原子差别极小, 故称为“ 假手性中心”。
根据手性C*的构型不同,聚合物分为三种结构:
RR R R HHHH
全同立构 Isotactic
RH RH HRHR
C O OCH2CH3
聚丙烯酸乙酯
[NH( CH2 )6NHOC ( CH2 )4CO ]n
尼龙-66
结构单元间的连接方式不同,又会产生序列异构。
例如首尾相接和首首相接的异构现象。其首尾相接、 首首相接和无规序列相接的聚合物,其化学组成相 同,连接方式不同,性能也是不一样的。
(2)聚合物的立体异构体
什么是同分异构? 化学组成相同,聚合物分子中原子或原子团相互连接
的次序不同而引起的异构叫做同分异构,又称结构 异构。
例如:通过相同单体和不同单体可以合成化学组 成相同、结构不同的聚合物。
如结构单元为-[ C2H4O-] n的聚合物可 以是聚乙烯醇、聚环氧乙烷等。
[ CH2 CH ] n OH
聚乙烯醇
7.1 引言
乙烯、丙烯在热力学上均具有聚合倾 向,但在很长一段时间内,用上述聚合方 法,却无法得到高分子量的聚合物。
为什么?
乙烯的自由基聚合必须在高温高压下进行,由 于较易向高分子的链转移,得到支化高分子,即 LDPE。
丙烯利用自由基聚合或离子聚合,由于其自阻聚 作用,都不能获得高分子量的聚合产物。
插入反应是配位阴离子机理 由于单体电子的作用,使原来的Ti-C键活化,极化 的Ti+-C -键断裂,完成单体的插入反应。
立构规整性成因 单体如果在空位(5)和空位(1)交替增长,所得聚合物 将是间同立构,实际上得到的是全同立构。
高分子化学第七章聚合物的化学反应

二、 化学因素
• 1. 几率因素
大分子链上相邻基团作无规成对反应时,往往有一 些孤立的基团残留下来,反应不能进行到底。
~~CH-CH2-CH-CH2-CH-CH2-CH-CH2-CH-CH2~~
O -CH2- O
OH
O -CH2- O
按反应的几率,羟基的最高转化率86.5%,实验
测得为85~87%。
二、 聚合物化学反应的影响因素
影响聚合物功能基反应能力的因素:
(1)物理因素 (2)化学因素
一、 物理因素
• 1. 结晶的影响(聚合物的聚集态)
对于部分结晶的聚合物,晶区分子的取向 度高,分子间作用力大,低分子试剂不易扩散 进去,反应往往只限于无定形区。无定形物处 于玻璃态时,链段被冻结,不利于低分子扩散 ,反应最好在Tg以上或使其适当溶胀后。
• CPE可用于电缆护套,耐热输送带,胶 辊,工业用胶管等。
2.聚氯乙烯的氯化(CPVC)
~CH2–CH~ + HCl Cl
~CH–CH~ + HCl Cl Cl
• 氯化聚氯乙烯的特点是耐热、耐老化 、耐化学腐蚀性好,基本性能于PVC 接近,但耐热性比PVC高。
三、聚乙烯醇的合成及其缩醛化
• 1.聚合
n CH2=CH BPO OCOCH3
-[ CH2-CH-] n
OCOCH3
控制合适条件,制备聚合度适当的产物
• 2.醇解
-[ CH2-CH-] n
CH3OH,OH–CH3COOCH3
OCOCH3
~~CH2-CH~~ OH
制备维尼纶纤维,醇解度要大于99% 悬浮聚合分散剂,醇解度大约为80%
• 3.缩醛化
化学分析。 (5)研究材料的老化和防老化
活性聚合

•
• • •
• ……
可控/“活性” 可控 “活性”自由基聚合 (CRP)
CRP成为当今高分子合成化学发展最迅速的领域 原因:大量可供聚合的单体,简单的反应装置,不苛刻的反应 条件对自由基的有效控制。 更重要的是,CRP产品具有巨大的市场潜力,不过要 充分发挥其潜力,在很多方面还需要研究。 今后的研究方向:开发新的引发/催化体系、 拓宽单体种类、合成结构清晰可控的新型 聚合物。更重要的是缩短工业化的进程。
三、对CRP的综合讨论与比较
所有可控自由基聚合具有一些共同的特征:链增长自由
基和各种休眠种达到动态平衡是所有可控自由基 聚合体系的关键。 聚合体系的关键。
四、CRP CRP的应用与前景 CRP
•
具有水溶性的双亲性嵌段共聚物已被成功用作表面活性剂,并且用于一 些高端产品,例如染料分散剂、添加剂、保健品和化妆品等。具有纳米形态 的嵌段共聚物可用作电子器件。接枝共聚物可用作聚合物共混增溶剂,并且 可以可以用到嵌段共聚物所能适用的许多领域。梯度共聚物非常有望用作表 面活性剂、噪音和振荡阻尼材料。 通过对支化度的调节,可以精确的控制聚合物加工过程中的熔融粘度。 这些聚合物(包括梳形和星形聚合物)可以用作黏度调节剂和润滑剂。大分 子拓扑结构控制的一个突出例子是大分子刷,这些聚合物经轻度交联可得到 超软弹性体。 CRP在链末端功能化方面也具有独特的优势 目前,结构规整的官能化聚合物与无机组分或者天然物质通过共价键结合成 的分子杂化材料受到了广泛关注,并且将会带来许多具有新功能的材料。( 分子纳米复合材料……) 潜在的应用包括微电子材料、软刻印刷技术、光电子元件、特种膜、传感器 和微流体组分
让我们坚强永不放弃 让我们勇敢面对困境 让我们对生活的爱和希望 燃烧在心里 付诸于行动 让我们微笑生活继续 让我们努力创造奇迹 让我们期待 这场属于我们的胜利
活性自由基聚合

链引发、链增长、链终止
传统的自由基聚合反应:
传统自由基聚合的问题:
在 20 世 纪50,60年代,自由基聚合达到了它 的鼎盛时期。但由于存在链转移和链终止反应,传 统自由基聚合不能较好地控制分子量及大分子结构。
活性/可控自由基聚合的提出:
1956年Szwarc等提出了活性聚合的概念,活 性聚合具有无终止、无转移、引发速率远远大于链 增长速率等特点,与传统自由基聚合相比能更好地 实现对分子结构的控制,是实现分子设计、合成具 有特定结构和性能聚合物的重要手段。
Chem. Commun. DOI: 10.1039/c2cc17780c
Transformation of Living Cationic Polymerization of Vinyl Ether into RAFT Polymerization
(A) Synthesis of Well-Defined Amphiphilic Block Copolymers of HPMA with a Thiol-Reactive Functional Monomer via the RAFT Polymerization
1. Iniferter法:
引发(initiator)-转移(transfer)-终止(terminator)活性自由基聚合
热引发和光引发:
常用的光活化型iniferter结构
单官能团
双官能团 多官能团
自由基聚合
Iniferter:
Iniferter法小结
☺ 用于苯乙烯(St)和甲基丙烯酸甲酯(MMA)的控
Mechansim of RAFT polymerization
Scheme 1 Homopolymerization ofEGDMAvia RAFT, and subsequent formation of a 3D single cyclized chain (graphic representation)election of RAFT agents for various polymerizations. For Z, addition rates decrease and fragmentation rates increase from left to right. For R, fragmentation rates decrease from left to right. A dashed line indicates partial control.
高分子化学7(阳离子聚合)

聚合反应分类
根据聚合机理,聚合反应可分为连锁 聚合和逐步聚合。阳离子聚合属于连 锁聚合的一种。
根据聚合物的结构和性能,聚合反应 可分为均聚合、共聚合和嵌段聚合等 。阳离子聚合可用于制备均聚物和共 聚物。
02
阳离子聚合基本原理
阳离子聚合的机理
01
02
03
链引发
引发剂在阳离子作用下形 成活性中心,引发单体聚 合。
高分子化学7(阳离子聚合)
• 引言 • 阳离子聚合基本原理 • 阳离子聚合的单体 • 阳离子聚合催化剂 • 阳离子聚合的影响因素 • 阳离子聚合的应用 • 结论
01
引言
主题简介
01
阳离子聚合是一种通过阳离子引 发的高分子聚合反应,其特点是 聚合过程中聚合物链带有正电荷 。
02
阳离子聚合在工业生产中具有重 要应用,如合成橡胶、塑料、涂 料等。
动力学参数的确定
通过实验数据拟合动力学方程,求得反应速率常数、活化能等动力 学参数。
阳离子聚合的活化能
活化能的概念
01
指引发聚合反应所需的最低能量。
活化能的影响因素
02
引发剂类型、单体类型、溶剂极性等。
活化能与聚合速率的关系
03
活化能越高,聚合速率越快;反之,活化能越低,聚合速率越
慢。
03
阳离子聚合的单体
选择依据
选择催化剂时应考虑其活性、稳定性、毒性等因素,以及聚合物的性能要求和 生产成本等因素。
05
阳离子聚合的影响因素
温度和压力的影响
温度
阳离子聚合反应是放热反应,温度升高会导致聚合速率增加,但同时也会加速聚 合物链断裂,影响聚合物分子量。因此,需要选择适当的温度以获得高分子量聚 合物。
高分子化学第七章

为了加速反应,往往另加酸作为聚酯化 的催化剂。反应速率将由自催化和酸催 化两项组成。则
外加酸催化聚酯化反应速率常数要比自 催化常数k大2个数量级。对酯化反应, 工业上总是外加酸作催化剂以加速反应, 自催化是不适用的。
7.4.2.2 平衡缩聚动力学
聚酯化反应是平衡常数较小的可逆反应。 如果小分子副产物不能及时排除,则逆 反应不能忽视。
无规共缩聚物 嵌段共缩聚物
共缩聚的作用
共缩聚使性能改变的一个重要方向是适当增加分子 链的柔性,降低结晶度,从而使玻璃化温度和熔点 适当降低。
7.3 线型缩聚反应的机理
缩聚速率和缩聚物的分子量是两大重要指标。 影响分子量的因素和分子量的控制是线型缩聚 中的核心问题。
7.3.1 线型缩聚与成环倾向
7.2.3
共缩聚
共缩聚或逐步共聚合不论在实际应用上,还是 理论上,都不如连锁或加成聚合成熟。 目前尚无接枝共缩聚物出现;交替共缩聚物合 成困难,应用也不广。对共缩聚的兴趣主要集 中在无规和嵌段上。
均缩聚 ; 杂缩聚 ; 共缩聚。
两种以上氨基酸的缩聚,或两种二元胺和一种 二元酸,一种二元胺和两种二元酸以及两种二 元胺和两种二元酸共同进行缩聚时,则可称做 共缩聚。以下两式为共缩聚物:
(2)化学降解
聚酯化和聚酰胺化的逆反应水解属于化学降解。
(3)链交换反应
大分子端基与另一大分子的弱键进行链交换反应, 如聚酯、聚酰胺等本身都可以进行链交换反应。
7.4 线型缩聚动力学
7.4.1 官能团等活性概念
逐步缩合须进行1—2百次。如果每步速率常数 都不相等,动力学将无法处理。 由实验研究得出了官能团等活性、与分子量大 小无关的重要概念,这一概念可用来处理缩聚 动力学。
活性聚合

活性聚合(living polymerization)的概念是1956年 Szwarc[1]提出的,即无终止、无转移、引发速率远大于增 长速率的聚合反应。由于没有链转移,聚合过程中聚合物 链的数目保持恒定;而没有链终止,直到体系中单体消耗完 ,聚合反应停止时,聚合物链仍然保持活性基。一旦加入 新的单体,聚合反应即可继续进行。所以Szwarc把这种聚 合方法叫做“活性聚合”(Living Polymerization) 。
典型的热引发转移终止剂是1,2-二取代四苯基乙烷类衍生物,研究发现[11, 12]这些 对称的碳一碳键热引发转移终止剂引发极性单体甲基丙烯酸甲酯(MMA )的聚合为活性聚 合,并且引发剂的活性顺序为PPE>TMPSN>TPSN。所得的PMMA可以作为大分子引发 剂引发第二单体苯乙烯(St)聚合,制备PMMA-b-PSt共聚物,但嵌段效率比较低。然而对 于引发非极性单体St的聚合来说,它们的作用与传统自由基聚合引发剂类似,没有活性 聚合的特征。Braun[13,14]认为,当1,2-二取代的四苯基乙烷衍生物引发苯乙烯聚合时,得 到的聚合物ω-端为五取代的C-C键,键能比较高,受热时不能再分解,为死端聚合;而在 引发MMA聚合时,得到的聚合物。一端为六取代的C-C键,键能较低,受热时仍能可逆 分解,实现活性自由基聚合。 由于文献中报道的热引发转移终止剂种类少,活性低, 只能在较高的温度(>800℃)下实现极性单体MMA的活性聚合,对非极性单体St的聚合是 传统的自由基聚合,无活性聚合特征。丘坤元等[I5, 16]研究了两种C-C键型热引发转移终 止剂:2,3-二氰基-2,3-二苯基丁二酸二乙酯(DCDPS )和2,3-二氰基-2,3-二(对-甲苯基)丁二 酸二乙酯 (DCDTS )引发乙烯基单体的聚合。结果发现,与Otsu和Braun所报道的四苯基 取代的乙烷衍生物类热引发转移终止剂相比较,DCDPS和DCDTS的活性较高,不但在 较低温度(50~ 100℃)下实现了MMA的活性聚合,而且首次在小分子热引发转移终止剂领 域实现了St的活性聚合。另外,他们还首次合成了一种氨酯型非对称性结构的小分子热 引发转移终止剂,用它引发MMA的本体聚合具有活性自由基聚合的特点;而在二甲基甲 酰胺 (DMF)溶剂中的溶液聚合却不是活性自由基聚合。但本体及溶液聚合产物PMMA 都能起大分子引发剂的作用可合成嵌段聚合物。
《高分子化学》张形欣主编,第七章逐步聚合

n
OH CH3
Cu O
O CH3
n
(2) -OCO- 聚酯,例:PET:
n HOCH2CH2OH + HOOC
COOH
H
OCH2CH2 (2n-1) H2O
(3) -NH-CO-
聚酰胺,例:尼龙-6,6: nHOOC(CH 2) 4 COOH + nH 2N(CH 2) 6NH 2 → HO-[-OC(CH 2) 4COHN(CH 2) 6NH -] n-H + (2n-1)H2 O
n
OR'OH
(3) Diels-Alder 反应,可溶性梯形聚合物合成:
O
O
O
+
O
O
O
n
(4) 氧化偶联反应,聚苯醚合成:
CH3 CH3
n
OH CH3
Cu O
O CH3
n
(5) 加成缩合反应,酚醛合成:
OH OH
+ CH 2O
CH 2OH
(加成)
OH OH OH OH
CH2OH +
CH2
(缩合) (6) 分解缩聚,聚甲撑合成: BF3
n HOCH2CH2OH + HOOC
COOH
H
OCH2CH2OOC
CO
n
O CH2CH2OH + (2n-1) H2O
(2) 逐步加聚反应(聚加成反应) ,聚氨酯合成:
n O C N R N C O + n HO R' OH O C N R N C H O OR'O C N R N C H H O O
n CH2=N2
物:
-[-CH7-]n- + n N2
高分子化学-第七章 聚合物的化学反应

(6)可回收单体和综合利用聚合物废料
(7)有助于了解聚合物的分子结构以及结 构与性能的关系。
(8)在高分子化学反应的基础上发展了功 能高分子 (9)聚合物的化学反应和缩聚、加聚反应 密切相关。
5
二、 聚合物化学反应的分类
根据聚合度和基团(侧基和端基)的变化,聚合物的 化学反应可分成:
• (1)聚合度相似的化学反应
OCOCH3
OCOCH3
控制合适条件,制备聚合度适当的产物
26
• 2.醇解 ]n [ CH2-CH- -
OCOCH3
CH3OH,OH–CH3COOCH3
~~CH2-CH~~ OH
制备维尼纶纤维,醇解度要大于99% 悬浮聚合分散剂,醇解度大约为80%
27
• 3.缩醛化
~~CH2– CH–CH2–CH–CH2 –CH~~ OH OH OH
15
二、 化学因素
• 1. 几率因素
大分子链上相邻基团作无规成对反应时,往往有一 些孤立的基团残留下来,反应不能进行到底。 ~~CH-CH2-CH-CH2-CH-CH2-CH-CH2-CH-CH2~~ O -CH2- O OH O -CH2- O 按反应的几率,羟基的最高转化率86.5%,实验 测得为85~87%。 若反应是可逆的,只要时间足够长,可以打破几 率的限制。 16
• 2. 邻近基团效应
由于大分子链上反应基团多,邻近基团相距很 近,因此,静电和位阻效应可使聚合物链上官能 团反应能力上升或下降。
~~CH2-CH-CH2-CH-CH2-CH~~ C=O C=O C=O O-• • • • • • H-N-H • • • • • • O-
OH-
17
18
一、聚二烯烃的加成与取代
高分子化学第七章

O C N R N
O C + HO R/ OH
O C N R NH
O C O R/ OH
O C NH R NH
O C O R/O
n
(2)根据聚合物主链结构分类 线型缩聚、体型缩聚 线型缩聚:参加反应的单体都只有两个反应 官能团(f=2),大分子向两个方向增长, 得到的聚合物是线型分子。 体型缩聚:参加缩聚反应的单体之一含有多 个反应官能团(f>2),在反应中分子向多 个方向增长,得到的是体型聚合物。
5. 缩聚过程中的副反应 环化反应、官能团的消去、化学降解、链 交换 1)环化反应 ω-羟基酸 HO-(CH2)n-COOH 为例 ① n=1,双分子缩合成环
- H2O
2 HO ( CH2 )n COOH
65 4 32 1 HOCH2COOCH2COOH
- H2O
O
4 C
3 O
2 CH2
1 C
O
2 官能度体系:同一单体带有两个不同且能 相互反应的官能团,得到线形聚合物; 如:
2-3、2-4官能度体系:体形缩聚物
e.g:苯酐和甘油反应
O HO—CH2——CH—CH2——OH + C O C O O—CH2—CH—CH2 —O——O O C O C O
OH
(2)单体的反应能力 以聚酯化反应为例:
§7.1
概述
1、缩合反应(Condensation) 许多有机官能团间的反应,除了形成主产物 外尚有低分子物产生称为缩合反应。
CH3COOH + CH3CH2OH
- H2O
CH3COOCH2CH3 O H N CH2CH3
CH3COOH + CH3CH2NH2
- H2O
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2020/6/11
高分子化学
CH3 O H2C C C OCH3 + R Li
CH3 OLi H2C C C OCH3
R
CH3 O H2C C C R + CH2O Li
CH3
CH3
CH2 C Li
+ H2C C
COOCH3
COOCH3
CH3
O CH3
CH2 C
C C CH2 + CH3O Li
COOCH3
第七章 活性聚合
2020/6/11
高分子化学
7.1 概述
7.1.1活性聚合概念
不存在链转移和链终止的聚合称为活性聚合。为了保证所有的活 性中心同步进行链增长反应而获得窄分子量分布的聚合物,活性聚 合一般还要求链引发速率大于链增长速率。
典型的活性聚合具备以下特征: (1)聚合产物的数均分子量与 单体转化率呈线性增长关系; (2)当单体转化率达100%后, 向聚合体系中加入新单体,聚合 反应继续进行,数均分子量进一 步增加,并仍与单体转化率成 正比;
配合物的作用机理被认为是它可以与引发活性种、链增长活性 种(包括阴离子和金属反离子)络合,形成单一而稳定的活性中心, 同时这种络合作用增大了活性链末端的空间位阻,可减少或避免活 性链的反咬终止等副反应的发生。
2020/6/11
高分子化学
7.3 活性阳离子聚合
在1956年Szwarc开发出活性阴离子聚合后,人们就开始向往实 现同是离子机理的活性阳离子聚合,但长期以来成效不大。直到 1985年,Higashimura、 Kennedy先后首先报导了乙烯基醚、异 丁烯的活性阳离子聚合,开辟了阳离子聚合研究的崭新篇章。
2020/6/11
高分子化学
7.1.2 活性聚合的动力学特征
在理想的活性聚合中,Rtr=Rt=0,且Ri>>Rp,即由链引发反应 很快定量形成活性中心,并同步发生链增长,体系中产生的聚合物 的浓度与活性中心浓度以及引发剂浓度相等,聚合速率与单体浓度 呈一级动力学关系:
R p d [M ]d t k p [M * ][M ] k p [I]0 [M ]
因此活性聚合又称计量聚合。
有些聚合体系并不是完全不存在链转移和链终止反应,但相对于 链增长反应而言可以忽略不计,分子量在一定范围内可控,明显具 有活性聚合的特征。为了与真正意义上的活性聚合相区别,把这类 聚合称为活性/可控聚合。这就大大扩展了活性聚合的概念。
2020/6/11
高分子化学
活性聚合是1956年美国科学家Szware首先发现:在无水、无氧、 无杂质、低温条件下,以THF为溶剂、萘钠为引发剂,进行苯乙 烯阴离子聚合,得到的聚合物溶液在低温、高真空条件下存放数 月后,再加入苯乙烯单体,聚合反应可继续进行,得到分子量更 高的聚苯乙烯。若加入第二种单体丁二烯,则得到苯乙烯-丁二烯 嵌段共聚物。根据以上实验结果,Szware等人第一次明确提出 了阴离子型无链终止、无链转移的聚合反应,即活性聚合的概念。 因为所得聚合物在单体全部耗尽后仍具有引发聚合活性,因此他 们同时提出了活性聚合物的概念。迄今为止活性聚合已从最早的 阴离子聚合扩展到其它如阳离子、自由基、配位等链式聚合。
C Li 三苯基甲基锂
2020/6/11
高分子化学
(2)在体系中添加配合物
将一些配合物如金属烷氧化合物(LiOR)、无机盐(LiCl)、 烷基铝(R3Al)以及冠醚等,添加到极性单体的阴离子聚合体系中, 可使引发活性中心和链增长活性中心稳定化,实现活性聚合。这种 在配合物存在下的阴离子活性聚合称为配体化阴离子聚合 (Ligated anionic polymerization),它是目前实现极性单体阴 离子活性聚合的最有力手段,较上途径(1)相比,单体适用范围 更广。
(1)使用立体阻碍较大的引发剂
1,1-二苯基已基锂、三苯基甲基锂等引发剂,立体阻碍大、反应 活性较低,用它们引发甲基丙烯酸甲酯阴离子聚合时,可以避免引 发剂与单体中羰基的亲核加成的副反应。同时选择较低的聚合温度 (如-78 ℃),还可完全避免活性端基“反咬”戊环而终止的副反 应,实现活性聚合。
1,1-二苯基已基锂 CH3 CH2 4 C Li
7.3.1活性阳离子聚合原理
在乙烯基单体的阳离子聚合中,链增长活性中心碳阳离子稳定性
极差,特别是β-位上质子氢酸性较强,易被单体或反离子夺取而发
生链转移:
阴离子聚合,尤其是非极性单体如苯乙烯、丁二烯等的聚合, 假若聚合体系很干净的话,本身是没有链转移和链终止反应的,即 是活性聚合。相对于其它链式聚合,阴离子聚合是比较容易实现活 性聚合的,这也是为什么活性聚合首先是通过阴离子聚合方法实现 的原因。
但是对于丙烯酸酯、甲基乙烯酮、丙烯腈等极性单体的阴离子 聚合,情况要复杂一些。这些单体中的极性取代基(酯基、酮基、 腈基)容易与聚合体系中的亲核性物质如引发剂或增长链阴离子等 发生副反应而导致链终止。以甲基丙烯酸甲酯的阴离子聚合为例, 已观察到以下几种亲核取代副反应:
将上式积分后可得:
ln(0 [/M [M ]kp ][)I0t]
ln([M与0]/[反M应])时间t呈线性关系,即聚合体系的链增长活性中心
浓度为一常数,即不存在链终止、链转移反应,这也可以作为一动 力学特征来判断聚合反应是否是活性聚合。
2020/6/11
高分子化学
7.2 活性阴离子聚合
7.3.1活性阴离子聚合的特点
CH3O
H3C CH2
C
O C
CH3
COOCH3
H3C CH2
O C C CH3
COOCH3
+ CH3O Li源自H3CCOOCH3
H3C
COOCH3
因此与非极性单体相比,极性单体难以实现活性阴离子聚合。
2020/6/11
高分子化学
7.3.2极性单体的活性阴离子聚合
为了实现极性单体的活性阴离子聚合,必须使活性中心稳定化 而清除以上介绍的副反应,主要途径有以下两种:
2020/6/11
高分子化学
(3)聚合产物分子量具有单分散性,即 Mw Mn →1
(4)聚合产物的数均聚合度应等于每个活性中心上加成的单体 数,即消耗掉的单体浓度与活性中心浓度之比:
Xn = f [M]已反应 / [I]0 = f [M]0 / [I]0(单体转化率100%)
f 为每个聚合物分子所消耗的引发剂分子数