金催化炔烃环异构化反应
金催化炔烃环异构化6-endo-dig cyclization of N-propargylaminoquinone

报告人: 王少仲 副教授 南京大学化学化工学院
2011年11月14日
Contents
1. Au(I)-Catalyzed 6-endo-dig Cycloisomerization of N-Propargylaminoquinones
Iodine-Induced 6-endo-dig Electrophilic Cyclization of N-Propargylaminoquinones CuCl2-Promoted 6-endo-dig Chlorocyclization of N-Propargylaminoquinones Au(I)-Catalyzed Cascade Reaction of NPropargylaminoquinones 2.
X-Ray Crystal Structure of 4a
Substrate Scope
Substrate Scope
Proposed Mechanism
Preparation of 3Clorosampangine
CuCl2-Promoted Cascade Cyclization
Au(I)-Catalyzed Cascade Reaction
1 2 3 4 5 6 7
a
5mol% Pd(OAc)2, CuCl2,CH3NO2, 80oC, 1h
5mol% Pd(OAc)2,FeCl3,CH3NO2, 80oC, 1h 1.0equiv CuCl2, CH3NO2, 80oC, 1h
0 50 17 60 66 0 85
Compound 1a as substrate and the amount of chlorine source is 3.0 equiv
lindlar催化剂催化炔烃反应

lindlar催化剂催化炔烃反应
Lindlar催化剂是一种常用于催化炔烃加氢反应的催化剂。
它由一种
稀有金属钯(Pd)支撑在石墨或氧化铝上制成,通常配合着少量的铅(Pb)和硫(S),能够在温和的反应条件下,将炔烃转化为烯烃,同
时避免过量还原导致副反应的发生。
使用Lindlar催化剂催化炔烃反应具有以下优点:
1. 可以将实验室中常用的炔烃转化为相应的烯烃,而无需使用高温和
高压等剧烈条件。
2. 与传统的加氢催化剂相比,Lindlar催化剂可以得到更选择性和高活
性的反应产物,而且只需要使用微量的钯,成本比较低廉。
3. Lindlar催化剂还可以在不需要氢气存在的情况下进行反应,这种反
应方式常常被用于有机合成中,方便实验操作。
但Lindlar催化剂催化炔烃反应也存在以下几个缺点:
1. 反应效率会受到催化剂的中毒作用影响。
因此,一旦催化剂被中毒,活性显著降低,进一步催化反应的可能性非常小。
2. Lindlar催化剂对某些炔烃的转化存在差异性,不同取代基和双键位
置的炔烃催化效果也会不同。
3. Lindlar催化剂会在催化过程中产生毒性物质,需要建立安全操作规程。
综上所述,Lindlar催化剂的应用广泛,但在具体的实验操作中要注意催化剂的中毒问题和毒性物质的处理。
廉价金属催化烯烃和炔烃自由基双官能团化反应

自由基双官能团化反应的机理和特点
• 自由基双官能团化反应的机理主要包括以下步骤:首先,一个分子中的自由基引发剂引发另一个分子的烯 烃或炔烃生成自由基;其次,生成的自由基与另一个分子中的烯烃或炔烃发生加成反应;最后,生成的新 的自由基与另一个分子中的烯烃或炔烃发生加成反应,生成最终的化合物。自由基双官能团化反应具有高 选择性、高效率和高原子经济性等特点。
烯烃和炔烃是常见的有机化合物,它 们具有丰富的化学反应性和生物活性 ,因此在有机合成、药物合成、材料 科学等领域具有广泛的应用。
传统的烯烃和炔烃官能团化反应主要 集中在氧化、还原、加成等反应,但 这些反应往往需要使用较为昂贵的催 化剂或条件较为苛刻,因此开发新的 、高效、环保的催化体系一直是化学 研究的重要方向。
03
官能团化试剂的研究
不同的官能团化试剂可以引入不同的 官能团,如羧基、羟基、氨基等,对 反应产物的结构和性质有重要影响。
廉金属催化的炔烃自由基双官能团化反应研究
炔烃自由基双官能团化反应的机理
在廉金属催化剂的作用下,炔烃分子中的π键被活化,生成自由基中间体,然后与不同的 官能团化试剂反应,实现双官能团化。
要点三
反应选择性和产物多 样性
目前对于烯烃和炔烃自由基双官能团 化反应的反应选择性和产物多样性仍 存在一定的限制,未来的研究将致力 于拓展反应的应用范围并提高产物多 样性。
06
参考文献
参考文献
01
参考文献1:作者1, 文章标题, 杂志名, 年份, 卷号(期号), 页码
• 引用内容1
02
参考文献2:作者2, 文章标题, 杂志名, 年份, 卷号(期号), 页码
温度对自由基双官能团化反应的速 率和产物分布有显著影响。在一定 范围内,提高温度可以促进反应速 率和产物纯度的提高。然而,过高 的温度可能导致副反应增多,降低 产物纯度。
05炔烃
sp的特点
① sp的形状类似于sp3和sp2,呈葫芦形 ②sp的能量介于s和p轨道之间; ③方向性:两个sp杂化轨道在同一条直线上;
④剩余的两个未参与杂化的p轨道,互相垂直,且均与两个sp 杂化轨道所在的直线垂直。
5)电负性: sp3 < sp2 < sp
(5) 总结 •碳碳叁键是由一个 键和两个 键 组成. •键能—乙炔的碳碳叁键的键能是:837 kJ/mol; 乙烯的碳碳双键键能是:611 kJ/mol; 乙烷的碳碳单键键能是:347 kJ/mol. •C-H键长—和p轨道比较, s轨道上的电子云更接近原 子核.一个杂化轨道的s成分越多,则在此杂化轨道上的 电子也越接近原子核.由sp杂化轨道参加组成共价键 , 所以乙炔的 C-H 键的键长 (0.106 nm) 比乙烯 (0.108 nm)和乙烷(0.110nm)的C-H键的键长要短. •碳碳叁键的键长—最短(0.120 nm),这是除了有两个 键,还由于 sp 杂化轨道参与碳碳键的组成.
CH3 Br C=C H H
(3) 和水的加成 CHCH + H2O
H2SO4 HgSO4
HO H2C=CH
RCCH + H2O
记住反应 条件!
H2SO4 HgSO4
H 分子重排 CH3-C=O 乙醛 OH O 分子重排 R-C=CH2 R-C-CH3 烯醇式化合物 酮
为什么发生重排? CH2=C-OH CH3-C=O H H •乙 醛 的 总 键 能 2 7 4 1 kJ/mol 比 乙 烯 醇 的 总 键 能 2678kJ/mol大,即乙醛比乙烯醇稳定. •由于两者能量差别不大 (63kJ/mol),在酸存在下 ,它们中 间相互变化的活化能很小.
(3) 乙炔的键
炔烃的亲核加成

炔烃的亲核加成炔烃是一类含有碳碳三键的有机化合物。
由于其特殊的碳碳三键结构,炔烃在有机合成中具有很高的反应活性,可以通过亲核加成反应引入各种官能团。
在有机合成中,炔烃的亲核加成反应是一种常用的合成方法,可以合成各种有机化合物。
炔烃的亲核加成反应可以分为酸催化和金属催化两种机制。
1. 酸催化亲核加成反应酸催化的机制是通过质子化炔烃中的碳碳三键使其亲电性增加,进而与亲核试剂发生反应,生成相应的加成产物。
例如,乙炔(C2H2)与水发生酸催化的亲核加成反应,生成乙醛(CH3CHO):C2H2 + H2O → CH3CHO2. 金属催化亲核加成反应金属催化的机制是利用金属离子作为中间体,促进炔烃与亲核试剂的反应。
常见的金属催化亲核加成反应包括氢化反应、卤化反应和环加成反应。
(1)氢化反应炔烃可以与氢气在金属催化剂的存在下发生氢化反应。
最经典的例子是乙炔与氢气在铂催化剂存在下反应生成乙烯:C2H2 + H2 → C2H4(2)卤化反应炔烃与卤素发生亲核加成反应,生成相应的卤代烃。
例如,乙炔与溴在存在铁为催化剂下反应生成1,2-二溴乙烷:C2H2 + Br2 → BrCH2CH2Br(3)环加成反应炔烃可以通过环加成反应形成环状化合物。
例如,乙炔与氢氰酸(HCN)在酸的存在下发生反应,生成丙烯腈(CH2=CHCN),即将乙炔中间的碳原子和氰基(CN)中的氮原子连接起来形成环状化合物:C2H2 + HCN → CH2=CHCN炔烃的亲核加成反应不仅可以生成单官能团的化合物,还可以进行多组分亲核加成反应,生成多官能团的化合物。
例如,炔烃可以与醛、酮等亲电试剂同时发生加成反应,生成α,β-不饱和酮、酸等化合物。
总结起来,炔烃的亲核加成反应是一种常用的有机合成手段,可以通过酸催化或金属催化的方式与亲核试剂发生反应,生成各种有机化合物。
这些反应为有机合成提供了丰富的化学方法学,广泛应用于药物合成、材料化学等领域。
铂催化剂在炔烃\烯炔类化合物成环反应中的应用

进 行 关环 反应 , 生成 一 个 高度 官能 化 的 环戊 烯 衍 生
以 1 1 PC 0mo% tI为催 化 剂 , 甲苯 溶 剂 中 1 0C 在 0  ̄下
物, 收率为 7 %。Shm ) 9 (ce e 5
P C2(.4 e ) t I 00 q
t ue oI ne. 8 O℃
反应 , 产物收率 中等 (6 7 %)(ce e ) 4 % ̄ 3 。S hm 9
P C 21 to% t I 0o 1
∞ z
2铂 催 化 炔 烃 \ 炔 构 成 六 元 环 的 反报道 了一个 联 烯 炔 的环 异 构 化 反应
作者简介 : 王倩(9 3 )女 , 1 8 一 , 浙江上业大学药物化学在读研究生 。
维普资讯
1 6一
Z E I N H MIALI D S R H JA G C E C U T Y N
Vo 3 o0 2 0 ) 1 9N . . 8(0 8
一
系列 高度 官 能 化 的 毗咯 衍 生 物 , 含苯 基 、 基 、 酯 羟
基、 长链烃基 的取代基 均不影响反应进行。 反应 以
PC 催 化剂 ,t H为 溶 剂 , 在 2 6 二 叔 丁基 一 tI为 EO 且 ,一 4 甲基 吡 啶存 在下 进 行 , 物 收率 为 5 %~ 8 此 一 产 5 8%。
环状结构简单分类。
1铂 催 化 炔 烃 \ 炔 构 成 五 元 环 的 反 烯
应
M n e 等… edz 报道 了一个 l6 ,~烯炔类化合物的 分 子 内环合 反 应 , 反应 在 P() 化 剂 存 在 下 进行 , tI I催
由炔 基与 金 属发 生 配位 作 用 . 丙 基 亲 核 基 团从 反 烯 方 向进攻 , 成 中问体 I, 由于亲 核 试剂 t O 的 形 I又 l H
过渡金属催化炔烃环化反应的研究进展

化学研究与应用第31卷第31卷第3期2019年3月化学研究与应用Chemical Research and Application Vol.31,No.3Mar.,2019文章编号:1004-1656(2019)03-0384-07过渡金属催化炔烃环化反应的研究进展刘兴燕1*,蒋瑶1,罗爽1,南柏鑫1,李高参2(1.重庆工商大学环境与资源学院,催化与环境新材料重庆市重点实验室,重庆400067;2.四川大学国家生物医学材料工程技术研究中心,四川成都610064)摘要:具有独特反应性和稳定性的炔烃类化合物一直以来是科研工作者们关注的热点之一,其中关于炔烃环化构筑一系列多环化合物的反应报道尤为突出。
本文根据不同金属催化剂,总结归纳了近几年铑、钌、钴、钯等过渡金属催化炔烃环化反应的研究进展,并简要讨论了催化转化的反应机理、优势以及局限性,最后展望了过渡金属催化炔烃环化反应的发展前景。
关键词:炔烃;环化反应;过渡金属催化;进展中图分类号:O621.3文献标志码:ARecent advances on transition -metal -catalyzed cyclization of alkynesLIU Xing-yan 1*,JIANG Yao 1,LUO Shuang 1,NAN Bo-xin 1,LI Gao-can 2(1.Chongqing Key Laboratory of Catalysis and New Environmental Materials ,College of Environment and Resources ,Chongqing Technology and Business University ,Chongqing 400067,China ;2.National Engineering Research Center for Biomaterials ,Sichuan University ,Chengdu 610064,China )Abstract :With the especial reactivity and stability ,the alkynes had been one of the research hotspots for constructing a series of poly-cyclic compounds.In this review ,the up-to-date summaries of transition-metal-catalyzed cyclization of alkynes on the basis of metal catalysts such as rhodium ,ruthenium ,cobalt and palladium were gave.It was pointed out that the plausible mechanisms ,advantages and limitations in these transformations.Finally ,the outlooks for transition-metal-catalyzed cyclization of alkynes was suggested.Key words :alkynes ;cyclization ;transition metal catalysis ;progress炔烃是一类含有碳碳三键的不饱和烃类,易于发生加成、环化等反应,反应活性的多样性使其成为有机合成化学中常见的关键组分,目前已被广泛作为反应的前体与多种碳、氮、氧原子等含活性氢的化合物进行[2+2]、[3+2]、[4+2]等加成、氧化环化或者串联环化反应,构筑一系列碳环或杂环骨架多环类化合物[1,2]。
炔烃复分解反应

炔烃复分解反应
炔烃复分解反应是指在金属催化下,碳碳三键的断裂和重组形成新的炔烃的反应。
这个反应通常是通过热化学或光化学方法进行。
在热化学活化体系中,催化活性物种的性质仍未知。
炔烃复分解反应可以分为炔烃关环复分解和炔烃交叉复分解。
炔烃关环复分解反应得到的环炔烃可以通过立体选择性的氢化还原为Z 构型或E构型的烯烃,也可进行硼氢化反应等。
该反应使用的催化剂及其使用量还有待改善,因此,未来该方面的发展值得期待。
如需更多信息,建议查阅相关文献或咨询化学家等专业人士。
过渡金属催化的炔烃碳金属化串联环化反应研究进展

第53卷第2期 辽 宁 化 工 Vol.53,No. 2 2024年2月 Liaoning Chemical Industry February,2024过渡金属催化的炔烃碳金属化串联环化反应研究进展聂敏铃(温州大学 化学与材料工程学院,浙江 温州 325035)摘 要:用过渡金属催化炔烃与有机硼试剂的加成反应,经过碳金属化串联环化可实现炔烃的不对称官能团化。
其中芳基硼酸因其高亲电性、高稳定性和来源广泛的特点最常被使用。
主要综述了近年来各种过渡金属催化的炔烃与芳基硼酸的碳金属化串联环化反应研究进展。
关 键 词:过渡金属;炔烃;芳基硼酸;碳金属化;串联环化中图分类号:O621 文献标识码:A 文章编号:1004-0935(2024)02-0289-05近年来,利用过渡金属催化转化构建碳碳键的方法已经被广泛研究。
特别是过渡金属催化的串联环化反应,可以从相对简单的原料出发,使用单一的催化剂,“一锅法”合成结构复杂的分子,具有效率高、原子经济性好、对环境保护友好等优点,因此受到许多科研人员的关注[1]。
用过渡金属催化炔烃与有机硼试剂的加成反应,经过碳金属化串联环化可实现炔烃的不对称官能团化。
其中芳基硼酸因其具有高亲电性、高稳定性和来源广泛的特点最常被使用[2-4]。
在过渡金属的使用中,主要以钯[5-11]、铑[12-19]等贵金属催化为主,后期也逐渐发展了 镍[20-25]、钴[26]、铜[27]等廉价金属催化的方法。
本文主要从贵金属催化和廉价金属催化2个方面综述近年来过渡金属催化的炔烃与芳基硼酸碳金属化串联环化反应研究进展。
1 贵金属催化的炔烃与芳基硼酸碳金属化串联环化反应2012年,陆熙炎[5]课题组报道了钯(Ⅱ)催化的烯酮-炔与芳基硼酸的对映选择性芳基化-环化反应,如图1所示。
该反应底物1.1经历碳钯化后先形成烯基钯阳离子中间体1.2,然后发生分子内碳碳双键插入得到中间体1.3,最后质子化得到产物1.4。
金催化的[4+2]环加成反应的最新进展
![金催化的[4+2]环加成反应的最新进展](https://img.taocdn.com/s3/m/4fc1870b312b3169a451a469.png)
第3期2019年6月No.3 June,2019作者简介:尚英伟(1993— ),男,河南灵宝人,硕士研究生;研究方向:计算化学。
摘 要:近年来,过渡金属催化的有机反应已经成为最常用的有机合成方法之一,而在过渡金属中,第一副族金属催化剂,如Cu ,Ag ,Au 等催化剂亦被大量应用在各类有机合成、药物以及天然产物的合成之中。
而在众多的有机合成反应之中,[4+2]环加成反应无疑是合成六元环有机产物最常用的一种方式;同时,金催化剂因为在近年来表现出的高催化活性、反应条件的温和性、催化反应的良好产率以及各种选择性的优势而受到人们的高度关注并被广泛地投入许多反应之中。
因此,对近3年来金催化的[4+2]环加成反应进行了综述,对这些反应的反应机理、产率、选择性等方面进行了综述。
关键词:金催化;[4+2]环加成反应;综述金催化的[4+2]环加成反应的最新进展尚英伟,程瑞姣,刘文竹,豆立娟(云南师范大学 化学化工学院,云南 昆明 650500)现代盐化工Modern Salt and Chemical Industry 过渡金属具有空的d 轨道,故能够与多种有机化合物进行配位作用,从而对有机化合物进行活化抑或构建手性中心,进而使其能够发生一些在常规条件下难以实现的化学反应,以合成相应的目标产物,如天然产物以及药物等。
因此,过渡金属时常被选用为有机合成反应的催化剂。
作为催化剂,过渡金属还具有反应条件温和、底物范围宽广、产率优良以及反应选择性好等优势。
在过渡金属中,金催化剂近年来被人们发现了具有高催化活性、温和的反应条件、良好的产率以及良好的区域选择性等特点。
这是从前人们所不曾意识到的,因为金催化剂作为生活中常见的贵金属,其稳定的化学性质以及高昂的价格是为人们所熟知的。
因此,在相当长的时间内,金催化剂被认为不具备或是具备相当低的催化活性。
但后来,Hashimi 、姜风超等课题组经研究发现了通过金催化的一系列化学反应,如环加成反应、氢烷氧基化反应、氢芳基化反应、氢羧化反应、氢胺化反应等。
有机金属化学-3

TiCl4
H2
R2Al2Cl3
COOH
1. O2
(CH2)10
2. HNO3
COOH
14.5.2 烯烃的高分子聚合反应 链增长反应:
R2AlC2H5 + (n−1)CH2=CH2 与脱氢铝反应相竞争:
R Al(C H ) H ⎯9⎯0 −⎯1⎯20⎯°⎯C→
100 bar
2
2 5n
R2AlCH2CH2R ⎯⎯→ R2AlH + CH2=CH2R'
14.4 烯烃的芳基/乙烯基化(Heck反应)
在Heck反应中,乙烯基氢原子被乙烯基,苯基或 芳基基团所取代。如:
Br
CO2Me
100
+
+ Et3N
1% Pd(OAc)2
CO2Me
2% PPh3
+ [Et3NH]Br
催化剂为Pd0配合物,由Pd(OAc)2, Et3N和 PPh3原位(in situ)形成。
Fe(CO)4
ΔT
_ CO
Fe(CO)3
14.3 不饱和分子的异构化 组成为LnMH或复合型(R3P)2NiCl2 + H2的催化剂,能
够加速到热力学更稳定分子的重排,就是说它们能够催化(端 内)烯烃和(孤立共轭)烯烃的转化。关键步骤是:
LnMH + CH2 CH CH2R
→ ←
H CH2
LnM CH CHR
变换不同的配体可精细调变催化剂,而且它们能被重复合成;催 化剂因此获得较高的专适性,并能在较低的温度下进行催化。配合物催 化剂不止在一种配位数下稳定,并且通过精细调整化学键的强度(变换配 体),能够选择性地“ 持住”低物分子,但又不至太紧。
金属卡宾配合物在有机合成中的应用-----烯烃的环丙烷化反应

金属配合物MLn中配体L的电子效应会影响金属卡宾 的稳定性,以及烯烃在金属上的配位,从而影响环丙 烷化产物的立体分布。烯烃的结构对环丙烷化产物的 立体分布有一定的影响但不大,主要是烯烃上取代基 的位阻效应所致。此外,反应温度下降导致立体选择 性提高,但同时反应速率大大下降。
影响选择性的因素
区域选择性
分子间的烯烃环化反应
分子间的烯烃环化反应
Casey 研究了Schrock型卡宾配合物的环丙烷化机理,发 现 对 于 (CO)5W=C(Ph)2 等 , 只 有 当 反 应 温 度 高 于 CO 从 (CO)5W=C(Ph)2 中解离的温度时才可进行,所以认为在反 应中烯烃取代了 CO配位,生成了卡宾 -金属 -烯配合物中间 体 ( 如下式 ) 。而另外一些卡宾配合物,如 (CO)5W=CHPh 等,具有很高的烯烃反应活性,反应可以在低于CO离解温 度下进行,该历程包含着直接的卡宾转移而无需生成卡宾 金属-烯三元配合物。
分子间的烯烃环化反应
烯烃与过渡金属卡宾配合物反应生成的过渡金属环丁烷 中间体,如发生还原消除反应,则得到环丙烷衍生物。
虽然两类过渡金属卡宾配合物都可以发生这个反应,但 对于 Fischer 型卡宾配合物,只有高度活化或高纯化的烯烃 (如乙烯基醚或 α,β 不饱和酯),环丙烷化才能进行。而对 于 Schrock 型卡宾配合物则没有这样的限制,反应只需要当 量的过渡金属卡宾配合物。
烯烃的环丙烷化反应
早期的烯烃环丙烷化反应采用 Cu 粉为催化剂,但 存在反应条件苛刻,反应温度高等缺点。之后,人们 开发金属卡宾配合物作为催化剂,不但可以提高其立 体、区域及对映选择性,而且可以大大提高产率,改 善反应条件。
金属卡宾催化剂与早期催化剂性能比较
分子间的烯烃环化反应
有机膦催化的异氰基乙酸酯与亚胺的加成反应

有机膦催化的异氰基乙酸酯与亚胺的加成反应摘要:本文从R构型的光学纯H8-BINOL出发,通过NBS溴代、Suzuki偶联、三氟甲磺酸酐酯化、与二苯基膦氧氢偶联和三氯硅烷还原等几步反应,得到了具有联萘骨架的3-苯基磷双官能团有机催化剂。
并初步探讨了其在异氰基乙酸酯与亚胺的加成反应中的应用。
关键词:有机膦催化剂;合成;异氰基乙酸酯;亚胺;加成反应一.前言有机膦化合物被广泛的应用在合成化学中,人们熟知的应用包括有:Wittig 反应,Staudinger反应,Mitsunobu反应,以及在过渡金属催化中使用的膦配体[1]。
上世纪六七十年代,关于有机膦化合物参与的亲核催化反应开始处于起步阶段。
直到最近的二十年里,许多叔膦参与的亲核催化反应才被大量报道。
1. 有机膦催化剂在不对称合成中的应用(1) 有机膦催化的Rauhut-Currier反应谈及亲核性膦催化的反应,Rauhut-Currier反应是必须被提到的,甚至有研究者认为Morita-Baylis-Hillman反应就是由该反应派生而来。
1963年,Rauhut和Currier[2a]在一篇专利中报道了膦催化的活化烯烃的二聚反应,就是我们在所知的Rauhut-Currier反应。
1965年该反应同时也被McClure[2b],Baizer和Anderson[2c]三位研究者独立报道。
这个过程通常被认为是:首先亲核性膦对活化烯烃发生可逆的共轭加成,随后前一步得到的烯醇负离子对另一分子的活化烯烃发生Michael 加成。
然后再发生分子内的质子迁移,随之发生E1cB消去过程得到活化烯烃的二聚体,并再生出膦。
1970年,McClure报道了首例交叉Rauhut-Currier反应[3](图2)。
图2 首例Rauhut-Currier反应和交叉Rauhut-Currier反应2002年,Krische小组[4a]和Roush小组[4b]分别报道了分子内的Rauhut-Currier反应(图3)。
有机化学中的炔烃的合成方法

有机化学中的炔烃的合成方法有机化学中炔烃的合成方法炔烃是一类具有三重键的有机化合物,具有广泛的应用领域,如医药、农药、材料科学等。
为了满足不同领域对炔烃的需求,有机化学家们通过多种方法开发出了一系列有效的炔烃合成方法。
本文将探讨有机化学中炔烃的合成方法,重点介绍一些常用的合成途径。
一、酸催化的炔烃合成方法酸催化是炔烃常用的合成方法之一,通过酸催化反应可以将适当的化合物转化为炔烃。
常用的酸催化反应有卤代烃的脱卤化反应、芳香醇的脱水反应和酯的热裂解反应等。
这些反应通常需要较高的温度和酸催化剂,反应条件较为苛刻。
二、金属催化的炔烃合成方法金属催化是炔烃合成的重要方法之一,在有机化学领域中得到了广泛的应用。
常见的金属催化炔烃合成方法有钯催化的交叉偶联反应、铜催化的瑞利大反应和铑催化的环加成反应等。
1. 钯催化的交叉偶联反应钯催化的交叉偶联反应是合成炔烃的重要方法之一。
该方法通过将含有卤素基团的化合物和含有炔基团的化合物进行反应,以产生新的炔烃化合物。
常见的交叉偶联反应有钯催化的Suzuki偶联反应、钯催化的Sonogashira偶联反应等。
2. 铜催化的瑞利大反应铜催化的瑞利大反应是一种常见的合成炔烃的方法。
这种反应可以将含有卤素基团的芳香化合物和含有炔基团的有机锌试剂反应,以合成炔烃。
瑞利大反应是高效、环境友好的方法,被广泛应用于炔烃的合成。
3. 铑催化的环加成反应铑催化的环加成反应是一类高效的合成炔烃的方法。
这种反应基于炔丙基金属中间体的形成,通过环外配体的替代反应来构建炔烃的碳链。
铑催化的环加成反应在有机合成中具有广泛的应用,能够合成多种不同结构的炔烃。
三、羰基化合物的脱羰基反应羰基化合物的脱羰基反应是一种合成炔烃的重要方法。
这种反应通过在适当条件下将含有羰基基团的化合物进行脱羰基反应,从而生成炔烃。
常见的脱羰基反应有马尔尼科夫反应、礼山氏反应等。
四、氧化嘧啶的还原氧化嘧啶化合物可通过还原反应合成炔烃。
林德拉催化剂与炔烃反应

林德拉催化剂与炔烃反应
林德拉催化剂是一种重要的有机金属化学催化剂,是由诺贝尔化学奖得主林德拉首次发现的。
它被广泛应用于有机合成领域中,尤其是在炔烃反应中。
炔烃是一类含有三键碳的有机化合物,在有机合成中具有重要的地位。
炔烃在存在着催化剂的情况下能够与许多化合物反应,形成不同的产物,其中最著名的是炔烃与卤代烃反应,可形成烷基卤化物;炔烃与铝烷反应,可得到相应的铝烷基炔。
而在这些反应中,林德拉催化剂则发挥着重要的作用。
具体而言,林德拉催化剂可以将烷基卤化物和亚烷基卤化物与炔烃反应,生成具有重要化学和生物活性的环烷烃。
此外,林德拉催化剂还可以将炔烃和醛、酮、酸酐等化合物反应,形成含有牵引结构的化合物。
这些产物具有广泛的应用领域,包括化学制药、涂料、染料等。
需要注意的是,林德拉催化剂虽然在炔烃反应中被广泛应用,但催化剂的选择和反应条件的优化非常重要。
在使用中需要根据具体的反应物选择适合的催化剂,并合理调节反应温度、反应时间等反应条件,以确保反应的高效性和选择性。
总之,林德拉催化剂是一种重要的有机金属催化剂,在炔烃反应中具有广泛应用。
通过优化催化剂的选择和反应条件,炔烃反应可以高效、选择性地进行,为有机合成提供了重要的工具和手段。
烯烃的环丙烷化反应

烯烃的环丙烷化反应是一种有机化学反应,通过该反应,烯烃可以转化为环丙烷化合物。
这种转化通常发生在催化剂的作用下,将烯烃的π键闭合形成三元环状结构。
环丙烷化反应在有机合成中非常重要,因为它能够构建环丙烷骨架,这是许多天然产物和药物分子的核心结构。
以下是几种类型的烯烃环丙烷化反应:
1.金属催化环丙烷化:
o Simmons-Smith环丙烷化反应是最著名的例子之一,它使用锌-碘化合物(如二碘甲烷)和一价铜盐作为催化剂,将烯烃转化为环丙烷。
o另一种是使用过渡金属如铑、铜、钯等催化剂,通过与有机金属试剂或其它适当配体的协同作用,实现烯烃的环丙烷化。
2.光化学环丙烷化:
o在光照射下,某些光敏试剂与烯烃反应,通过光诱导的过程将烯烃转化为环丙烷。
这种反应通常需要特定的光催化剂,并且在某些情况下
可以实现区域选择性和立体选择性的环丙烷化。
3.亲电环丙烷化:
o在某些情况下,烯烃可以与亲电试剂如卤代环丙烷衍生物反应,通过SN2历程形成环丙烷。
4.催化氢化环丙烷化:
o通过氢气与过渡金属催化剂(如雷尼镍、铂或钯)的协同作用,烯烃可在氢气气氛下进行氢化环丙烷化反应。
这些反应的效率和选择性很大程度上取决于催化剂、底物和反应条件的选择。
在实际应用中,研究者往往会设计和优化催化剂体系,以提高环丙烷化产物的产率和立体选择性。
钌催化1 6-烯炔环化反应的研究进展
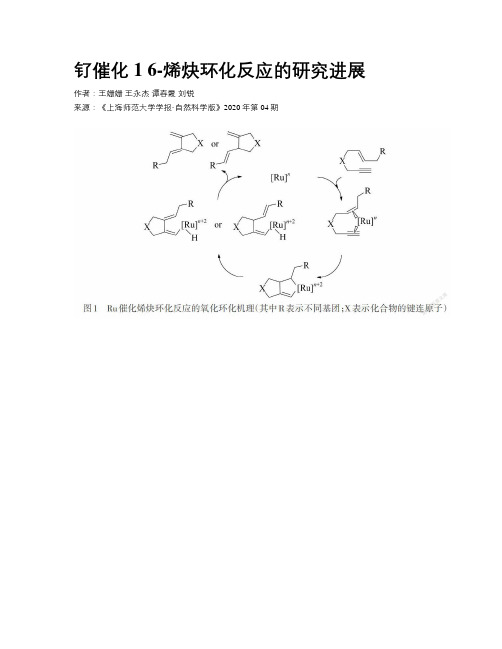
钌催化1 6-烯炔环化反应的研究进展作者:王姗姗王永杰谭春霞刘锐来源:《上海师范大学学报·自然科学版》2020年第04期摘要:五元環和六元环是许多药物及生物活性分子中的基本结构单元,广泛存在于各种天然产物中.因此,开发高效的合成方法构筑五元/六元环一直是有机合成的热点课题之一.其中,由于过渡金属钌(Ru)催化1,6-烯炔的环化反应具备原子经济、反应条件温和、对官能团兼容性好、产率和选择性高等特点,近年来得到了研究者的广泛青睐.基于反应机理及产物的多样性,简要综述了Ru催化1,6-烯炔环化反应的发展历程和最新进展.关键词:钌(Ru); 1,6-烯炔; 催化; 环化反应; 原子经济性中图分类号: O 6-1 文献标志码: A 文章编号: 1000-5137(2020)04-0443-11Abstract: As the fundamental building blocks of medicines and bioactive molecules,five- or six-membered rings could be easily found in a series of natural products.Therefore,the development of methodologies toward the formation of five- or six-membered rings-containing compounds is still a hot topic in synthetic chemistry.Of all significant methods,ruthenium(Ru)-catalyzed 1,6-enyne cyclization reactions are especially attractive because of the atom-economic fashion,mild reaction conditions,good functional group tolerance and excellent selectivity.This review will focus on the development and recent process of Ru-catalyzed 1,6-enyne cyclization reactions on the basis of the different mechanisms and product diversity.Key words: ruthenium(Ru); 1,6-enyne; catalysis; cyclization; atom-economy0 引言1985年,著名有机化学家TROST[1]利用金属钯(Pd)催化烯炔的环化反应实现了烯炔的阿尔德-烯(Alder-ene)环异构化反应.尽管当时这个反应对有机官能团的兼容性较差,但是这个反应的发现为烯炔环化反应奠定了坚实的基础.从那时起,科学家们相继发展了许多其他过渡金属络合物催化的烯炔环化反应,合成了系列重要的五元环/六元环化合物.近年来,过渡金属络合物催化烯炔的环异构化反应再次激发了人们的兴趣,主要原因是人们发现它们在系列重要的天然产物合成中有重要的应用,例如愈伤组织A和呋喃喹啉A/B/C[2].其中,利用过渡金属催化的1,6-烯炔的环化反应构筑五元/六元环状化合物则是一种非常高效的方法.这种方法的优点在于反应条件温和、对各种官能团兼容性好、原料简单易得及原子经济性等.本文作者简要介绍了金属钌(Ru)催化1,6-烯炔环化反应的通用机理,再结合具体示例综述Ru催化烯炔环化反应的研究进展.1 Ru催化烯炔环化反应的机理根据Ru络合物和1,6-烯炔的配位方式,可以将Ru催化1,6-烯炔的反应机理分为以下几种:氧化环化机理、烯丙基C-H活化机理、碳碳三键C≡C活化机理和复分解机理[3-4].1.1 氧化环化机理如图1所示,当金属Ru含有富电子配体时,首先在反应溶液中经历配体解离过程,产生金属Ru配位不饱和中心[Ru]n.然后金属Ru的配位不饱和中心可以同时与碳碳双键C=C及C≡C配位,形成含有18个电子的金属Ru活性中间体,再通过氧化环化的过程生成环戊烯Ru 中间体,与此同时金属Ru的化合价由+2变为+4.在环戊烯钌中间体中,当金属Ru β位上含有氢(H)原子时,环戊烯钌中间体可以通过β-H消除的方法产生乙烯基钌中间体.最后通过金属还原消除反应得到相应的Alder-ene产物[5].1.2 烯丙基C-H活化机理然而,金属Ru络合物与1,6-烯炔也可以通过烯丙基C-H活化机理同样制备出含有1,3-二烯或1,4-二烯的环状化合物,如图2所示.这种机理的关键在于当金属Ru与1,6-烯炔的C=C双键配位之后,直接通过烯丙基C-H活化的方式产生烯丙基钌中间体;然后再通过氢金属化的策略得到相应的乙烯基钌中间体,最后再通过金属还原消除的方法制备出二烯类化合物,其中产物的类型与1,6-烯炔的结构有重要的关系[6].1.3 C≡C活化机理一般情况下,如果在烯炔环化反应中所采用的金属Ru络合物含有强吸电子配体,那么这种金属Ru络合物可以作为一种较强的路易斯酸,通过C≡C活化机理将1,6-烯炔转化为一系列重要的环状化合物.如图3所示,采用强亲电/亲三键的金属Ru络合物为催化剂,那么金属Ru络合物首先能够与1,6-烯炔配位形成η2-配合物,然后通过6-endo-dig或5-exo-dig环化反应的方式分别产生金属Ru活性中间体,再通过分子内重排和金属消除等步骤最后制备出双环[4.1.0]庚烯类化合物或者复分解产物[7].1.4 复分解机理当卡宾型的金属Ru络合物与1,6-烯炔反应时,一般情况下则通过复分解反应产生相应的1,3-二烯类环状产物(也称为复分解产物),这种反应通常包含2种机理.如图4所示,当卡宾钌络合物与1,6-烯炔的C≡C首先结合时,称为Yne-then-ene型机理.在这种反应机理中,Ru配合物与C≡C通过复分解反应形成环丁烯钌中间体;然后通过分子内重排反应分别产生2种乙烯基卡宾钌中间体;最后再通过卡宾钌中间体与独立的C=C的复分解反应释放出对应的五/六元复分解产物,与此同时产生金属Ru活性中间体.然而,针对Ru催化1,6-烯炔的复分解反应的影响因素目前并未明确,因为动力学研究表明卡宾钌也可以与1,6-烯炔的C=C首先结合,通过Ene-then-yne的路线合成出类似的五/六元复分解产物,如图5所示[8].2 Ru催化1,6-烯炔环化反应根据Ru催化1,6-烯炔环化反应中反应原料的多样性,可以将其分为单分子环异构化反应和双分子环化反应.2.1 单分子环异构化反应1995年,TROST等[9]首次发现了金属Ru络合物可以催化1,6-烯炔的Alder-ene环异构化反应,如图6所示.在这个反应中,TROST采用在空气不稳定的环戊二烯基三(乙腈)钌(II)六氟磷酸盐(CpRu(MeCN)3PF6)为催化剂,以丙酮和甲苯作为反应溶剂,以O/N键连的1,6-烯炔为反应原料,合成了系列重要的环状1,4-二烯化合物.值得注意的是,在这个反应中,1,6-烯炔烯丙位上取代基的类型和数目对产物的类型有重要的影响.2000年,TOSTE等[5]对CpRu(CH3CN)3PF6催化1,6-烯炔环异构化反应制备五元杂环化合物的反应进行了详细的研究.结果表明:催化剂的用量及反应物的浓度与反应速率、产率有重要的关系.例如,当烯炔的炔丙位含有OBMP基团时,对应目标产物的产率高达83%,如图7所示.这个反应在丙酮或N,N-二甲基甲酰胺都有可以顺利的进行,且对1,7-烯炔也具有良好的兼容性.2001年,TROST等[10]提出利用Ru催化分子内[5+2]环加成反应的策略合成七元环的方法.采用摩尔分数为10%的CpRu(CH3CN)3PF6为催化剂,可以在30 min之内将含有环丙烷的1,6-烯炔骨架结构转化为对应的双环化合物,其产量高达80%,如图8所示.此外,研究表明可以通过选择不同的取代基对产物的相对构型进行调节,系统地控制反应的区域选择性.这种反应方法已经在天然产物合成中有重要的应用.2003年,TROST等[11]发现该反应对烯丙位含有取代基的1,6-烯炔也有良好的兼容性,这进一步拓宽了Ru催化1,6-烯炔的底物范围.结果表明底物中取代基的类型和位置对环丙烷结构的键裂解方向有一定影响.因此可以通过调节取代基的策略来控制环加成过程的非对映选择性.该反应涉及3个C-C键的形成和2个C-C键的裂解,这进一步验证了Ru催化的分子内[5+2]环加成反应的机理.首先金属Ru活性中间体与烯炔的不饱和键进行配位,形成钌环戊烯中间体,然后通过环丙烷的插入反应形成了环辛烯钌中间体;最后通过还原消除反应生成对应的七元环产物.2010年,TROST等[12]发现摩尔分数为5%的CpRu(MeCN)3PF6可以将物质的量浓度为0.1 mol·L-1的戊烯基烯炔转化为5,5-稠环化合物.这个反应的优点在于反应时间短(23 ℃下反应3 h)、产率和选择性高等.如图9所示,当取代基R为叔丁基二甲基硅氧基(OTBS)时,产率高达85%,非对映体过量(dr)大于19/1.当取代基为酯基时,这个反应依然具有良好的非对映选择性.基于以上研究成果,TROST等[12]在类似的反应条件下也实现了从1,7-烯炔到氢化萘化合物的转化,如图10所示.在这个研究工作中,发现这种双环化反应对各种官能团的兼容性均较好,包括醛、酰胺和羧酸.尽管对含有伯醇结构底物的反应性相对较差,但可以通过增加催化剂负载量、延长反应时间和升高温度的策略提高对应化合物的产率.此外,当炔烃末端含有大位阻取代基時,例如TMS,反应在常规优化条件下较难进行,这表明取代基空间位阻对反应有一定的影响.与此同时,TROST等[12]发现也可以利用基于环庚烯的烯炔在丙酮中合成一系列含有六元环和七元环的环状化合物,如图11所示.这个反应也对各种官能团有较好的耐受性,例如含有酯(99%产率)、醛(87%产率)和酰胺(99%产率)等.但是当烯炔底物中含有氰基时,双环化反应无法顺利进行,因为氰基可以导致催化剂失活.同年,TROST等[13]报道了利用Ru催化烯炔醇合成双环[3.1.0]庚烷酮类化合物的环异构化反应,如图12所示.与经典反应机理不同,烯炔醇首先通过炔丙醇的氧化过程产生含有羰基的卡宾钌中间体,然后卡宾钌中间体与C=C通过[2+2]环加成/金属消除的策略释放出相对应的双环[3.1.0]庚烷酮产物.在这个反应中,添加剂三氟甲磺酸铟(In(OTf)3)、樟脑磺酸(CSA),及反应体系的浓度对提高反应产率有重要的促进作用.此外,底物的拓展研究表明这个催化过程对1,7-烯炔也有良好的实用性.2017年,TROST等[14]在以前的研究基础上开发了一种新型的带有卤素转移的环异构化反应,如图13所示.该反应对且对各种官能团也有很好的兼容性.通过对反应溶剂的优化和仔细筛选,发现当使用0.5 mol·L-1的四氢呋喃(THF)溶液为反应体系,以摩尔分数为5%的CpRu(MeCN)3PF6作为催化剂,在50 ℃下反应18 h时,反应产率高、选择性好.对1,6-卤代烯炔的底物拓展实验表明端炔位含有甲基时,对应目标产物具有较高的立体选择性和产率;其次,利用甲硅烷基醚取代的烯炔也可以高效合成相应的产物,表明空间位阻对反应的影响较小.然而,当炔烃末端位置含有芳香取代基时,对应目标化合物的顺反(Z/E)选择性较差,并且这些反应也需要较高的反应温度来实现底物的完全转化.值得注意的是烯炔中双键的构型对反应没有明显的影响.当卡宾型的金属Ru络合物与1,6-烯炔反应时,一般情况下则通过复分解反应产生相应的1,3-二烯类环状产物(也称为复分解产物),这种反应通常包含2种机理.如图4所示,当卡宾钌络合物与1,6-烯炔的C≡C首先结合时,称为Yne-then-ene型机理.在这种反应机理中,Ru配合物与C≡C通过复分解反应形成环丁烯钌中间体;然后通过分子内重排反应分别产生2种乙烯基卡宾钌中间体;最后再通过卡宾钌中间体与独立的C=C的复分解反应释放出对应的五/六元复分解产物,与此同时产生金属Ru活性中间体.然而,针对Ru催化1,6-烯炔的复分解反应的影响因素目前并未明确,因为动力学研究表明卡宾钌也可以与1,6-烯炔的C=C首先结合,通过Ene-then-yne的路线合成出类似的五/六元复分解产物,如图5所示[8].2 Ru催化1,6-烯炔环化反应根据Ru催化1,6-烯炔环化反应中反应原料的多样性,可以将其分为单分子环异构化反应和双分子环化反应.2.1 单分子环异构化反应1995年,TROST等[9]首次发现了金属Ru络合物可以催化1,6-烯炔的Alder-ene环异构化反应,如图6所示.在这个反应中,TROST采用在空气不稳定的环戊二烯基三(乙腈)钌(II)六氟磷酸盐(CpRu(MeCN)3PF6)为催化剂,以丙酮和甲苯作为反应溶剂,以O/N键连的1,6-烯炔为反应原料,合成了系列重要的环状1,4-二烯化合物.值得注意的是,在这个反应中,1,6-烯炔烯丙位上取代基的类型和数目对产物的类型有重要的影响.2000年,TOSTE等[5]对CpRu(CH3CN)3PF6催化1,6-烯炔环异构化反应制备五元杂环化合物的反应进行了详细的研究.结果表明:催化剂的用量及反应物的浓度与反应速率、产率有重要的关系.例如,当烯炔的炔丙位含有OBMP基团时,对应目标产物的产率高达83%,如图7所示.这个反应在丙酮或N,N-二甲基甲酰胺都有可以顺利的进行,且对1,7-烯炔也具有良好的兼容性.2001年,TROST等[10]提出利用Ru催化分子内[5+2]环加成反应的策略合成七元环的方法.采用摩尔分数为10%的CpRu(CH3CN)3PF6为催化剂,可以在30 min之内将含有环丙烷的1,6-烯炔骨架结构转化为对应的双环化合物,其产量高达80%,如图8所示.此外,研究表明可以通过选择不同的取代基对产物的相对构型进行调节,系统地控制反应的区域选择性.这种反应方法已经在天然产物合成中有重要的应用.2003年,TROST等[11]发现该反应对烯丙位含有取代基的1,6-烯炔也有良好的兼容性,这进一步拓宽了Ru催化1,6-烯炔的底物范围.结果表明底物中取代基的类型和位置对环丙烷结构的键裂解方向有一定影响.因此可以通过调节取代基的策略来控制环加成过程的非对映选择性.该反应涉及3个C-C键的形成和2个C-C键的裂解,这进一步验证了Ru催化的分子内[5+2]环加成反应的机理.首先金属Ru活性中间体与烯炔的不饱和键进行配位,形成钌环戊烯中间体,然后通过环丙烷的插入反应形成了环辛烯钌中间体;最后通过还原消除反应生成对应的七元环产物.2010年,TROST等[12]发现摩尔分数为5%的CpRu(MeCN)3PF6可以将物质的量浓度为0.1 mol·L-1的戊烯基烯炔转化为5,5-稠环化合物.这个反应的优点在于反应时间短(23 ℃下反应3 h)、产率和选择性高等.如图9所示,当取代基R为叔丁基二甲基硅氧基(OTBS)时,产率高达85%,非对映体过量(dr)大于19/1.当取代基为酯基时,这个反应依然具有良好的非对映选择性.基于以上研究成果,TROST等[12]在类似的反应条件下也实现了从1,7-烯炔到氢化萘化合物的转化,如图10所示.在这个研究工作中,发现这种双环化反应对各种官能团的兼容性均较好,包括醛、酰胺和羧酸.尽管对含有伯醇结构底物的反应性相对较差,但可以通过增加催化剂负载量、延长反应时间和升高温度的策略提高对应化合物的产率.此外,当炔烃末端含有大位阻取代基时,例如TMS,反应在常规优化条件下较难进行,这表明取代基空间位阻对反应有一定的影响.与此同时,TROST等[12]发现也可以利用基于环庚烯的烯炔在丙酮中合成一系列含有六元环和七元环的环状化合物,如图11所示.这个反应也对各种官能团有较好的耐受性,例如含有酯(99%产率)、醛(87%产率)和酰胺(99%产率)等.但是当烯炔底物中含有氰基时,双环化反应无法顺利进行,因为氰基可以导致催化剂失活.同年,TROST等[13]報道了利用Ru催化烯炔醇合成双环[3.1.0]庚烷酮类化合物的环异构化反应,如图12所示.与经典反应机理不同,烯炔醇首先通过炔丙醇的氧化过程产生含有羰基的卡宾钌中间体,然后卡宾钌中间体与C=C通过[2+2]环加成/金属消除的策略释放出相对应的双环[3.1.0]庚烷酮产物.在这个反应中,添加剂三氟甲磺酸铟(In(OTf)3)、樟脑磺酸(CSA),及反应体系的浓度对提高反应产率有重要的促进作用.此外,底物的拓展研究表明这个催化过程对1,7-烯炔也有良好的实用性.2017年,TROST等[14]在以前的研究基础上开发了一种新型的带有卤素转移的环异构化反应,如图13所示.该反应对且对各种官能团也有很好的兼容性.通过对反应溶剂的优化和仔细筛选,发现当使用0.5 mol·L-1的四氢呋喃(THF)溶液为反应体系,以摩尔分数为5%的CpRu(MeCN)3PF6作为催化剂,在50 ℃下反应18 h时,反应产率高、选择性好.对1,6-卤代烯炔的底物拓展实验表明端炔位含有甲基时,对应目标产物具有较高的立体选择性和产率;其次,利用甲硅烷基醚取代的烯炔也可以高效合成相应的产物,表明空间位阻对反应的影响较小.然而,当炔烃末端位置含有芳香取代基时,对应目标化合物的顺反(Z/E)选择性较差,并且这些反应也需要较高的反应温度来实现底物的完全转化.值得注意的是烯炔中双键的构型对反应没有明显的影响.当卡宾型的金属Ru络合物与1,6-烯炔反应时,一般情况下则通过复分解反应产生相应的1,3-二烯类环状产物(也称为复分解产物),这种反应通常包含2种机理.如图4所示,当卡宾钌络合物与1,6-烯炔的C≡C首先结合时,称为Yne-then-ene型机理.在这种反应机理中,Ru配合物与C≡C通过复分解反应形成环丁烯钌中间体;然后通过分子内重排反应分别产生2种乙烯基卡宾钌中间体;最后再通过卡宾钌中间体与独立的C=C的复分解反应释放出对应的五/六元复分解产物,与此同时产生金属Ru活性中间体.然而,针对Ru催化1,6-烯炔的复分解反应的影响因素目前并未明确,因为动力学研究表明卡宾钌也可以与1,6-烯炔的C=C首先结合,通过Ene-then-yne的路线合成出类似的五/六元复分解产物,如图5所示[8].2 Ru催化1,6-烯炔环化反应根据Ru催化1,6-烯炔环化反应中反应原料的多样性,可以将其分为单分子环异构化反应和双分子环化反应.2.1 单分子环异构化反应1995年,TROST等[9]首次发现了金属Ru络合物可以催化1,6-烯炔的Alder-ene环异构化反应,如图6所示.在这个反应中,TROST采用在空气不稳定的环戊二烯基三(乙腈)钌(II)六氟磷酸盐(CpRu(MeCN)3PF6)为催化剂,以丙酮和甲苯作为反应溶剂,以O/N键连的1,6-烯炔为反应原料,合成了系列重要的环状1,4-二烯化合物.值得注意的是,在这个反应中,1,6-烯炔烯丙位上取代基的类型和数目对产物的类型有重要的影响.2000年,TOSTE等[5]对CpRu(CH3CN)3PF6催化1,6-烯炔环异构化反应制备五元杂环化合物的反应进行了详细的研究.结果表明:催化剂的用量及反应物的浓度与反应速率、产率有重要的关系.例如,当烯炔的炔丙位含有OBMP基团时,对应目标产物的产率高达83%,如图7所示.这个反应在丙酮或N,N-二甲基甲酰胺都有可以顺利的进行,且对1,7-烯炔也具有良好的兼容性.2001年,TROST等[10]提出利用Ru催化分子内[5+2]环加成反应的策略合成七元环的方法.采用摩尔分数为10%的CpRu(CH3CN)3PF6為催化剂,可以在30 min之内将含有环丙烷的1,6-烯炔骨架结构转化为对应的双环化合物,其产量高达80%,如图8所示.此外,研究表明可以通过选择不同的取代基对产物的相对构型进行调节,系统地控制反应的区域选择性.这种反应方法已经在天然产物合成中有重要的应用.2003年,TROST等[11]发现该反应对烯丙位含有取代基的1,6-烯炔也有良好的兼容性,这进一步拓宽了Ru催化1,6-烯炔的底物范围.结果表明底物中取代基的类型和位置对环丙烷结构的键裂解方向有一定影响.因此可以通过调节取代基的策略来控制环加成过程的非对映选择性.该反应涉及3个C-C键的形成和2个C-C键的裂解,这进一步验证了Ru催化的分子内[5+2]环加成反应的机理.首先金属Ru活性中间体与烯炔的不饱和键进行配位,形成钌环戊烯中间体,然后通过环丙烷的插入反应形成了环辛烯钌中间体;最后通过还原消除反应生成对应的七元环产物.2010年,TROST等[12]发现摩尔分数为5%的CpRu(MeCN)3PF6可以将物质的量浓度为0.1 mol·L-1的戊烯基烯炔转化为5,5-稠环化合物.这个反应的优点在于反应时间短(23 ℃下反应3 h)、产率和选择性高等.如图9所示,当取代基R为叔丁基二甲基硅氧基(OTBS)时,产率高达85%,非对映体过量(dr)大于19/1.当取代基为酯基时,这个反应依然具有良好的非对映选择性.基于以上研究成果,TROST等[12]在类似的反应条件下也实现了从1,7-烯炔到氢化萘化合物的转化,如图10所示.在这个研究工作中,发现这种双环化反应对各种官能团的兼容性均较好,包括醛、酰胺和羧酸.尽管对含有伯醇结构底物的反应性相对较差,但可以通过增加催化剂负载量、延长反应时间和升高温度的策略提高对应化合物的产率.此外,当炔烃末端含有大位阻取代基时,例如TMS,反应在常规优化条件下较难进行,这表明取代基空间位阻对反应有一定的影响.与此同时,TROST等[12]发现也可以利用基于环庚烯的烯炔在丙酮中合成一系列含有六元环和七元环的环状化合物,如图11所示.这个反应也对各种官能团有较好的耐受性,例如含有酯(99%产率)、醛(87%产率)和酰胺(99%产率)等.但是当烯炔底物中含有氰基时,双环化反应无法顺利进行,因为氰基可以导致催化剂失活.同年,TROST等[13]报道了利用Ru催化烯炔醇合成双环[3.1.0]庚烷酮类化合物的环异构化反应,如图12所示.与经典反应机理不同,烯炔醇首先通过炔丙醇的氧化过程产生含有羰基的卡宾钌中间体,然后卡宾钌中间体与C=C通过[2+2]环加成/金属消除的策略释放出相对应的双环[3.1.0]庚烷酮产物.在这个反应中,添加剂三氟甲磺酸铟(In(OTf)3)、樟脑磺酸(CSA),及反应体系的浓度对提高反应产率有重要的促进作用.此外,底物的拓展研究表明这个催化过程对1,7-烯炔也有良好的实用性.2017年,TROST等[14]在以前的研究基础上开发了一种新型的带有卤素转移的环异构化反应,如图13所示.该反应对且对各种官能团也有很好的兼容性.通过对反应溶剂的优化和仔细筛选,发现当使用0.5 mol·L-1的四氢呋喃(THF)溶液为反应体系,以摩尔分数为5%的CpRu(MeCN)3PF6作为催化剂,在50 ℃下反应18 h时,反应产率高、选择性好.对1,6-卤代烯炔的底物拓展实验表明端炔位含有甲基时,对应目标产物具有较高的立体选择性和产率;其次,利用甲硅烷基醚取代的烯炔也可以高效合成相应的产物,表明空间位阻对反应的影响较小.然而,当炔烃末端位置含有芳香取代基时,对应目标化合物的顺反(Z/E)选择性较差,并且这些反应也需要较高的反应温度来实现底物的完全转化.值得注意的是烯炔中双键的构型对反应没有明显的影响.当卡宾型的金属Ru络合物与1,6-烯炔反应时,一般情况下则通过复分解反应产生相应的1,3-二烯类环状产物(也称为复分解产物),这种反应通常包含2种机理.如图4所示,当卡宾钌络合物与1,6-烯炔的C≡C首先结合时,称为Yne-then-ene型机理.在这种反应机理中,Ru配合物与C≡C通过复分解反应形成环丁烯钌中间体;然后通过分子内重排反应分别产生2种乙烯基卡宾钌中间体;最后再通过卡宾钌中间体与独立的C=C的复分解反应释放出对应的五/六元复分解产物,与此同时产生金属Ru活性中间体.然而,针对Ru催化1,6-烯炔的复分解反应的影响因素目前并未明确,因为动力学研究表明卡宾钌也可以与1,6-烯炔的C=C首先结合,通过Ene-then-yne的路线合成出类似的五/六元复分解产物,如图5所示[8].2 Ru催化1,6-烯炔环化反应根据Ru催化1,6-烯炔环化反应中反应原料的多样性,可以将其分为单分子环异构化反应和双分子环化反应.2.1 单分子环异构化反应1995年,TROST等[9]首次发现了金属Ru络合物可以催化1,6-烯炔的Alder-ene环异构化反应,如图6所示.在这个反应中,TROST采用在空气不稳定的环戊二烯基三(乙腈)钌(II)六氟磷酸盐(CpRu(MeCN)3PF6)为催化剂,以丙酮和甲苯作为反应溶剂,以O/N键连的1,6-烯炔为反应原料,合成了系列重要的环状1,4-二烯化合物.值得注意的是,在这个反应中,1,6-烯炔烯丙位上取代基的类型和数目对产物的类型有重要的影响.。
金催化炔烃环化反应

金催化炔烃环化反应嘿,你知道金催化炔烃环化反应吗?这就像是化学世界里一场超级酷炫的魔法秀呢!金,这个平时总是和富贵、奢华联系在一起的元素,在化学舞台上摇身一变,成了一个神奇的魔法师。
炔烃就像是一群调皮的小精灵,它们平时到处乱窜,自由得很。
可是当金这个魔法师出现的时候,就像是孙悟空遇到了紧箍咒一样。
金轻轻一挥它的魔法棒,那些炔烃小精灵就开始乖乖听话,按照金的指示开始环化。
这环化的过程就像是在编织一个超级复杂又精美的花环,每个炔烃都找到了自己的位置,一环扣着一环,那场面,比世界上最精密的齿轮组合还要奇妙。
你要是把这个反应想象成一场舞会,金就是那个最有魅力的主持人。
炔烃们原本杂乱无章地在舞池里晃悠,金主持人一登场,就把这些炔烃们两两配对,然后让它们优雅地旋转起来,最后形成一个完美的环状结构,就像舞者们组成了一个绚丽的圆形图案。
金催化炔烃环化反应还有点像搭积木。
炔烃是那些形状各异的小积木块,金则是那个有着无限创意的小建筑师。
它把那些炔烃积木巧妙地拼接在一起,一会儿功夫,就搭建出了一个独特的环状建筑,这个建筑在化学的世界里可是有着独一无二的功能和意义呢。
有时候我觉得这个反应就像一场魔法烹饪。
炔烃是食材,金就是大厨手中的神奇调味料。
大厨金把炔烃食材放进化学的大锅里,稍微施展一下催化魔法,嘿,一道美味的环状化合物大餐就出锅了。
而且这道菜还不是普通的菜,它在医药、材料等各个领域都是备受欢迎的珍馐。
从微观的角度看,这金催化的过程简直就是一场星际大战。
炔烃分子就像是宇宙中的小行星,金原子就像拥有超能力的外星文明。
外星文明金用它神秘的力量,让那些小行星改变轨迹,相互碰撞融合,最后形成了一个稳定的环形星球,也就是环化后的产物。
这个反应还像一场神秘的变形记。
炔烃原本有着自己简单的形态,在金的催化下,就像毛毛虫变成蝴蝶一样,华丽转身成为了复杂而迷人的环状化合物。
这新的形态不仅外观上让人惊艳,在化学性质上也是有了质的飞跃。
金催化炔烃环化反应就像是化学森林里的一种神秘魔法。
金催化羟胺和炔烃加成反应选择性问题和(co)_5分子解离途径的理论计算研究

金催化羟胺和炔烃加成反应选择性问题和(co)_5分子解离途径的理论计算研究
金催化的羟胺和炔烃加成反应是一种金催化的有机反应,
在这种反应中,羟胺与炔烃在金催化剂的作用下,发生加成
反应,生成二维炔烷酮。
这种反应具有很高的选择性,能够有效地生成所需的产物,并且在反应过程中产生的副产物很少。
在理论计算研究中,人们可以使用量子化学方法来研究这
种反应的机理。
通过对反应中参与物质的分子轨道能量和激发能级的研究,可以更好地理解反应的过程。
另外,还可以研究(CO)_5分子解离的途径,以便更好地解释反应的选择性。
总的来说,通过理论计算研究,人们可以更好地了解金催
化的羟胺和炔烃加成反应的机理和选择性,为进一步提高反
应的效率和生产产品提供理论依据。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
article info
Article history: Received 5 September 2012 Revised 11 October 2012 Accepted 16 October 2012 Available online 23 October 2012
Keywords: Pentacyclic pyrido[4,3,2-mn]acridin-8-one Domino reaction Gold catalysis 6-endo-dig Cycloisomerization N-Propargylaminoquinone
abstract
A novel methodology taking advantage of a domino reaction initiated by an Au(I)-catalyzed 6-endo-dig cycloisomerization under silver-free condition was developed to prepare pentacyclic pyrido[4,3,2mn]acridin-8-ones from N-propargylaminoquinones. Triphenylphosphinegold(I) chloride in combination with TFA was firstly employed and displayed excellent catalytic efficiency in the domino reaction.
Ó 2012 Elsevier Ltd. All rights reserved.
8H-Pyrido[4,3,2-mn]acridin-8-one is a basic skeleton embedded in marine alkaloids isolated from sponges and ascidians.1 These alkaloids have attracted considerable attention because of their significant cytotoxicity.2 The isolated pentacyclic pyrido[4,3,2mn]acridin-8-one alkaloids (amphimedine,3 neoamphimedine,4,5 meridine,6 isocystodamine,7,8 and ecionine A9) feature fused pyridone, pyridine, or tetrahydropyridine heterocycle at C9, 10 position of the pyridoacridine ring system and display certain specific biological activities such as inhibition of topoisomerase II and induction of neuronal differentiation (Fig. 1). The pronounced biological properties make them to be potential drug candidates for the treatment of cancers. However, the scarcity of the alkaloids has hampered further biological evaluations. Efforts to obtain pentacyclic pyrido[4,3,2-mn]acridin-8-one alkaloids as well as relevant analogs by means of chemical synthesis still remain in urgent need.10
N
N O 8H -Pyrido[4,3,2-mn]acrid-8-one
N O
N H3C
N O
Amphimedine
N
N
H3C
N
OO
Neoamphimedine
OH N
N N
HN N
N
N
O
Meridine
N NH2 O
Isocystodamine
N O NH
Ecionine A
Figure 1. Pentacyclic pyrido[4,3,2-mn]acridin-8-one alkaloids.
Hao Yin, Fanji Kong, Shaozhong Wang ⇑, Zhu-Jun Yao
School of Chemistry and Chemical Engineering, State Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, PR China
o-nitro or o-trifluoroacetamidocinnamaldehyde-N,N-dimethylhydrazone as diene to synthesize meridine and cystodamine, even though the yield of the hetero-Diels–Alder cycloaddition from the 1-azadiene was low and side products involving dihydroanthraquinone and regioisomer of the desired adduct were accompanied seriously.
Assembly of pentacyclic pyrido[4,3,2-mn]acridin-8-ones via a domino reaction initiated by Au(I)-catalyzed 6-endo-dig cycloisomerization of N-propargylaminoquinones
Tetrahedron Letters 53 (2012) 7078–7082
Contents lists available at SciVerse ScienceDirect
Tetrahedron Letters
journal homepage: /locate/tetlet
The classic strategy to assemble pentacyclic pyrido[4,3,2mn]acridin-8-one skeleton is closely associated with the construction of tricyclic diazaanthraquinone. Always, 4-arylated 1,6-, 1,8-, and 1,9-diazaanthraquinones were elaborated as straightforward precursors of pyrido[4,3,2-mn]acridin-8-ones, and the final formation of pyridine ring was established by an intramolecular reductive cyclization or hydrolytic cyclization from the diazaanthraquinone (Scheme 1). Conventionally, the 4-arylated diazaanthraquinone
Aiming at discovering new methodology to construct pentacyclic pyrido[4,3,2-mn]acridin-8-one heterocycles, we envisioned a domino reaction initiated by Au(I)-catalyzed 6-endo-dig cycloisomerization of N-propargylaminoquinone,16 which is distinct from the stepwise methodology based on hetero-Diels–Alder cycloaddition. As outlined in Scheme 2, N-propargylaminoquinone containing free aniline functionality attached on alkyne terminus was designed as the substrate. Under the gold catalysis,17 two possible reaction pathways should take place in principle. The activated triple bond would be attacked either by the enamine moiety of aminoquinone in a 6-endo manner (pathway a) or by the amine in a 5-endo manner (pathway b). The former cycloisomerization will definitely afford the tricyclic 4-(2-aminophenyl)-azaanthraquinone, which might be spontaneously converted into pentacyclic pyrido[4,3,2-mn]acridin-8-one by an intramolecular condensation between carbonyl of quinone and aniline group.