配位化学讲义第四章(1)价键理论、晶体场理论(完整)
第四章 配位化学

配分子: Ni(CO) 4 , 配离子: 4 , BF
CoCl3 ( NH3 )3 Fe(CN)6 4- , Co(NH3 )5 (H 2O)3 , Cu(NH 3 ) 4 2 Co(NH3 )5 (H 2O)Cl3
Cu2[SiF6] 六氟合硅酸(IV)铜(I),
或六氟合硅酸(2-)铜 H4[Fe(CN)6] 六氰合铁(II)酸
4-1 配合物的基本概念
含配阳离子的配合物命名
命名时,阴离子在前,阳离子在后,与无机盐、无 机碱的命名同, 如: [Co(NH3)6]Cl3 三氯化六氨合钴(III) [Cu(NH3)4]SO4 硫酸四氨合铜(II) [Ag(NH3)2](OH) 氢氧化二氨合银(1+)
以罗马数 字Ⅱ、Ⅲ、 Ⅳ表示
4-1 配合物的基本概念
当配体是一很长名称的有机化合物或复杂配体时, 给该配体加一圆括号。如果中心离子有多种价态,则 应在中心离子名称后的括弧中以罗马数字表示其氧化 态。也可在配离子名称后头的圆括号内以阿拉伯数字 (如+1,-2)表明配离子所带的电荷。例如
[Co(NH3)6]3+ 六氨合钴(III)离子;
Cl
NH3 NH3 Cl
瑞士无机化学家.因创立配位化学而获得1913 年诺贝尔化学奖
Co
NH3
1923年,英国化学家西奇维克Sidgwick) 提出有效原子序数(EAN)法则….
XRD
③ 配位化学的蓬勃发展时期
20世纪40年代前后,第二次世界大战期间,无机化学家在围绕耕耘周期 表中某些元素化合物的合成中得到发展,在工业上,美国实行原子核裂变 曼哈顿(Manhattan)工程基础上所发展的铀和超铀元素溶液配合物的研究。 以及在学科上,195l年Panson和Miler对二茂铁的合成打破了传统无机和 有机化合物的界限。从而开始了无机化学的复兴。 Ziegler和Natta的金属烯烃催化剂,Eigen的快速反应。Lipscomb的硼烷 理论,Wnkinson和Fischer发展的有机金属化学,Hoffmann的等瓣理论 Taube研究配合物和固氮反应机理,Cram,Lehn和Pedersen在超分子化学 方面的贡献,Marcus的电子传递过程。在这些开创性成就的基础上,配 位化学在其合成、结构、性质和理论的研究方面取得了一系列进展。 我国配位化学的研究在中华人民共和国成立前几乎属于空白. 80年代后, 我国配位化学研究已步入国际先进行列。特别在下列方面取得了重要进 展:(1)新型配合物、簇合物、有机金属化合物和生物无机配合物,特别是 配位超分子化合物的合成及其结构研究取得丰硕成果,丰富了配合物的内 涵;(2)开展了热力学、动力学和反应机理方面的研究,特别在溶液中离子 萃取分离和均向催化等应用方面取得了成果;(3)现代溶液结构的谱学研究 及其分析方法以及配合物的结构和性质的基础研究;(4)随着高新技术的 发展,具有光、电、热、磁特性和生物功能配合物的研究正在取得进展。
第4章 配合物的晶体场理论及配合物的光谱-4h

•
由磁矩可判断内轨或外轨型配合物
s n(n 2) B
(n—分子中未成对电子数)
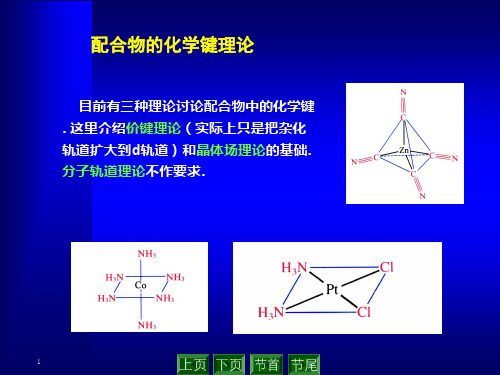
价键理论
价键理论的局限性:
(1)定性理论:不能定量或半定量的说明配合物的性质;
( 2 )不能解释配合物的吸收光谱 ( 颜色)、反应机理等:只 能解释配合物处于基态时的性质,如配位数、几何构型。但 涉及到激发态的问题等就无能为力; (3)无法解释某些配合物稳定性规律和不正常氧化态配合物
4.1 晶体场理论(CFT)
4.1.2 d轨道的能级分裂能
设自由离子中每个d轨道的能量为E0(这时5个d轨道的能量相 同),球形场中每个d轨道的能量上升为Es。(这时5个d轨道的 能量仍然相同)。 以Es为零点,将分裂后轨道的能量差称为分裂能。
4.1 晶体场理论(CFT)
•八面体场:分裂能的大小用10Dq或△0表示
4.1 晶体场理论(CFT)
•正四面体场:分裂能相对于八面体场较小,分裂能的大小为4/9△0,用 △t表示。
3d 2d 0 4 d d 4.45Dq 0 t 9 8 2 4 3 d 1.78Dq 0 t d 2.67Dq 0 t 45 5 15 5
反之则称为弱场配体。
同样可计算出d1~10金属离子在四面体及八面体、正方形场中的 CFSE。
4.1 晶体场理论(CFT)
d1~10金属离子在四面体及八面体、正方形场中的CFSE
dn d0 d1 d2 d3 d4 d5 d6 d7 正八面体场 弱场 0 -4Dq -8Dq -12Dq -6Dq 0Dq -4Dq -8Dq 强场 0 -4Dq -8Dq -12Dq -16Dq+P -20Dq+2P -24Dq+2P -18Dq+P 正四面体场 弱场 0Dq -2.67Dq -5.34Dq -3.56Dq -1.78Dq 0Dq -2.67Dq -5.34Dq 强场 0 -2.67Dq -5.34Dq -8.01Dq+P -10.68Dq+2P -8.9Dq+2P -7.12Dq+P -5.34Dq 平面正方形场 弱场 0 -5.14Dq -10.28Dq -14.56Dq -12.28Dq 0Dq -5.14Dq -10.28Dq 强场 0Dq -5.14Dq -10.28Dq -14.56Dq -19.70Dq+P -24.82Dq+2P -29.12Dq+2P -26.84Dq+P
价键理论和晶体场理论
67.524 ×10-20 35.250 ×10-20 强 3d6 t2g6 eg0 0 0 低自旋 内轨型 d2sp3
八面体场中电子在t 八面体场中电子在 2g和eg轨道中的分布
只 有 一 种 排 列 1 d4 2 d5 3 d6 2 d7 1 d1 d2 d3 d8 d9
高 自 旋
4
5
4
3
低 自 旋
+ [Cr (H2O)6]3+ [Cr (H2O)6]2+ [CrCl6]3-
[MoCl6]319200
∆o /cm-1
17600
14000
13600
配位体的影响: ● 配位体的影响:光谱化学序列 (ectrochemical series) [Co(H2O)6]3+ [CoF6]3[Co(NH3)6]3+ [Co(CN)6]313000 18600 22900 34000 ∆o /cm-1 各种配体对同一M产生的晶体场分裂能的值由小到大的顺序 产生的晶体场分裂能的值由小到大的顺序: 各种配体对同一 产生的晶体场分裂能的值由小到大的顺序 I-<Br-<Cl-,SCN-<F-<OH-<C2O42<H2O<NCS-<edta<NH3<en<bipy <phen<SO32-<NO2<CO, CN初步看作是配位原子电负性的排列: 初步看作是配位原子电负性的排列: 卤素 < 氧 < 氮 < 碳 电负性
直 线 形
平面 三角形
正四 面体
平面 正方形
三角 双锥 形四方 锥形(2) 配 Nhomakorabea物的磁性
配合物磁性的测定是判断配合物结构的一个重要手段. 配合物磁性的测定是判断配合物结构的一个重要手段 物质在磁场中表现出来的性质. 磁 性:物质在磁场中表现出来的性质 顺磁性: 顺磁性:被磁场吸引 n > 0 , µ > 0,如O2, NO, NO2. , 反磁性: 反磁性:被磁场排斥 n =0 , µ = 0. 铁磁性:被磁场强烈吸引 铁磁性:被磁场强烈吸引. 例:Fe,Co,Ni. , , 磁 矩: µ=[n(n+2)]1/2 (B.M.)玻尔磁子 玻尔磁子. 玻尔磁子
配位化学讲义

配位化学Coordination Compounds内容提要1.基本概念①配合物的定义②配合物的组成③配合物的命名2.化学键理论①配合物的价键理论②晶体场理论3.配位平衡①配位平衡常数②配位平衡的移动4.鳌合物和生物配体①鳌合效应②影响鳌合物稳定性的因素第一节配位化合的基本概念一、配位化合物的定义•配合物是以具有接受电子对的离子或原子(统称中心原子)为中心,与一组可以给出电子对的离子或分子(统称配体),以一定的空间排列方式在中心原子周围所组成的质点(配离子或配分子)为特征的化合物。
CuSO 4Solution adding NaOH Cu(OH)2Precipitation [Cu(NH 3)4]SO 4Complex第一节基本概念二、配合物的组成•多数配合物由配离子与带相反电荷的离子组成,•带正电荷的配离子称为配阳离子,带负电荷的配离子称为配阴离子,配合物也可以是电中性的配位分子,•含配离子的化合物和配位分子统称为配合物,•习惯上把配离子也称为配合物。
[Ag(NH 3)2]+; [HgI 4]2-; [Fe(NCS)4]-; Pt(NH 3)2Cl 2]第一节基本概念1.配合物的内层(inner sphere)和外层(outer sphere)[Cu ( NH 3 )4 ]SO 4Central Ligands (‘赖跟的)atomInner sphere Outer sphereCoordination compound电中性的配位分子只有内层,没有外层。
第一节基本概念2.中心原子(central atom)•配合物中接受孤对电子的阳离子或原子统称为中心原子。
•中心原子一般是金属离子,大多为过渡元素,特别是第ⅧB族元素以及相邻近的一些副族元素。
•某些副族元素的原子和高氧化值非金属元素的原子也是较常见的中心原子,如[Ni(CO)4]中的Ni(0)、[SiF6]2-中的Si(Ⅳ)第一节基本概念3.配体(ligand)和配位原子(ligating atom)•与中心原子以配位键结合的阴离子或中性分子称为配体[Ag(NH3)2]+中NH3、[Ni(CO)4]中CO 、[SiF6]2-中F-•配体中直接向中心原子提供孤对电子形成配位键的原子称为配位原子NH3中的N、CO中的C、F-中的F •配位原子的最外电子层都有孤对电子,常见的是电负性较大的非金属的原子N、O、C、S、F、Cl、Br、I第一节基本概念3.配体(ligand)和配位原子(ligating atom)单齿配体(monodentate ligand)多齿配体(multidentate ligand)•单齿配体NH3、H2O、F-、Cl-少数配体虽有两个配位原子,由于两个配位原子靠得太近,只能选择其中一个与中心原子成键,故仍属单齿配体-、ONO-、SCN-、NCS-如CN-、NC-、NO2第一节基本概念3.配体(ligand)和配位原子(ligating atom)•多齿配体¾双齿配体:H2N-CH2-CH2-NH2(乙二胺,简写为en)¾三齿配体:H2NCH2CH2NHCH2CH2NH2(二亚乙基三胺,简写为DEN) ¾六齿配体:乙二胺四乙酸根Ethylenediaminetetraacetic ion , EDTA C H 2C H 2N N H 2C C H 2H 2C C H 2C O -O C O -OC C O -O -O O第一节基本概念第一节基本概念4.配位数(coordination number)•配合物中直接与中心原子键合的配位原子数目。
配位化学讲义 第四章(1) 价键理论、晶体场理论

第三章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A2″:p zE″:(d xz、d yz)2、σ轨道杂化方案1)四面体分子AB4(Td)[CoCl4]2-以四个杂化轨道的集合作为分子点群(Td)表示的基,确定该表示的特征标:E 2 -1 2 0 0 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -1 1 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+ 6×(-1)×2]=0a(E)=1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+ 6×0×2]=0a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24 [3×4+8×0×1+3×(-1)×0+6×(-1)×0 +6×1×2]=1约化结果Γ=A1+T2由特征标表:A1T2s (p x、p y、p z)(d xy、d xz、d yz)可有两种组合:sp3(s、p x、p y、p z)、sd3(s、d xy、d xz、d yz)* 以一组杂化轨道为基的表示的特征标的简化计算规则:Γ 5 2 1 3 0 3约化结果:Γ= 2A1′+A2〞+E′A1′A2〞E′s p z (p x、p y)d z2(d xy、d x2-y2)两种可能的组合:(s、d z2、p z 、p x、p y)( s、d z2、p z、d xy、d x2-y2)约化得:Γ=A1g+B1g+E uA1g B1g E us d x2-y2(p x、p y)d z2两种类型:dsp2(d x2-y2、s、p x、p y)、d2p2(d z2、d x2-y2、p x、p y)5)八面体AB6(O h) 例:[Fe(H2O)6]3+(d z2、d x2-y2、s、p x、p y、p z) 3、π成键杂化方案在AB n分子中,原子A上要有2n个π型杂化轨道和在B原子上的2n个π原子轨道成键。
基础化学:第四章 配位化合物-1

配位药物化学
顺铂 (Cisplatin)
卡铂
奥沙利铂
奈达铂
乐铂
铂类抗癌药物
❖ 4.1 配合物 (coordination compound) 的基本概念
一、配合物的定义、组成和命名
1. 定义
➢ 由配体和中心原子以配位键结合,按一定组成和 空间构型所形成的化合物
➢ 配体(L):能够给出示有 SO42-
未能查出Cu2+ X射线晶体衍射证明为
[Cu(NH3)4]SO4
配合物
配合物的应用广泛
➢ 人体内胰岛素:锌的配合物 ➢ 植物的固氮作用:铁、钼的配合物 ➢ 植物光合作用:镁的配合物 ➢ 维生素B12:钴的配合物 ➢ 人体血液中传送氧气的血红蛋白:铁的配合物 ……
血红蛋白
H2OO2
第四章 配位化合物
❖ 4.1 配合物的基本概念 ❖ 4.2 价键理论 ❖ 4.3 晶体场理论
CuSO4溶液 CuSO4溶液 CuSO4溶液 +过量氨水 CuSO4溶液 +过量氨水 CuSO4溶液 +过量氨水
NaOH溶液 BaCl2溶液 BaCl2溶液 NaOH溶液 无水乙醇
示有Cu2+
示有 SO42-
(3). 配体的数目用一、二、三、四等表示
(4). 在最后一个配体名称之后、金属前缀以“合”字 (5). 当中心离子具有多种氧化态时,在该原子
后用括号注明(罗马数字) (6). 若配体名称较长或为复杂配体时,配体名
称写在配体数目后的括号中
[Co(NH3)3(H2O)Cl2]+ 二氯•三氨•水合钴(Ⅲ)离子
➢ 中心原子(M):具有接受孤对电子或多个不定域 电子的空轨道的离子或原子
配位键
价键理论-晶体场-配位化合物

四、 离域π键及反馈π键
以 [Ni(CN)4]2- 为例:
-NC
CN-
Ni
-NC
CN-
dsp2 杂化,用了2个 P轨道,还剩下一个Pz 空轨道 9个原子在同一平面上,可以和CN- 离子充满电子的
π2pz轨道重叠,而形成9原子8电子的离域π键,
因而增强了[Ni(CN)4]2- 的稳定性。
反馈π键: 当配位体给出电子对与中心元素形成σ键时,如果中心元素的某 些d 轨道有孤电子对 ( 如dxy, dyz, dxz ),有的配位体有空的π分子轨道 (如CO 有空的π* 轨道) 或空的p或d轨道,而二者的对称性又合适时,则中心元素的孤 对d电子也可以反过来给予配位体形成“反馈π键”。
5
或
d4s 正方锥型 VO(AcAc)2
d2sp3
6
或
正八面体 Co(NH3)63+
sp3d2
实例
3d
4s 4p
dsp3
3d
4s 4p
d4s
3d
4s 4p
d2sp3
三、 外轨型配合物和内轨型配合物(高自旋型配合物和低自 旋型配合物)
电子排布遵循三个原则:能量最低原理、保里不相容原理和洪特规则( 最多轨道原则),即在等价轨道中,自旋单电子数要最大,状态最稳定。
第三章 配合物的 化学键理论
配合物的定义
❖ 1 配合物的特点: 中心离子(或原子)有空的价电子轨道
❖
❖
配体分子或离子含有孤对电子或π键电子
❖
配合物形成体与配体可形成具有一定空间
构型和一定特性的复杂(化学质点)离子或分子。
❖ 配合物是由可以给出孤对电子或多个不定域电子的一定 数目的离子或分子(统称配体)和具有接受孤对电子或多个 不定域电子的空轨道的原子或离子(统称中心原子),按一 定的组成和空间构型所形成的化合物。
配位化学讲义 第四章(1) 价键理论、晶体场理论

配位化学讲义第四章(1)价键理论、晶体场理论第三章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valencebond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[Fe(CN)6]4-sp杂化直线型[AgCl2]-二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A 2″:p zE″:(d xz、d yz)2、σ轨道杂化方案1)四面体分子AB4(Td)[CoCl4]2-以四个杂化轨道的集合作为分子点群(Td)表示的基,确定该表示的特征标:r1r4r2r3恒等操作,χ(E)=4 C3操作,χ(C3)=1对C2、S4和σd用同样方法处理,得T d E 8C3 3C2 6S46σdΓ 4 1 00 2约化:T d E 8C3 3C2 6S4 6σdA1 1 1 1 11A2 1 1 1 -1 - 1E 2 -1 2 00 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -11 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+6×(-1)×2]=0a(E)=1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+6×0×2]=0a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24 [3×4+8×0×1+3×(-1)×0+6×(-1)×0+6×1×2]=1约化结果Γ=A1+T2由特征标表:A1T2s(p x、p y、p z)(d xy、d xz、d yz)可有两种组合:sp3(s、p x、p y、p z)、sd3(s、d xy、d xz、d yz)* 以一组杂化轨道为基的表示的特征标的简化计算规则:①不变(1)②改变符号(-1)③与其他函数变换(0)2)再以[CdCI5]3-三角双锥(D3h)为例:41325D3h E 2C33C2σh2S3 3σvΓ 5 2 13 0 3约化结果:Γ= 2A1′+A2〞+E′A1′A2〞E′s p z (p x、p y)d z2(d xy、d x2-y2)两种可能的组合:(s、d z2、p z 、p x、p y)( s、d z2、p z、d xy、d x2-y2)3)[HgI3]- ( D3h)123D3h E 2C3 3C2σh2S33σvΓ 3 0 13 0 1约化得:Γ=A1′+E′A1′E′s (p x、p y)d z2(d xy、d x2-y2)可能的组合有:(s、p x、p y)、(s、d xy、d x2-y2)、(d z2、p x、p y)、(d z2、d xy、d x2-y2)4)平面AB4型分子(D4h)例:[PtCl4]2-C2′C2″D4h E 2C4(C41,C43) C2(C42) 2C2′2C2″i 2S4σh 2σv2σdΓ 4 0 0 20 0 0 4 2 0约化得:Γ=A1g+B1g+E uA1g B1g E us d x2-y2(p x、p y)d z2两种类型:dsp2(d x2-y2、s、p x、p y)、d2p2(d z2、d x2-y2、p x、p y)5)八面体AB6(O h) 例:[Fe(H2O)6]3+O h E 8C3 6C26C4 3C2i 6S4′8S6 3σh 6σdΓ 6 0 0 2 2 0 0 0 4 2约化得:Γ=A1g+E g+T1u A1g E gT1us (d z2、d x2-y2) (p x、p y、p z)只有唯一的d2sp3杂化(d z2、d x2-y2、s、p x、p y、p z)3、π成键杂化方案在AB n分子中,原子A上要有2n个π型杂化轨道和在B原子上的2n个π原子轨道成键。
配位化学理论

1. 轨道分裂 C.N.=6
在球型场中 在球型场中
八面体场
在八面体场中 在八面体场中
eg: dz2
dx2-y2
t2g: dxz dyz dxy Eeg-Et2g=10Dq 4Eeg+ 6Et2g =0 Eeg=6Dq Et2g=-4Dq
d 轨道示意图
1. 轨道分裂
四面体场
在球型场中 在球型场中 在四面体场中 在似 最大重叠
配体场理论
金属轨道 s px、py、pz dxy、dxz、dyz dz2、dx2-y2
σ键作用
对称性符号 a1g (非简并)
1u
分子轨道类型 σ σ 和π π σ
(三重简并)
t2g (三重简并) eg (二重简并)
配位体的哪些轨道能用于形成σ键?
简单直观的方法: 金属离子价轨道的对称性 (形状)→ 可以与之重叠的配体轨道!
cfse6043p24cfse304cfse6043p123p影响cfse的因素电子数目配位体的强弱晶体场的类型晶体场理论的应用颜色磁性稳定性配合物离子的颜色所吸收光子的频率与分裂能大小有关颜色的深浅与跃迁电子数目有关此类跃迁为自旋禁阻跃迁因此配离子颜色均较浅许多过渡金属配合物的颜色产生于d电子在晶体场分裂而得的两组d轨道之间的跃迁即通常所谓的dd跃迁的吸收光谱图水溶液中的ti离子以ti形式存在晶体场分裂能等于20300与其对应的波长为500nm左右相应于可见光的绿色波段
两组轨道的能量与八面体场中正好相反。其能量差用符号 △T表示: △T = E(t2g) - E(eg)
1. 轨道分裂
在球型场中
平面正方形体场
在平面四边形场中
dx2–y2
dx2–y2
dz2
Δ
配位化合物的价键理论 配合物的晶体场理论

.配位化合物的价键理论配合物的晶体场理论一.配合物的构型与中心的杂化方式二中心杂化轨道的形成1. ns np nd 杂化1 个 4s 空轨道,3 个 4p 空轨道和2 个 4d 空轨道形成 sp3d2杂化轨道,正八面体分布。
6 个F-的 6 对孤对电子配入sp3d2空轨道中,形成正八面体构型的配合单元。
例 2 Ni(CO)4的成键情况在配体 CO 的作用下,Ni 的价层电子重排成 3d104s0形成 sp3杂化轨道,正四面体分布,4 个CO 配体与 sp3杂化轨道成配键,形成的 Ni(CO)4构型为正四面体。
例 1 和例 2 的相同点是,配体的孤对电子配入中心的外层空轨道, 即 ns np nd 杂化轨道, 形成的配合物称外轨型配合物. 所成的键称为电价配键. 电价配键不是很强.例 1 和例 2 的不同点是,CO 配体使中心的价电子发生重排,这样的配体称为强配体。
常见的强配体有 CO、 CN-、NO2-等;例1 中 F-不能使中心的价电子重排,称为弱配体。
常见的弱配体有 F-、Cl-、H2O 等。
而 NH3等则为中等强度配体。
对于不同的中心,相同的配体其强度也是不同的。
2. (n-1) d ns np 杂化例 3 讨论的成键情况形成 d2sp3杂化,使用 2 个 3d 轨道, 1 个 4s 轨道,3个4p 轨道。
用的内层 d 轨道。
形成的配离子为正八面体构型。
空出 1 个内层 d 轨道,形成 dsp2杂化轨道,呈正方形分布。
故构型为正方形。
例 3 和例 4 中,杂化轨道均用到了 ( n - 1 ) d 内层轨道,配体的孤对电子进入内层,能量低,称为内轨配合物,较外轨配合物稳定。
所成的配位键称为共价配键。
三价键理论中的能量问题内轨配合物稳定,说明其键能 E内大,大于外轨的 E外,那么怎样解释有时要形成外轨配合物呢?其能量因素如何?上面的例题中我们看到,形成内轨配合物时发生电子重排,使原来平行自旋的 d 电子进入成对状态,违反洪特规则,能量升高。
无机化学-配位化学基础-配合物的化学键理论

解得: Et2 = + 1.78 Dq Ee = - 2.67 Dq
dxy ,dxz 和 dyz 轨道(即t 轨道) d x2-y2和 d z2轨道(即e 轨道)
( 3 ) 正方形场
sq = 17.42 Dq
四面体、八面体或正方形场中,中心金属离子5个d 轨道的能级分裂
t = 4.45 Dq
sq = 17.42 Dq
中心离子
电荷↑,半径↑, △ ↑
同一几何构型配合物的 △ : 八面体场△o
第二过渡系列中心离子 > 第一过渡系列(40 - 50%)
第三过渡系列中心离子 > 第二过渡系列(20 - 25%)
正八面体配合物ML6的△o (cm-1)
1 cm-1 = 1.23977 10-4 eV = 1.19 10-2 kJ.mol-1
电荷迁移跃迁: KMnO4 , K2CrO4 , HgO 等
(中心离子为d 0 或d 10的化合物)
互相极化 e(荷移跃迁) Mn+ ——— O2- ———→ Mn+
h
→ 显示互补色
E
hν e
O2-
1951年,几位化学家用CFT解释了 [Ti(H2O)6]3+的吸收光谱,应用于配合物,迅 速发展。
9.3.2.1 要点
1. 静电模型:配合物中Mn+ - L纯粹是静电作用,均
为点电荷,L是阴离子成偶极分子.
2. d 轨道能量分裂:
中心离子的d 轨道的能量在非球形对称的配位体形成
的晶体场中都升高,且发生分裂,分离能为 △ :
d4 – d7 构型中心离子在 八面体强场和弱场中d电子的排布
弱 场 ( △o < P )
d4
晶体场理论

价键理论的优点和局限性
价键理论的优点:
★ 能简明解释配位化合物几何构型和磁性等性质; ★ 可以解释[Co(CN)6]4-存在高能态电子,非常容易被 氧化,是很强的还原剂,能把水中的H+还原为H2。
价键理论的不足:
★ 无法解释[Cu(NH3)4]2+稳定的事实 ★ 对配合物产生高低自旋的解释过于牵强. ★ 无法解释配离子的稳定性与中心离子电子构
在自由的过渡金属离子中,d轨道是五重简并的,但五个d轨 道的空间取向不同 。所以在具有不同对称性的配位体静电场的 作用下,将受到不同的影响, 使原来简并的5个d轨道产生能级 分裂。
(三)正八面体场 中d轨道的能级分 裂
八面体配位离子中,6个配位体沿± x,± y,± z 坐标接近M,
L的负电荷对 dZ2 、d x2y2 道的电子排斥作用大,使这两轨道能级上 升较多,而夹在两坐标之间 d xy、d xz、d yz受到推斥较小,能级上升较
提供的孤对电子,形成相应数目的配位键。配位键的 数目就是中心离子的配位数。
配位键的类型及配合物的空间构型
配位离子
3d
Fe(CN)64-
C+ o(NH3)63
Co(CN)64Ni(CN)42-
3-
4s 4p 5s
杂化轨道 几何形状
d2sp3 d2sp3 d2sp3 dsp2
—
八面体 八面体 八面体 平面四方 八面体
• 实验证明,对于第一过渡系金属离子的四面体配合物,因Δt = (4/9)Δo , 即Δ 较小,常常不易超过Ep,尚未发现低自旋配合物。
(3) 高自旋态即是Δ较小的弱场排列,不够稳定,未成对电子多而磁矩高, 具顺磁性。低自旋态即是Δ较大的强场排列,较稳定,未成对电子少而磁 矩低。
第四章 配位化学-2

要点:
(1)中心离子(或原子)空轨道杂化,配体提 供孤对电子。
(2)配位键的本质为共价键。
(3)配合物(配离子)的空间构型取决于中心 离子的杂化方式。
2020/8/16
精品课件
3
杂化轨道形式与配合物的空间构型
配 空间构型 杂化轨
位
道类型
数
实例
2 直线形 3 平面三角形 4 正四面体 4 四方形 5 三角双锥 5 四方锥 6 八面体
故可用磁矩的降低判断内轨型配合物的生成
2020/8/16
精品课件
21
例:Co(NH3)63+的μ =0 B.M,判断该配
离子的空间构型,中心离子所采用的杂化 轨道和内、外轨类型
解:
Co3+: 3d6
μ =0 B.M, 推出n=0ቤተ መጻሕፍቲ ባይዱ即单电子数为零,
如NiCl42-(正四面体),Ni(CN)42-(平面正方形)。
sp3杂化
dsp2杂化
2020/8/16
精品课件
7
4) 配位数为5(较少见):呈三角双锥和四方锥
如Ni(CN)53- (四方锥),[CuCl5]3- (三角双 锥)。
3-
N
2020/8/16
N C
C N
C
N
C
Ni
C N
精品课件
dsp3杂化
外轨型
配位键的键能:内轨型 > 外轨型
配合物的稳定性:内轨型 > 外轨型 稳定常数: 内轨型 > 外轨型
2020/8/16
精品课件
19
2)配合物的磁性(价键理论的实验依据) 磁性:磁矩(磁天平测出)
n(n2) B.M.
第四章(本1) 价键理论、晶体场理论

第三章配合物的化学键理论目标:解释性质,如磁学性质、光谱等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-二、对配合物磁性的解释1)配合物磁性与配合物中成单电子数的关系配合物的分子磁矩μ与配合物中未成对电子数n 有关。
如:对某些配合物:µ=[n(n+2)]1/2 B.M.1B.M. = 9.27×10-21erg·G-12)实验发现:如:K4[Fe(CN)6] µ=0.00 B.M.FeSO4.7H2Oµ=5.10 B.M.②内、外轨型配合物磁性③继承了传统的价键概念(配位共价键),简明易于理解。
2)不足A. 定量程度差。
B. 无法说明Cu2+平面正方形内轨型配合物的稳定性如[Cu(NH3)4]2+:要点:①把配体视为点电荷或偶极子(不考虑其结构);②配体与中心离子间的作用是纯静电相互作用,不存在电子交换,即不形成任何共价键。
二、d轨道能级分裂(单电子能级的分裂)1、定义:由于d轨道空间取向不同,与非球形对称静电场的作用则不相同,引起d轨道能级发生分裂。
Δt = E t2 - E e = =4/9Δo 40/9Dq---------(1)同理,若选Es为能量零点,则3E t2+2E e=5E s=0---------(2)联立(1)和(2),解出:E t2=1.78Dq,E e=-2.67D q三、d电子排布及配合物磁性4、四面体配合物由于Δt=4/9Δo, 大多为弱场高自旋排布。
第四章 配位化合物的理论

Co 3d74s2: Co3+ 3d6: 在配位后, CoF63-:
6F-
sp3d2 在CoF63-中, 杂化轨道的类型为sp3d2, 配离子有4个单电子, 显 顺磁性, 为外轨型配合物(也叫电价配合物)。 6CN- Co(CN)63-: d2sp3 在Co(CN)63-中, Co3+中心离子以d2sp3杂化轨道成键, 配离子 没有成单电子, 显抗磁性, 为内轨型配合物(也叫共价型配合物)。
●当P<△时, 电子成对耗能较少, 此时将取低自旋状态。 根据P和△的相对大小可以对配合物的高、低自旋进行预言: ①在弱场时, 由于△值较小, 配合物将取高自旋构型, 相反, 在 强场时, 由于△值较大, 配合物将取低自旋构型。
②对于四面体配合物, 由于△t=(4/9)△0, 这样小的△t值, 通常 都不能超过成对能值, 所以四面体配合物通常都是高自旋的。
d 轨道的分裂并非纯粹的静电效应,
其中的共价因素也不可忽略。
2. 配合物高低自旋的预言
对于一个处于配位场中的金属离子, 其电子排布究竟采用高自 旋, 还是低自旋的状态, 可以根据成对能和分裂能的相对大小来进 行判断: ●当P>△时, 因电子成对需要的能量高, 电子将尽量以单电子 排布分占不同的轨道, 取高自旋状态;
d 轨道能级在不同配位场中的分裂
4 平面正方形场 设四个配体只在x、y平面上沿±x和±y 轴方向趋近于中心原 子, 因dx2-y2轨道的极大值正好处于与配体迎头相撞的位置, 受排斥 作用最强, 能级升高最多。其次是在xy平面上的dxy轨道。而dz2仅 轨道的环形部分在xy平面上, 受配体排斥作用稍小, 能量稍低, 简并 的dxz、dyz的极大值与xy平面成45°角, 受配体排斥作用最弱, 能量 最低。总之, 5条d轨道在Sq场中分裂为四组, 由高到低的顺序是: ①dx2-y2, ②dxy, ③dz2, ④dxz和dyz。 5. Jahn-Teller效应 非直线形分子的简并轨道的不对称占据会导致分子畸变,结果 降低了分子的对称性和轨道的简并度,使体系能量进一步降低, 这一现象叫做Jahn-Teller效应。
第4章 配合物的晶体场理论及配合物的光谱-4h
价键理论
价键理论的要点:
(1)形成配位键的必要条件是:配体L至少含有一对孤对电
子对,而中心体M必须有空的价轨道;
(2)在形成配合物(或配离子)时,中心体所提供的空轨道 (s、p,d、s、p或s、p、d)必须首先进行杂化,形成能量相同 的与配位原子数目相等的新的杂化轨道; (3)配合物的中心体 M与配体 L之间的结合,一般是靠配体 单方面提供孤对电子对与 M共用,形成配键 M ←∶L,这种 键的本质是共价性质的,称为σ配键。
4.1 晶体场理论(CFT)
由计算可知, CFSE = 2× (-10Dq+P)
故实际采取哪种方式应取决于 10Dq( 轨道分裂能 ) 与 P( 电子成 对能 ) 的相对大小。如果 10Dq > P ,则以低自旋方式存在,反 之则以高自旋存在。 把晶体场足够强,以致于使得10Dq>P的配体称为强场配体,
由于各轨道总能量保持不变,轨道能量的升高总值必然等于轨道能量下降的 总值。这就是所谓的重心守恒原理。 原简并的轨道在外电场作用下如果发生分裂,则分裂后所有轨道能量改变值 的代数和为0。
2d 3d 0 d d 10Dq 0
3 d 0 6Dq 5
2 d 0 4Dq 5
dxy dxz dyz
t2 2 5
t t=
球形对称静电场 自由离子 简并轨道
dz2 dx2-y2
3 5
4 9
0
t
e
相对于八面体 而言,四面体 场中的排斥作 用较小。
在四面体场中 d轨道能级分裂
4.1 晶体场理论(CFT)
三、平面正方形场
设四个配体分别沿x和y轴正、负方向趋近中心离子。因dx2-y2轨 道极大值正好处于与配体迎头相接的位置,受排斥作用最强, 能级升高最多;其次是xy平面上的dxy轨道。而dz2轨道的环形部 分在xy平面上,受配体排斥作用较小,能量较低; 简并的 dxz 、dyz 的极大值与 xy 平面成 450 角,受斥作用最弱,能 组最低。
配位化学:配合物的晶体场理论和配位场理论

(五) d 轨道在平面正方形场中的分裂
dx2-y2
dxy
dz2
x
y
x
y
dxz, dyz
D4h场
平面正方形场中d轨道能级分裂图
(六) d轨道中电子的排布—高自旋态和低自旋态
1、分裂能Δ和成对能P
(1) 八面体场中d轨道的分裂能
• 高能量的dx2-y2和dz2二重简并轨道,称为dγ能级 • 低能量的dxy、dxz和dyz三重简并轨道,称为dε
Eeg Et2g 10Dq 2Eeg 3Et2g 0
得:
Eeg
6Dq
(或0.6
)
0
Et 2 g
4Dq
(或
0.4
)
0
可见,在八面体场中, d轨道分裂的结果是:与Es能级相比较, eg轨道能量上升了6Dq, t2g轨道能量下降了4Dq。
影响Δ大小的因素: ①配体:中心离子固定,配体构型一定,Δ值大小与配体有关, 大致为:
2、d电子排布
电子在d轨道中的排布与Δ和P相对大小有关。
Δ< P
Δ> P
Ea E0 (E0 ) 2E0 Eb E0 (E0 P) 2E0 P
讨论:
(1) 0 P, Ea Eb , (A)态稳定,弱场时高自旋排布稳定。 (2) 0 P, Ea Eb , (B)态稳定,强场时低自旋排布稳定。
(3) 高自旋态即是Δ较小的弱场排列,不够稳定,未成对电子多而磁矩高, 具顺磁性。低自旋态即是Δ较大的强场排列,较稳定,未成对电子少而磁 矩低。
(4) 对比稳定性时,高自旋与外轨型,低自旋与内轨型似有对应关系,但二 者是有区别的。高、低自旋是从稳定化能出发的,内、外轨型是从内外 层轨道的能量不同出发的。