IPTG诱导蛋白质表达实验步骤总结
维真生物-IPTG诱导蛋白表达的实验方法

IPTG诱导蛋白表达的实验方法实验原理:E.coli的乳糖操纵子(元)含Z、Y及A三个结构基因,分别编码半乳糖苷酶、透酶和乙酰基转移酶,此外还有一个操纵序列O、一个启动序列P及一个调节基因I。
I基因编码一种阻遏蛋白,后者与O序列结合,使操纵子(元)受阻遏而处于关闭状态。
在启动序列P上游还有一个分解(代谢)物基因激活蛋白(CAP)结合位点。
由P序列、O序列和CAP结合位点共同构成lac操纵子的调控区,三个酶的编码基因即由同一调控区调节,实现基因产物的协调表达。
在没有乳糖存在时,lac操纵子(元)处于阻遏状态。
此时,I序列在PI启动序列操纵下表达的Lac阻遏蛋白与O序列结合,阻碍RNA 聚合酶与P序列结合,抑制转录起动。
当有乳糖存在时,lac操纵子(元)即可被诱导。
在这个操纵子(元)体系中,真正的诱导剂并非乳糖本身。
乳糖进入细胞,经β-半乳糖苷酶催化,转变为异乳糖。
后者作为一种诱导剂分子结合阻遏蛋白,使蛋白构象变化,导致阻遏蛋白与O序列解离、发生转录。
异丙基硫代半乳糖苷(IPTG)的作用与异乳糖相同,是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
实验试剂:1、LB(Luria-Bertani)培养基:酵母膏(Yeast extract)5g 、蛋白胨(Peptone) 10g 、NaCl 10g 、琼脂(Agar) 1-2% 、蒸馏水(Distilled water) 1000ml 、pH 7.0。
2、IPTG 贮备液:2 g IPTG溶于10 mL 蒸馏水中,0 . 22 μm 滤膜过滤除菌,分装成1 mL /份,-20 ℃保存。
3、凝胶电泳加样缓冲液:50 mmol / L Tris -CI ( pH 6 . 8 ) 、50 mmol / L DTT、2 % SDS (电泳级)、0.1 %溴酚蓝、10 %甘油。
实验步骤:1、用适当的限制性内切核酸酶消化载体DNA和目的基因;2、按连接步骤连接目的基因和载体,并转化到转化DH5α感受态细胞中;3、分别挑取单菌落接种于5 mL LB ( 0.1 g·L-1 氨苄青霉素)中,37℃培养过夜;4、以2 %体积比转接于2 ml LB( 0.1 g·L-1 氨苄青霉素)中,37℃继续培养2.5 hr,至细菌对数生长期,加0.1 mmol/L IPTG 2 µl诱导3-4 hr,同时设立不加IPTG诱导作为对照;5、取上述菌液1 mL,12000 rpm离心10min,收菌沉淀;6、重悬于冰的100 µl PBS中,加入PMSF至终浓度为10 mmol/L。
IPTG诱导蛋白表达全面介绍

IPTG诱导蛋白表达全面介绍IPTG全面介绍:优点·使用·配制·特点摘要: 异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
IPTG 常用于需要诱导β-半乳糖苷酶活性的克隆实验。
它常与X-Gal 或Bluo-Gal 结合使用,用于重组细菌菌落的蓝白筛选,这些菌落可以诱导lac 操纵子在大肠杆菌中的表达。
IPTG 与lacI 阻遏蛋白结合并改变其构象而发挥作用,防止β-半乳糖苷酶编码基因lacZ 的抑制。
中文名:异丙基-β-D-硫代半乳糖苷( IPTG)英文名称:Isopropyl-beta-D-thiogalactopyranoside用途:异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
IPTG 常用于需要诱导β-半乳糖苷酶活性的克隆实验。
它常与X-Gal 或Bluo-Gal 结合使用,用于重组细菌菌落的蓝白筛选,这些菌落可以诱导lac 操纵子在大肠杆菌中的表达。
IPTG 与lacI 阻遏蛋白结合并改变其构象而发挥作用,防止β-半乳糖苷酶编码基因lacZ 的抑制。
使用方法:首先把IPTG配制成23.83 mg/ml(100 mM)的水溶液,并进行过滤除菌后保存。
然后在100 ml的琼脂培养基中,加入100 μl 的上述溶液、200 μl的X-Gal(20 mg/ml的二甲基甲酰胺(DMF)溶液)和100 μl的Amp(100 mg/ml),制作成IPTG、X-Gal、Amp平板培养基。
当dna 片段插入至pUC系列载体(或其他带有lacZ、Amp基因载体),然后转化至lacZ缺失细胞中后,涂布上述的IPTG、X–Gal、Amp平板培养基,可根据长出菌体的蓝白色,而方便地挑选出基因重组体(白色为具有DNA插入片段的基因重组体)。
配制方法:在8ml蒸馏水中溶解2g IPTG后,用蒸馏水定容至10ml,用0.22μm滤器过滤除菌,分装成1ml小份贮存于-20℃。
IPTG诱导蛋白表达全面介绍

IPTG全面介绍:优点·使用·配制·特点摘要: 异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
IPTG 常用于需要诱导β-半乳糖苷酶活性的克隆实验。
它常与X-Gal 或Bluo-Gal 结合使用,用于重组细菌菌落的蓝白筛选,这些菌落可以诱导lac 操纵子在大肠杆菌中的表达。
IPTG 与lacI 阻遏蛋白结合并改变其构象而发挥作用,防止β-半乳糖苷酶编码基因lacZ 的抑制。
中文名:异丙基-β-D-硫代半乳糖苷( IPTG)英文名称:Isopropyl-beta-D-thiogalactopyranoside用途:异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
IPTG 常用于需要诱导β-半乳糖苷酶活性的克隆实验。
它常与X-Gal 或Bluo-Gal 结合使用,用于重组细菌菌落的蓝白筛选,这些菌落可以诱导lac 操纵子在大肠杆菌中的表达。
IPTG 与lacI 阻遏蛋白结合并改变其构象而发挥作用,防止β-半乳糖苷酶编码基因lacZ 的抑制。
使用方法:首先把IPTG配制成23.83 mg/ml(100 mM)的水溶液,并进行过滤除菌后保存。
然后在100 ml的琼脂培养基中,加入100 μl 的上述溶液、200 μl的X-Gal(20 mg/ml的二甲基甲酰胺(DMF)溶液)和100 μl的Amp(100 mg/ml),制作成IPTG、X-Gal、Amp平板培养基。
当dna 片段插入至pUC系列载体(或其他带有lacZ、Amp基因载体),然后转化至lacZ缺失细胞中后,涂布上述的IPTG、X–Gal、Amp平板培养基,可根据长出菌体的蓝白色,而方便地挑选出基因重组体(白色为具有DNA插入片段的基因重组体)。
配制方法:在8ml蒸馏水中溶解2g IPTG后,用蒸馏水定容至10ml,用0.22μm滤器过滤除菌,分装成1ml小份贮存于-20℃。
蛋白质纯化方法

含组氨酸标签的蛋白的诱导表达及纯化一.用IPTG诱导启动子在大肠杆菌中表达克隆化基因所需特殊试剂:1M IPTG1.将目的基因与IPTG诱导表达载体连接,构成重组质粒并转化相应的表达用的大肠杆菌。
将转化体铺于含相应抗生素的LB平板,37℃培养过夜。
通过酶切序列分析等筛选带有插入片段的转化体。
2.分别挑取对照菌和重组菌1个菌落,接种于1ml含有相应抗生素的LB培养液中,37℃通气培养过夜。
3.取100微升过夜培养物接种于5ml含有相应抗生素的LB培养液中(各10份),适当的温度(20-37℃)震荡培养4小时,至对数中期(A550=0.1-1.0)。
4.对照菌和重组菌各取1ml未经诱导的培养物于离心管中,剩余培养物中加入IPTG至终浓度分别为0.5,1.0,1.5,2.5,3.0,3.5,4.0,4.5,5.0mM相同的温度继续通气培养。
5.在诱导的1,2,3,4,5个小时取1ml样品于Ep管中。
细菌的生长速率严重影响外源蛋白的表达,因此必须对接种菌量,诱导前细菌生长时间和诱导后细菌密度进行控制。
生长过度或过速会加重细菌合成系统的负担,导致包涵体的形成。
生长温度可能是影响大肠杆菌高度表达目的蛋白的最重要因素。
低温培养能在一定程度上抑制包涵体的形成。
IPTG的浓度对表达水平的影响也非常大。
所以通过试验确定最佳的培养条件是很必要的。
6.将所有样本室温最高速度离心1分钟,弃上清,沉淀重悬于100微升1×SDS蛋白上样缓冲液中,100℃加热5分钟,室温最高速度离心1分钟,取15微升样品上样于SDS聚丙烯酰胺凝胶,用SDS-PAGE观察表达产物条带,从而确定优化的培养条件。
二.大量表达靶蛋白1.取保存的重组大肠杆菌菌液150微升接种于30毫升含相应抗生素的LB培养液中,在100毫升锥形瓶中,300rpm,37℃通气过夜培养。
2.取16毫升过夜培养物接种于800ml含相应抗生素的LB培养液,在2L的锥形瓶中用预实验所确定的最佳诱导温度,300rpm,通气培养4小时,至对数生长中期。
IPTG诱导蛋白表达

IPTG诱导蛋白表达E.coli的乳糖操纵子(元)含Z、Y及A三个结构基因,分别编码半乳糖苷酶、透酶和乙酰基转移酶,此外还有一个操纵序列O、一个启动序列P及一个调节基因I。
I基因编码一种阻遏蛋白,后者与O序列结合,使操纵子(元)受阻遏而处于关闭状态。
在启动序列P上游还有一个分解(代谢)物基因激活蛋白(C AP)结合位点。
由P序列、O序列和CAP结合位点共同构成lac操纵子的调控区,三个酶的编码基因即由同一调控区调节,实现基因产物的协调表达。
在没有乳糖存在时,lac操纵子(元)处于阻遏状态。
此时,I序列在PI启动序列操纵下表达的Lac阻遏蛋白与O序列结合,阻碍RNA聚合酶与P序列结合,抑制转录起动。
当有乳糖存在时,lac操纵子(元)即可被诱导。
在这个操纵子(元)体系中,真正的诱导剂并非乳糖本身。
乳糖进入细胞,经b-半乳糖苷酶催化,转变为半乳糖。
后者作为一种诱导剂分子结合阻遏蛋白,使蛋白构象变化,导致阻遏蛋白与O序列解离、发生转录。
异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定,因此被实验室广泛应用。
材料1、诱导表达材料( 1 ) LB (Luria—Bertani))培养基酵母膏 (Yeast extract) 5g 蛋白胨 (Peptone) 10gNaCl 10g 琼脂 (Agar) 1-2%蒸馏水 (Distilled water) 1000ml pH 7.0适用范围:大肠杆菌( 2 ) IPTG 贮备液:2 g IPTG溶于10 mL 蒸馏水中,0 . 22 μm 滤膜过滤除菌,分装成1 mL /份,-20 ℃ 保存。
( 3 ) l× 凝胶电泳加样缓冲液:50 mmol / L Tris -CI ( pH 6 . 8 )50 mmol / L DTT2 % SDS (电泳级)0.1 %溴酚蓝10 %甘油2、大肠杆菌包涵体的分离与蛋白纯化材料1 )酶溶法(1)裂解缓冲液:50 mmol / L Tris-CI ( pH 8 . 0 )1 mmol / L EDTA100 mmol / LNaCI(2)50 mmol / L 苯甲基磺酰氟(PMSF )。
IPTG诱导的外源蛋白表达

1. 课题起源:
试验环节:
接种 扩大培养 IPTG诱导 提取、纯化
蛋白
思索:
IPTG(异丙基硫代半乳糖苷)旳作用原理?
2. IPTG旳作用原理
IPTG (isopropylthiog- alactoside,异丙基硫代 半乳糖苷): 化学合成旳乳糖类似物,很强旳β -半乳糖苷酶诱导剂,诱导乳糖操纵元(lac operon)旳开放,本身不被催化分解。类似旳 X-gal(5-溴-4-氯-3-吲哚-β-半乳糖苷)也是一种 人工化学合成旳半乳糖苷,可被β-半乳糖苷 酶水解产生兰色化合物,所以能够用作 β-半 乳糖苷酶活性旳指示剂。IPTG和X-gal都被广 泛应用在分子生物学和基因工程旳工作中。
2.3 乳糖操纵元(lac operon)旳正调控
不难看出:CAP结合位点就是一种起正性调控 作用旳操纵子,CAP则是对转录起正性作用旳 调控蛋白──激活蛋白,编码CRP旳基因也是 一种调控基因,但是它并不在lac操纵元旳附 近,CAP能够对几种操纵元都起作用。
从上所述,乳糖操纵元属于可诱导操纵元 (inducible operon),此类操纵元一般是关 闭旳,当受效应物作用后诱导开放转录。此类 操纵元使细菌能适应环境旳变化,最有效地利 用环境能提供旳能源底物。
2.2 乳糖操纵元(lac operon)旳负调控
在这过程中乳糖(实际起作用旳是 别乳糖)就是诱导剂,与R结合起到去 阻遏作用(derepression),诱导了利 用乳糖旳酶类基因转录开放
2.3 乳糖操纵元(lac operon)旳正调控
细菌中旳cAMP含量与葡萄糖旳分解 代谢有关,当细菌利用葡萄糖分解产 生能量时,cAMP生成少而分解多, cAMP含量低;相反,当环境中无葡 萄糖可供利用时,cAMP含量就升高。
实验 蛋白质的诱导表达

一般融合载体图解
阅读框架的保证
原核表达使用的宿主菌
一般可使用的菌株有: JM109, BL21(DE3), Y1090等。
本实验使用的菌株为:BL21(DE3溶原菌) 噬菌体DE3 : 21抗性区,
LacI基因, LacUV5启动子 T7 RNA聚合酶基因
T7 RNA聚合酶:提高T7启动子启动 的蛋白质的表达水平
稀有密码Biblioteka :多数氨基酸都有一个以上的密码子, 但有一些 E.Coli很少 使用, 当异源目的基因的mRNA过表达时, tRNA 的数量直接 反应密码子的偏倚性, 一个或多个tRNA的稀有或缺少会导 致翻译的停止
毒性基因和质粒稳定性:
氨苄青霉素的使用 补充葡萄糖
提高表达水平的措施
提高翻译水平: 调整SD序列与AUG间的距离、点突变改变碱基、增 加mRNA稳定性 减轻细胞的代谢负荷,提高表达水平: 细菌的生长和外源基因的诱导表达分开。 化学诱导,温度诱导。 提高蛋白质稳定性,防止降解。 表达融合蛋白,表达分泌蛋白(防止降解,减轻代 谢负荷、恢复天然构象)
蛋白表达系统操作流程
主要步骤
制备表达载体
操作
1. 酶切, 去磷酸化
2. 胶回收纯化
制备目的DNA 1. 质粒纯化/PCR 2. 酶切 3. 胶回收纯化 将目的DNA 克隆到载体上 1. 目的片断与载体连接
2. 转化非表达型宿主菌
3. 筛选阳性克隆: 菌落PCR, 质粒 提取酶,测序确定阅读框
转化表达宿主菌
实验
蛋白质的诱导表达
背景知识介绍
基因工程的最终目的:外源基因的高效 表达,获得有重要价值的蛋白质产品 蛋白质的表达是指为获取大量的目标蛋 白而进行的有目的性的蛋白质合成 需将编码所需蛋白的基因插入质粒或其 他载体,然后导入活细胞。 包括外源基因的克隆、转录、翻译、加 工、分离纯化
IPTG-诱导表达
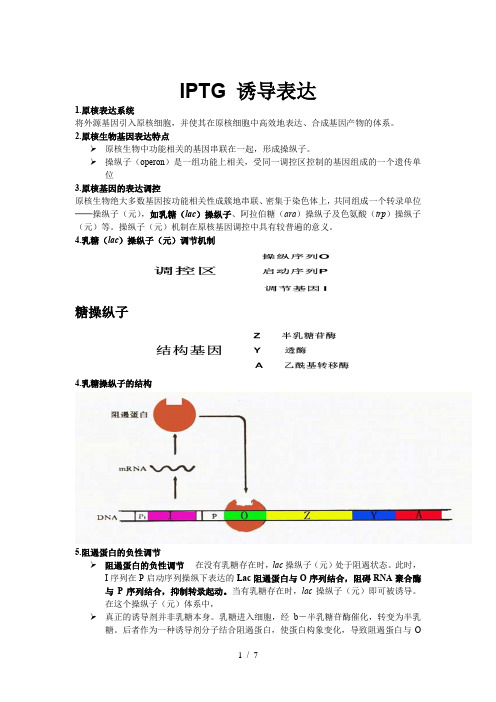
IPTG 诱导表达1.原核表达系统将外源基因引入原核细胞,并使其在原核细胞中高效地表达、合成基因产物的体系。
2.原核生物基因表达特点➢原核生物中功能相关的基因串联在一起,形成操纵子。
➢操纵子(operon)是一组功能上相关,受同一调控区控制的基因组成的一个遗传单位3.原核基因的表达调控原核生物绝大多数基因按功能相关性成簇地串联、密集于染色体上,共同组成一个转录单位──操纵子(元),如乳糖(lac)操纵子、阿拉伯糖(ara)操纵子及色氨酸(trp)操纵子(元)等。
操纵子(元)机制在原核基因调控中具有较普遍的意义。
4.乳糖(lac)操纵子(元)调节机制糖操纵子4.乳糖操纵子的结构5.阻遏蛋白的负性调节➢阻遏蛋白的负性调节在没有乳糖存在时,lac操纵子(元)处于阻遏状态。
此时,I序列在P启动序列操纵下表达的Lac阻遏蛋白与O序列结合,阻碍RNA聚合酶与P序列结合,抑制转录起动。
当有乳糖存在时,lac操纵子(元)即可被诱导。
在这个操纵子(元)体系中,➢真正的诱导剂并非乳糖本身。
乳糖进入细胞,经b-半乳糖苷酶催化,转变为半乳糖。
后者作为一种诱导剂分子结合阻遏蛋白,使蛋白构象变化,导致阻遏蛋白与O序列解离、发生转录。
➢异丙基硫代半乳糖苷(IPTG)是一种作用极强的诱导剂,不被细菌代谢而十分稳定.6.乳糖操纵子的结构及阻遏作用7.外源基因在原核表达系统中表达的必要条件1.删除内含子和5’非编码区2.外源基因置于强启动子和SD顺序控制下3.维持正确开放阅读框架(ORF)4.mRNA稳定且可有效转译,形成的蛋白质不被降解8.影响外源基因在原核细胞中表达效率的因素1、启动子:建立表达载体时,选择强启动子2、基因剂量3、核糖体结合位点9.常见原核强启动子•Plac:受Lac阻遏蛋白负调,受IPTG的诱导•Ptrp:取自大肠杆菌色氨酸操纵子。
•Ptac :Lac启动子和Trp启动子的杂合启动子。
•P L和P R启动子:噬菌体早期左/右向启动子,受λ噬菌体CI基因负调控。
IPTG诱导表达修订版

周三下午提质粒电泳之后
1 把昨天做好的转化转到有盖的试管中,每个菌准备三个有盖的试管,做好标记,每个试管加入3ml液体LB培养基,3ul 50mg/ml的氨苄青霉素,至50ug/ml,从培养皿上挑一个比较圆的单菌进试管,盖上盖子,放上摇床,250rpm,37℃摇菌,12h。
周四早上
2 把上面做好的试管培养一晚取出,重新再做第一步,即每个试管再准备一个新的有盖试管,在该试管加入3mlLB培养基,3ul 50mg/ml的氨苄,取50ul第一步的菌液接到新试管中盖上盖子,标记,放上摇床,250rpm,37℃摇菌,OD600=0.8~1.2(1.0)
周四中午
3 往菌液中各加入IPTG液,终浓度为1mM,留一管做未诱导对照,全部放在22℃,250rpm,摇菌,OD600= 1.5~1.8 4h后开始测OD
4 诱导后拿出来,将菌液全部离心至1.5mlEP管中,4000rpm,10min,去上清,沉淀-20℃保存。
周六 5 向沉淀中加入400ul破菌液,超声波破碎(3s,3s,)7~8次
6 将上一步菌液加40ul到1全,2全,3全的EP管中,然后把2,3到4℃,12000转,10min 离心,把离心后的上清40ul加入到2上,3上EP管中。
7 把1全,2全,3全,2上,3上的EP管加入8ul上样缓冲液,沸水浴5min。
8 L0027,V100,F0043跑蛋白胶,点样10ul。
IPTG 诱导表达

阻遏蛋白的负性调节 真正的诱导剂是半乳糖 异丙基硫代半乳糖苷(IPTG)
pGEX-4T载体图谱
IPTG诱导表达实验步骤
第一天 在超净台中将含有PGEX-4T-2空载体的甘油菌 5ul接种在5ml的LB培养液中,摇床过夜。 第二天 • 将含有PGEX-4T-2空载体的过夜菌按照1:200比 例接种至100mlLB中, 37℃振荡培养2.5小时。 • 留取1.5ml菌液作为未诱导培养的对照。 • 在超净台中加入终浓度为0.4-1mM的IPTG于30℃ 诱导表达3.5小时。
蛋白样品制备
取1.5ml诱导后菌液及预先准备的未诱导培 养的菌液离心,将沉淀部分加入相同体积的蒸 馏水与上样buffer,将蛋白样品于沸水中加热10 分钟,离心取上清进行SDS-PAGE,检测融合蛋 白的表达情况。
RNA聚合酶保护区 结构基因
5 3 5
-50 -40 -30 -20 -10 1 10
3 5 3 5 开始转录 -10 区 T A T A A T Pu A T A T T A Py (Pribnow box)
3 -35 区 TTGACA AA C T G T RNA-pol辨认位点 (recognition site)
原核生物启动子保守序列
-35区 trp
tRNATyr
IPTG 诱导表达
原核基因的表达调控
原核生物绝大多数基因按功能相关性成簇地 串联、密集于染色体上,共同组成一个转录单位 ──操纵子,如乳糖(lac)操纵子、阿拉伯糖 (ara)操纵子及色氨酸操纵子。操纵子机制在原 核基因调控中具有较普遍的意义。
原核生物的转录单位是操纵子,操纵子包括 相关结构基因及其上游的调控序列。
调控序列
结构基因
实验五 用IPTG诱导大肠杆菌表达重组绿色荧光蛋白

实验五用IPTG诱导大肠杆菌表达重组绿色荧光蛋白一、目的要求1.学习和掌握重组蛋白在大肠杆菌中表达的原理和实验技术;2.观察大肠杆菌在诱导过程中的形态。
二、原理pET 系统是有史以来在E.coli 中克隆表达重组蛋白的功能最强大的系统。
目的基因被克隆到pET 质粒载体上,受噬菌体T7强转录及翻译(可选择)信号控制;表达由宿主细胞提供的T7 RNA 聚合酶诱导。
T7 RNA 聚合酶机制十分有效并具选择性:充分诱导时,几乎所有的细胞资源都用于表达目的蛋白;诱导表达后仅几个小时,目的蛋白通常可以占到细胞总蛋白的50%以上。
尽管该系统极为强大,却仍能很容易地通过降低诱导物的浓度来削弱蛋白表达。
降低表达水平可能可以提高某些目的蛋白的可溶部分产量。
该系统的另一个重要优点是在非诱导条件下,可以使目的基因完全处于沉默状态而不转录。
用不含T7 RNA聚合酶的宿主菌克隆目的基因,即可避免因目的蛋白对宿主细胞的可能毒性造成的质粒不稳定 (详见I. F.部分)。
如果用非表达型宿主细胞克隆,可以通过两种方法启动目的蛋白的表达:用带有受λpL 和 pI 启动子控制的T7 RNA 聚合酶的λCE6噬菌体侵染宿主细胞,或者将质粒转入带有受lacUV5 控制的T7 RNA聚合酶基因的表达型细胞。
在第二种情形下,可以通过在细菌培养基中加入IPTG 来启动表达。
尽管有时(例如非毒性目的蛋白) 可以直接将目的基因克隆到表达型宿主细胞中,但这种策略并不是通用做法。
两种T7 启动子以及多种拥有不同抑制本底表达水平的宿主细胞共同构成了一个极为灵活而有效的系统,使各种目的蛋白得以最优化表达。
三、试剂与器材试剂:LB培养基、IPTG、氨苄青霉素、草酸铵结晶紫染色液,路哥氏(Lugol)碘液,95%乙醇,0.5%沙黄染色液。
器材:摇床、显微镜等。
四、操作步骤A、诱导许多研究表明细胞生长速率严重影响外源蛋白的表达,因此必须对接种菌量、诱导前细胞生长时间和诱导后细胞密度进行控制。
IPTG诱导表达步骤

大肠杆菌中蛋白质诱导表达
(一)材料
1. IPTG(用去离子水配成1 00mM,用0.22μm滤器过滤除菌,-20℃保存)
2. 10xPBS缓冲液: NaCl 1.37 M
KCl 27 mM
Na2HPO4 100 mM
KH2PO420 mM
3. PMSF(用异丙醇配成10mM,-20℃保存)
4. 10% TritonX-100
(二)方法
1. 挑取单克隆接种于少量含有抗生素的LB液体培养基中,37℃, 250rpm震荡培养过夜;
2. 将过夜培养物按1:100的比例接种到新鲜的含有抗生素的LB液体培养基中,37℃,
250rpm震荡培养2-4h至OD600为0.5;
3. 加入IPTG至终浓度为0.3mM,继续37℃, 250rpm震荡培养2-3h;
4. 4℃, 4000rpm离心10min后,去上清,加入适量IxPBS重悬,在加入PMSF至终浓度为
0.1 mM,冰上预冷10min;
5. 进行冰浴超声破碎5-10min (10-20sec/次),至菌液较为澄清;
6. 加入10% TritonX-100至终浓度为0.5%, 4 ℃震荡15min;
7. 4℃, 12000rpm离心10min后,取上清:
8. 用相同体积的1 xPBS重悬沉淀,将诱导前及诱导后所取的样品,以及超声破碎后的上清
和沉淀分别进行蛋白电泳检测,考马斯亮蓝染色,脱色:
9. 根据脱色结果,可确定是否表达出目的蛋白,并且判断所表达的蛋白是可溶性还是以包
涵体形式存在。
诱导纯化蛋白实验报告

一、实验名称:诱导纯化蛋白实验二、实验目的:1. 掌握蛋白质的诱导表达方法;2. 学习蛋白质的纯化技术;3. 了解蛋白质纯度的鉴定方法。
三、实验原理:蛋白质的诱导表达是指在合适的诱导剂作用下,使蛋白质在细胞内大量合成。
本实验采用IPTG(异丙基-β-D-硫代半乳糖苷)作为诱导剂,诱导表达目的蛋白。
蛋白质的纯化主要包括亲和纯化、离子交换层析、凝胶过滤层析等方法。
本实验采用亲和纯化法,利用目的蛋白与特定配体的特异性结合,实现蛋白质的纯化。
蛋白质纯度的鉴定主要通过SDS-PAGE(十二烷基硫酸钠-聚丙烯酰胺凝胶电泳)和Western Blot(蛋白质印迹)等方法。
四、实验材料与仪器:1. 材料:(1)大肠杆菌菌株:表达载体pET-28a+;(2)IPTG(异丙基-β-D-硫代半乳糖苷);(3)诱导剂;(4)蛋白质纯化柱;(5)SDS-PAGE试剂;(6)Western Blot试剂;(7)其他试剂。
2. 仪器:(1)电泳仪;(2)凝胶成像系统;(3)Western Blot成像系统;(4)离心机;(5)恒温水浴锅;(6)其他实验仪器。
五、实验步骤:1. 转化:将pET-28a+表达载体转化到大肠杆菌菌株中,筛选阳性克隆。
2. 培养与诱导:将阳性克隆接种到LB液体培养基中,37℃、200 rpm培养过夜。
按1:100的比例将过夜培养物接种到新鲜的LB液体培养基中,37℃、200 rpm培养至OD600=0.6时,加入IPTG诱导表达目的蛋白。
3. 收集细胞:诱导表达6小时后,离心收集细胞,用磷酸盐缓冲液(PBS)洗涤细胞两次。
4. 蛋白质裂解:将细胞重悬于裂解缓冲液中,冰浴30分钟,超声破碎细胞。
5. 亲和纯化:将裂解液上样到蛋白质纯化柱,收集流出液和结合液。
用洗脱缓冲液洗脱结合蛋白,收集洗脱液。
6. SDS-PAGE:取适量洗脱液进行SDS-PAGE电泳,观察目的蛋白的纯度。
7. Western Blot:将SDS-PAGE电泳后的凝胶进行转膜,用一抗和二抗进行Western Blot检测,观察目的蛋白的表达。
蛋白表达纯化实验步骤

蛋白表达纯化实验步骤(待改良)1、取适当相应蛋白高表达的动物组织提total-RNA。
2、设计蛋白表达引物。
引物要去除信号肽,要加上适当的酶切位点和保护碱基。
3、RT-PCR,KOD酶扩增获取目的基因c DNA.4、双酶切,将cDNA.克隆入PET28/32等表达载体。
5、转化到DH5α感受态细菌中扩增,提质粒。
6、将质粒转化入表达菌株,挑菌检测并保种。
表达菌株如Bl21(DE3)、Rosetta gami(DE3)、Bl21 codon(DE3)等。
7、蛋白的诱导表达。
1)将表达菌株在3ml LB培养基中摇至OD=0.6左右,参加IPTG,浓度梯度从25μM到1m M。
37度诱导过夜(一般3h以上即有大量表达)。
2)SDS-PAGE电泳检测目的蛋白的表达。
注:目的蛋白包涵体表达量一般会到达菌体蛋白的50%以上,在胶上可以看到明显的粗大的条带。
3)将有表达的菌株10%甘油保种,保存1ml左右就足够了,并记录IPTG浓度范围。
甘油是用0.22μm过滤除菌的,储存浓度一般是30%-60%,使用时自己计算用量。
4)用上述IPTG浓度范围的最低值诱导10ml表达菌,18度,低转速(140-180rpm),诱导过夜作为包涵体检测样品。
注意:1.如果表达的蛋白对菌体有毒性,可以在加IPTG之前的培养基中参加1%的葡萄糖用来抑制本底表达。
葡萄糖会随着细菌的繁殖消耗殆尽,不会影响后面的表达。
2.保种可以取一局部分成50μl一管,每次用一管,防止反复冻融。
8、包涵体检测。
方案见附件29、如有上清表达,则扩大摇菌。
1)取保种的表达菌株先摇10ml,37度,300rpm摇至OD>=1.5,约5h左右,视菌种的活性而异,也可过夜摇菌。
2)将上一步中的8ml参加300ml培养基中37度,250rpm摇至OD= 1.0左右(约2.5h~3h),然后加IPTG(浓度同包涵体检测中使用的浓度。
)注:菌液浓度要适当的浓一些,否则第二天收集不到足够的菌体,因为低温低转速细菌生长非常缓慢。
7.29IPTG诱导蛋白表达

7.29IPTG诱导蛋白表达
IPTG诱导蛋白表达
1.将两个工程菌的种子液取1ml转接至50ml的离心管中,37度揺菌2到3小时,至OD 值为0.6,每个菌做3个重复。
2.将转接后的两种菌液IPTG诱导表达,分别设置3个浓度梯度,GLCNASE(周质空间表达蛋白)的浓度梯度分别为0.01mM(每10ml 菌液加1M的IPTG0.1μL),0.05mM(每10ml菌液加1M的IPTG0.5μL),0.1mM(每10ml菌液加1M的IPTG1μL)。
GLCNACASE (胞内表达蛋白)的浓度梯度分别为0.1 mM(每10ml菌液加1M的IPTG1μL),0.5mM(每10ml菌液加1M的IPTG5μL),1mM(每10ml菌液加1M的IPTG10μL).
3.每管加10mlLB培养基和10μl浓度为100mg/ml的AMP,使混合溶液的终浓度为
100μg/ml.GLCNACASE在30度揺菌4小时,6小时,8小时分别取样,GLCNASE分别揺菌2小时4小时6小时分别取样保存。
4.配制SDS_PAGE蛋白电泳用胶,存放在4度冰箱备用。
IPTG诱导方法

ITPG诱导方法1、诱导将BL-pET-FAT/CD36菌液接种到4 mL LA液体培养基中,37 ℃,200 rpm振摇培养过夜。
(IPTG浓度)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,共接8管,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1 M的IPTG,使IPTG 终浓度分别为使IPTG终浓度分别为0,0.01,0.1,0.2,0.5,1,5,10 mM,置37 ℃摇床继续培养4 h,置4 ℃保存备用。
另取不含外源基因片段的质粒pET-32a(+)转化BL21感受态细胞作相同处理,加IPTG诱导4 h作为对照。
样品处理后进行SDS-PAGE分析。
(诱导时间)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,共接8管,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1 M的IPTG,使IPTG 终浓度为0.5 mM,进行诱导表达,分别在诱导0、1、2、3、4、6、8、12 h时,每次取出一个离心管,置4 ℃保存备用。
另取不含外源基因片段的质粒pET-32a(+)转化BL21感受态细胞,加IPTG诱导4 h作为对照。
样品处理后进行SDS-PAGE分析。
2、样品处理(0.5 mM、6h找出最佳IPTG浓度和诱导时间)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1 M的IPTG,使IPTG终浓度为mM,诱导h,10000 rpm离心1 min收集菌体,弃上清。
在细菌沉淀中加入1 mL 1×结合缓冲液,冰浴超声50次,每次4s,间隔9s,超声结束后13000 rpm离心30 min,分离上清和沉淀。
取上清50 µL,加入50 µL 2×SDS上样缓冲液;沉淀加入500 µL 1×SDS上样缓冲液,均在100 ℃水浴加热5 min,用于SDS-PAGE电泳3、SDS-PAGE分析(1)样品的处理取1 mL菌液,10000 rpm离心1 min,弃上清,重悬于100 µL 去离子水,加入100 µL 2×SDS凝胶上样缓冲液,涡旋混匀,100 ℃加热3-5分钟,10000 rpm 离心3 min备用。
IPTG诱导方法

ITPG诱导方法1、诱导将BL-pET-FAT/CD36菌液接种到4 mL LA液体培养基中,37 ℃,200 rpm振摇培养过夜。
(IPTG浓度)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,共接8管,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1M的IPTG,使IPTG 终浓度分别为使IPTG终浓度分别为0,0.01,0.1,0.2,0.5,1,5,10mM,置37 ℃摇床继续培养4 h,置4 ℃保存备用。
另取不含外源基因片段的质粒pET-32a(+)转化BL21感受态细胞作相同处理,加IPTG诱导4 h作为对照。
样品处理后进行SDS-PAGE分析。
(诱导时间)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,共接8管,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1M的IPTG,使IPTG 终浓度为0.5mM,进行诱导表达,分别在诱导0、1、2、3、4、6、8、12 h时,每次取出一个离心管,置4 ℃保存备用。
另取不含外源基因片段的质粒pET-32a(+)转化BL21感受态细胞,加IPTG诱导4 h作为对照。
样品处理后进行SDS-PAGE分析。
2、样品处理(0.5 mM、6h找出最佳IPTG浓度和诱导时间)取100 µL培养过夜的菌液接种到5 mL LA液体培养液中,37 ℃,200 rpm振摇培养至OD600至0.6~0.8时,加入1M的IPTG,使IPTG终浓度为mM,诱导h,10000 rpm离心1 min收集菌体,弃上清。
在细菌沉淀中加入1 mL1×结合缓冲液,冰浴超声50次,每次4s,间隔9s,超声结束后13000 rpm离心30 min,分离上清和沉淀。
取上清50 µL,加入50 µL 2×SDS上样缓冲液;沉淀加入500 µL 1×SDS上样缓冲液,均在100 ℃水浴加热5 min,用于SDS-PAGE电泳3、SDS-PAGE分析(1)样品的处理取1 mL菌液,10000 rpm离心1 min,弃上清,重悬于100 µL 去离子水,加入100 µL 2×SDS凝胶上样缓冲液,涡旋混匀,100 ℃加热3-5分钟,10000 rpm 离心3 min备用。
IPTG诱导表达

原核基ห้องสมุดไป่ตู้的表达调控
原核生物绝大多数基因按功能相关性成簇地 串联、密集于染色体上,共同组成一个转录单位 ──操纵子,如乳糖(lac)操纵子、阿拉伯糖 (ara)操纵子及色氨酸操纵子。操纵子机制在原 核基因调控中具有较普遍的意义。
原核生物的转录单位是操纵子,操纵子包括 相关结构基因及其上游的调控序列。
蛋白样品制备
取1.5ml诱导后菌液及预先准备的未诱导培 养的菌液离心,将沉淀部分加入相同体积的蒸 馏水与上样buffer,将蛋白样品于沸水中加热10 分钟,离心取上清进行SDS-PAGE,检测融合蛋 白的表达情况。
RNA聚合酶保护区 结构基因
5 3 5 3 -35 区 TTGACA AA C T G T RNA-pol辨认位点 (recognition site) 开始转录 -10 区 T A T A A T Pu A T A T T A Py (Pribnow box) 3 5 3 5
调控序列
结构基因
乳糖操纵子(元)调节机制
操纵序列O
调控区
乳糖操纵子 结构基因
启动序列P 调节基因 I Z 半乳糖苷酶 Y A 透酶 乙酰基转移酶
操纵子
I
CAP P O
Z
调控区
P:启动序列 O:操纵序列
Y 结构基因
X
I:调节序列
乳糖(-)
I
诱导物
CAP
P
O
Z
Y
X
乳糖(+)
I
CAP
P
O
Z
Y
X
要点
-50
-40
-30
-20
-10
1
10
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验总结
IPTG诱导表达蛋白
15-11-3
配固体培养基和液体培养基,灭菌。
配Binding Buffer和脱盐液各500ml。
在400ml体积时调节溶液pH值至7.4,定容后转移到试剂瓶中。
溶液使用前要抽滤(滤除溶液内的气泡和不溶性杂质)。
倒板:(5个灭菌的培养皿)
①微波加热溶解固体培养基(每次加热30s,待有沸腾的迹象时,减少加热的时间)。
②超净台操作,以1˸1000的比例(1ml培养基~1μl抗生素)向培养基中加入Kan+抗生素(50mg/ml),摇匀。
③倒板。
④培养皿放置在超净台上凝固,凝固后将培养皿倒着放入培养箱中。
15-11-4
热转化:(将质粒导入大肠杆菌中)
①提前将大肠杆菌(冰上融化)和质粒拿出来融化,混合(冰上操作)后冰上放置30min。
②42℃热水浴1min(超过90s会损伤细菌),取出后立即放在冰上冷却2~3min,加入1mlSOC培养基。
③摇床上摇45min,150rpm,37℃。
(使菌体复苏)
④离心,3000rpm,3min。
⑤涂板:取200μl上述液体,喷到板上,用玻璃涂布棒涂匀,放入培养箱中30min晾干后倒着放置。
(12~16h)
15-11-5
配IPTG(0.2g/ml),用灭菌水,超净台操作。
挑单克隆:(将细菌放入培养液中,以便进行扩大培养和后续实验)
①提前准备好几支装有3ml培养基的试管,每管加入3μl Kan+抗生素(50mg/ml),塞好塞子,试管倾斜,是抗生素溶入培养液中。
(塞子打开前和塞好后都要在酒精灯上过火灭菌)。
②小镊子过火灭菌后,夹取一个枪头,在培养皿上挑一个单个的菌落,轻轻划过。
将带有菌落的枪头放入试管中,塞好塞子
【试管中培养基液澄清】
③将试管放在摇床中培养一晚,37℃,250rpm。
(尽量不超过12h)【试管中培养基液变混浊】
注意:操作尽量在靠近酒精灯的地方进行。
15-11-6 扩大培养、诱导表达
①以(抗生素˸培养基)1˸1000的比例向培养基中加入Kan+抗生素,摇匀后再加入(每100ml培养基)1ml单克隆溶液,摇匀。
②摇床上放置2.5h,37℃,180rpm。
③以48μl IPTG/100ml培养基的比例加入IPTG诱导,移去牛皮纸,恒温摇床上摇6h,30℃,180rpm。
冰上放置一晚(抑制细胞生长速率,有利于蛋白质充分折叠)
15-11-6
离心收集细菌:将培养液倒入50ml离心管中,8000rpm,离心5min,弃上清。
沉淀加水冲洗混匀,再离心,弃上清。
加PBS冲洗混匀,离心,弃上清。
加PBS混匀,将溶液移入PU管中,-20℃保存。
15-11-7
破碎、离心细胞
①4ml细胞液用超声破碎3~4次(探头不能碰到管底和管壁,且离管底1cm左右,4~7ml),至溶液透明。
②将溶液分装到1.5ml离心管中,(10000rpm,4℃,10~15min)离心2~3次至没有白色沉淀。
蛋白质纯化脱盐
①离心后得到的上清液用针筒抽取过膜,先后用镍柱纯化、脱盐柱脱盐(柱子不能干,不能有气泡)。
镍柱:水洗→Binding→加蛋白→Binding→Elution→水洗→乙醇。