第四章羰基化合物的反应及碳负离子
合集下载
04第4章:羰基化合物的反应B2

2014-5-29 15
(2)电子效应 ①诱导效应 当羰基连有吸电子基时,使羰基碳上 的正电性增加,有利于亲核加成的进行; 强吸电子基团
H2O Cl3C
CCl3CHO
H
C
OH OH
Cl Cl C Cl H C H O O H
2014-5-29
形成分子内氢键 产物稳定,平衡向右移动
16
②共轭效应 羰基上连有与其形成共轭体系的基团时, 由于共轭作用可使羰基稳定化,因而亲核加成速 度减慢。 • 使羰基碳正电性加强的因素都有利于反应。
2,4-二硝基苯腙
C=N— NH-C-NH2 (白)
31
H—NH—NH-C-NH2
氨基脲
缩氨脲
第三节:羰基加成反应的立体化学
羰基具有平面结构,Nu可从任何一面进攻羰基碳。在 下列的3中,加成后引入了第二个C*,生成的两个非对映 体的量不相等,既亲核加成具有一定的立体选择性。 ① 当R=R′时,加成产物为同一物。 ② 当R≠R′时,加成产物为外消旋体(Nu从羰基两面进 攻的几率相等)。 ③ 当羰基与手性碳原子相连时,Nu从两面进攻的几率就 不一定相等,加成后引入第二个手性碳原子,生成的 两个非对映体的量也不一定相等。
(H3C)3C
K<<1
13
Nu
R R'
C
O
R R'
C
O Nu
sp2杂化 平面三角型 键角:120° 109°28 ′
sp3杂化 四面体
产物中基团拥挤程度增大。
2014-5-29
R 越大,妨碍Nu:进攻C原子。
14
缓解角张力
O
HCN
OH CN
K=10000
O + HCN
(2)电子效应 ①诱导效应 当羰基连有吸电子基时,使羰基碳上 的正电性增加,有利于亲核加成的进行; 强吸电子基团
H2O Cl3C
CCl3CHO
H
C
OH OH
Cl Cl C Cl H C H O O H
2014-5-29
形成分子内氢键 产物稳定,平衡向右移动
16
②共轭效应 羰基上连有与其形成共轭体系的基团时, 由于共轭作用可使羰基稳定化,因而亲核加成速 度减慢。 • 使羰基碳正电性加强的因素都有利于反应。
2,4-二硝基苯腙
C=N— NH-C-NH2 (白)
31
H—NH—NH-C-NH2
氨基脲
缩氨脲
第三节:羰基加成反应的立体化学
羰基具有平面结构,Nu可从任何一面进攻羰基碳。在 下列的3中,加成后引入了第二个C*,生成的两个非对映 体的量不相等,既亲核加成具有一定的立体选择性。 ① 当R=R′时,加成产物为同一物。 ② 当R≠R′时,加成产物为外消旋体(Nu从羰基两面进 攻的几率相等)。 ③ 当羰基与手性碳原子相连时,Nu从两面进攻的几率就 不一定相等,加成后引入第二个手性碳原子,生成的 两个非对映体的量也不一定相等。
(H3C)3C
K<<1
13
Nu
R R'
C
O
R R'
C
O Nu
sp2杂化 平面三角型 键角:120° 109°28 ′
sp3杂化 四面体
产物中基团拥挤程度增大。
2014-5-29
R 越大,妨碍Nu:进攻C原子。
14
缓解角张力
O
HCN
OH CN
K=10000
O + HCN
第四章:缩合反应(1节)-1

CH3OCH2Cl + ZnCl2 ArH + CH2Cl
H3C
O
CH2Cl
CH3 OZnCl2 + CH2Cl
ZnCl2 ArCH2Cl H3 C
O H ZnCl2
+ H CH3OH + ZnCl2
CH3OZnCl2 + H
芳环上有释电子基,有利于反应进行;吸电子基 芳环上有释电子基,有利于反应进行; 则不利于反应进行。 则不利于反应进行。
4).分子内的醇醛缩合反应 4).分子内的醇醛缩合反应
具有α 具有α活性氢的二羰基化合物在催化剂碱的 作用下,发生分子内的醇醛缩合反应 作用下,发生分子内的醇醛缩合反应 ,生产五 六元环状化合物。 元、六元环状化合物。
O LDA O O O O HO O
H 3O
O
LDA : lithium diisopropylmide(二异丙胺锂) 二异丙胺锂) 二异丙胺锂
CH3 .HCl RCOCH3 + HCHO + HN CH3 H2O CH3 RCOCH2CH2N .HCl CH3
• 含活泼氢原子的化合物有:酮、醛、酸、酯、腈、硝基烷、 含活泼氢原子的化合物有: 硝基烷、 炔、酚及杂环化合物 • 醛可以是:甲醛、三聚甲醛、多聚甲醛及活性大的脂肪醛和 醛可以是:甲醛、三聚甲醛、 芳香醛 • 胺可以是:仲胺、伯胺及氨 胺可以是:仲胺、
芳醛的α 羟烷基化(安息香缩合) 2. 芳醛的α-羟烷基化(安息香缩合)
芳醛在含水乙醇中,以氰化钠为催化剂, 芳醛在含水乙醇中,以氰化钠为催化剂,加热后 发生双分子缩合生成α 羟基酮的反应。 发生双分子缩合生成α-羟基酮的反应。
2 C6H5CHO NaCN / EtOH / H2 O pH 7~8, C6H5C O CHC6 H5 OH 96%
第四章碳负离子型延伸碳链反应

② 尤其适用于醛的α-C烃化,用酸做催化剂,避免Aldol缩合
③ 无多烃化产物,只有单烃化产物
④ 不对称酮进行烃化时,取代产物发生在取代较少的 C上
19
O
N H
1)CH2=CHCH2Br O
N
CH2CH=CH2
2)H2O
O
CH3 +
N H
N
+
CH3
90%
N CH3
10%
20
4.3 酰基化反应
具有活泼亚甲基的化合物(如乙酰乙酸乙酯、丙二酸酯、
15
3. 含—个羰基的化合物和腈的α-碳烷基化
(1)弱酸性活泼亚甲基或甲基化合物有: ① 醛、酮以及羧酸衍生物(含—个羰基的化合物); ② 腈(含—个腈基的化合物)。
16
(2)反应条件与特征:
① 弱酸性的化合物进行碳烷化反应时,常出现如自身缩合、 多烷基化等多种副反应 ; ② 当醛、酮化合物在进行C-烷基化反应时,为了避免酯缩 合反应,应选用很强的碱或位阻较大的碱 ,如:
(2)制备:醛、酮 + 胺缩合
(3)性质
R'2' N C C R2' + R'''-X
R'
R2N C C R ''' X
R
R R'
O R'
H2O
R C C R'''
R'
羰基α-C、β-C烯胺烃化
18
O
(4)影响因素
N
H
N H
N
脱水剂:Na2CO3 、K2CO3时用苯带水
优点:
① 操作简单,原料易得,收率较高
(优选)碳负离子反应ppt讲解

羰基化合物切断:
O
OH
R
O
OH
+ R
O OH R
OM 烯醇负离子
O
O
B
常用碱的性能:
强亲质子和亲核能力的碱:HO- , CH3O-, C2H5O-, RS-, CN-等。
强亲质子弱亲核能力的碱:H- , NH2-。 具有强亲质子和相当弱亲核性的碱:Et2N- ,C6H5N- ,
Me3Si-N-。
例 醛、酯的α-H烷基化时,不能用OH-去脱 质子。如用OH-时则:醛发生醇醛缩合:
(优选)第一章碳负离子反应 ppt讲解
1.1 基本原理
1.1.1碳负离子的形成
碳负离子:是以一个带有负电荷的三价碳为中心原 子的中间体,是有机化学反应中常见的活性中间
体。如甲基负离子
、烯丙基负离子
苄基负离子
、三苯甲基负离子
制备:金属有机化合物 异裂
RLi,RMgX,RC CNa
碳负离子是有机分子中的碳氢键失去质子后 所形成的共轭碱 :
OH CC
H NCC
H NO2 C
强碱
O CC
强碱 NCC
强碱
NO2 C
O CC
NCC O NC O
1.1.2碳负离子的形成条件
从结构上讲,至少含有一个氢的碳原子的邻位要有 一个活化基团,一些带有双键或三键的吸电子基 团 C O 、—NO2、—SO3H、—CN、—C≡CH等 都是活化基团。活化基团有两个作用:
化下,丁二酸酯或α-羟基取代的丁二酸酯却能和酮
羰基反应(Stobbe反应),机理:
EtO2C
CH2CH2 CO2Et
EtO EtOH
EtO2C
CH CH2 COOEt
第四章_有机合成

影响烯醇负离子形成的区域选择性的因素包括: (1)α-H的酸性
(2) Bronsted 碱的碱性
(3)非质子性溶剂可以减少烯醇负离子的逆反应,有 利于动力学控制产物的形成。
O S CH3 CH3 DM SO
O HCCH3
CH3 DM F
N(CH3)2
CH2OCH3
O P N(CH3)2
O N
2 与亲电烯的反应 带有酯基、氰基、硝基等吸电子基团的烯烃称为亲电烯,
其能与硫叶立德发生环丙烯化反应
硫叶立德在低温下与α,β-不饱和酮反应生成环氧化 合物,而亚砜型硫叶立德则在较高的温度下与α,β-不饱 和酮反应生成三员碳环化合物。
热力学控制
动力学控制
2 二甲亚砜负离子作为中间体的化学反应
二甲亚砜是非常弱的酸,必须用强碱如NaH除去质子形 成负离子。
a. Wittig 反应可以用于制备合成能量上不利的环外双键化合物 的合成。
b. 当与α,β-不饱和羰基化合物作用时,不会发生1,4-加成。 十分适合于合成萜类、多烯类化合物
c. 反应具有立体选择性。在非极性溶剂中,稳定的磷叶立德
与醛反应优先生成反式烯烃;而活泼的磷叶立德则优先生成 顺式烯烃。
动力学控制 热力学控制
4 与卤代烃的偶合反应
C 2 H 5 M g B rC H 2 C H C H 2 B r C 2 H 5 C H 2 C H C H 2 9 4 %
三 有机铜化合物在合成中的应用
有机铜锂试剂较相应的烷基锂碱性、亲核性弱,与孤立的羰基 酯基、羧基等不反应,但其可以与卤代烃或磺酸酯发生亲核取代 反应,并使环氧乙烷开环,而且还可以与α,β-不饱和羰基化合 物发生共轭加成。
在惰性溶剂(如氯仿、二氯甲烷、乙醚、苯)中,烯烃与 过氧酸(如过氧苯甲酸、间氯过氧苯甲酸、过氧三氯乙酸、过 氧乙酸)反应生成环氧化合物,反应具有立体选择性。过酸通 常优先作用于空间障碍小的一边,并得到顺式加成物。
羰基化合物的亲核加成和亲核取代反应

Nu E
2) 羰基氧被N取代 C O
H2NY
C OE Nu
Y CN
3) 羰基氧被C取代
CO
4) 亲核取代反应
CO Y
Ph3P CR1R2
R1 CC
R2
Nu
CO + Y
Nu
一、羰基的结构及反应特性
4、亲核反应活性 烷基给电子作用和体积位阻效应
一、羰基的结构及反应特性
4、 亲核反应活性
空间位阻影响
一、羰基的结构及反应特性
MO 位阻较小
Nu
E
L
S
R
OE
M
S
+
R
Nu
L
主要产物
OE
M
S
Nu
R
L
次要产物
二、羰基的亲核加成
2. 含O, S亲核试剂
包括H2O, ROH, RSH, 和NaHSO3 (1) H2O
水合物
酸或碱催化,加快平衡的达到,但不影响平衡移动。
二、羰基的亲核加成
(1) H2O
O +
H3C CH3 99.8%
Et
CH3
1,4-加成 100%
O CH CH C
1.
MgBr
2.H3O
O CH CH2 C
1,4-加成 92%
O
1.
MgBr
CH CH C CH3
2.H3O
O CH CH2 C CH3 +
OH CH CH C H
1,4-加成 12%
1,2-加成 80%
三、羧酸及其衍生物的亲核取代反应
1.亲核取代反应的机理
4、亲核反应活性
O C RX
碳负离子的反应

含有α氢原子的酮与酯之间也可以进行缩合 反应主要产物为β-二酮。
例如:
第二节 β-二羰基化合物的烷基化、酰基 化及其在合成中的应用
两个羰基被一个碳原子隔开的化合物称 为β-二羰基化合物。
β-二羰基化合物一般泛指β-二酮、β-酮 酸酯、丙二酸酯等含活泼亚甲基化合物 。
这类化合物主要的反应类型是亚甲基碳 上的烷基化、酰基化反应。
一、乙酰乙酸乙酯
无色,具有水果香味,沸点118℃, 微溶于水,易溶于多种有机溶剂。
反应可在不同的酯之间进行,称为交叉 酯缩合。
Claisen 缩合举例
混合酯缩合
反应机理
狄克曼(Dieckmann)缩合(也叫酯分子内 缩合)
含6个或7个碳的二元酸酯,在碱性催化 剂作用下环化生成五元或六元环为主的 -酮酯称为狄克曼反应。
分子内羟醛缩合
羟醛缩合反应不仅可以在分子间进行,含有α-氢原子的 二元醛或酮也可以进行分子内缩合,生成环状化合物, 是制备5~7元环化合物的常用方法之一。
ห้องสมุดไป่ตู้
β-羟基醛在加热时即失去一分子水,生成 α,β-不饱和醛
常用的碱性催化剂除了氢氧化钠、氢氧化钾外,还有叔 丁醇铝、醇钠等。 由此可见,通过羟醛缩合反应可以制备α,β-不饱和醛,进 一步还可以转变为其它化合物。所以羟醛缩合反应是有 机合成中用于增长碳链的重要方法之一。
含有α-氢原子的酮在稀碱作用下也 可以发生类似反应,即羟酮缩合反应, 但是反应的平衡偏向反应物一侧,例如 ,丙酮在氢氧化钡催化下,发生反应。
碳负离子的结构与碳正离子或碳自由基 不同,因为带负电荷的碳原子最外层有3对 成键电子和1对未成键电子,这样的4对电子 需要采取相互远离的方式排列,因此碳负离 子采用sp3杂化轨道成键,未成键电子对与3 个共价键形成一个四面体结构。碳正离子、 碳自由基和碳负离子的结构对比如下图所示 。
高等有机化学羰基化合物的反应

R
R
Nu CCCO H
R
Nu C C C OH
R
2) 影响加成方式的因素
a.羰基活性小;b.试剂的亲核性弱时 c.两者空间位阻大时, 一般按1,4 -加成;反之按1,2 -加成:
O PhCH CHC R EtMgBr
Et PhCHCH2COR
Et
PhCH2 CH C R OH
R
H Me Et i-Pr
无a-H的芳香醛在CN-的作用下生成a-羟基酮
OδCH
δ+
Ⅰ
O CH CN Ⅱ
H迁移 极性反转
O OH H C C
CN
OH O CC CN H
Ⅳ
H迁移
O OH
-CN
CC CN H
Ⅴ
O OH C CH
Ⅵ
六、羰基与 Wittig 试剂反应
1、Wittig 试剂制备: 膦的叶立德 (ylid)
Ph3P + X CH R Ph3P CH R
*羰基上空阻大小(空间因素)
*共轭作用存在时羰基稳定,反应活性减少
结论:脂肪醛>甲基酮>环已酮>芳香酮 芳醛>芳香酮
二、 加成-消去反应 OH
R C NHR
C=O + NH2 Y
R
C=NH
*反应机理:亲核加成-
Y
消去反应
NH2 Y 亚胺
*酸催化:控pH=6,
NH2 NH2
R 取代亚胺
(希夫碱) OH
反应需要 过量的强碱
O
O
CH3C CH COEt
无α-H的酯可和有α-H的酯或酮可交叉缩合:
O
O
1. NaH
HCOOEt
高等有机化学第四章有机反应中间体解析

正电荷分散程度大
共轭体系的数目越多,正碳离子越稳定:
CH2 CH 3C > CH2 CH 2CH > CH2 CHCH2
当共轭体系上连有取代基时,供电子基团使正碳离子 稳定性增加;吸电子基团使其稳定性减弱:
CH3
CH2 >
CH2 > O2N
CH2
环丙甲基正离子比苄基正离子还稳定:
3C >
2CH > CH2 >
含有带负电荷的三价碳原子的原子团。 是最早被确认的活性中间体
1、碳负离子的结构
两种构型: 未共用电子对占据p轨道
未共用电子对占据sp3杂化轨道
有利构型!
桥头碳负离子 角锥结构可以快速翻转,不具有手性
三元环碳负离子难于翻转 得到构型保持的氘代产物
当碳负离子与相邻的不饱和体系共轭时,平面结 构变为有利结构
CH2
环丙甲基正离子的结构:
C
其结果是使正电荷分散
CH2
空的 p 轨道与弯曲轨道的交盖
随着环丙基的数目增多,
CH2
CH2
正碳离子稳定性提高。
直接与杂原子相连的碳正离子结构:
氧上未共有电子对所 占 p 轨道 与中心碳原子上的空的 p轨道 侧面交盖,未共有电子对离域, 正电荷分散。
CH3 O CH2
CH3O CH2
HC CH
NaNH3 液 NH3
HC CNa
NH3
Ph3C H
NaNH3 液 NH3
Ph3CNa
NH3
CH3COCH2COOEt NaOEt CH3COCHCOOEt
常用的碱 ■ 有机锂试剂:n-BuLi, PhLi, MeLi ■ KOBut ■ LDA
碳负离子的经典反应.

碳负离子
DESIGN BY2014.12
经典反应
碳负离子 碳原子的性质 在有机化合物中, 碳原子常采取SP、SP2和SP3三种杂化形式。我们知道与P轨道 相比较S轨道更靠近原子核, 所以S轨道中的电子离核较近,受原子核束缚的较紧, 因此当杂化轨道S 成份增加时, 其吸电子的能力增强, 电负性增大, 接纳负离子中的电子的能力增加, 从而导致负离子 的稳定性增大。
O
O
该反应的净结果是二甲氨甲基取代了α-H ,故又称为氨甲基化反应,产物 为β-氨基酮。
=
=
C C H3 + HC HO + (C H3)2NH
HCl
C C H2 C H2N(C H 3)2 HC l
Mannich反应机理
(i)
R O C H CHR' H R OH C CHR' H R OH C CHR'
O
-
ClCH2CH2COEt
CH3C-CH-COOEt CH2CH2COOC2H5 O
OH H2O
-
H
+
- CO2
O
CH3CCH2CH2CH2COH
O O CH3CCH2CH2CH2CCH3
O EtO O
O
CH3CCH2COEt
-
ClCH2CH2CCH3
O CH3C-CH-COOEt CH2CH2CCH3 O
OH H2O
-
H
+
- CO2
O
O
CH3CCH2CH2CH2CCH3 + CH3COOH
用乙酰乙酸乙酯合成二羰基化合物
O O O CH3CCH2COC2H5 NaOC2H5 O O
DESIGN BY2014.12
经典反应
碳负离子 碳原子的性质 在有机化合物中, 碳原子常采取SP、SP2和SP3三种杂化形式。我们知道与P轨道 相比较S轨道更靠近原子核, 所以S轨道中的电子离核较近,受原子核束缚的较紧, 因此当杂化轨道S 成份增加时, 其吸电子的能力增强, 电负性增大, 接纳负离子中的电子的能力增加, 从而导致负离子 的稳定性增大。
O
O
该反应的净结果是二甲氨甲基取代了α-H ,故又称为氨甲基化反应,产物 为β-氨基酮。
=
=
C C H3 + HC HO + (C H3)2NH
HCl
C C H2 C H2N(C H 3)2 HC l
Mannich反应机理
(i)
R O C H CHR' H R OH C CHR' H R OH C CHR'
O
-
ClCH2CH2COEt
CH3C-CH-COOEt CH2CH2COOC2H5 O
OH H2O
-
H
+
- CO2
O
CH3CCH2CH2CH2COH
O O CH3CCH2CH2CH2CCH3
O EtO O
O
CH3CCH2COEt
-
ClCH2CH2CCH3
O CH3C-CH-COOEt CH2CH2CCH3 O
OH H2O
-
H
+
- CO2
O
O
CH3CCH2CH2CH2CCH3 + CH3COOH
用乙酰乙酸乙酯合成二羰基化合物
O O O CH3CCH2COC2H5 NaOC2H5 O O
第四章 碱催化缩合反应和烃基化反应(6)-1

8
4.1.2 酯缩合反应
1. Claisen酯缩合反应 Claisen酯缩合反应:酯和含活泼甲基或亚甲基的羰基化合物在强
碱作用下缩合,生成β-羰基化合物的反应称之。
◆ 机理:
H CH2COOC2H5
乙酸乙酯
O
NaOC 2H5
O
-
O-
CH2-C-OC2H5
O-
CH2=C-OC2H5
CH3-C-O-C 2H5 + -CH COOC H 2 2 5
◆ 讨论:
① 羰基使α-H的酸性大增,在强碱(碱性大于OH-)作用下,发 生亲核加成-消除反应,最终得到β-二羰基化合物。
② 酮的酸性一般大于酯,所以在乙醇钠的作用下,酮更易生 成碳负离子。例如:
O CH3-C-CH3 O CH3-C-CH2
O OCH3-C-CH2-C-CH3 OCH2CH3
O-
第四章 碱催化缩合反应和烃基化反应
4.1.4 Stobbe 反应 丁二酸酯或α-烃基取代的丁二酸酯在碱作用下,与羰基化合物 (常用酮)进行缩合而得α-烷烃(或芳烃)亚甲基丁二酸酯的反
应称为Stobbe 反应。
CH2COOC2H5 (C6H5)2C O H+ + CH2COOC2H5 C COOC2H5 CH2COOH (CH3)3C OK (CH3)3C OH, (C6H5)2C C COOC2H5 CH2COO
C O
R'
R
C H
C O
R'
醛的缩合反应,生成碳负离子为决速步骤;酮的缩合反应,碳 负离子对酮羰基的加成是决速步骤。
第四章 碱催化缩合反应和烃基化反应 2
R
H2 C
药物合成反应_第四章_缩合反应

O
CH(COOC2H5)2 CH-CH2COCH3
O O δN δ+
加成方式跟底物酮的结构以及格氏试剂所带烷基的位阻有较大关系!
①PhMgBr 1 4
O
2 3
②H2O
位阻2小4大或使用RLi 主要发生1,2加成 Ph 位阻2大4小或使用R2CuLi,亦或 在格氏试剂中添加卤化亚铜 主要发生1,4加成 Ph
OH Ph O
Ph
①C2H5MgBr ②H2O
有 机 金 属 化 合 物 的 反 应
Strecker氨基酸合成反应。
反应机理:
反 应
该反应是制备氨基酸的方便方法。
3
Strecher
应用特点:
可以用氯化铵和氰化钾替代HCN-NH3,还可用氰化三甲基硅Me3SiCN替换氰化钾。
CHO NH2 TMSCN/MeOH 盐酸胍 CN
O O Cl CHO NH2 H3C 催化剂 CN CH2Cl2 NC N CH3
末端烯烃的反应收率相对较好,且主要产物为环状缩醛;
2
Prins
应用特点:
制备1,3-二醇
HCHO HCOOH OOCH H2O OH OH
OOCH
制备1,3-二氧六环
HCHO 酸性树脂 O
反 应
O
2
反应机理:碳负离子对羰基的加成消除。
芳醛在含水乙醇中,以氰化钠(钾)为催化剂,加热后发生双分子缩合生成α -羟基酮。 关键在于如何产生碳负离子。
HO OHC
应用特点: 不同醛可得不同苄氯烃基
HO ClCH2OCH3 AlCl3 NO2 OHC
CH2Cl
卤 烷 基 化
(Blanc)
NO2
CH3CHO O HCl O
CH(COOC2H5)2 CH-CH2COCH3
O O δN δ+
加成方式跟底物酮的结构以及格氏试剂所带烷基的位阻有较大关系!
①PhMgBr 1 4
O
2 3
②H2O
位阻2小4大或使用RLi 主要发生1,2加成 Ph 位阻2大4小或使用R2CuLi,亦或 在格氏试剂中添加卤化亚铜 主要发生1,4加成 Ph
OH Ph O
Ph
①C2H5MgBr ②H2O
有 机 金 属 化 合 物 的 反 应
Strecker氨基酸合成反应。
反应机理:
反 应
该反应是制备氨基酸的方便方法。
3
Strecher
应用特点:
可以用氯化铵和氰化钾替代HCN-NH3,还可用氰化三甲基硅Me3SiCN替换氰化钾。
CHO NH2 TMSCN/MeOH 盐酸胍 CN
O O Cl CHO NH2 H3C 催化剂 CN CH2Cl2 NC N CH3
末端烯烃的反应收率相对较好,且主要产物为环状缩醛;
2
Prins
应用特点:
制备1,3-二醇
HCHO HCOOH OOCH H2O OH OH
OOCH
制备1,3-二氧六环
HCHO 酸性树脂 O
反 应
O
2
反应机理:碳负离子对羰基的加成消除。
芳醛在含水乙醇中,以氰化钠(钾)为催化剂,加热后发生双分子缩合生成α -羟基酮。 关键在于如何产生碳负离子。
HO OHC
应用特点: 不同醛可得不同苄氯烃基
HO ClCH2OCH3 AlCl3 NO2 OHC
CH2Cl
卤 烷 基 化
(Blanc)
NO2
CH3CHO O HCl O
药物合成反应第四章 缩合反应

2 2 2 2
CH2
HC
CH2Leabharlann CH2CH2 [4+2]环加成反应 1,3-偶极环加成反应
20
1. [4+2]环加成反应 共轭二烯烃与烯烃、炔烃进行环加成,生成 环己烯衍生物的反应属[4+2]环加成反应,该反应 称为Diels-Alder反应,也称“双烯加成”。 最简单的[4+2]环化加成是1,3-丁二烯与乙 烯加成反应,假定丁二烯分子与乙烯分子面对面 互相接近,丁二烯的最高占有轨道与乙烯的最低 空轨道或丁二烯的最低空轨道与乙烯的最高占有 轨道都可以重叠成键,因此,[4+2]环加成是对称 允许反应。
CH2CH3
31
催化剂的影响: 醛或酮的自身缩合反应常用碱作催化剂,酸催化剂 应用较少。
32
④应用特点 制备长链醛(醇)
例:2-乙基己醇(异辛醇)的生产 2 CH3CH2CH2CHO
CH3CH2CH2CH2-CHCHO OH CH2CH3
CH3CH2CH2CH2=CCHO CH2CH3
33
CH3CH2CH2CH2-CHCH2OH CH2CH3
性条件下可以离解,生成烯醇负离子,从而成为亲核试 剂,进攻羰基碳或卤代烃,发生亲核加成反应、亲核取 代反应。
O C C H
B
C
O C
C
O C
烯醇负离子由于羰基的共轭作用得以稳定 烯醇负离子
4
烯醇中的C=C双键接受亲电试剂进攻,发生α-卤代 反应,醛酮、羧酸和酰卤可以发生该反应。 烯醇负离子作为亲核试剂,进攻卤代烃的缺电子 碳,则发生亲核取代反应;进攻羰基碳则发生亲核 加成反应。
R' +
R
HC
C OH
CH2
HC
CH2Leabharlann CH2CH2 [4+2]环加成反应 1,3-偶极环加成反应
20
1. [4+2]环加成反应 共轭二烯烃与烯烃、炔烃进行环加成,生成 环己烯衍生物的反应属[4+2]环加成反应,该反应 称为Diels-Alder反应,也称“双烯加成”。 最简单的[4+2]环化加成是1,3-丁二烯与乙 烯加成反应,假定丁二烯分子与乙烯分子面对面 互相接近,丁二烯的最高占有轨道与乙烯的最低 空轨道或丁二烯的最低空轨道与乙烯的最高占有 轨道都可以重叠成键,因此,[4+2]环加成是对称 允许反应。
CH2CH3
31
催化剂的影响: 醛或酮的自身缩合反应常用碱作催化剂,酸催化剂 应用较少。
32
④应用特点 制备长链醛(醇)
例:2-乙基己醇(异辛醇)的生产 2 CH3CH2CH2CHO
CH3CH2CH2CH2-CHCHO OH CH2CH3
CH3CH2CH2CH2=CCHO CH2CH3
33
CH3CH2CH2CH2-CHCH2OH CH2CH3
性条件下可以离解,生成烯醇负离子,从而成为亲核试 剂,进攻羰基碳或卤代烃,发生亲核加成反应、亲核取 代反应。
O C C H
B
C
O C
C
O C
烯醇负离子由于羰基的共轭作用得以稳定 烯醇负离子
4
烯醇中的C=C双键接受亲电试剂进攻,发生α-卤代 反应,醛酮、羧酸和酰卤可以发生该反应。 烯醇负离子作为亲核试剂,进攻卤代烃的缺电子 碳,则发生亲核取代反应;进攻羰基碳则发生亲核 加成反应。
R' +
R
HC
C OH
第四章 缩合反应

OH
OH-
HO
Ar C H
Ar C
CN
CN
CN
O C Ar H
亲核加成
HO O Ar C C
H2O
HO HO
Ar
Ar C C Ar
-CN
OH OH
-H+
Ar C C Ar
O OH Ar C C Ar
NC H
CN H
H
H
一、α-羟烷基化
中间体
OH
C
R
CN
当R为吸电子基团时有利于反应,但不能生成对称的α -羟基 酮, 能与苯甲醛反应生成不对称的α -羟基酮.如:
ArCH2Cl
NaOH ArCH2OH
KCN ArCH2CN
[O] ArCHO
H2O(H+) ArCH2COOH
NH3 ArCH2NH2
R3N ArCH2NR3Cl
二、α-卤烷基化(Blanc反应,氯甲基化反应)
Blanc氯甲基化反应可用于延长碳链
HCHO/HCl/ZnCl2
CH2Cl KCN
CH2CN H2O H+
β 羟基醛或β 羟基酮的反应(醛、酮之间的缩合)
OH 或 H 2 R CH2 C R'
O
H2 R ' R O RC C C C
OH H
R' - H2O
R' R O RH2C C C CR'
一、α-羟烷基化
1 Aldol缩合 机 理 a: 碱催化
无机碱: NaOH, Na2CO3 有机碱: EtONa, NaH
O
H3C C CH2CH2
O
N CH3
H3C C CH2CH2
羰基化合物的反应

(79%)
醛、酮的混合缩合(又称交叉缩合),即克莱森-施密特缩合,要得到单一的产物,
其中一个组分应无 α-H。
C6H5CHO + CH3CHO 羰基组分 α-H组分
NaOH 20oC
OH
C6H5CH CH2CHO ∆
C6H5CH=CHCHO
由于大共轭体系的形成,使乙醛的自缩合反应成为次要的
O C6H5CHO +
R1CH H
OH
C NR2 R2
-H2O
R1CH C NR2 R2
R1CH C NR2 β R2
烯胺是重要的有机合成中间体,其 β-C 有很强的亲核性,具有碳负离子的性
质。
O
+ N H
N β
RX
NX R
H3O
O R
+ N
HH
RX 可以是 α-卤代酮,α-卤代酯,卤代苄,酰氯等。
烯胺的烃基化产物水解后即脱去仲胺,恢复原来的羰基。
CN O + H2C
NH4OAc
COOH
CN
C
+ H2O
COOH
CH3NO2 NaOH
CH=CHNO2 + H2O
CHO
CH3COCH2COOEt Et3N
COCH3 CH=C
COOEt
+ H2O
CHO + CH2(COOH)2 吡啶
CH=CHCOOH + CO2 + H2O
O2N
O2N
4.柏金反应,芳香醛与酸酐在相应的羧酸盐(钾盐或钠盐)催化下缩合生成 α,
H
O H3C
H
B
(过量 )
3.克诺文诺盖尔反应,醛酮与其它含活泼氢化合物的缩合。反应历程和羟醛缩
羰基化合物的亲核加成和取代反应

OH
2.H3O
酮
R
R C R'
O
低温和空间位阻作用 3°醇
R''
R CR' 使用不活泼的金属试剂
3°醇
酮 可能将反应控制在酮的阶段
二、羰基的亲核加成
4. 含C亲核试剂
(4) Ylide试剂 An ylide is a compound that has opposite charges on adjacent, covalently bonded atoms with complete octets of valence electrons, for example, a phosphonium ylide:
O CH CH C
1.
MgBr
2.H3O
O CH CH2 C
1,4-加成 92%
O
1.
MgBr
CH CH C CH3
2.H3O
O CH CH2 C CH3 +
OH CH CH C H
1,4-加成 12%
1,2-加成 80%
三、羧酸及其衍生物的亲核取代反应
1. Mechanism for nucleophilic substitution
关于亲核加成的小结 1. 概述 —— 结构、机理、反应活性、立体化学 2. 含O, S亲核试剂 包括H2O, ROH, RSH, 和NaHSO3
羰基的保护 醛酮的分离纯化
3. 含N亲核试剂 (1) RNH2 and YNH2 (2) R2NH
亚胺及其衍生物的形成 烯胺的形成
4. 含C亲核试剂 —— C-C键的形成
第十三章
羰基化合物的亲核加成和取代反应
Nucleophilic Additions and Substitutions of Carbonyl Compounds
第四节 羰基化合物的卤取代反应3版
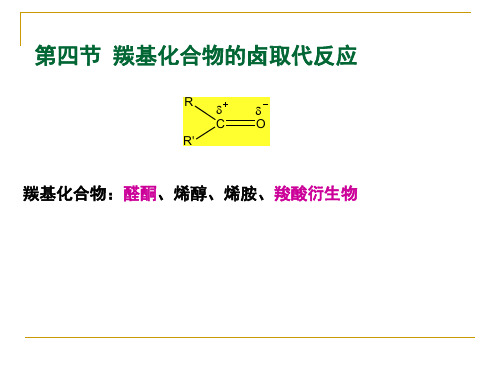
1、丙二酸酯的α-卤取代反应
COOC2H5 COOC2H5
Br2/CCl4
COOC2H5
Br
75%
COOC2H5
强碱作用,极性溶剂DMSO与CuCl2反应
COOC 2 H 5 COOC 2 H5
1 )NaH/DMS O 2)CuBr2
COOC 2 H5
Br
90%
COOC 2 H5
形成烯醇β-碳负离子的羧酸酯
Br
+
OH
CH2
CH2
CH3
-Br2
H2C
OH
CH2
CH2
CH3
OH
H3C
Br
Br
Br2 CH3
O
Br CH2
O
H3C
CH2
1. 5%
CH2
CH3
CH2
CH
CH3 58%
Br
热力学产物
*溴化剂或碘化剂/醋酸钠或吡啶等碱性物质(动力学控制产物)
*③ 羰基α-位取代基的电性效应的影响
酸催化反应:α-位有供电子基,利于反应。
CH2 H
X 2 or NBS (NCS ) H3C
O H3C
CH2X H
AcO
AcO
X=Br 70% X=Cl 25%
提高不对称酮区域选择性的一种方法 反应后生成的丙酮易于蒸馏除去 溴代产率高 烯醇酯形成
*2 烯醇硅烷醚的卤化反应
(1) 通式
R1 OSiMe 3 X2
R2
R3
(2)反应机理
X R1
(1)反应通式
O
H3C H3C CH3
CH3
1 )I 2 /Na OH/H2 O 2 )H +
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
共轭碱
举例:
1)E1cb消除
.. B H-CH2-CH-CH2CH3 X E1cb CH2-CH-CH2CH3 X CH2=CH-CH2CH3
2)羰基化合物的烯醇负离子互变 在碱性条件下:
O B R-C-CH-R' O O -
R-C-CH2R'
R-C=CHR'
3)通过加成-消除的亲核芳香取代反应
..
C
C6H13 H
CH3
但对三苯甲基负离子,中心碳sp2杂化,碳负离 子呈平面型,有利于三个苯环共轭,稳定碳负 离子。
桥头碳负离子,能形成稳定的棱锥形构型。
1) CO2
Cl
..
2) H3O
+ COOH
碳负离子构型证明:
BuLi 1 CO 2
C6H12 CH CH3 I
C6H12CH CH3
2 H3O
思考:
H
CH3 Li
H
CH3
. . 1)
H 2)
H
CO2 H3O +
CH3 COOH
H H
H -
H H
H
H
H
3、碳负离子稳定性及测定方法
将烃类视为碳氢酸,测定其酸离解常数, 由pK来判定碳负离子的稳定性。
R-H
+
B
-
R
-
+
HB
可由两个酸性烃类化合物与其金属盐之间建立平 衡,求得pKa值。
R-H
H + N
ClCH2CH CH2 CH3CH2 O H2 O C H C CH3 CH2
Cl
CH2
CH2CH
CH3CH2CCHCH2CH
+
N H
+
HCl
CH3
三、羰基化合物的反应
1、羰基的亲核加成反应
机理1:
Nu A
O
C B
慢
O A C B Nu
快
H
O A C B Nu OH
A C B Nu
① 试剂进攻羰基上C原子,生成氧负离子的一步 是决定反应速率的一步。 ② 为使亲核试剂的负电荷裸露出来,增加亲核性, 常需碱催化。
Li
C6H12 CH CH3 COOH
0º C , 生成dl外消旋体 -70º C, 生成80% 外消旋体, 20%构型保持。
..
C6H13 C
..
H
C6H13 C H C6H13
CO2
CH3
CH3
CO2
CH3
C
H
..
CH3
H3 O +
COOH C6H13 C H
H3O+
C6H13 H
C
CH3
COOH
Ph3P CH2
(n-Bu)3P CHR
Wittig 试剂与羰基发生亲核加成反应,生成烯烃:
C O
Ph3P C
R R'
C
C R R'
Georg Wittig
反应机理:
C O + Ph3P
C O C O
CR2
C O R2C PPh3
O C C + PPh3 R
R2C PPh3 R2C PPh3
氧膦环丁烷中间体
第四章 碳负离子及羰基化合物的反应 (Carbanions and Reactions of Carbonyl Groups)
一、碳负离子的产生、结构和稳定性
1、产生
碳负离子:有机分子通过碳上失去质子形成的带负 电荷的活性中间体。 1)强碱作用下; 2)邻近有使电子离域之基团。
C H+ B
共轭酸
C + HB
+ +
CH2
-
Ph3 P (CH3)2S
CH2 C H2
(CH3)2S PhLi
C H2 CH2
季铵盐:(CH ) N + Cl 反应活性:
(CH3)2S
+ (CH3)3 N
+
C H2 > Ph3P
-
+
-
CH2
+ > > (CH3)3 N
CH2
2) S电子成分效应
RC
C
-
> R2C
CH ≈ Ar
-
-
> R3C
CH2
-
sp杂化
S成分: 50%
sp2
33.3%
sp3
25%
3)芳香性
符合休克尔规则的C- 稳定
B: R B: R -
反芳香性,反应速度慢
R
H
4n+2=6, 芳香性,反应速度快
H
R
4) 溶剂
非质子性的溶剂利于C- 的裸露
δδ+
R
M δ-
+ H
OH
-
O
δ+
HC
N
CH3 CH3
+ H R + H
去质子速度为反应速度控制步骤。
去质子活性与电子效应和空间效应有关。
O CH3CCH2CH3 O B CH3CCH2C(CH3) 3 B O CH3CCH-CH3 H3 C H3 C
O
C CH C CH3
相对速度 41.5
CH3
< 0.1
空间位阻大,去质子速度慢; 烷基取代基多,烯醇负离子稳定性好。
sp3杂化 四面体
羰基加成的立体化学:
2)若羰基的邻近碳原子有手性,亲核加成遵循Cram规则
Nu
C M C OH
Cram规则-1
S
L
C M C
R
+
O
+ H Nu
L S
R
Et Ph
O
H
1) LiAlH4 2) H2O
Et HO
H
H CH3
CH3
Ph
羰基加成的立体化学:
Cram规则-2
L O R O
2)酸催化
O R + R C OH C CH2 R'+ H + 快 R OH R C
+
OH
C
CH2
R'
慢
CH2
R'
CH R'
+
H
+
例:
O
OH CH3C CH2 H + Cl
-HCl
OH CH3C CH2
CH3CCH3 + H Cl
质子化酮去除α-质子形成烯醇的速度是反应速度控制步骤。
3)酸催化与碱催化比较 (1) 酸催化,形成多取代烯醇, 碱催化,形成取代基较少烯醇负离子。 (2) 酸催化,形成烯醇, 碱催化,形成烯醇盐。 (3) 烷基取代基对活性的影响在碱催化时比在 酸催化时明显。 (4) 无论酸或碱催化,含有单独一个酮基、醛 基、酯基的化合物,平衡时几乎不存在烯 醇式,以酮式存在。
+ + OH H OH M OH -
非质子性溶剂,正极埋在溶剂分子内部,负极与亲核试剂的 正离子形成离子-偶极键,将亲核试剂正负离子隔开,使碳负 离子裸露,反应速度加快。 质子性溶剂使C- 溶剂化,反应速度降低。
冠醚的加入可将负离子亲核试剂裸露在外,促进反应。
O O
O + O M O O
R
-
二、烯醇和烯胺
(5)形成烯醇时的动力学与热力学控制
O R2CHCCH2R' B ka O kb O B -
R2C=CCH2R'
R2CHC=CHR' B
[A] = [B]
ka
kb
A
动力学控制:取决于去质子速度(非质子溶剂、强碱、 无过量酮存在) 位阻小的H易被夺去 热力学控制:取决于产物的稳定性(过量酮存在、质子 性溶剂稳定C- ),C=C上取代基多的烯 醇稳定
NO2 O2N OR NO2 NO2
+
-
OR'
O2N
-
OR OR ' NO2 NO2
O2N
OR '
NO2
4)通过金属有机化合物异裂
RLi、RMgX、RC = CNa
2、结构
两种合理构型推测: ..
..
109°28′
C
sp3 杂化 棱锥型
C
sp2杂化 平面三角型
90°
举例:
碳负离子的空间构型取决于所连基团。 通常情形,棱锥型碳负离子的孤对电子处于sp3 杂化轨道上,斥力小,稳定。
CH
H
COOC2H5 OC2H5
CH2
COOC2H5 H5C2OOCCH2CH2
O CH2CH2COOC2H5
HCl,H2O,C2H5OH 60%
O
O CH3CH2CCHCH2CH CH3 CH2
例3:
CH3CH2CCH2CH3
O CH3CH2CCH2CH3+ N CH3CH2
+ N C
C
H
CH3
O R2CHC-R'
B R2 C
O C
-
O
R'
R2 C O
C
R'
O
R2 C C
R'
+
S
D
R2 C D
C
R'
(2)动力学证明
O ' R2CHC-R O
R2 C C R' 慢 反应 B O R2 C C R' R2 C O
C
R'
O
+
O
快反应 X2 R2 C X
举例:
1)E1cb消除
.. B H-CH2-CH-CH2CH3 X E1cb CH2-CH-CH2CH3 X CH2=CH-CH2CH3
2)羰基化合物的烯醇负离子互变 在碱性条件下:
O B R-C-CH-R' O O -
R-C-CH2R'
R-C=CHR'
3)通过加成-消除的亲核芳香取代反应
..
C
C6H13 H
CH3
但对三苯甲基负离子,中心碳sp2杂化,碳负离 子呈平面型,有利于三个苯环共轭,稳定碳负 离子。
桥头碳负离子,能形成稳定的棱锥形构型。
1) CO2
Cl
..
2) H3O
+ COOH
碳负离子构型证明:
BuLi 1 CO 2
C6H12 CH CH3 I
C6H12CH CH3
2 H3O
思考:
H
CH3 Li
H
CH3
. . 1)
H 2)
H
CO2 H3O +
CH3 COOH
H H
H -
H H
H
H
H
3、碳负离子稳定性及测定方法
将烃类视为碳氢酸,测定其酸离解常数, 由pK来判定碳负离子的稳定性。
R-H
+
B
-
R
-
+
HB
可由两个酸性烃类化合物与其金属盐之间建立平 衡,求得pKa值。
R-H
H + N
ClCH2CH CH2 CH3CH2 O H2 O C H C CH3 CH2
Cl
CH2
CH2CH
CH3CH2CCHCH2CH
+
N H
+
HCl
CH3
三、羰基化合物的反应
1、羰基的亲核加成反应
机理1:
Nu A
O
C B
慢
O A C B Nu
快
H
O A C B Nu OH
A C B Nu
① 试剂进攻羰基上C原子,生成氧负离子的一步 是决定反应速率的一步。 ② 为使亲核试剂的负电荷裸露出来,增加亲核性, 常需碱催化。
Li
C6H12 CH CH3 COOH
0º C , 生成dl外消旋体 -70º C, 生成80% 外消旋体, 20%构型保持。
..
C6H13 C
..
H
C6H13 C H C6H13
CO2
CH3
CH3
CO2
CH3
C
H
..
CH3
H3 O +
COOH C6H13 C H
H3O+
C6H13 H
C
CH3
COOH
Ph3P CH2
(n-Bu)3P CHR
Wittig 试剂与羰基发生亲核加成反应,生成烯烃:
C O
Ph3P C
R R'
C
C R R'
Georg Wittig
反应机理:
C O + Ph3P
C O C O
CR2
C O R2C PPh3
O C C + PPh3 R
R2C PPh3 R2C PPh3
氧膦环丁烷中间体
第四章 碳负离子及羰基化合物的反应 (Carbanions and Reactions of Carbonyl Groups)
一、碳负离子的产生、结构和稳定性
1、产生
碳负离子:有机分子通过碳上失去质子形成的带负 电荷的活性中间体。 1)强碱作用下; 2)邻近有使电子离域之基团。
C H+ B
共轭酸
C + HB
+ +
CH2
-
Ph3 P (CH3)2S
CH2 C H2
(CH3)2S PhLi
C H2 CH2
季铵盐:(CH ) N + Cl 反应活性:
(CH3)2S
+ (CH3)3 N
+
C H2 > Ph3P
-
+
-
CH2
+ > > (CH3)3 N
CH2
2) S电子成分效应
RC
C
-
> R2C
CH ≈ Ar
-
-
> R3C
CH2
-
sp杂化
S成分: 50%
sp2
33.3%
sp3
25%
3)芳香性
符合休克尔规则的C- 稳定
B: R B: R -
反芳香性,反应速度慢
R
H
4n+2=6, 芳香性,反应速度快
H
R
4) 溶剂
非质子性的溶剂利于C- 的裸露
δδ+
R
M δ-
+ H
OH
-
O
δ+
HC
N
CH3 CH3
+ H R + H
去质子速度为反应速度控制步骤。
去质子活性与电子效应和空间效应有关。
O CH3CCH2CH3 O B CH3CCH2C(CH3) 3 B O CH3CCH-CH3 H3 C H3 C
O
C CH C CH3
相对速度 41.5
CH3
< 0.1
空间位阻大,去质子速度慢; 烷基取代基多,烯醇负离子稳定性好。
sp3杂化 四面体
羰基加成的立体化学:
2)若羰基的邻近碳原子有手性,亲核加成遵循Cram规则
Nu
C M C OH
Cram规则-1
S
L
C M C
R
+
O
+ H Nu
L S
R
Et Ph
O
H
1) LiAlH4 2) H2O
Et HO
H
H CH3
CH3
Ph
羰基加成的立体化学:
Cram规则-2
L O R O
2)酸催化
O R + R C OH C CH2 R'+ H + 快 R OH R C
+
OH
C
CH2
R'
慢
CH2
R'
CH R'
+
H
+
例:
O
OH CH3C CH2 H + Cl
-HCl
OH CH3C CH2
CH3CCH3 + H Cl
质子化酮去除α-质子形成烯醇的速度是反应速度控制步骤。
3)酸催化与碱催化比较 (1) 酸催化,形成多取代烯醇, 碱催化,形成取代基较少烯醇负离子。 (2) 酸催化,形成烯醇, 碱催化,形成烯醇盐。 (3) 烷基取代基对活性的影响在碱催化时比在 酸催化时明显。 (4) 无论酸或碱催化,含有单独一个酮基、醛 基、酯基的化合物,平衡时几乎不存在烯 醇式,以酮式存在。
+ + OH H OH M OH -
非质子性溶剂,正极埋在溶剂分子内部,负极与亲核试剂的 正离子形成离子-偶极键,将亲核试剂正负离子隔开,使碳负 离子裸露,反应速度加快。 质子性溶剂使C- 溶剂化,反应速度降低。
冠醚的加入可将负离子亲核试剂裸露在外,促进反应。
O O
O + O M O O
R
-
二、烯醇和烯胺
(5)形成烯醇时的动力学与热力学控制
O R2CHCCH2R' B ka O kb O B -
R2C=CCH2R'
R2CHC=CHR' B
[A] = [B]
ka
kb
A
动力学控制:取决于去质子速度(非质子溶剂、强碱、 无过量酮存在) 位阻小的H易被夺去 热力学控制:取决于产物的稳定性(过量酮存在、质子 性溶剂稳定C- ),C=C上取代基多的烯 醇稳定
NO2 O2N OR NO2 NO2
+
-
OR'
O2N
-
OR OR ' NO2 NO2
O2N
OR '
NO2
4)通过金属有机化合物异裂
RLi、RMgX、RC = CNa
2、结构
两种合理构型推测: ..
..
109°28′
C
sp3 杂化 棱锥型
C
sp2杂化 平面三角型
90°
举例:
碳负离子的空间构型取决于所连基团。 通常情形,棱锥型碳负离子的孤对电子处于sp3 杂化轨道上,斥力小,稳定。
CH
H
COOC2H5 OC2H5
CH2
COOC2H5 H5C2OOCCH2CH2
O CH2CH2COOC2H5
HCl,H2O,C2H5OH 60%
O
O CH3CH2CCHCH2CH CH3 CH2
例3:
CH3CH2CCH2CH3
O CH3CH2CCH2CH3+ N CH3CH2
+ N C
C
H
CH3
O R2CHC-R'
B R2 C
O C
-
O
R'
R2 C O
C
R'
O
R2 C C
R'
+
S
D
R2 C D
C
R'
(2)动力学证明
O ' R2CHC-R O
R2 C C R' 慢 反应 B O R2 C C R' R2 C O
C
R'
O
+
O
快反应 X2 R2 C X