五大波谱解析步骤简述 (一) 紫外光谱 解析UV应用时顾及吸收带的
紫外光谱讲解

计算举例:
应用实例:
当存在环张力或立体结构影响到共轭时,
计算值与真实值误差较大。
2.3.3 α , β-不饱和醛酮
2.3 共轭有机化合物的紫外吸收
2.3.1 共轭体系的形成使吸收移向长波方向
共轭烯烃的π π*跃迁 均为强吸收带, ≥10000, 称为K带。
共轭体系越长,其最大吸收越移往长波方向, 强度也增加,且出现多条谱带。
2.3.2 共轭烯烃及其衍生物
Woodward-Fieser 规则
取代基对共轭双烯 λmax的影响具有加和性。
L:液层厚度(光程长度),通常以cm为单位; c:溶液的浓度;
ε:吸光系数;
εmax表明了该吸收物质最大限度的吸光能力,也反 映了光度法测定该物质可能达到的最大灵敏度。 εmax越大表明该物质的吸光能力越强,用光度法测 定该物质的灵敏度越高。
ε>104:
102<ε< 10 4 :
强吸收;
中强吸收;
不同生色团的红移顺序为:
NO2 > Ph >CHO > COCH3 > COOH > COO- >CN
> SO2NH2 ( > NH3+)
应用实例:
酚酞指示剂
3). 双取代苯
1) 对位取代
两个取代基属于同类型时, λmax 红移值近似为
两者单取代时的最长 波长 。 两个取代基类型不同时, λmax 的红移值远大于两 者单取代时的红移值之和 。(共轭效应)
紫外光谱分析法分析

max 1480 150 200 365 600
π→π*跃迁
Unsaturated alkane π→π*
s*
Chromophore (生色团): 含有π键 的不饱和基团称为生色团。 —C=C—、 —C=O、 —N=O、 —N=N—、 —C三C 、—C三N
E
p*
K E ,B R
n
p
s
n→π*跃迁
ห้องสมุดไป่ตู้
杂芳环化合物 五元杂芳环按照呋喃、吡咯、噻吩的顺序 增强芳香性,其紫外吸收也沿此顺序逐渐接近 于苯的吸收。在上述三种杂芳环中,硫的电子 较氮、氧能更好地和二烯的π 电子共轭,因此 噻吩的紫外吸收在最长波长。生色团、助色团 的取代,导致五元杂芳环的紫外吸收发生较大 的变化(深色位移和增色效应)。 吡啶的共轭体系和苯环相类似,故吡啶的 紫外吸收类似于苯的紫外吸收, 吡啶在251nm 处的吸收强,ε =2000,也显示精细结构。
(1)含有O、N、S、卤素等杂 原子的饱和基团,
如—OH、—OR、—NH2、
s* p*
K R
—NHR、—X 等,与生色团相 连时,就会发生n →π*跃迁。
E
n
p
s
E ,B
(2)含有杂原子的不饱和基团, 如-C=O、-N=N-等,在杂原子 上有未成键的n 电子,发生n →π*跃迁。
Absorption band 吸收带
A.饱和的有机化合物
a.饱和的碳氢化合物 唯一可发生的跃迁为σ → σ * ,能级差 很大,紫外吸收的波长很短,属远紫外范围。如 甲烷、乙烷的最大吸收分别为125nm、135nm。 b.含杂原子的饱和化合物 杂原子具有孤电子对,一般为助色团,这 样的化合物有n → σ *跃迁。但大多数情况,它 们在近紫外区仍无明显吸收。硫醚、二硫化物、 硫醇、胺、溴化物、碘化物在近紫外有弱吸收, 但其大多数均不明显。
有机波谱分析 第一章-紫外光谱

电子
§2.1 紫外光谱的基本知识
2. 分子轨道 根据分子轨道理论,当两个原子结合成分子时,两个原子的 原子轨道线性组合成两个分子轨道,其中一个具有较低的能量叫 做成键轨道,另一个具有较高的能量叫做反键轨道。
2= - 2 1
反键轨道
能量
2
成键轨道
1
1
= + 1
2
电子通常在成键轨道上,当分子吸收能量后可以激发到反键轨道上
三、紫外光谱表示法
1.图示法 紫外光谱可用图表示,也可用数据表示 紫外光谱图是由横坐标、纵坐标和吸收曲线组成的。
对甲苯乙酮的紫外光谱图
loge loge1
loge2
lmax1
lmax2
l/nm
横坐标表示吸收光的波长,用nm(纳米)为单位。 纵坐标表示吸收光的吸收强度,可以用A(吸光度)、T(透射比或透光率或 透过率)、1-T(吸收率)、(吸收系数) 中的任何一个来表示。
(3)吸光度具有加和性,即在某一波长λ ,当溶液中含有多种吸光 物质时,该溶液的吸光度等于溶液中每一成分的吸光度之和,这一 性质是紫外光谱进行多组分测定的依据。 (4)理论上,朗伯-比尔定律只适用于单色光,而实际应用的入射 光往往有一定的波长宽度,因此要求入射光的波长范围越窄越好。朗 伯-比尔定律表明在一定的测定条件下,吸光度与溶液的浓度成正比, 但通常样品只在一定的低浓度范围才成线性关系,因此,定量测定时 必须注意浓度范围。同时,温度、放置时间、pH等因素也会对样品 的光谱产生影响,测定时也必须注意。
于远紫外区,一般出现在200nm附近,受杂原子性质的影响较大。
(4) *跃迁
是单键中的σ电子在σ成键和反键轨道间的跃迁。因σ和σ*之间的能级差最大,所以 σ→σ*跃迁需要较高的能量,相应的激发光波长较短,在150~160nm范围,落在远紫 外光区域,超出了一般紫外分光光度计的检测范围。
有机化合物波谱解析 第一章 紫外光谱(UV)

第一节 基础知识
一、 电磁波的基本性质及分类
1.电磁辐射(电磁波,光) :以巨大速度通过空 间、不需要任何物质作为传播媒介的一种能量。
2.电磁辐射的性质:具有波、粒二向性。
• 波动性:
c
,
104
(m
(cm
)
1() 式(31-11)
• 粒子性: E h h c ( (式1-33)- 2)
光的波长越短(频率越高),其能量越大。
能级跃迁
能级跃迁
(1)转动能级间的能量差ΔEr:0.005~0.050eV,跃迁产
生吸收光谱位于远红外区。远红外光谱或分子转动光谱;
(2)振动能级的能量差ΔEv约为:0.05~1eV,跃迁产生
的吸收光谱位于红外区,红外光谱或分子振动光谱;
(3)电子能级的能量差ΔEe较大1~20eV。电子跃迁产生
仪器分析:测定复杂结构的化合物 样品用量少
• 四谱同时用或联用技术 • 四谱比较: • 灵敏度:MS>UV>IR>1HNMR>13CNMR
MS: 微克级
UV: ppb级
IR:毫克级(可微克级,FTIR)
1HNMR:0.5mg }可回收
13CNMR: 0.5mg
四谱的信息量比较:
1HNMR及13CNMR
• 广泛应用于石油化工,高分子化工,精细化工,环境分 析,生物化工,皮革化工,生物药品分析,新药品的结 构表征,天然有机,生物有机,金属有机化学,化学, 医学,生理病理
• 概论
波谱分析:UV,IR,NMR,MS(有机)----结构分析
四谱提供的信息:
质谱(MS)—— 分子量及部分结构信息 红外光谱(IR) —— 官能团种类 紫外—可见光谱(UV / Vis)—— 共轭结构 核磁共振谱(NMR)—— C-H骨架及所处化学环境
波谱分析-UV

③若在260nm、300nm、330nm附近有强 吸收带,则相应于有三个、四个、五个共 轭双键。若化合物本身有颜色,则所含共 轭双键数应在五个以上。
④若在230-270nm波长区有中强吸收带, 有时有精细结构,则可能是苯环。
⑤在200-250nm无吸收,但在270-350nm 有弱(ε10-100)吸收,可能的基团是未共 轭的C=O、C=N、N=N。
Λmax 247nm
(环己烷)
ε 17000
253 19000
237 10250
231 5600
227
(肩峰)
--
空间结构二面角的影响
O
两面角 λmax
O
0-10 466nm
O
O
90 370nm
二、分子离子化的影响
苯酚和苯胺在不同酸度条件下的UV谱
ph-NH2 + H+ → ph-NH3+ ph-OH + OH- → ph-O-
第一章 紫外吸收光谱
(Ultraviolet -Visible)
第一节 基本原理
紫外-可见分区
真空紫外 普通紫外 可见光区
100nm 200nm 400nm
800nm
电子光 谱的产 生
分子吸收光谱的表达
UV:A~λ;IR:T~ v
电子跃迁和所产生的吸收带
E
σ*
π*
π
* 4
π
* 3
n
π
π2
π1
三、溶剂的影响
1. 溶剂吸收
几种溶剂的极限波长
溶剂 极限波长/nm 溶剂 极限波长/nm 溶剂 极限波长/nm
95%乙醇 210
水
210
正己烷
波谱分析第六章UV谱

b.二取代苯:
Ⅰ.对位二取代苯
若两个取代基属同一类型,则E2带红移值由红移效应 最大的基团决定;若两个取代基属不同类型,则E2 带红移值由二者协同作用决定,且红移增值大于二 者单取代的红移增值之和。
Ⅱ.邻位和间位二取代苯
此类二取代苯不论取代基是何类型,对E2带红移值的 贡献大致等于两个取代基红移值增值之和。
根据量子理论,光子(或电磁辐射)的能量为: E=hν=hc/λ=hcσ 紫外光的能量与化学键的能量相仿,有足够的能量
使分子进行光化学反应。
6.1.2.紫外吸收光谱的产生及其表示方法
1.分子中价电子在电子能级间跃迁产生紫外吸收光 谱;
分子和原子一样,也有它的特征分子能级,这些能 级是由分子内部运动决定的。分子内部运动包括① 电子围绕原子核的运动;②分子内原子在平衡位置 附近的振动;③分子绕其重心的转动;④分子重心 的平移;⑤分子中各基团的内旋转。
b. Woodward-Fieser规则只适合成串共轭的分子, 不适合交叉共轭的分子。交叉共轭体系只能选取一 个,分叉上的双键不算延长双键。
同环双键母体 253
五个取代烷基5×5 两个环外双键5×2
288nm(285nm)
c.选择较长共轭体系作为母体。若同时存在同环双 键和异环双键时,应选取同环双键作为母体。
(3)含不饱和杂原子化合物:例如:醛、酮、酯、酰胺、 酰氯、睛、重氮、硝基、亚硝基、亚砜等。此类化合物可发 生σ→σ*,n→σ*,π→π*和n→π*其中n→π*(R吸收带) 跃迁所需能量较低,吸收带处在近紫外区,易于检测,可用 于结构分析,只不过吸收强度弱。
对于醛、酮类化合物,R带在270~300 nm,εmax=10~ 20L·mol-1·cm-1
分享五大波谱解析步骤简述一紫外光谱解析UV应用时顾及吸收带

分享:五大波谱解析步骤简述(一) 紫外光谱解析UV应用时顾及吸收带的位置,强度和形状三个方面。
从吸收带(K带)位置可估计产生该吸收共轭体系的大小;从吸收带的强度有助于K带,B带和R带的识别;从吸收带的形状可帮助判断产生紫外吸收的基团,如某些芳香化合物,在峰形上可显示一定程度的精细结构。
一般紫外吸收光谱都比较简单,大多数化合物只有一、两个吸收带,因此解析较为容易。
可粗略归纳为以下几点:①如果化合物在220~800nm区间无吸收,表明该化合物是脂肪烃、脂环烃或它们的简单衍生物。
②如果在220~250nm间显示强吸收(ε近10000或更大),表明有R带吸收,即分子结构存在共轭双烯或α,β—不饱和醛、酮。
③如果在250~290nm间显示中等强度(ε为200~1000)的吸收带,且常显示不同程度精细结构,表明结构中有苯环或某些杂芳环的存在。
④如果在290nm附近有弱吸收带(ε<100),则表明分子结构中非共轭羰基。
⑤如果在300nm上有***度吸收,说明该化合物有较大的共轭体系;若***度吸收具有明显的精细结构,说明为稠环芳、稠环杂芳烃或其衍生物。
(二)红外光谱1. 解析红外光谱的三要素(位置、强度和峰形)在解析红外光谱时,要同时注意红外吸收峰的位置,强度和峰形。
吸收位置是红外吸收最重要的特点,但在鉴定化合物分子结构时,应将吸收峰的位置辅以吸收峰强度和峰形综合分析。
每种有机化合物均显示若干吸收峰,对大量红外图谱中各吸收峰强度相互比较,归纳出各种官能团红外吸收强度的变化范围。
只有熟悉各官能团红外吸收的位置和强度处于一定范围时,才能准确推断出官能团的存在2 .确定官能团的方法对于任何有机化合物的红外光谱,均存在红外吸收的伸缩振动和多种弯曲振动。
因此,每一个化合物的官能团的红外光谱图在不同区域显示一组相关吸收峰。
只有当几处相关吸收峰得到确认时,才能确定该官能团的存在。
例1. 甲基(CH3):2960cm-1和2870cm-1为伸缩振动,1460cm-1和1380cm-1为其弯曲振动。
五大波谱解析步骤简述(一)紫外光谱解析UV应用时顾及吸收带的

五大波谱解析步骤简述(一)紫外光谱解析UV应用时顾及吸收带的五大波谱解析步骤简述(一)紫外光谱解析UV应用时顾及吸收带的位置,强度和形状三个方面。
从吸收带(K带)位置可估计产生该吸收共轭体系的大小;从吸收带的强度有助于K带,B带和R带的识别;从吸收带的形状可帮助判断产生紫外吸收的基团,如某些芳香化合物,在峰形上可显示一定程度的精细结构。
一般紫外吸收光谱都比较简单,大多数化合物只有一、两个吸收带,因此解析较为容易。
可粗略归纳为以下几点:①如果化合物在220~800nm区间无吸收,表明该化合物是脂肪烃、脂环烃或它们的简单衍生物。
②如果在220~250nm间显示强吸收(ε近10000或更大),表明有R带吸收,即分子结构存在共轭双烯或α,β—不饱和醛、酮。
③如果在250~290nm间显示中等强度(ε为200~1000)的吸收带,且常显示不同程度精细结构,表明结构中有苯环或某些杂芳环的存在。
④如果在290nm附近有弱吸收带(ε<100),则表明分子结构中非共轭羰基。
⑤如果在300nm上有***度吸收,说明该化合物有较大的共轭体系;若***度吸收具有明显的精细结构,说明为稠环芳、稠环杂芳烃或其衍生物。
(二)红外光谱1. 解析红外光谱的三要素(位置、强度和峰形)在解析红外光谱时,要同时注意红外吸收峰的位置,强度和峰形。
吸收位置是红外吸收最重要的特点,但在鉴定化合物分子结构时,应将吸收峰的位置辅以吸收峰强度和峰形综合分析。
每种有机化合物均显示若干吸收峰,对大量红外图谱中各吸收峰强度相互比较,归纳出各种官能团红外吸收强度的变化范围。
只有熟悉各官能团红外吸收的位置和强度处于一定范围时,才能准确推断出官能团的存在2 .确定官能团的方法对于任何有机化合物的红外光谱,均存在红外吸收的伸缩振动和多种弯曲振动。
因此,每一个化合物的官能团的红外光谱图在不同区域显示一组相关吸收峰。
只有当几处相关吸收峰得到确认时,才能确定该官能团的存在。
简述五种光谱法的原理

简述五种光谱法的原理光谱法是一种常用的分析技术,常常应用于化学、物理和生物学等领域。
根据不同原理和应用领域的不同,可将光谱法分为多种类型。
下面就详细介绍五种常见的光谱法及其原理。
一、紫外-可见吸收光谱紫外-可见吸收光谱是一种测量样品在可见光和紫外光区域吸收的技术。
在该技术中,用一束具有连续波长的光照射样品,然后检测透射光,通过计算样品吸收的光量,可以推断出样品分子的化学结构。
紫外-可见吸收光谱利用的原理是,当样品中的分子吸收可见光或紫外光时,其电子能级会发生跃迁,这个跃迁与分子的化学成分有关,因此,可以通过测量样品吸收的光谱来推断其化学成分。
二、荧光光谱荧光光谱是一种利用样品在受到特定波长激发后发出荧光的技术。
在该技术中,样品收到特定波长的激发光后,会发生电子从基态跃迁到激发态,然后再跃迁回原来的基态时发出荧光。
样品发出的荧光光谱与其分子结构有关,可以用来分析样品的成分和活性。
荧光光谱利用的原理是,荧光发生的条件是样品中存在能级差异,当分子处于激发态时,电子具有更高的能量,可以通过荧光现象发射短波长的光,从而生成荧光光谱。
三、原子吸收光谱原子吸收光谱是一种测量样品中金属和金属离子浓度的技术。
在该技术中,根据不同原子的能级结构,通过特定波长的光激发分子中的特定原子,然后测量样品透射光的强度,从而推断样品中特定原子的浓度。
原子吸收光谱利用的原理是,输入特定波长的光激发样品中的原子,当样品中的特定原子吸收更多的光时,其原子的能级结构会发生变化,从而改变吸收光的强度,因此可以通过测量吸收光的强度来推断样品中特定原子的浓度。
四、红外光谱红外光谱是一种基于样品吸收红外光的技术。
在该技术中,样品收到具有一定波长的红外光后,吸收光的振动能量与样品中的官能团的振动能量有关。
从而,可以通过分析样品吸收红外光的振动频率,推断出样品中所包含的官能团。
红外光谱利用的原理是,各种原子或原子团具有强烈的吸收红外辐射的振动能力,这种振动能力取决于其分子结构的特定配置,因此可以通过测量样品吸收的红外辐射的振动频率和强度来推断样品中的分子结构。
有机波谱解析-第二章 紫外光谱

C
Hale Waihona Puke n<pOC
C
p*
n > p p*
n
n C
p* p
p*
n
p n 非极性
p
O 非极性
C C
p
极性
极性
n → p*跃迁:兰移; ; pp np
(4)尽量和文献中所用的溶剂一致。
(5)溶剂挥发性小、不易燃、无毒性、价格便宜。
5. 电子跃迁的类型
紫外吸收光谱是由价电子的能级跃迁而产生的,有机化 合物的紫外—可见吸收光谱是三种电子跃迁的结果:σ电子、 π电子、n电子。 s* n p* H C O
s
p E 分子轨道理论:成键轨道—反键轨道。
各种电子能级的能量及电子跃迁类型如右图
3. 紫 外 光 谱 图
横坐标:波长或频率 纵坐标:吸光度(A) 或 透过率(T)
紫外光谱(图)的特点: 吸收谱带少; 吸收谱带宽; 通常以谱带吸收最强的波长表示谱带位置,称 为最大吸收波长(λmax) ,是分子的特征常数, 与分子电子结构相关,可推测化合物中生色团类 型和共轭大小; 吸收强度以最大吸收波长处的摩尔吸光系数 (εmax)表示,也是分子特征常数和鉴定化合物 的重要依据。
H H c H
取代基 红移距离 -SR 45(nm)
c H
max=162nm 助色基团取代 p
-NR2 40(nm) -OR 30(nm)
p*(K带)发生红移。
-Cl 5(nm) CH3 5(nm)
(2)共轭烯烃中的
p → p*
波谱分析第二章03,04有机化合物紫外光谱解析

邻 3 7 0 2 13 20
间 3 7 0 2 13 20
对 10 25 10 15 58 85
例1
CO O H
Y= OH X=NH2
基值 取代值
230 nm 58 nm 288 nm
NH2
实测 288 nm
例2 计算4-乙酰氨基苯甲醛 CH3CONH 的K吸收带。
基值 对位NHAc取代 计算值 实测值 250nm 45nm 295nm 292nm
10
35 35 6 15 25
β
12
30 30 6 12 30 80 95
γ
18
17 50 6 12 25
18
31 50 6 12 25
溶剂校正:
二氧六环 +5nm 乙醚 +7nm
氯仿 水
+1nm -8nm
例1
例2
O
基值 烷基取代 α β
215nm 10 nm(=1*10) 12 nm(=1*12) 计算 实测
10~1000
π4 π*
*
π* π3 n π2 π π
*
εmax
n
随着与羰基共轭数目的增加, π→π* 跃迁能量不断降低,K带 迅速红移,且吸收强度增加。 n→π* 跃迁因共轭链的增加影 响较小。
π1
C=C
C=O
C=C
C=O
不饱和羰基分子轨道和电子跃迁
α,β不饱和羧酸及其衍生物
R带比相应醛酮显著地紫移。 RCO-X中,X基团中n占据p轨道与 羰基π轨道发生p-π共轭效应, 形成多电子的大π体系。 X基团的亲电诱导效应羰基n轨道 能级轨道略有降低。 由于反键轨道能级较高,K带也 发生紫移。 烷基取代的α,β不饱和羧酸及 其衍生物,由于 -π共轭效应, K带向红移动。
紫外可见吸收光谱法基本原理和解析

变。在温度和波长等条件一定时,ε 仅与吸收物质本身的性质有关,与待 测物浓度无关。 (3)可作为定性鉴定的参数。
2020/5/24
18
(4)同一吸收物质在不同波长下的ε值 是不同的。在最大吸收波长λmax 处的摩尔吸光系数以εmax表示。
εmax表明了该吸收物质最大限度的 吸光能力,也反映了光度法测定该
不饱和基团称为生色团。简单的生色团由双 键或叁键体系组成,如乙烯基、羰基、亚硝
基、偶氮基-N=N-、乙炔基、腈基—C三N
等。
2020/5/24
33
生色团 烯 炔 羧基 酰胺基 羰基 偶氮基 硝基 亚硝基 硝酸酯
某些常见生色团的吸收光谱
溶剂 正庚烷 正庚烷 乙醇 水 正己烷 乙醇 异辛酯 乙醚
二氧杂环己烷
光区,在200nm左右,ε很大,为强吸收带。 共轭程度越大,所需能量越低,λmax增大。 不饱和烃、共轭烯烃和芳香烃类均可发生该
类跃迁。
如:乙烯、丁二烯、己二烯的λmax分别为
162、217、268nm。
2020/5/24
31
(4)n*跃迁(200~400nm 弱吸收)
(含杂原子的双键不饱和有机化合物) 所需能量最低,吸收波长200~400nm。ε较 小,弱吸收。
紫外可见吸收光谱法基本原理 和解析
2020/5/24 1
一、概述 1.定义:在光谱分析中,依据物质的分子对
光的选择性吸收而建立起来的分析方法称为 吸光光度法。 2.分类 ①红外吸收光谱(IR)
分子振动光谱,吸收光波长范围0.251000 m ,主要用于有机化合物结构鉴定。
(主要应用中红外2.5 25 m )
★用不同波长的单色光照射,测吸光度-- 吸
四、紫外吸收光谱分析

苯: E1带180184nm; e=47000 E2带200204 nm e=7000 苯环上三个共扼双键的 p → p*跃迁特征吸收带; B带230-270 nm
e=200
苯 甲苯 间二甲苯
p → p*与苯环振动引起;
max(nm) 254 261 263
e max 200 300 300
含有未共用电子对的杂原 子(N、O、S、X)的饱和 化合物发生n→σ* 跃迁;
含-NH2 、-OH、-X CH3Br λmax=204nm
例:CH3OH λmax=184nm
(3)π→π*跃迁
π电子跃迁到反键π* 轨道所产生的跃迁,这类跃 迁所需能量比σ→σ*跃迁小,若无共轭,与n→σ* 跃迁差不多。200nm左右 吸收强度大,在104~105范围内,强吸收
四、紫外吸收光谱分析
一、紫外吸收光谱的产生
formation of UV
1.概述
紫外吸收光谱:分子价电子能级跃迁。 波长范围:100-800 nm. (1) 远紫外光区: 100-200nm
(2) 近紫外光区: 200-400nm
(3)可见光区:400-800nm 可用于结构鉴定和定量分析。
电子跃迁的同时,伴随着振
5.溶剂效应
(1 )对最大吸收波长的影响
随着溶剂极性的增大 ——π→π*跃迁吸收峰向长波方向移动,即发生红移 —— n→π*跃迁吸收峰向短波方向移动,即发生蓝移 例:异亚丙基丙酮 O
CH3 C—— 孤立双键的化合物 双键和含杂原子的双 键化合物产生π→π* 、 n→π*、 n→σ*
—— 共轭双键的化合物 使π→π* 所需能量降低, 吸收峰长移,吸收强度增强。
——羰基化合物
紫外吸收光谱分析法紫外吸收光谱基本原理详解演示文稿

E=hv= hc/nλ。h 为Plank常数,h=6.626×10-34Js。
一、紫外吸收光谱的产生
formation of UV
紫• 外吸收光谱:分子价电子能级跃迁。
波长范围:100-800 nm.
(1) 远紫外光区: 100-200nm
最灵敏。吸收曲线是定量分析中选择入射光波长的重要 依据。
3.电子跃迁与分子吸收光谱
物质分子内部三种运动形式: (1)电子相对于原子核的运动; (2)原子核在其平衡位置附近的相对振动; (3)分子本身绕其重心的转动。
分子具有三种不同能级:电子能级、振动能级和转动能级 三种能级都是量子化的,且各自具有相应的能量。 分子的内能:电子能量Ee 、振动能量Ev 、转动能量Er
n→σ* < σ→σ*
2 σ→σ*跃迁
所需能量最大;σ电子只有吸收远紫外光的能量才能发
生跃迁;
饱和烷烃的分子吸收光谱出现在远紫外区;
吸收波长λ<200 nm;
例:甲烷的λmax为125nm , 乙烷λmax为135nm。
只能被真空紫外分光光度计检测到;
s*
作为溶剂使用;
p*
E K
R
E,B
n
p
s
3 n→σ*跃迁
1.紫外—可见吸收光谱
有机化合物的紫外—可见吸收光谱是三种电子跃迁的结果:
σ电子、π电子、n电子。
s*
HC O
s
Hp
n
p*
E K
R
n
E,B
p
s
。 分子轨道理论:成键轨道—反键轨道
当外层电子吸收紫外或可见辐射后,就从基态向激发态(反键轨道)跃 迁。主要有四种跃迁所需能量ΔΕ大小顺序为:n→π* < π→π* <
紫外 波普分析
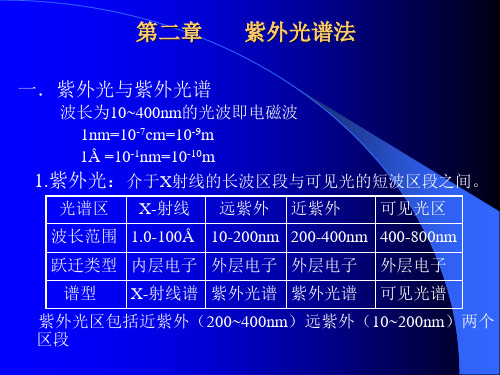
第二章紫外光谱法一.紫外光与紫外光谱波长为10~400nm的光波即电磁波1nm=10-7cm=10-9m1Å=10-1nm=10-10m1.紫外光:介于X射线的长波区段与可见光的短波区段之间。
光谱区X-射线远紫外近紫外可见光区波长范围 1.0-100Å 10-200nm200-400nm400-800nm跃迁类型内层电子外层电子外层电子外层电子谱型X-射线谱紫外光谱紫外光谱可见光谱紫外光区包括近紫外(200~400nm)远紫外(10~200nm)两个区段1.1 紫外光与紫外光谱●远紫外区(10~200nm):在此波长范围内,大气有吸收,必须在真空条件下操作,普通仪器观察不到,对仪器要求高,远紫外也叫真空紫外区,所以远紫外区在普通有机化合物机构分析上没有应用。
●近紫外区(200~400nm):在此波长范围内,玻璃有吸收,一般用石英比色器,因此称近紫外区为石英紫外区,近紫外区最为有用,通常所谓的紫外光谱就是指近紫外区的光谱。
●2.紫外光谱:以波长10~400nm的电磁波照射物质分子,即以紫外光照射物质分子,由分子的电子能级跃迁而产生的光谱叫紫外光谱。
紫外光谱是电子光谱的一部分,可见光谱也是电子光谱,电子光谱是由电子跃迁而产生的吸收光谱的总称。
1.2紫外光谱的产生、特征及表示法●二.紫外光谱的产生、特征及表示法●1.紫外光谱的产生主要是因为物质分子的能量具有量子化的特征(即物质分子的能量具有不连续的特征)。
一个分子有一系列能阶,其中包括许多电子能阶,分子震动能阶以及分子转动能阶。
1.2.1n=24 纯电子跃迁3 1-20ev21纯转动跃迁纯振动跃迁0.05-1ev n=1 0.05ev以下电子,振动,转动能级示意图1.2.2●当分子在入射光的作用下发生了阶电子跃迁,也就是说分子中阶电子由低能级E 0跃迁到高能级E 1(激发态),根据量子理论电子在跃迁时所吸收的能量不是连续的,而是量子化的,即所吸收的光子能量等于两个能级的差值:●△E=E 1-E 0=hv=h·c/λ(v=c/λ)●式中:h=Plank 常数=6.62×10-27尔格·秒c=光速3×1010cmλ=波长用nm 表示v=频率用周/秒(Cps )或赫兹(Hz )E=能量单位为尔格,电子伏特ev 或卡/摩尔1.2.3●分子的内部运动包括有转动、振动和电子运动。
紫外光谱的解析

紫外光谱的解析一、紫外光谱的基本原理1. 概念•紫外光谱(UV)是分子吸收紫外•可见光区(200•800nm)的电磁波而产生的吸收光谱。
它反映了分子中的电子跃迁情况。
当分子吸收紫外光时,分子中的价电子从低能级跃迁到高能级。
•例如,在一些有机化合物中,存在着π电子和n电子(非键电子)。
这些电子可以发生π• π跃迁、n• π跃迁等。
其中,π• π跃迁通常所需能量较高,对应的吸收波长相对较短,多在200nm左右;而n• π跃迁所需能量较低,吸收波长相对较长,一般在270• 350nm范围。
2. Lambert - Beer定律•这是紫外光谱分析的基本定律,其表达式为 A = εbc。
其中,A是吸光度,表示物质对光的吸收程度;ε是摩尔吸光系数,它与物质的性质有关,反映了物质对特定波长光的吸收能力,单位为L/(mol·cm);b是光程长度,即样品池的厚度,单位为cm;c是溶液中物质的摩尔浓度,单位为mol/L。
•例如,在测定某一化合物的浓度时,如果已知其摩尔吸光系数和光程长度,通过测量吸光度就可以计算出溶液中的物质浓度。
假设某物质的摩尔吸光系数为1000L/(mol·cm),光程长度为1cm,测得吸光度为0.5,根据Lambert• Beer定律,可算出该物质的浓度c = A/(εb)=0.5/(1000×1)= 5×10⁻⁴mol/L。
二、紫外光谱中的特征吸收带1. R带• R带是由n•π跃迁产生的吸收带。
其特点是吸收强度较弱,摩尔吸光系数一般在10• 100L/(mol·cm)范围内,吸收峰波长较长,多在270• 350nm。
•在醛、酮、硝基化合物等分子中常常可以观察到R带。
例如,丙酮分子中的羰基(C = O)上的n电子可以发生n• π跃迁,在约279nm处有一个R带吸收峰。
2. K带• K带是由共轭体系中的π• π跃迁产生的吸收带。
其吸收强度较大,摩尔吸光系数通常大于10000L/(mol·cm),吸收峰波长与共轭体系的大小有关。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
五大波谱解析步骤简述
(一)紫外光谱
解析UV应用时顾及吸收带的位置,强度和形状三个方面。
从吸收带(K带)位置可估计产生该吸收共轭体系的大小;从吸收带的强度有助于K带,B带和R带的识别;从吸收带的形状可帮助判断产生紫外吸收的基团,如某些芳香化合物,在峰形上可显示一定程度的精细结构。
一般紫外吸收光谱都比较简单,大多数化合物只有一、两个吸收带,因此解析较为容易。
可粗略归纳为以下几点:
①如果化合物在220~800nm区间无吸收,表明该化合物是脂肪烃、脂环烃或它们的简单衍生物。
②如果在220~250nm间显示强吸收(ε近10000或更大),表明有R带吸收,即分子结构存在共轭双烯或α,β—不饱和醛、酮。
③如果在250~290nm间显示中等强度(ε为200~1000)的吸收带,且常显示不同程度精细结构,表明结构中有苯环或某些杂芳环的存在。
④如果在290nm附近有弱吸收带(ε<100),则表明分子结构中非共轭羰基。
⑤如果在300nm上有***度吸收,说明该化合物有较大的共轭体系;若***度吸收具有明显的精细结构,说明为稠环芳、稠环杂芳烃或其衍生物。
(二)红外光谱
1. 解析红外光谱的三要素(位置、强度和峰形)
在解析红外光谱时,要同时注意红外吸收峰的位置,强度和峰形。
吸收位置是红外吸收最重要的特点,但在鉴定化合物分子结构时,应将吸收峰的位置辅以吸收峰强度和峰形综合分析。
每种有机化合物均显示若干吸收峰,对大量红外图谱中各吸收峰强度相互比较,归纳出各种官能团红外吸收强度的变化范围。
只有熟悉各官能团红外吸收的位置和强度处于一定范围时,才能准确推断出官能团的存在
2 .确定官能团的方法
对于任何有机化合物的红外光谱,均存在红外吸收的伸缩振动和多种弯曲振动。
因此,每一个化合物的官能团的红外光谱图在不同区域显示一组相关吸收峰。
只有当几处相关吸收峰得到确认时,才能确定该官能团的存在。
例1. 甲基(CH3):2960cm-1和2870cm-1为伸缩振动,1460cm-1和1380cm-1为其弯曲振动。
例2. 亚甲基(CH2):2920cm-1和2850cm-1为其伸缩振动,1470cm-1和720cm-1为其弯曲振动。
例3. 酯基:νC=O为1750~1725cm-1,νC-O在1300~1050cm-1有两个吸收谱带。
l3.3 红外光谱解析的顺序
(1)根据确定的分子,计算不饱和度,预测可能的官能团。
(2)首先观察红外光谱的官能团区,找出该化合物可能存在的官能团。
(3)查看红外光谱的指纹区,找出官能团的相关吸收峰,最后才确定该化合物存在某官能团。
(4)判断是否芳香族化合物,若为芳香化合物,找出苯的取代位置。
(5)根据红外光谱指纹区的吸收峰与已知化合物的红外光谱或标准图谱对照,确定是否为已知化合物。
(三)核磁共振氢谱
核磁共振技术发展较早,20世纪70年代以前,主要是核磁共振氢谱的研究和应用。
70年代以后,随着傅里叶变换波谱仪的诞生,13C —NMR的研究迅速开展。
由于1H—NMR的灵敏度高,而且积累的研究资料丰富,因此在结构解析方面1H—NMR的重要性仍强于13C —NMR。
解析图谱的步骤
1.先观察图谱是否符合要求;①四甲基硅烷的信号是否正常;②杂音大不大;③基线是否平;④积分曲线中没有吸收信号的地方是否平整。
如果有问题,解析时要引起注意,最好重新测试图谱。
2.区分杂质峰、溶剂峰、旋转边峰(spinning side bands)、13C卫星峰(13C satellite peaks)
(1)杂质峰:杂质含量相对样品比例很小,因此杂质峰的峰面积很小,且杂质峰与样品峰之间没有简单整数比的关系,容易区别。
(2)溶剂峰:氘代试剂不可能达到100%的同位素纯度(大部分试剂的氘代率为99-99.8%),因此谱图中往往呈现相应的溶剂峰,如CDCL3中的溶剂峰的δ值约为7.27 ppm处。
(3)旋转边峰:在测试样品时,样品管在1H-NMR仪中快速旋转,当仪器调节未达到良好工作状态时,会出现旋转边带,即以强谱线为
中心,呈现出一对对称的弱峰,称为旋转边峰。
(4)13C卫星峰:13C具有磁距,可以与1H偶合产生裂分,称之为13C卫星峰,但由13C的天然丰度只为1.1%,只有氢的强峰才能观察到,一般不会对氢的谱图造成干扰。
3.根据积分曲线,观察各信号的相对高度,计算样品化合物分子式中的氢原子数目。
可利用可靠的甲基信号或孤立的次甲基信号为标准计算各信号峰的质子数目。
4.先解析图中CH3O、CH3N、、CH3C=O、CH3C=C、CH3-C等孤立的甲基质子信号,然后再解析偶合的甲基质子信号。
5.解析羧基、醛基、分子内氢键等低磁场的质子信号。
6.解析芳香核上的质子信号。
7.比较滴加重水前后测定的图谱,观察有无信号峰消失的现象,了解分子结构中所连活泼氢官能团。
8.根据图谱提供信号峰数目、化学位移和偶合常数,解析一级类型图谱。
9.解析高级类型图谱峰信号,如黄酮类化合物B环仅4,-位取代时,呈现AA,BB,系统峰信号,二氢黄酮则呈现ABX系统峰信号。
10. 如果一维1H-NMR难以解析分子结构,可考虑测试二维核磁共振谱配合解析结构。
11. 组合可能的结构式,根据图谱的解析,组合几种可能的结构式。
12. 对推出的结构进行指认,即每个官能团上的氢在图谱中都应有相应的归属信号。
(四)核磁共振碳谱(13C—NMR)
解析图谱的步骤
1.鉴别谱图中的非真实信号峰
(1)溶剂峰:虽然碳谱不受溶剂中氢的干扰,但为兼顾氢谱的测定及磁场需要,仍常采用氘代试剂作为溶剂,氘代试剂中的碳原子均有相应的峰。
(2)杂质峰:杂质含量相对于样品少得多,其峰面积极小,与样品化合物中的碳呈现的峰不成比例。
(3)测试条件的影响:测试条件会对所测谱图有较大影响。
如脉冲倾斜角较大而脉冲间隔不够长时,往往导致季碳不出峰;扫描宽度不够大时,扫描宽度以外的谱线会折叠到图谱中来;等等,均造成解析图谱的困难。
2.不饱和度的计算
根据分子式计算的不饱和度,推测图谱烯碳的情况。
3.分子对称性的分析
若谱线数目等于分子式中碳原子数目,说明分子结构无对称性;若谱线数目小于分子式中碳原子数目,说明分子结构有一定的对称性。
此外,化合物中碳原子数目较多时,有些核的化学环境相似,可能δ值产生重叠现象,应予以注意。
4.碳原子δ值的分区
碳原子大致可分为三个区
(1)高δ值区δ>165ppm,属于羰基和叠烯区:①分子结构中,如
存在叠峰,除叠烯中有高δ值信号峰外,叠烯两端碳在双键区域还应有信号峰,两种峰同时存在才说明叠烯存在;②δ>200 ppm的信号,只能属于醛、酮类化合物;③160-180ppm的信号峰,则归属于酸、酯、酸酐等类化合物的羰基。
(2)中δ值区δ90-160ppm(一般情况δ为100-150ppm)烯、芳环、除叠烯中央碳原子外的其他SP2杂化碳原子、碳氮三键碳原子都在这个区域出峰。
(3)低δ值区δ<100ppm,主要脂肪链碳原子区:①不与氧、氮、氟等杂原子相连的饱和的δ值小于55ppm;
②炔碳原子δ值在70-100ppm,这是不饱和碳原子的特例。
5.碳原子级数的确定
由低共振或APT(attached proton test)、DEPT(distortionless enhancement by polarization transfer)等技术可确定碳原子的级数,由此可计算化合物中与碳原子相连的氢原子数。
若此数目小于分子式中的氢原子数,二者之差值为化合物中活泼氢的原子数。
6.推导可能的结构式
先推导出结构单元,并进一步组合成若干可能的结构式。
7.对碳谱的指认
将碳谱中各信号峰在推出的可能结构式上进行指认,找出各碳谱信号相应的归属,从而在被推导的可能结构式中找出最合理的结构式,即正确的结构式。