药物分析1.7含量测定
高效液相色谱法测定福多司坦原料药的含量

高效液相色谱法测定福多司坦原料药的含量目的建立福多司坦原料药含量的测定方法。
方法采用高效液相色谱法,以十八烷基硅烷键合硅胶柱C18(250 mm×4.6 mm,5mm)为色谱柱,水-甲醇(99.5 :0.5)为流动相,流速为0.8 mL/min,检测波长:220 nm,温度:30 ℃。
结果福多司坦的线性范围为y=1 575.2x-1 319,r=0.989 242,平均回收率为95.89%(RSD=1.7%)。
结论该法精确可靠,简单方便,建立了HPLC法测定福多司坦原料药中的福多司坦含量的方法。
标签:福多司坦;高效液相色谱法;含量测定福多司坦为S-(3-羟基丙基)-L-半胱氨酸是无臭,无味的类白色至微黄色结晶或结晶性粉末,由SSP公司和三菱Welfide公司生产的一种新型的祛痰药[1]。
福多司坦结构中有非游离密封的巯基,对局部黏蛋白无活性作用,口服后经代谢产生含有游离巯基的代谢产物而发挥药理作用,因而用药后无明显胃肠道副作用[2]。
实验证明,福多司坦可以改变分泌物的组成和流变学性质,体内代谢物导致支气管分泌物中黏蛋白的二硫键断裂,降低痰液黏度,使受抑制的呼吸功能得到改善;还可以改善和增强抗生素对支气管黏膜的渗透作用,利于呼吸道各种炎症的治疗[3]。
国外临床研究表明与羧甲司坦、乙酰半胱氨酸和氨溴索等同类产品比较,本品对支气管扩张、慢性支气管炎、支气管哮喘、弥漫性支气管炎、尘肺症、肺气肿、肺结核和非典型耐酸杆菌感染症具有很好的疗效[4]。
作为新型作用的去痰剂,该药对气道上皮杯细胞的过量形成起抑制作用,大剂量给药没有发生药物蓄积,肝和肾功能中度障碍[5]。
为了确保该药质量,本研究建立了HPLC 测定福多司坦原料药含量的方法,具有快速、简便、专属性好、结果测定准确、可靠等优点[6]。
1 材料与方法1.1 仪器Agilnt1100型高效液相色谱仪;自动进样器;DAD检测器;Agilent Chemstation色谱工作站。
药物分析含量测定方法与验证

药物分析含量测定方法与验证选择合适的测定方法是药物分析含量测定的首要任务。
常用的测定方法包括色谱法、光谱法、电化学法和滴定法等。
在选择方法时,需要考虑以下几个因素:1.应用对象:考虑药物的理化性质和活性成分的特点,选择适合的分析方法。
例如,对于具有荧光特性的活性成分,可以选择荧光光谱法进行分析。
2.灵敏度和准确度:选取具有良好灵敏度和准确度的方法,以确保对样品中微量活性成分的准确测定。
例如,高效液相色谱法(HPLC)和气相色谱法(GC)是常用的灵敏度高的色谱法。
3.可行性和经济性:考虑方法的可行性和经济性,选择合适的仪器设备和试剂,以确保分析成本和操作难度的控制。
在药物分析含量测定方法选择后,需要对方法进行验证以确保其准确性和可靠性。
药物分析含量测定方法的验证主要包括以下几个方面:1.精密度和重复性:重复测定多个样品,并计算其相对标准偏差(RSD)来评估方法的精密度。
RSD越小,说明方法的重复性越好。
2.准确度:与已知浓度的标准样品进行比较,计算方法的准确度。
一般使用回收率作为评价指标,回收率越接近100%,说明方法的准确度越高。
3.灵敏度和线性:测定一系列不同浓度的样品,并计算测定结果的线性相关性。
通常使用相关系数和回归方程来评估方法的线性。
4.特异性和选择性:分析样品中可能存在的干扰物质,并验证方法对活性成分的选择性。
一般通过添加干扰物质来测试方法的特异性。
5.稳定性:测试方法在不同条件下的稳定性,包括温度、湿度、光照等。
确保方法在不同条件下的测定结果一致。
除了以上验证的基本步骤,还需要根据具体的药物分析含量测定方法的特点和要求,进行其他适当的验证参数的测试。
在实际操作中,应制定详细的验证方案和实施计划,并按照规定的方案进行验证实验。
实验结果需要进行统计分析,并按规定的标准判断方法的验证结果。
综上所述,药物分析含量测定方法的选择和验证是确保药物质量稳定性和一致性的重要环节。
通过选择合适的方法和进行严格的验证,可以保证药物分析结果的准确性和可靠性,并为药物研发和生产提供科学依据。
药物分析含量测定练习题

药物分析含量测定练习题药物分析含量测定练习题药物分析是药学中的一项重要内容,它关乎到药物的质量和安全性。
而药物分析含量测定是药物分析中的一个关键环节。
下面,我们来做一些药物分析含量测定的练习题,以加深对这个重要概念的理解。
1. 问题描述:某药厂生产的某种药物,标签上标明每片含有100毫克的活性成分A。
现从该批药物中取出10片,经适当处理后,得到溶液B。
取溶液B的适量,加入适量的试剂C,反应得到产物D。
将产物D经适当稀释后,用紫外分光光度计测定其吸收值为0.6。
已知标准曲线的方程为A=0.02C+0.1,其中A为吸光度,C为活性成分A的浓度。
请计算该批药物中活性成分A的含量。
解析:首先,我们需要计算产物D的浓度。
根据标准曲线方程,吸光度为0.6时,活性成分A的浓度C为(0.6-0.1)/0.02=25毫克/升。
由于我们取的是10片药物,所以溶液B中的活性成分A的浓度为25毫克/升*10片=250毫克/升。
而每片药物中的活性成分A的含量为250毫克/升*1000毫升/升=250000微克/片。
2. 问题描述:某药厂生产的某种药物,标签上标明每粒含有50毫克的活性成分B。
现从该批药物中取出5粒,经适当处理后,得到溶液E。
取溶液E的适量,加入适量的试剂F,反应得到产物G。
将产物G经适当稀释后,用高效液相色谱法测定其峰面积为1000。
已知标准曲线的方程为B=0.005A,其中B为活性成分B的浓度,A为峰面积。
请计算该批药物中活性成分B的含量。
解析:根据标准曲线方程,峰面积为1000时,活性成分B的浓度B为0.005*1000=5毫克/升。
由于我们取的是5粒药物,所以溶液E中的活性成分B的浓度为5毫克/升*5粒=25毫克/升。
而每粒药物中的活性成分B的含量为25毫克/升*1000毫升/升=25000微克/粒。
3. 问题描述:某药厂生产的某种药物,标签上标明每毫升含有10毫克的活性成分C。
现从该批药物中取出10毫升,经适当处理后,得到溶液H。
UPLC法测定生长抑素原料药的含量

超高效液相色谱法测定生长抑素原料药的含量摘要:建立快速,准确,灵敏的测定生长抑素原料药的方法。
方法采用沃特世ACQUITY UPLC超高液相色谱系统,以磷酸(取磷酸11Ml,加水900 mL,用三乙胺调PH至2.3,用水稀释至刻度1000)为流动相,(A)—乙腈(B)梯度洗脱,流速:0.3ml/min;检测波长为215nm; 柱温45℃;进样体积2μL。
结果生长抑素在0.0051492mg/ml—0.12873mg /ml范围内有良好的线性关系(R2 = 0.9993),平均回收率(n=9)为99.1%。
该检测快速,准确,灵敏度高,重复性好,为生长抑素原料药的质量控制提供了一种快速准确的方法。
关键词:超高效液相色谱生长抑素原料药生长抑素(Somatostatin)主要用于严重急性食道静脉曲张出血、急性胃或十二指肠溃疡出血、并发性急性糜烂性胃炎或出血性胃炎、胰、胆和肠瘘的辅助治疗,胰腺手术后并发症的预防和治疗,糖尿病酮症酸中毒的辅助治疗[1-3]。
超高效液相色谱法是一个新兴的领域,Waters ACQUITY UPLC™系统也出现不久,目前已发表的应用资料还很不足。
与传统的HPLC相比,超高效液相色谱法的速度、灵敏度及分离度分别是HPLC的9倍、3倍及1.7倍[4]。
目前,UPLC多应用于代谢组学分析及其他一些生化领域,在天然产物的分析方面运用也逐渐兴起,因为在这些领域深入研究需要更高的分析精度。
使用超高效液相色谱法对天然产物分析,特别是药物分析研究领域的发展将是一个极大的促进[5]。
本实验选择采用超高效液相色谱法(UPLC)进行生长抑素原料药含量的测定,为提升生长抑素原料药的质量标准提供方法依据。
2 材料与方法2.1材料2.1.1仪器沃特世ACQUITY UPLC超高液相色谱系统,配置ACQUITY PDA检测器及Empower2色谱管理软件(Mitford,Boston,MA,USA),十万分之一电子天平(型号: XP 205),超纯水制备仪(型号:arium 611UV),pH计(型号:PHS-3C)2.1.2 试剂乙腈为色谱纯(上海星可生化有限公司,批号:200912112),磷酸为分析纯(广东光华化学厂有限公司,批号:20070123),三乙胺为分析纯(东光华化学厂有限公司,批号:220081010),生长抑素原料药对照品(深圳市翰宇药业股份有限公司,批号:20090101 含量:8.3% ),生长抑素原料药样品(深圳市翰宇药业股份有限公司,批号:20090801)。
双标多测法在柏子养心丸5个成分含量测定中的应用
药 物 分 析 杂 志 Chin J Pharm Anal 2021,41(1)
·1 3 5·
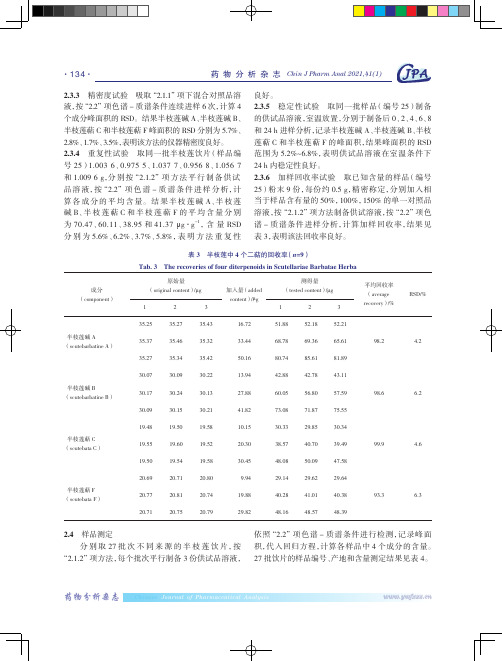
表 4 27 批国内市售半枝莲饮片中 4 个二萜的含量(平均值 ±SD,n=3)
Tab. 4 Contents of four diterpenoids in S. barbata of 27 batches collected from domestic origins(mean±SD,n=3)
成分 (component)
表 3 半枝莲中 4 个二萜的回收率(n=9) Tab. 3 The recoveries of four diterpenoids in Scutellariae Barbatae Herba
原始量 (original content)/μg
1
2
3
加入量(added content)/μg
明 2 种溶剂对分析物的提取率没有差别,不过甲醇作 统,来提高离子化效率。采用线性梯度洗脱,让各成
为溶剂的供试品溶液的色谱峰形明显好于乙醇,因此 分均在流动相中的有机相比例较低时出峰,利于保证
最终选择甲醇作为提取溶剂。
较高的离子化效率。在本文选择的流动相的溶剂系
超声波提取法具有提取效率高,提取时间短,提 统和梯度洗脱程序下,4 个成分的色谱峰形好,获得
10 批、7 批和 4 批,可以看到,即使相同产地的样品,
3.2 质谱参数的优化
4 个二萜类成分的含量也有明显差异,可见产地因素
因此选择超声波提取法。
不同来源饮片中均能检出并定量,但含量波动范围都
为使各成分提取完全,固定超声提取时间为 1 h。 较大,4 个成分在饮片中的最大含量分别约是最低含
由于半枝莲中二萜类成分含量较低,另外参考《中华 量的 10、8、5、10 倍。其中,在 22 批次饮片中半枝莲
药物分析含量测定结果计算

药物分析含量测定结果计算药物分析中,含量测定是非常重要的一项工作。
本文将从实验步骤和结果分析两个方面,介绍药物分析含量测定结果的计算方法。
实验步骤1. 确定药物的纯度首先,需要确定药物的纯度。
这可以通过比色法、化学分析法及物理分析法等多种方法来确定。
确定药物纯度的目的是为了将含量测定的样品溶解度确定为已知量。
2. 药物样品预处理在进行含量测定前,需要对药物样品进行预处理。
具体步骤如下:1.将药物样品称取一定量,加入清洗过的烧杯中。
2.加入适量的溶剂,使药物充分溶解。
3.插入磁子,将烧杯放在磁子上,加热搅拌使药物充分溶解。
3. 确定含量测定方法在选择含量测定方法时,需要根据药物的性质和目的来选择。
比如,对于含有羰基、羧基等官能团的药物,可以选择紫外分光光度法进行含量测定。
4. 构建标准曲线通过测定一系列药物浓度下的吸光度值,可以构建出一条标准曲线,用于测定实验样品中药物的浓度。
5. 测定实验样品浓度测定实验样品浓度的方法与构建标准曲线类似。
首先需要将样品溶解,然后根据已经构建好的标准曲线中吸光度与药物浓度的对应关系,计算出样品中药物的浓度。
结果分析1. 常用计算公式在分析含量测定结果时,需要掌握一些常用的计算公式,如下:1.药物的含量计算公式:含量(%)= 实验样品中药物的浓度(mg/mL) / 药物纯度 × 100%2.相对标准偏差计算公式: RSD(%)= 标准偏差 / 平均值 × 100%2. 结果解读根据药物的含量测定结果,可以进行一些结果解读。
例如,当实验样品中药物浓度接近标准品时,说明样品处理的过程中没有发生重大误差。
当样品处理过程中出现大量的杂质和干扰时,样品测定结果会偏高,因此需要严格按照实验步骤进行操作。
药物分析含量测定的结果计算需要掌握一些基本的实验步骤和计算方法。
通过合理的样品预处理、选择合适的测定方法和构建标准曲线等措施,可以获得准确可靠的含量测定结果。
药物分析-含量测定结果计算

第四节 含量测定结果计算
22.10 × 8.806×1.025 标示量 = % ×100% = 99.7% 0.1 4 × ×1000 2
习题: 习题: 安乃近注射液含量测定方法如下,精密量取本品( 安乃近注射液含量测定方法如下,精密量取本品(规 格为1ml:0.25g 10ml, 100ml量瓶中 加乙醇80ml 1ml:0.25g) 量瓶中, 80ml, 格为1ml:0.25g)10ml,置100ml量瓶中,加乙醇80ml, 再加水稀释至刻度,摇匀,立即精密量取10ml 10ml, 再加水稀释至刻度,摇匀,立即精密量取10ml,置锥 形瓶中,加乙醇20ml 20ml, 6.5ml与甲醛溶液0.5ml, 与甲醛溶液0.5ml 形瓶中,加乙醇20ml,水6.5ml与甲醛溶液0.5ml,放 分钟,加盐酸溶液(9→1000)1.0ml,摇匀, 置1分钟,加盐酸溶液(9→1000)1.0ml,摇匀,用碘 滴定液(0.1mol/L, =1.010)滴定, 滴定液(0.1mol/L,F =1.010)滴定,至溶液所系显 的淡黄色在35秒内不腿,消耗碘滴定液(0.1mol/L) 35秒内不腿 的淡黄色在35秒内不腿,消耗碘滴定液(0.1mol/L) 14.09ml, 1ml的碘滴定液 0.1mol/L) 的碘滴定液( 14.09ml,每1ml的碘滴定液(0.1mol/L)相当于 17.57mg的 17.57mg的C13H16N3NaO4S H2O。计算本品注射液中安乃 近(C13H16N3NaO4S H2O)含量相当于标示量的百分含 的百分数(标示量% 的百分数(标示量%)
第四节 含量测定结果计算
平均片重 V × T × F ×平均片重 标示量%= 标示量%= W ×标示量 21.95× 21.95×18.02/1000 ×0.9850 ×10.2114/20 = 1.0321 ×0.2 =96.4% ×100 ×100
《药物分析》第6章:药物的含量测定方法.

化学键合相色谱:最广泛的
将固定相的官能团键合在载体上,形成的固定相 ,一般属于 分配色谱法,不易流失
19:46
(一)基本概念:
1.色谱图:信号---时间曲线 2.基线:无样品时,用流动相冲洗检测出的流出曲线,一般平行于时间 轴 3.噪声:基线信号的波动 4.漂移:基线随时间的变化 5.色谱峰: 6.拖尾因子:衡量色谱峰的对称性, 应为0.95-1.05 7.峰宽: 8.半峰宽 9.标准偏差 10.峰面积 11.保留时间 12.理论塔板数(柱效):表示色谱柱的分离效率
19:46
沉淀滴定法:以沉淀反应为基础,多以硝酸银为
滴定液,也称银量法。按所用指示剂的不同分为 铬酸钾指示剂法 铁铵矾指示剂法 吸附指示剂法
非水滴定法:在非水溶剂(有机溶剂与不含水的
无机溶剂)中进行滴定分析的方法。 非水碱量法:是以冰醋酸为溶剂,高氯酸为滴定液, 甲紫微指示剂,测定弱碱性药物及其盐类 非水酸量法:是在碱性溶液中,以甲醇钠为滴定液, 麝香草酚蓝为指示剂,二甲基甲酰胺等为溶剂, 滴定弱酸性药物
19:46
(二)基本原理
待分离物质在两相间进行分配时,在固定相中溶解度较小 的组分,在色谱柱中向前迁移速度较快;在固定相中溶解 度较大的组分,在色谱柱中向前迁移速度较慢,从而达到 分离的目的。 依固定相与流动相极性的不同,分为正相色谱和反相色谱 1.正相色谱法 流动相极性小于固定相
一般用极性物质作固定相,非极性溶剂(如苯、正己烷等)作流动相,主要 用于分离极性化合物,极性小的组分先流出,极性大的组分后流出。
V F T 100 % W 1000
19:46
3.滴定的分类
按反应方式分类: 直接滴定法:滴定液与被测物质直接反应
药物分析之西药分析——含量均匀度检查法

量均匀度系指小剂量片剂、膜剂、胶囊剂或注射用无菌粉末等制剂中的每片(个)含量偏离标示量的程度。
凡检查含量均匀度的制剂,不再检查重(装)量差异。
除另有规定外,取供试品10片(个),照各药品项下规定的方法,分别测定每片以标示量为100的相对含量X,求其均值X和标准差S①以及标示量与均值之差的绝对值A(A=│100-X|);如A+1.80S≤15.0,即供试品的含量均匀度符合规定;若A+S>15.0,则不符合规定;若A+ 1.80S >15.0,且A+S≤15.0,则应另取20片(个)复试。
根据初、复试结果,计算30片(个)的均值X、标准差S和标示量与均值之差的绝对值A;如A+1.45S≤15.0,即供试品的含量均匀度符合规定;若A+1.45S>15.0,则不符合规定。
如该药品项下规定含量均匀度的限度为±20%或其他百分数时,应将上述各判断式中的15.0改为20.0或其他相应的数值,但各判断式中的系数不变。
在含量测定与含量均匀度检查所用方法不同时,而且含量均匀度未能从响应值(如吸收度)求出每片含量情况下,可取供试品10片(个),照该药品含量均匀度项下规定的方法,分别测定,得仪器测定法的响应值Y(可为吸收度、峰面积等),求其均值Y。
另由含量测定法测得以标示量为100的含量X<[A] >,由X<[A]>除以响应值的均值Y,得比例系数K(K=X<[A]>/Y)。
将上述诸响应值Y与K相乘,求得每片标示量为100的相对百分含量X(X=KY),同上法求其均值X和S以及A,计算,判定结果,即得。
药物分析含量测定结果计算

药物分析含量测定结果的计算原料药 以实际百分含量表示:片剂 片剂的含量测定结果常用含量占标示量的百分比表示:标示量%═%100⨯标示量每片的实际含量═%100⨯⨯标示量平均片重取样量测得量m m注射液 注射液的含量测定结果一般用实测浓度占标示浓度的百分比表示:1. 原料药含量测定结果的计算 (1)滴定分析法① 直接滴定法: (无空白)T ——滴定度(g/mL),每毫升滴定液相当于被测组分的克数 V ——滴定时,供试品消耗滴定液的体积(mL ) F ——浓度校正因子W ——供试品的质量 (g)例1:P 93 例题例2:非那西丁含量测定:精密称取本品0.3630g 加稀盐酸回流1小时后,放冷,用亚硝酸钠液(0.1010mol/L )滴定,用去20.00mL 。
每1mL 亚硝酸钠液(0.1mol/L )相当于17.92mg 的C 10H 13O 2N 。
计算非那西丁的含量为(E )A. 95.55%B. 96.55%C. 97.55%D. 98.55%E. 99.72% %100⨯=WTVF百分含量%72.99%1003630.0101.01010.000.2092.17%3=⨯⨯⨯⨯=-非那西丁%100⨯=取样量测得量百分含量m m %100%⨯=标示实测标示量c c 标准实际c c F =② 剩余滴定法 (做空白)V 0——滴定时,空白消耗滴定液的体积(mL ) 其他符号的意义同直接滴定法含量计算公式例1:P 94 例题例2:精密称取青霉素钾供试品0.4021g ,按药典规定用剩余碱量法测定含量。
先加入氢氧化钠液(0.1mol/L)25.00mL ,回滴时消0.1015mol/L 的盐酸液14.20mL ,空白试验消耗0.1015mol/L 的盐酸液24.68mL 。
求供试品的含量,每1mL 氢氧化钠液(0.1mol/L)相当于37.25mg 的青霉素钾。
(2)紫外分光光度法① 吸收系数法例——P 122:(1)对乙酰氨基酚的含量测定方法为:取本品约40mg ,精密称定,置250mL 量瓶中,加0.4%氢氧化钠溶液50mL 溶解后,加水至刻度,摇匀,精密量取5mL ,置100mL 量瓶中,加0.4%氢氧化钠溶液10mL ,加水至刻度,摇匀,照分光光度法,在257nm 的波长处测定吸收度,按C 8H 9NO 2的吸收系数( )为715计算,即得。
药物分析关于含量测定

方法选择药物的含量测定所采用的分析方法一般要求操作简便,结果准确重现。
但对于药物的不同形式,其含量测定方法的选择依据有所侧重。
对于化学原料药的含量测定,因为纯度较高、所含杂质较少、故强调测定结果的准确和重现。
通常要求方法具有更高的准确度和精密度,首选容量分析法,如用容量法不适宜时,可考虑选用HPLC法,尤其在有关物质干扰,或多组份物质时,具有特殊优势。
因仪器或操作间的偏差较大,一般不选用UV法,尤其不首选“吸收系数法”定量,不选用末端吸收峰作测定波长 须用UV法时,可采用不受仪器及其它变化影响的“对照品比较法”定量,测定溶液的吸收度宜在0.3—0.7间。
元素测定法如定氮法,在其它方法均不适宜时可采用,但因不能反映化合物含量的变化情况,此法不可用于稳定性考察中。
对于药物制剂的含量测定 则因为制剂组分复杂、干扰物之多,且含量限度一般较宽,故更加强调方法的灵敏度和专属性或则选择性,选用方法时应考虑辅料等的干扰,首选方法一般为HPLC法,在有关物质、辅料不干扰的情况下,也可选用UV法或原料药项下的容量法,复方制剂首选HPLC法较为适宜。
而对于药物制剂的定量检查,如溶出度、含量均匀度检查中药物的溶出量或则含量测定,因为分析样本量较大、且限度亦较宽,在辅料不干扰测定时宜选用管谱分析法。
药品的含量或效价是评定药品的主要指标之一,设计其测定方法时,应根据药品特性、剂型、处方、鉴别试验和纯度检查综合考虑,当鉴别试验和纯度检查保证了专属性和纯度的情况下,含量测定方法的选择要着眼于准确性、稳定性和可重复性。
原料药含量的测定一选用原则:对于组份单一的原料药,首选精密度高,操作简便、快速的容量法测定含量,可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件1反应须按一个方向完全进行2反应要迅速,必要时可通过加热或加入催化剂等方法提高反应速度3共存物不得干扰主要反应,或能用适当方法消除4确定等当点的方法要简单、灵敏5标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定,标化时不发生副反应等要求。
药物分析第四章药物的含量测定

1.7 非水溶液滴定法:。。。。。
1.6 非水滴定法
p228
非水滴定法是在非水溶液中进行的滴 定分析方法。 非水溶剂指的是有机溶剂和不含水的 无机溶剂。 以非水溶剂为介质,不仅能增大有机 化合物的溶解度,而且能使在水中进 行不完全的反应进行完全,从而扩大 了滴定分析的范围
1.7 非水滴定法
第二节 容量分析法
容量分析也称滴定分析,是经典的分析方 法 特点:准确度高;操作简便、快速;仪器 简单,价廉; 缺点:取样量较大和专属性较差,
容量分析多用于API的含量测定。
第四章 药物的含量测定
第一节 第二节 第三节 第四节 第五节 第六节 概述 容量分析法 分光光度法 色谱法 原子吸收法 其他方法
方法: 取本品0.4g,精密称定,精密加氢氧化钠滴 定液(0.1mol/L)25ml,微温溶解,放冷, 加中性乙醇100ml,加溴麝香草酚蓝指示液 10滴,用盐酸滴定液(0.1mol/L)滴定,并 将滴定的结果用空白试验校正。每1ml的氢 氧化钠滴定液(0.1mol/L)相当于35.14mg 的C14H13N3O4S2。
m 滴定液的摩尔浓度 mol/L M 被测物的毫摩尔质量 mg/mmol
制剂的含量计算
每单元制品中的含有量=
V×F×T W ×平均单剂重量
相当于标示量的百分含量=
V×F×T W × 平均单剂重量 ×100% 标示量
常用滴定剂与相应的基准物质
反应类型 酸碱滴定 酸碱滴定 非水酸碱滴定 碘量法 碘量法 络合滴定 滴定剂 氢氧化钠滴定液 盐酸/硫酸滴定液 基准物质 指示剂 邻苯二甲酸氢钾 酚酞 无水碳酸钠 甲基红-溴甲酚绿
巴比妥类药物具有 弱酸性 巴比妥类药物分子结构中都有 1,3二酰亚胺基团,能发生酮式和烯醇式的
药物分析技术 第五章 药物含量测定

2.滴定液的配制和标定
配制方法
▪ 1.直接法:准确称取--溶解--定容 ▪ 2.间接法:配制---用基准物质标定 ▪ 标定:用基准物质或标准溶液准确测定滴
定液浓度的过程 浓度校正因子F:比值应该在0.95-1.05之间
加催化剂来加速; ④必须有适当简便的方法确定滴定终点。
容量分析的计算问题
应该将滴定度(T )乘以滴定液的浓度校
正因数(F),换算成实际的滴定度(T'
=T*F)。其中, 滴定液实际配制浓度
F=
滴定液规定的浓度
加上,有时溶液需要稀释,所以被测药物 的含量公式可表示为:
含量%= F T V D 100% W
▪ 4.方法:具有一定的分辨力、专属性、稳定性和 灵敏度,其准确度和精密度均高。
重量分析
化学分析法
容量分析(滴定)
含量测定
仪器分析法
光谱法
色谱法 电泳法
效价测定(生物学法)
重量分析法
▪ 以质量为测量值的分析方法。 称取一定重量的供试品,用适当的方法
将被测组分与试样中其它组分分离,根据 被测组分和供试品的重量以计算组分的含 量的百分数的定量方法。
注意事项
▪参考课本
五、碘量法
以碘为氧化剂,或以碘化物为还 原剂的滴定法。
直接碘量法 间接碘量法
剩余碘量法 置换碘量法
碘滴定液的配制:加大量碘化钾
增加I- 浓度 A、增加 I2溶解度,并减少 I2 挥发
KI
I2
KI
2 3
B、降低 I2/I -氧化电位,反应完全
滴定液的标定 碘滴定液
药物分析 药物的含量测定方法——色谱分析法

Ais = cX + cX
AX
cX
cX
=
cX
(Ais / AX ) - 1
R=
2(tR2 - tR1 )
1.5
1.70(W1,h/2 +W2,h/2 )
• 定量(LOQ), S/N≥10; 定性(LOD), S/N≥3
(4)拖尾因子 • 以峰髙定量时, T 值应在0.95~1.05之间
T=W0.05h/2d1
(5)重复性 • 峰面积RSD≤2.0% (n=5)
3. 测定法 (1) 内标法
2. 系统适用性试验 (1) 色谱柱理论板数(n) (2) 分离度(R) (3) 灵敏度 (4) 拖尾因子(T) (5) 重复性
2. 系统适用性试验 (1)色谱柱理论板数 n=16(tR/W)2; n=5.54(tR/Wh/2)2 (2)分离度
R
=
2(tR2
- tR1
) ;
W1 +W2
(3)灵敏度
(2) 外标法
f As CR AR CS
Cx
Ax AS'
C
' S
f
Cx
Ax AR
CR
(二) 气相色谱法(GC)
主要应用于具有挥发性或其衍生物具有挥发性的药物及 其相关物质的分析。 《中国药典》中被广泛用于残留溶剂的测定中。
• 载气:氦气、氮气、氢气 • 色谱柱:常用毛细管柱——熔融石英或玻璃
HPLC
• 采用高压输液泵将规定的流动相泵入装有 填充剂的色谱柱,对供试品进行分离测定 的色谱方法。
特点 (1) 高灵敏度:ng~µg/ml (2) 高专属性:选择性检测 (3) 高效能与高速度 • 复方制剂含量测定的首选方法
药物分析药物的含量测定方法——色谱分析法

药物分析药物的含量测定方法——色谱分析法药物的含量测定是药物分析中的重要内容之一,对于药物的质量控制和剂型的稳定性评估具有重要的意义。
而色谱分析法是一种常用的药物含量测定方法,它基于药物分子与色谱柱固定相之间的相互作用原理,通过药物分子在色谱柱上的分离和检测来测定药物的含量。
本文将介绍色谱分析法在药物含量测定中的应用,并重点介绍了高效液相色谱法(HPLC)和气相色谱法(GC)两种常用的分析方法。
高效液相色谱法(HPLC)是一种常用的药物含量测定方法,它常用于测定水溶性药物和中、大分子化合物。
HPLC的原理是利用高压将样品流动相推送到固定相填充的色谱柱中,样品在固定相上分离,再通过检测器进行药物含量的测定。
HPLC的优点是分析速度快、分离效果好、灵敏度高,可以同时测定多种组分。
在药物含量测定中,HPLC可用于测定药物的纯度、含量、杂质和分解产物等重要指标。
例如,可以利用HPLC测定药物中杂质的含量,通过测定无机离子、有机杂质和重金属等指标,评估药物的安全性和纯度。
另外,还可以利用HPLC测定药物中活性成分的含量,用于质量控制和剂型的稳定性评估。
气相色谱法(GC)是一种常用的药物含量测定方法,主要用于测定揮发性物质和热稳定性物质。
GC的原理是利用气相载气将样品蒸发并传递到柱中,再通过柱内固定相的分离,最后通过检测器进行含量测定。
GC的优点是分离效果好、分析速度快、灵敏度高。
在药物含量测定中,GC可用于测定药物中揮发性成分的含量,如挥发油和有机溶剂等。
常用的应用包括测定中药提取物中的挥发油、测定药物中的有机溶剂残留等。
此外,GC还可用于测定药物中的稳定性物质,通过测定反应产物和分解产物的含量来评估药物的稳定性。
总结来说,色谱分析法是一种常用的药物含量测定方法,高效液相色谱法(HPLC)和气相色谱法(GC)是其中两种常用的分析方法。
HPLC适用于测定水溶性药物和中、大分子化合物的含量,GC适用于测定揮发性物质和热稳定性物质的含量。
药物分析中的药物含量分析方法

药物分析中的药物含量分析方法药物含量分析是药物分析领域中一项十分重要的技术手段。
药物的含量分析主要用于确定药物制剂中活性成分的含量,以保证药物的质量和疗效。
本文将介绍常见的药物含量分析方法,包括定量分析法、滴定分析法、色谱分析法和光谱分析法。
1. 定量分析法定量分析法是药物含量分析的基础方法之一。
它基于物质的定量分析原理,通过实验测定药物含量的多少。
常用的定量分析方法有重量法、容量法和电位滴定法。
(1)重量法:将一定质量的药物样品称取,并进行溶解、稀释等处理后,通过质量差计算出药物的含量。
(2)容量法:通过向药物样品中滴加标准溶液,使溶液达到等量点(终点),从而推算出药物的含量。
(3)电位滴定法:利用反应溶液中的特定药物含量与溶液电压的关系,通过电位滴定仪进行电位滴定,从而确定药物的含量。
2. 滴定分析法滴定分析法是一种通过滴定试剂与药物样品反应来确定药物含量的方法。
常用的滴定法有酸碱滴定法、氧化还原滴定法和络合滴定法。
(1)酸碱滴定法:根据药物样品的酸碱性质,采用适当的滴定试剂进行滴定,并通过滴定量计算出药物的含量。
(2)氧化还原滴定法:利用药物与氧化剂或还原剂反应的氧化还原过程,通过滴定试剂的耗量推算出药物含量。
(3)络合滴定法:利用药物与滴定试剂之间形成络合物的特性,通过滴定试剂的耗量计算出药物的含量。
3. 色谱分析法色谱分析法是一种基于化学试剂在固定相上的吸附、分离和检测的方法。
常用的色谱法有气相色谱法(GC)、液相色谱法(HPLC)和薄层色谱法(TLC)。
(1)气相色谱法(GC):将药物样品挥发成气态,通过在固定相上的分离和检测,确定药物的含量。
(2)液相色谱法(HPLC):将药物样品溶解在溶剂中,通过在固定相上的分离和检测,确定药物的含量。
(3)薄层色谱法(TLC):将药物样品涂抹在薄层板上,通过吸附、分离和检测,确定药物的含量。
4. 光谱分析法光谱分析法是一种根据药物与光的相互作用,通过测量药物对光的吸收、散射和发射等光学性质,来确定药物含量的方法。
高良姜药材中4种有效成分的含量测定

高良姜药材中4种有效成分的含量测定【摘要】目的建立评价高良姜药材质量的含量测定方法。
方法采用DiamonsilTMC18柱(4.6 mm×200 mm,5 μm),流动相为乙腈磷酸溶液,梯度洗脱,流速1 mL/min,柱温30 ℃,检测波长为220 nm。
结果:高良姜素、山柰素甲醚、5羟基羟基甲氧苯基)苯庚酮、5羟基双苯庚酮的线性范围分别为0.267~1.328 μg、0.103 4~0.517 μg、0.310 8~1.554 0 μg、0.247 3~1.236 μg,平均回收率分别为100.9%、97.0%、96.8%、99.0%,RSD值分别为1.7%、1.4%、1.8%、0.6%。
结论该方法准确、简便,具有良好的重复性和稳定性,可用于高良姜药材的质量控制。
【关键词】高良姜;高效液相色谱法;黄酮;庚烷;质量控制Abstract:Objective To develop an HPLC method for characterizing the active constituents, namely, galangin, kaempferoldiphenylheptane C, in Alpinia officinarum Hance. Method Samples were analyzed on an HPLC Diamonsil C18 column(200 mm×4.6 mm,5 μm) by using gradient elution of acetonitrile (A) and 0.2% aqueous phosphori c acid (B) at a flow rate of 1.0 mL·min-1.The diodephenylheptane A and diphenylheptane C had good linearity over the ranges of 0.267-1.328,0.1034-0.517,0.3108-1.554 and0.2473-1.236 μg,respectively, and the average recoveries were100.9%(RSD=1.7%)、97.0%(RSD=1.4%),96.8% (RSD=1.8%) and99.0%(RSD=0.6%),respectively.ConclusionThe method is rapid,accurate and reproducible, which is helpful in the quality control of Alpinia offic inarum Hance.Key words:Alpinia officinarumHance;HPLC;flavonoids;diarylheptanoids;quality evaluation高良姜为姜科山姜属植物高良姜Alpinia officinarum Hance的干燥根茎,其性热味辛,归脾、胃经,具温胃散寒、消食止痛的功效,用于脘腹冷痛、胃寒呕吐、嗳气吞酸等[1]。
药物分析 药物的含量测定方法——滴定分析法

I2
+
2
S
2O
23
2
I-
+
S4O
26
n=1
y=2
T m 1 M 0.1 1 121.76 6.088 (mg/ml)
yn
21
• 每1ml硫代硫酸钠滴定液(0.1mol/L)相当过量滴定液A, 用滴定液B滴定剩余的A
含量 (%) (VA FA VB FB ) TA 100 W
其中: VA FA VBO FB
含量 (%) = (VBO -VBS ) FB TA ×100 W
• 注意:浓度校正因子F为滴定液B的FB 滴定度T为滴定液A的TA
示例:司可巴比妥钠(m=260.27)含量测定
• 本品0.1g, 加水10ml溶解, 精密加溴滴定液 (0.05mol/L) 25ml, 再加盐酸5ml, 放置15分钟, 加碘化钾试液10ml, 用硫代硫酸钠滴定液 (0.1mol/L) 滴定, 滴定结果用空白试验校正
1
• 每1ml碘滴定液(0.05mol/L)相当于8.806mg的C6H8O6
2. 含量的计算
(1)直接滴定法 (2) 生成物滴定法 (3) 剩余量滴定法
(1) 直接滴定法
1 含量 (%) V T 100 W
2 F = 实际摩尔浓度 规定摩尔浓度
含量
(%)
=
V T' W
100
=
V' T W
100
O
Br Br O
N
N
ONa + Br2
R
N
OH
ONa
R
N
OH
Br2 + 2 I- 2 Br- + I2
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.7 含量测定药品的含量测定是指准确测定药品有效成分或者指标性成分的测定。
含量测定时评价药品质量、判断药物优劣和保证药品疗效的的重要手段。
含量测定需在鉴别无误和杂质检查符合规定的基础上进行。
除个别品种不收载含量测定项外,原则上均按药品质量标准进行含量测定。
药品的含量测定应根据侧成分的物理化学性质选择相应的测定方法。
测定药物含量时,可选用容量分析法和仪器分析法。
容量分析法(滴定分析法)的仪器设备简单、易于操作,不需要使用化学对照品,成本低、速度较快,结果准确、方法耐用性高,但方法缺乏专属性,主要适用于对结果准确度和精密度要求较高的样品测定。
虽然其专属性不如先分离后分析的仪器分析法高,但仍作为国内外药典中药物的含量测定首选含量分析法;光谱分析法简便、快速,灵敏度高、并具有一定的准确度,但方法专属性稍差,主要适用于对灵敏度要求较高、样本量较大的分析项目;色谱分析法则具有高灵敏度与高专属性、并具有一定的准确度,但其结果计算需要对照品,本法主要适用于对方法的专属性与灵敏度要求较高的组分复杂、干扰成分较多样品的含量测定。
随着仪器和检测技术的快速发展,仪器分析法的准确度和精密度越来越高。
国内外药典中应用仪器分析法进行药物含量测定日益普及。
采用仪器分析方法测定含量时,需要采用化学对照品。
1.7.1方法选择药物的含量测定所采用的分析方法一般要求操作简便,结果准确重现。
但对于药物的不同形式,其含量测定方法的选择依据有所侧重。
对于化学原料药的含量测定,因为纯度较高、所含杂质较少,故强调测定结果的准确和重现,通常要求方法具有更高的准确度和精密度,首选容量分析法,如用容量法不适宜时,可考虑选用HPLC法,尤其在有关物质干扰,或多组份物质时,具有特殊优势。
因仪器或操作间的偏差较大,一般不选用UV法,尤其不首选“吸收系数法”定量,不选用末端吸收峰作测定波长;须用UV法时,可采用不受仪器及其它变化影响的“对照品比较法”定量,测定溶液的吸收度宜在0.3~0.7间。
元素测定法如定氮法,在其它方法均不适宜时可采用,但因不能反映化合物含量的变化情况,此法不可用于稳定性考察中。
对于药物制剂的含量测定,则因为制剂组分复杂、干扰物之多,且含量限度一般较宽,故更加强调方法的灵敏度和专属性或则选择性,选用方法时应考虑辅料等的干扰,首选方法一般为HPLC法,在有关物质、辅料不干扰的情况下,也可选用UV法或原料药项下的容量法,复方制剂首选HPLC法较为适宜。
而对于药物制剂的定量检查,如溶出度、含量均匀度检查中药物的溶出量或则含量测定,因为分析样本量较大、且限度亦较宽,在辅料不干扰测定时宜选用管谱分析法。
药品的含量或效价是评定药品的主要指标之一,设计其测定方法时,应根据药品特性、剂型、处方、鉴别试验和纯度检查综合考虑,当鉴别试验和纯度检查保证了专属性和纯度的情况下,含量测定方法的选择要着眼于准确性、稳定性和可重复性。
1.7.1.1原料药含量的测定一.选用原则对于组份单一的原料药,首选精密度高,操作简便、快速的容量法测定含量,可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件:(1)反应须按一个方向完全进行;(2)反应要迅速,必要时可通过加热或加入催化剂等方法提高反应速度;(3)共存物不得干扰主要反应,或能用适当方法消除;(4)确定等当点的方法要简单、灵敏;(5)标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定(标化时不发生副反应)等要求。
方法叙述中要强调:(1)供试品的取用量应满足滴定精度的要求(消耗滴定液约20ml);(2)滴定终点的判断要明确,如选用指示剂法,应考虑其变色敏锐,提供滴定曲线,并用电位法校准其终点颜色。
(3)为排除因加入其它试剂或混入杂质对测定结果的影响,或便于剩余滴定法的计算,可采用“将滴定的结果用空白试验校正”的办法;(4)最后要给出滴定度(采用四位有效数字)。
容量法测定含量要注意参加反应的应是药物分子活性部分,而不应是次要的酸根或碱基部分。
如盐酸西替立嗪、盐酸氨溴索,有研究者采用氢氧化钠滴定液滴定法直接测定其含量,事实上这种方法测定的是其中盐酸的含量,由于其成盐工艺中的酸、碱配比不当时,可严重影响成品酸碱度,故此法的测定结果并不能代表其活性成分有机碱的含量,这种方法不能准确把握其有效成分含量。
应采用非水滴定法,以无水冰醋酸为溶剂屏蔽掉盐酸的干扰,用高氯酸滴定其有机碱的含量。
高氯酸非水滴定法因适应性广(适用于有机弱碱及其盐类)、方法简便和测定结果精密等优点,在化学原料药的含量测定中较为常用。
二.化学分析法1.酸碱滴定法酸碱滴定法是在水溶液中以酸碱中和反应来测定物质含量的方法,可用来测定酸、碱、弱酸盐、弱碱盐等。
芳酸及其酯类原料药的含量测定多采用酸碱滴定法,如萘普生、阿司匹林。
2.非水溶液滴定法在非水溶剂(有机溶剂与不含水的无机溶剂)中进行滴定分析的方法。
在非水溶剂中滴定,可使原来在水中不能进行完全的反应顺利进行,还能使在水中不能溶解的药物溶解在非水溶液中,增大药物的溶解度,扩大滴定分析的应用范围。
非水滴定法包括非水碱量法和非水酸量法。
其两者的比较参看表1。
表1.非水碱量法和非水酸量法的比较(1)非水碱量法 通常是以冰醋酸为溶剂,高氯酸为滴定液,测定弱碱性药物及其盐类的分析方法,在药物含量测定中应用非常广泛。
溶剂:碱的滴定宜选择酸性溶剂,冰醋酸是滴定弱碱性物质最常用的溶剂。
市售的冰醋酸中含有水分,水分的存在可影响滴定突跃,故一般按计算量加入醋酐,以除去水分。
滴定液:非水碱量法通常使用高氯酸的冰醋酸溶液作滴定液,因为高氯酸在冰醋酸中有较强的酸性,且绝大多数有机碱的高氯酸盐易溶于有机溶剂,有利于滴定的进行。
市售高氯酸为含HCl04 70.0%~72.O%的水溶液,故需加入计算量的醋酐除去水分。
高氯酸滴定液受温度影响较大,因此样品的测定与标定应在同一温度进行,若温度差超过2℃以上,应重新标定或带温度补偿值加以校正。
校正公式:)(0011.01001T T C C I -+= 指示剂:非水碱量法可用指示剂或电位法指示终点,常用的指示剂为结晶紫。
(2)非水酸量法 通常是在碱性溶液中,以甲醇钠为滴定液,麝香草酚蓝或偶氮紫为指示剂,二甲基甲酰胺、乙二胺等为溶剂,滴定弱酸性药物的分析方法。
主要用于滴定极弱的酸类如酚类、酰亚胺类药物的含量测定,如乙虎胺。
非水溶液酸碱滴定法常用于氨基酸类、有机碱及其盐类等的原料药的含量测定,如尼可刹米、地西泮。
3.银量法对于易水解或则机构中含有卤族元素的药物,能与Ag +生成AgX 沉淀,可选用银量法测定药品含量,即可用指示剂判定终点,也可用电位滴定法判定。
银量法按所用指示剂的不同分为铬酸钾指示剂法、铁铵矾指示剂法和吸附指示剂法。
(1)铬酸钾指示剂法 是在中性溶液中,用硝酸银滴定液滴定氯化物或溴化物,以K 2CrO 4作指示剂,Ag +和CrO 42- 形成砖红色沉淀指示终点。
多用于Cl -、Br - 的测定。
(2)铁铵矾指示剂法用NH4SCN为滴定剂,以硫酸铁铵为指示剂,在硝酸酸性(防止出现Fe(OH)3红棕色沉淀)溶液中测定Ag+的滴定方法,Fe3+和SCN-形成红色配合物指示终点。
(3)吸附指示剂法用硝酸银滴定液滴定,以吸附指示剂(荧光黄)确定终点的滴定方法,一般用以测定卤化物,滴定时避免阳光直射,因卤化银遇光易分解,使沉淀变为灰黑色。
4.碘量法是以碘为氧化剂,或以碘化物为还原剂进行滴定的方法。
按照滴定的方式分为直接碘量法、剩余碘量法和置换碘量法。
(1)直接碘量法是用碘滴定液直接滴定还原性物质的方法。
在滴定过程中I2被还原为I-,该法只能在酸性、中性或弱碱性溶液中进行,一般用淀粉指示剂指示终点,淀粉遇碘变蓝色,反应极其灵敏。
也可用碘自身的颜色指示终点,化学计量点后,溶液中稍过量的碘即显黄色而指示终点。
如维生素C的含量测定。
(2)剩余碘量法是在供试品(还原性物质)溶液中,先加入定量过量的碘滴定液,待I2与待测组分反应完全后,再用硫代硫酸钠滴定液滴定剩余的碘,来求出待测组分含量的方法。
滴定时用淀粉作指示剂,在近终点时加入,因为当溶液中有大量碘存在时,碘易吸附在淀粉表面,影响终点的判断。
如复方对乙酰氨基酚片中咖啡因的含量测定。
(3)置换碘量法如硫代硫酸钠滴定液的标定即采用该法,以重铬酸钾为基准物,加入碘化钾置换出定量的碘,碘再用硫代硫酸钠滴定液滴定。
5.亚硝酸钠法是利用亚硝酸钠滴定液在盐酸溶液中与具有芳伯氨基的化合物发生重氮化反应,定量生成重氮盐,根据消耗亚硝酸钠的量来计算药物含量的方法,可选用永停滴定法判定终点。
如盐酸普鲁卡因的含量测定。
6.铈量法也称硫酸铈法,是以硫酸铈[Ce(S04)2]作为滴定液,在酸性条件下测定还原性物质的滴定方法。
采用邻二氮菲作指示剂,化学计量点后,Fe2+被氧化成Fe3+,生成邻二氮菲铁,由红色转变为淡蓝色而指示终点。
铈量法由于不受制剂中淀粉、糖类的干扰,因此特别适合片剂、糖浆剂等制剂的测定。
《中国药典》2005年版采用铈量法测定的药物有:硫酸亚铁片及硫酸亚铁缓释片、葡萄糖酸亚铁及其制剂、富马酸亚铁及其制剂等。
7.配位滴定法是以形成稳定配合物的配位反应为基础的滴定分析法。
主要用于金属离子的测定,目前应用最广泛的配位滴定剂是EDTA(乙二胺四醋酸二钠),因此通常所谓的配位滴定法,主要是指使用EDTA滴定液的滴定法,一般选用金属指示剂指示滴定终点。
如葡萄糖酸钙、硫酸锌的含量测定均采用此法,用铬黑T作指示剂。
8.重量分析法重量分析法是通过称量物质的某种称量形式的质量来确定被测组分含量的一种定量分析方法。
当无合适的容量分析法时,可通过测定药品中有效含量或则是其衍生物重量对药品含量进行测定。
二.仪器分析法1.紫外-可见分光光度法通常分子结构中含有共轭体系结构、苯环和杂环的药物都可以通过紫外分光光度法检测其特征吸收光谱,计算出其含量。
有色药物或与显色剂反应后在可见区有特征吸收的药物,可选用可见分光光度发测定含量。
此法准确度与精密度均较高,灵敏度亦较高,仪器价格低廉,仪器操作简便快捷,应用广泛,是原料药含量测定首选的仪器分析方法。
2.色谱法(1)高效液相色谱法本法具有高灵敏度,高选择性,高效能,高速度及应用广泛的特点,HPLC法使用高效固定相,流动相采用高压泵输送,在线进行检测,是杂质或则其他干扰因素较多的品种其含量测定的常用方法,色谱柱的填充剂通常用十八烷基硅烷键合硅胶,检测器通常用紫外检测器,定量通常采用外标法,有时也可选用内标法加校正因子法、面积归一化法等。
(2)气相色谱法气象色谱法采用气体为流动相流进装有填充剂的色谱柱进行分离测定的,主要用于分离分析易挥发的物质。
本法是一种高效能、高选择性、高灵敏度、操作简单、应用广泛的分离分析方法。