JP日本药典(药局方)标准品汇总信息-2016-update
第十七次修订版日本药典第二增补本

・稠度试验法 透度计(稠度计) →测定软硬度
15
新收录试验法④
6.17 蛋白质药品针剂的不溶性微粒子试验法
“6.07 针剂的不溶性微粒子试验法” 第1法 利用光遮蔽粒子计数法测定蛋白质药品时的课题
• 在许多情况下容量较少 →试验时需混合许多容器内的溶液后进行试验
分类
概要
将药品各条中的制剂有效期作为参考
修订 明确了实时发布试验的定位
删除有关有效期的最终有效年月的标示规定
参考信息《确保药品原药和制剂质量的基本思路》
10
制剂总则(修订方案)
3.1.
项目 针剂
3.1.4. 核糖体针剂
分类
备注
修订 引用一般试验法(新的核糖体针剂)
新制定 增加有关DDS的形状
DDS:Drug Delivery system
6
日本药典的构成
基于法第41条的公布内容
・通则 ・生药总则 ・制剂总则 ・一般试验法 ・药品各条 ・药品各条 生药等 ・参考紫外可见吸收光谱 ・参考红外吸收光谱
参考信息 附录 etc
8
第二增补本概略
通则 生药总则 制剂总则:[1]制剂总则
:[2]制剂包装总则 :[3]制剂各条 :[4]生药相关制剂各条 一般试验法 药品各条
○英文版网页
https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000066597.ht ml
6
第十八次修订版日本药典制作基本方针
1.全面收录重要的医疗保健药物 2.积极引进最新的学术和技术 3.推进国际化 4.根据需要及时进行部分修订并通过行政部门顺利执行 5.确保透明性并推进日本药典的普及
多西他赛日本药典JP16
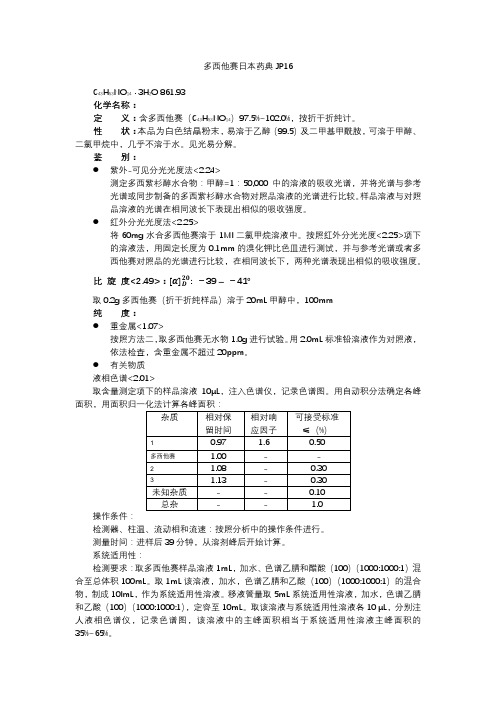
多西他赛日本药典JP16C43H53NO14· 3H2O 861.93化学名称:定义:含多西他赛(C43H53NO14)97.5%~102.0%,按折干折纯计。
性状:本品为白色结晶粉末,易溶于乙醇(99.5)及二甲基甲酰胺,可溶于甲醇、二氯甲烷中,几乎不溶于水。
见光易分解。
鉴别:●紫外-可见分光光度法<2.24>测定多西紫杉醇水合物:甲醇=1:50,000中的溶液的吸收光谱,并将光谱与参考光谱或同步制备的多西紫杉醇水合物对照品溶液的光谱进行比较。
样品溶液与对照品溶液的光谱在相同波长下表现出相似的吸收强度。
●红外分光光度法<2.25>将60mg水合多西他赛溶于1Ml二氯甲烷溶液中。
按照红外分光光度<2.25>项下的溶液法,用固定长度为0.1mm的溴化钾比色皿进行测试,并与参考光谱或者多西他赛对照品的光谱进行比较,在相同波长下,两种光谱表现出相似的吸收强度。
比旋度<2.49>:: -39 –-41°取0.2g多西他赛(折干折纯样品)溶于20mL甲醇中,100mm纯度:●重金属<1.07>按照方法二,取多西他赛无水物1.0g进行试验。
用2.0mL标准铅溶液作为对照液,依法检查,含重金属不超过20ppm。
●有关物质液相色谱<2.01>取含量测定项下的样品溶液10μL,注入色谱仪,记录色谱图。
用自动积分法确定各峰面积,用面积归一化法计算各峰面积:操作条件:检测器、柱温、流动相和流速:按照分析中的操作条件进行。
测量时间:进样后39分钟,从溶剂峰后开始计算。
系统适用性:检测要求:取多西他赛样品溶液1mL,加水、色谱乙腈和醋酸(100)(1000:1000:1)混合至总体积100mL。
取1mL该溶液,加水,色谱乙腈和乙酸(100)(1000:1000:1)的混合物,制成10lmL,作为系统适用性溶液。
移液管量取5mL系统适用性溶液,加水,色谱乙腈和乙酸(100)(1000:1000:1),定容至10mL。
【推荐下载】中、美、欧、日4种药典收载10种基原植物相同生药的质量检测标

中、美、欧、日4种药典收载10种基原植物相同生药的质量检测标 【编者按】医药论文是科技论文的一种是用来进行医药科学研究和描述研究成果的论说性文章。
论文网为您提供医药论文范文参考,以及论文写作指导和格式排版要求,解决您在论文写作中的难题。
中、美、欧、日4种药典收载10种基原植物相同生药的质量检测标 【关键词】药典;质量标准;植物药 植物药在历史上曾是世界各国医疗的主要药物,如中国的应用历史至少有2000年。
近代伴随化学、制药工业的飞速发展,植物药的主流地位在世界范围内逐渐被化学合成药物所替代。
然而,随着近年来回归自然、绿色产品消费市场的不断扩大,以及植物药在某些方面具有化学合成药物无法相比的优势而重新获得各国注意,植物药及其精制剂、提取物的质量标准、安全性等问题日益受到关注。
1 中国、日本药典植物药收载标准概况 在中国传统医学中,植物药作为临床使用的主要药物,很早就被应用于汤、散剂等成方制剂,形成了一套独特的体系。
植物药被收载于历代各类药物学(本草)著作中,唐代由政府编修、颁布的药典性质著作《新修本草》收载的大部分是植物药,可以说是药典收载植物药的开始。
中医学及其应用植物药的技术在日本、朝鲜、越南等国也被广泛传播,影响深远。
目前,这些国家的药典都收载了很多植物药,并针对其安全性和质量制定了一定的标准。
以有代表性的日本为例,《日本药局方》(JP)第14改正版(含追补版和局外生药规格)共收载生药(含粉末)249种,据统计,其中基原和药用部位与2000年版《中华人民共和国药典》(CP)一致的有83个品种[1]。
植物生药作为中药材收载于CP一部。
在鉴别方面,CP过去版本常用显色和试管反应,专属性较差,而现在已全面转向薄层色谱法(TLC)为主的鉴别手段。
2005年版CP新增了专属性TLC鉴别662项。
在检查项目方面,普遍增加了杂质、水分、灰分和酸不溶性灰分等项目检查,以保证中药材的纯净度,新增杂质检查34个品种,水分检查178个品种,灰分有135个品种,酸不溶性灰分有130个品种。
日本药典 (JP)和日本的辅料标准

3. 推动国际协调的进程 (1)
通过药典讨论小组(PDG)与欧洲药典和美国药典合作 → 带来了日本药典第15版中的各种协调最终文本 20个一般章节中有19个 35个辅料专论中有24个
3. 推动国际协调的进程 (2)
在每个协调一般测试方法和辅料中均包括了协调 现状陈述 未说明的区别和当地要求均采用了通用符号(黑色 菱形 ◆ ) 一般信息中的信息补遗 -日本药典中实施的国际协调
2004年4月1日依法建立的一个新的半政府性 机构(公司性质的管理机构)。 进行几乎所有药品和医疗器械方面的科学和 技术评估和检查 在确定日本药典中起着实质的办公 室
日本药品和医疗器械局 6 楼-11楼 Shin-kasumigaseki大 厦 Chiyoda-ku, 东京
http://www.mhlw.go.jp/topics/bukyoku/iyaku/
yakkyoku/index.html
英文版的日本药典
http://www.mhlw.go.jp/topics/bukyoku/iyaku/ yakkyoku/english.html
5.最新分析方法的引入和参考标准中统 一系统的确立。
日本药典(JP)和日本的辅料标准
Kiyomi UENO
日本药品和医疗器械局(PMDA), 日本药
典
辅料标准
日本药典 (JP) 日本厚生劳动省 部门公告 日本的药物辅料(JPE)(医薬品添加物規格) 公告 医薬食品局長通知 散装准药品成分的日本标准 (医薬部外品原料規格) 公告 医薬食品局長通知 食品添加剂的日本规范和标准(食品添加物規格) 日本厚生劳动省 部门公告 厚生労働大臣告示
日本药典委员会/药政管理和食品卫生委员会(日 本厚生劳动省) 15个专家委员会,1个附设委员会和3个工作小组 (药品和医疗器械局) 成员: -大约150个专家,来自国家研究机构、大学及其它 部门 -东京、大阪药品生产商联合会以及其它所示各方 的代表 频率:每个委员会负责两个月
甘草四国药典比较

甘草四国药典比较班级:51 学号:1045114 姓名:陈多清一、质量标准比较1.中国药典(CHP2010)来源:本品为豆科植物甘草Radix Glycyrrhiza uralensis Fisch.、胀果甘草Glycyrrhiza in flataBat.或光果甘草Glycyrrhiza glabra L.的干燥根及根茎。
春、秋二季采挖,除去须根,晒干。
性状:1)根呈圆柱形,长25~100cm,直径0.6~3.5cm。
外皮松紧不一。
表面红棕色或灰棕色,具显著的纵皱纹、沟纹、皮孔及稀疏的细根痕。
质坚实,断面略显纤维性,黄白色,粉性,形成层环明显,射线放射状,有的有裂隙。
根茎呈圆柱形,表面有芽痕,断面中部有髓。
气微,味甜而特殊。
2)胀果甘草根及根茎木质粗壮,有的分枝,外皮粗糙,多灰棕色或灰褐色。
质坚硬,木质纤维多,粉性小。
根茎不定芽多而粗大。
3)光果甘草根及根茎质地较坚实,有的分枝,外皮不粗糙,多灰棕色,皮孔细而不明显。
鉴别:1)本品横切面:木栓层为数列棕色细胞。
栓内层较窄。
韧皮部射线宽广,多弯曲,常现裂隙;纤维多成束,非木化或微木化,周围薄壁细胞常含草酸钙方晶;筛管群常因压缩而变形。
束内形成层明显。
木质部射线宽3~5列细胞;导管较多,直径约至160μm;木纤维成束,周围薄壁细胞亦含草酸钙方晶。
根中心无髓;根茎中心有髓粉末淡棕黄色。
纤维成束,直径8~14μm,壁厚,微木化,周围薄壁细胞含草酸钙方晶,形成晶纤维。
草酸钙方晶多见。
具缘纹孔导管较大,稀有网纹导管。
木栓细胞红棕色,多角形,微木化。
2)取本品粉末1g,加乙醚40ml,加热回流1小时,滤过,药渣加甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣加水40ml使溶解,用正丁醇提取3次,每次20ml,合并正丁醇液,用水洗涤3次,蒸干,残渣加甲醇5ml使溶解,作为供试品溶液。
另取甘草对照药材1g,同法制成对照药材溶液。
再取甘草酸铵对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。
草药标准品一览表
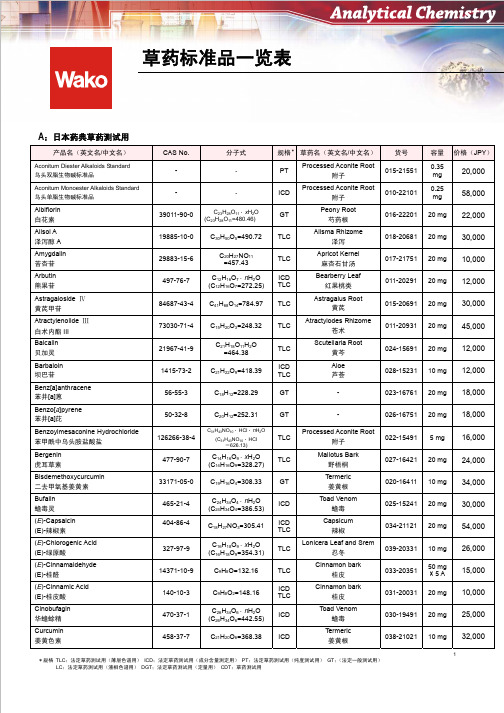
容量 0.35 mg 0.25 mg 20 mg 20 mg 20 mg 20 mg 20 mg 20 mg 20 mg 10 mg 20 mg 20 mg 5 mg 20 mg 10 mg 20 mg 20 mg 10 mg 50 mg ×5A 20 mg 20 mg 10 mg
价格(JPY)
20,000 58,000 22,000 30,000 10,000 12,000 30,000 45,000 12,000 12,000 18,000 18,000 16,000 24,000 34,000 30,000 54,000 26,000 15,000 10,000 25,000 32,000
C31H43NO10 (C31H43NO10 HCl =626.13)
HCl nH2O
C14H16O9 xH2O (C14H16O9=328.27) C19H16O4=308.33 C24H34O4 nH2O (C24H34O4=386.53) C18H27NO3=305.41 C16H18O9 xH2O (C16H18O9=354.31) C9H8O=132.16 C9H8O2=148.16 C26H34O6 nH2O (C26H34O6=442.55) C21H20O6=368.38
产品名(英文名/中文名) Dehydrocorydaline Nitrate 硝酸脱氢紫堇碱 Demethoxycurcumin 去甲氧基姜黄素
CAS No. 13005-09-9 22608-11-3 53-70-3
分子式 C22H24N2O7 =428.44 C20H18O5=338.35 C22H14=278.35 C17H24O9 xH2O (C17H24O9=372.37) C10H10O4=194.18 C17H24O10=388.37 C16H20O9=356.32 C17H26O4=294.39
日本药典JP17电子版JP17e_4-3

JP XVII
Infrared Reference Spectra
2093
Emedastine Fumarate
Emorfazone
Enalapril Maleate
2094
Infrared Reference Spectra
JP XVII
Enflurane
JP XVII
Infrared Reference Spectra
2115
Concentrated Glycerin
Glycine
Gonadorelin Acetate
2116
Infrared Reference Spectra
JP XVII
Guaifenesin
Guanabenz Acetate
Hydroxypropylcellulose
Hymecromone
2122
Infrared Reference Spectra
JP XVII
Hypromellose Phthalate (200731)
Hypromellose Phthalate (220824)
Ibudilast
JP XVII
Glibenclamide
JP XVII
Infrared Reference Spectra
2113
Gliclazide
Glimepiride
L-Glutamic
Байду номын сангаасAcid
2114
Infrared Reference Spectra
L-Glutamine
JP XVII
Glutathione
日本药局方(12版)简介

日本药局方(12版)简介奉辐白,鹌药典之页?一f占日本药局方(12版)简介黑龙江省药品检验所(哈尔滨150001)型:三苎靛f.;J3 El本药局方(12版)已于1991年4月1El公布施行;本版对凡例,收载品种,般检查法,生药及生药制剂规格等作了修订现将修订的主要内容介绍如下.1凡例凡例中,增加了第19和2l两项第19项是关于检验数据的处理?第21项是关于检验操作”立即”含义的解释”立即”是指在前一个实验操作完了后,下一个操作应在3Os内进行的意思.在本版中,将原凡例的20项改为22项;37项改为39项.修改了第2,3,j,6,7项.第2项药品名鞯的规定:药品名称应按药品各条所载的El文名或别名.但制剂总则中的散剂项下将散称作细粒.第3项药品合格与否的判定:药品合格与吾应按药品各条及凡例,生药总则,一般检查法的有关规定进行判定.性状项下的晶形,溶液的酸碱性,稳定性,吸收度,凝点,折光率,旋光度,牯度,相对密度,沸点,熔点及生药的含有量等只供参考不作合格与否的判定依据.第5项原子量采用1989年最新原子量表.第6项增加计量单位g,pg, OSYI],r/lOSl~l及内毒素EU;第7项增加表示重量对重量的百万分率ppm删去了原11舨凡例第38项关于药品极量的规定.2收载品种本版共收载1221个品种.其中一部收载750个品种(含新品种170个),二部收载471个品种.继续收载的原l1版品种一部是580个-二部是471个删去的品种一部是3个,二部是9个(附表).新收载的品种中,有机原料药117种.有机制剂26种,抗生紊25种,放射性药2种.2l新收载的有机原料药的品名氮乙酸二羟铝,异山梨醇,雌三醇,利尿酸,酚抻宁,腾喜龙,盐酸金刚烷胺,盐酸精氨酸,心得舒,盐酸异丙肾上腺素,盐酸心得平,盐酸喹诺酮心安,盐酸可乐定,盐酸环戊醇胺酯,盐酸地芬多尼,盐酸地拉齐普克雹革,盐酸羟哌噻唑酮,盐酸多拉普I仑,盐酸抗酞嗪, 盐酸多巴胺,盐酸三甲双酮,盐酸去甲替林,盐酸安太乐,盐酸比哌立登,盐酸布库洛尔,盐酸布非洛尔,盐酸1994年3月第29卷甭3附表收载品种舟类表收载品种类别一部二部无机药品有机药品制剂药品生药植物与动物药抗生素生物制剂放射性药卫生材料总计布拉洛尔,盐酸氟胺安定,盐酸舒心通,盐酸丙卡巴肼, 盐酸甲氯酚酯,肾上腺素色素缩氨脲磺酸钠,氮基甲陵氯甘油醚,坎利酸钾,枸橼酸芬太尼,枸橼酸维静宁,优降糖,氯硝安定,酮布洛芬,醋酸氯地孕酮,酰胺,环扁桃酯,双氯灭酸钠,二氯苯磺胺,安太布星,达舒平,阿糖胞苷,6一去氢逆孕酮,前列腺素Fsj0,潘生丁,溴化异丙阿托品,溴化丁基东莨菪碱,酒石酸烯丙左吗哺,硝酸双氯苯咪唑,肉桂嗪,安妥明,硫糖铝,硫噻嗪, 苯磺唑酮,硝基呋海目钠,磺甲硝咪唑,眭哺氟尿嘧啶, 甲磺氮革脲,乐可安,磷酸二氯乙酯钠,发癣退,达哌啶醇,甲氧萘丙酸,烟酸环己醇酯,淮氯苄烷胺液,18?甲基炔诺酮,氯苯氨丁酸,二苯基醋酸钠,哈洛沙佐姆,吡_苯氧磺钠,双醋苯啶,吡哌酸,安嗽灵,羟甲香豆素,丁苯乙肟,富马酸苄环庚烷,布美他尼,击氧异雄甾酮,丙安定,氟羟甲基睾丸紊,醋酸肤轻松,氟安定,丙酸轻甲雄酮,三羟苯丙酸,丙酸氯地米松,溴吡二氯革,苯蝗香豆酮,己酮可可碱,己烯雌酚,聚维酮碘,聚苯乙烯磺酸钠,亚叶酸钙,甲磺酸二氢麦角胺,甲磺酸去铁胺,甲磺酸溴麦角隐亭,甲磺酸甲胺乙吡啶,炔雌醇甲醚,甲基地高辛,甲氧补固脂紊,甲环硫甾烷,甲呋缩尿,苯阿胺酸氮芥,碘化二乙氧磷酰硫胆碱,硫酸丁酚胺,硫酸异丙喘宁,硫酸长春新碱,硫酸长春碱,硫酸二甲苄哌, 磷酸可的松钠,磷酸哌嗪,磷酸倍他米松钠,氯羟安定.2.2新收载的有机药品制剂雌三醇片,雌三醇混悬注射液,利尿酸片,氰化腾喜龙注射液,盐酸精氨酸注射液,二氯苯磺胺片,6-去氢逆孕酮片,酒石酸烯丙左吗喃片,苯磺唑酮片,磷酸183?舯Ⅲ.,勰,0跎...三氯乙酯钠糖浆,发癣遢溶液,烟酸双己醇醇片,18一甲基炔诺酮,炔雌醇片,氯苯胺丁酸片,双酷苯啶栓,双噻替培吡啶片,丁苯乙肟乳膏,富马酸苄环庚烷片,环苯安定片,氟安定胶囊,丙酸羟甲雄酮注射液,己烯雌酚二磷酸醇片,注射用硫酸长春新碱,硫酸二甲苄哌片, 磷酸哌嚷片.2.3新收载的抗生素更生霉素,盐酸阿柔素比星,盐酸头孢替安,盐酸头孢甲肟,盐酸阿霉素,盐酸巴卡西林,盐酸万古霉素, 克拉维酸钾盐,头孢克罗,头孢羟氮苄,头孢羟唑钠,头孢替坦,头孢哌酮钠,头孢磺啶钠,头孢唑肟钠,头孢抄定,头孢氨呋肟钠,磷霉素钠,拉他头孢钠,硫酸西梭霉素,硫酸博来霉素,硫酸培来霉素,硫酸小诺米星,磷酸氯洁霉素.2.4新收载的放射药氯化铟(“In)注射液,碘化钠(I)胶囊.2.5唰去的品种乳酸心可定,乳酸心可定片,苯氧乙基青霉素,苄青霉素普鲁卡因,胶体金(“Au)注射液,锌,伊克度软膏,水杨酸.酚软膏,缬草酊,远志,桔梗水,麝香,干燥气性坏疽抗毒素.非活性狂犬疫苗,百日咳.白喉混合疫苗,百日咳,白喉,破伤风混合疫苗3关于制剂总则增加两个新剂型.即气雾剂与溶液荆.制剂的外文名,第¨版为拉丁文名,第l2版改用英文名.4一般检童法第12版增加棱磁共振法和铁盐检查法.修订的主要内容:4.1吸收度测定法紫外光区测定的光源,删去了第11版补充本规定的氙灯,改用氚放电管.可见光区测定用光源仍用钨灯及卤钨灯.4.2荧光光度测定法收载了激光和碱卤灯光源.4.3酮,异丙醇,第三丁醇检查法这版规定凡原料药进行此项检查者.用此原料制备制剂时不需再作此项检查.4.4渗透压测定法美国药典xxiI渗透压是用Osmolarity表示,是指wt/vol的意思.如用Osmolarity表示时.则是wt/wt的意思.由于r与1的含义不同.是区别容量与重量浓度的第n版增补本英文版采用了美国药典Osmo—larity用语,操作时应按wt/vol进行但严密的渗透压溶液的浓度与湿度有关.这版排除了温度的影响而采184?用了OsmoIarity这一概念.限于不太浓的溶液,在常温下wt/vo[与wt/wt的差不大,所检验操作按Osmo—larity的概念进行.4.5卡氏水分测定法第11版称水分定量法,本版改为水分测定法”定量法”的概念是测定主要成分的量,测定法”是测定数据.本版对测定法的内窖作了修改,除测定甲醇外也可测定其它低级醇.反应试剂的盐基除用吡啶外也可用咪唑与a一氨基呲啶.4.6粘度测定法本版增加了关于特殊粘度的记述,并考虑与《日本工业标准》的一致性对内窖作了修订.5关于生药的修订内容5.1收载品种删去麝香.5.2基原对黄蓍,甘草,枳实,香附,细辛,山茉萸,地黄,萍蓬草,苍术,桑白皮,泽泻,茯苓,茅根等品种的基原作了修订.5.3对生药含有成分的规格值5.3.1熊果:熊果甙应在7以上,测定方法采用HPLC,对照品:J{Il果甙.5.3.2黄柏:盐酸小檗碱应在1.2以上,测定方法采用HPLC.对照品:盐酸小檗碱5.3.3黄柏粉:同黄柏5.3.4黄连:盐酸小檗碱应在4.2以上:测定方法采用HPLCt对照品:盐酸小檗碱5.3.5黄连粉:同黄连.5.3.6蟾酥:蟾酥甾体(蟾酥灵,华蟾酥毒基,脂蟾毒配基的总量)应在5.8以上.测定方法采用ttPLC,对照品:蟾酥灵,华蟾酥毒基,脂蟾酥毒配基.5.3.7薄荷油:薄荷脑应在30以上.测定方法采用GC法.对照品:L一薄荷脑.5.3.8马钱子:士的宁应在1.07以上.测定方法采用HPLC,对照品:硝酸士的宁5.3.9桉叶油:桉油精应在70以上测定方法采用GC法t对照品:桉油精.5.3.10东莨菪根:总生物碱(莨菪碱,东莨菪碱总量)应在0.29以上.测定方法采用ttPLC,对照品:硫酸阿托品,溴氢酸东莨菪碱.5.4性状5.4.1黄芩粉:将镜检项下的”柔细胞”改为”含少量淀粉粒的柔细胞.5.4.2黄连:册}去性状中通常多为烤焦”.1994年3月第29卷第35.4.3甘草:甘草酸应为2~6,测定方法采用HPLC,对照品:甘草酸5.4.4甘草粉:与甘草同.5.4.5苦参:对横断面的描述作部分修改.5.4.6桂皮粉:将淀粉粒直径6~15Fro改为6~20Fro.5.4.7欧龙胆:对镜检描述作了部分修改5.4.8欧龙胆粉:增加导管径为20~80Fm5.4.9非洲防己:对形状描述作了部分修改5.4.10柴胡:增加淀粉粒直径为2~10m.54.儿栀子:将”果肉中”改为”胎座”.5.4.12栀子粉:将”果皮要素改为”花房与果皮要素”.5.4.13山茱萸:将”果肉去掉种子的裂孔……”.改为“……去掉真正果实的裂孔……”.5.4.14芍药:芍药甙应为2~6测定方法采用HPLC.对照品芍药甙5.4.15芍药粉:与芍药阿.5.4.16生姜:删去”外皮”的描违=5.4.17美远志:对外观和镜检的描述作了调整,部分作r改写5.4.18七节人参:删去对”残茎”和”根”的描述.5.4.19当药粉:将镜捡描述的各要点作了综合. 5.4.20知母:对”中心柱”的描述作了简化.5.4.21当归:对镜检描述的”树脂室”改为”油室”,淀粉糙直径改为i9~20Fm.删去”形成层”的描述. 5.4.22吐根:对性状描述进行了简化.54.23麦冬:增加镜幢描述.5.4.24北抄参:对性状描述作了调整和改写.5.4.25白芷:对性状描述作了部分简化.5.4.26白术:对镜检描述增加了对外皮的描述. 5.4.27白术粉:对味的描述增加了”微苦=5.4.28茯苓;对性状描述作了调整和改写.5.4.29茯苓粉:对菌丝作了详细描述.5.430防己:对外观和镜检的描述作了调整.一部分作了改写5.4.31牡丹皮:对复粒淀粉数2~1o个改为2~10 数个.5.4.32术通:对外观与镜检描述作了调整.一部分进行改写.5.4.33术香:对性状描述进行了部分改写.5.4.34薏苡仁:对性状描述进行了简化.54.35东莨菪:增加镜检的描述.5.5鉴别1994年3月第29嚣第35.5.I梓实,山椒车前草,橙皮,龙胆等品种增加TLC鉴别法.5.5.2紫草:鉴别(2)增加显色反应的描述5.6物理常数5.6.I胡麻油:碘价103~116改为1.3~1185.6.2硬脂醇:对熔点测定法作了改进.5.6.3鲸蜡醇:用硬脂醇的熔点测定法.6中药制剂品种的修订6.I删去的制剂品种缬草酊,递志桔梗水.6.2中药制剂含有成分的规格值62.I熊果流浸膏:熊果甙含量应在3O以上测定方法与熊果叶同.2.2甘草浸膏:甘草酸含量应在5卜.测定方法与甘草同.6.2.3马钱子浸膏:士的宁含量应在6.8i以上.测定方法与马钱子同6.2.4马钱子酊:士的宁音量为0.(9~u.116wv,测定方法与马钱予同6.2.5马钱子浸膏散:士的宁含量应在o.68上测定方法与马钱子同6.2.6东莨菪浸膏:包生物碱(蓖菪碱东莨若碱总量)应为0.90~1.09测定方法与东莨菪碱同6.3鉴别6.3.I苦味酊:增加显色反应和TLC鉴别法6.3.2当药,碳酸氢钠散:增加TLC鉴别洼.6.3.3橙皮糖浆:增加TLC鉴别法.6.3.4橙皮酊:增加TLC鉴别法.6.3.5东莨菪浸膏;修改了鉴别(1)显色反应的操作.6.4纯度检查甘草粗浸膏:删去重金属艟查项目6.5灰分甘草粗浸膏的灰分改为127第12版含量测定采用的分析方法7.I仪器分析方法7.I.I高效液相色谱法测定的品种共101个,其中属二部的品种25个.7.I.2气相色谱法测定的品种33个.其中属二部的品种18个.7.I.3分光光度法测定的品种12个,其中届二部的品种14个.7.I.4吸收度比法测定的品种5个.7.I.5原子吸收法测定的品种9个,其中属二部的品种1个.l857.2化学分析方法7.2.1非水溶媒法测定的品种210个.其中属二部的品种2个.72.2中和法测定的品种117个,其中属二部的品种37十.7.2.3银量法测定的品种34个,其中属二部的品种2十.72.4醇制氢氧化钾法测定的品种l3个,其中属二部的品种1个7.2.s络量法测定品种37个,其中属二部的品种15 ,7.2.6四甲羟胺法测定的品种13个.7.2.7碘量法测定的品种17个.其中属二部的品种5 个.7.2.8溴量法测定的品种1o个,其中属二部的品种5 个:7.2.9四苯硼钠法测定的品种5个.7.210硫代硫酸钠法测定的品种18个.其中属二部的7个7.2.11碘酸钾法测定品种7个.7.2.12三氯化钛法测定的品种3个.7.2.13甲醇钠,二氧六围法测定的品种2个7.2.14硫代硫酸铵法测定的品种4个,其中属二部的品种3个.7.2.15二氯吲哚酚法测定的品种2个.我院药品消耗信息的ABC分析北京军区医院(100700)问善张维旗王建民赵汉臣ABC分析俗称重点分析法,是确定主次关系的可靠方法.它将研究对象分为ABC3类,A类是主要困紊,B类是次主要因索,C类是次要因索.根据该分类找出关键性环节,抓住主要矛盾,采取相应的管理措施,即能提高管理效率.我们在药品采购,供应,管理计算机应用软件的设计中.引入了ABC分析法数学模型, 对我皖近年来的药品消耗数据,进行盒额,数量,价格, 发放频率等多方面分析,收到了较好的效果1应用项目爰数学模型设药品的单价为a(批发价);年消耗量为b;发放频率为P.11消耗数量分析:Ai--b}1.2消耗金额分析:Ai—a×b;1.3价格分析:Ai—a;1.4发放频率分析:Ai竺p}1.5根据上述4个分析目的.分别请取数据,按ABC分析运算步骤求解:(1)以A值大小排序}(2)求各个序号与序号累加值的百分值}(3)求各品种Aj值与_Aj 累加值的百分值(4)求各品种\/A与V Aj累加值的百分值;(j)在At与累加值的百分比值项下的33.3及66.6处划线,分成A,B,C3类,分别得出各类分析结果.2分析目的爰意义l862.1消耗数量的分析可得出消耗量最多及最少的品种,提示台理确定存放货位及库存贮备量A娄药品, 由于用量大.为保证医疗需要,应建立稳定而畅通的供货槊道.适当加大贮备量.在仓库中应提供足够的储藏面积.而c类品种,建立货架就可满足储备要求.2.2通过消耗金额的分析可掌握资金投向.A娄药品占用资金较多,对这部分品种应格控制购买置,避免固定资金占用过多.以有限资金发挥最大的作用,实现最佳经济教益.2.3价格的分析结果可作为药品分级管理的重要依据.由于贵重药品主要根据价格确定,国家无统一标准.不步单位只能凭经验或习晴确定贵重药品品种,随意性大,科学性差,无法反映药品的内在竹值规律.钱院通过逐年价格的ABC分析,确定贵重药品品种,井定期调整,提高了管理的台理性,科学性.2.4发放频率的分析结果可明了药品的发放慨况.提示药库的货位布局在面积大.品种多的仓库内,确定药品货位时,应将发放次数多的A类品种放在易取易运的位置.利于工作.减轻劳动强度.2.5ABC分析可为药品更新换代提供准确信息.肖耗数量,消耗金额及发放次数均为0的药品.如果连续几年无消耗,即应列为淘汰品种对于这些药品采取停止采购.消耗现有库存的方法.减少积压和浪费1994年3月第20卷第3。
人参表格版

标准项目Item 欧洲药典7.0版EP7.0美国药典36版USP36日本药局方16版JP16日本药局方16版JP16(红参)韩国药典9.0版KP9.0含量限度(基于干燥品)Content Limit 人参皂苷Rg1+人参皂苷Rb1总量≥0.4%。
人参皂苷Rg1≧0.20 %人参皂苷Rb1≧0.10 %人参皂苷Rg1≧0.10 %人参皂苷Rb1≧0.20 %人参皂苷Rg1≧0.10 %人参皂苷Rb1≧0.20 %人参皂苷Rg1≧0.10 %人参皂苷Rb1≧0.20 %植物来源Origin 整根或经切片干燥,称为白参;蒸制后干燥,称为红参为亚洲五加科植物人参Panax ginseng C.A. Mey的干燥根部五加科植物人参Panaxginseng C. A. Mey的去除须根或用热水快速烫过的根部为五加科人参的根茎经蒸制后成为五加科植物人参Panax ginseng C. A.Mey的去除须根和木栓层的根部性状Description主根呈纺锤形或圆柱形,或有支根,长约20cm,直径 2.5cm,多拘挛而弯曲。
白参表面为淡黄色,红参是棕红色并有纵纹。
芦碗可见茎痕。
断口小。
横切面可见黄棕色的点状树脂道及放射状裂隙。
白参下部可见若干须根,红参一般没有。
梭形或圆柱形的根,具有独特的芳香气味,有分枝,通常为1—10cm,有时可达20cm,直径约2.5cm,有一个或多个茎痕。
表面淡黄色至金色,下部有多支支根呈粗糙质感,支根多有明显的突起。
主根多与根茎等长或较短,断面呈象牙白,且有环纹。
横断面为多层次的细胞面。
次生韧皮部多空隙,有少量的筛管组织和分泌细胞环。
木质部特点是导管单个散在或数个相聚,薄壁细胞含草酸钙簇晶。
主根呈细长圆柱形,从中部分出2—5条支根。
人参主根长5—20 cm,直径0.5—3 cm,表面呈浅棕黄色到浅灰棕色不等,通常布有纵皱和须根的茎痕,有时布有疣状突起和短小残留根茎部分。
断面表面通常平坦,为淡黄棕色,近形成层部分为棕色。
JP日本药典(药局方)标准品汇总信息-2016-update

23541-50-6
65
他唑巴坦酸标准品及杂质对照品
Tazobactam
89786-04-9
66
酞氨西林盐酸盐标准品及杂质对照品
TalampicillinHydrochloride
39878-70-1
67
Teicoplanin标准品及杂质对照品
Teicoplanin(混合物)
3
阿扑西林标准品及杂质对照品
Aspoxicillin
63358-49-6
4
硫酸阿米卡星标准品及杂质对照品
AmikacinSulfate
39831-55-5
5
阿莫西林标准品及杂质对照品
日本药典(药局方)标准品咨询中心
何工136-o9o9-2o29
Amoxicillin
61336-70-7
6
硫酸阿贝卡星标准品及杂质对照品
53
头孢妥仑匹酯标准品及杂质对照品
CefditorenPivoxil
117467-28-4
54
头孢地尼标准品及杂质对照品
Cefdinir
91832-40-5
55
头孢磺啶钠标准品及杂质对照品
CefsulodinSodium
52152-93-9
56
头孢他啶标准品及杂质对照品
Ceftazidime
72558-82-8
13614-98-7
94
美罗培南标准品及杂质对照品
Meropenem
119478-56-7
95
利福平标准品及杂质对照品
Rifampicin
13292-46-1
96
硫酸核糖霉素标准品及杂质对照品
世界各国药典大汇总

世界各国药典大汇总中国药典(CHP):介绍就省了,大家都比较熟悉。
美国药典/国家处方集(简称USP/NF)U.S. Pharmacopeia / National Formulary:由美国政府所属的美国药典委员会(The United States Pharmacopeial Convention)编辑出版。
USP于1820年出第一版,1950年以后每5年出一次修订版,到2005年已出至第28版。
NF1883年第一版, 1980年15版起并入USP,但仍分两部分,前面为USP,后面为NF。
美国药典是美国政府对药品质量标准和检定方法作出的技术规定,也是药品生产、使用、管理、检验的法律依据。
NF收载了美国药典(USP)尚未收入的新药和新制剂。
美国药典正文药品名录分别按法定药名字母顺序排列,各药品条目大都列有药名、结构式、分子式、CAS登记号、成分和含量说明、包装和贮藏规格、鉴定方法、干燥失重、炽灼残渣、检测方法等常规项目,正文之后还有对各种药品进行测试的方法和要求的通用章节及对各种药物的一般要求的通则。
可根据书后所附的USP 和NF的联合索引查阅本书。
英国药典(BP):/《英国药典》是英国药品委员会(British Pharmacopoeia Commission)的正式出版物,是英国制药标准的重要来源。
英国药典不仅为读者提供了药用和成药配方标准以及公式配药标准,而且也向读者展示了许多明确分类并可参照的欧洲药典专著。
英国药典出版周期不定。
BP2004该药典由三卷本组成。
其中两卷为英国药典、一卷为英国兽药典(兽医药品部分)。
各条目均以药品名称字母顺序排列,内容包括药品性质、制法、血产品、免疫产品、电磁药品制法及外科材料等部分。
英国药典书后附有全部内容关键词索引。
欧洲药典(EP):/欧洲药典委员会1964年成立。
1977年出版第一版《欧洲药典》。
从1980年到1996年期间,每年将增修订的项目与新增品种出一本活页本,汇集为第二版《欧洲药典》各分册,未经修订的仍按照第一版执行。
药典标准

。
[ 收稿日期] 2 0 0 9 0 3 0 5 [ 通信作者] 赵中振, 教授。T e l : ( 0 0 8 5 2 ) 3 4 1 1 2 4 2 4 , E m a i l : z z z h a o @h k b u . e d u . h k
·2 1 1 9 ·
第3 4卷第 1 6期 2 0 0 9年 8月
V o l . 3 4 ,I s s u e 1 6 A u g u s t , 2 0 0 9
5 ] 卷和第 3卷正文中, 包括 3 4种药用植物 [ 。
疗用途; 剂量。 1 . 1 . 6 越南药典 越南是传统医药大国, 受中国文化影响 至深, 在迅速崛起的东盟中具有重要影响。目前最新的《 越 南药典》 是2 0 0 5年出版的第 3版, 该版药典包括了 3 4 2个化 学药物、 2 7 6个生药, 3 7个越南传统药物以及 4 7个生物制
[ 2 ]
前, 最新版本的《 英国药典》 为2 0 0 9年版, 其中第 3卷收录了 h e r b a l a n dc o m p l e m e n t a r ym e d i c i n e s ) 植物医学和补充医学( h e r b a l d r u g ) , 将2 0 0 7版中收载的中药甘草 使用的植物药( ( l i q u o r i c er o o t f o r j s ei nt r a d i t i o n a l C h i n e s em e d i c i n e s ) 并入条 i q u o r i c eR o o t f o r u s ei nt r a d i t i o n a l h e r b a l m e d i c i n e s ( 传统 目L 植物药甘草) 。《 英国药典》 2 0 0 9版首次将传统植物药( 包括 中药) 的饮片单列条目, 如经洗净和软化后切成横片或纵片 的 黄 芪, 以 P r o c e s s e dA s t r a g a l u sR o o tf o rU s ei nT r a d i t i o n a l
Contents-日本药典目录英文版

CONTENTSPreface (i)The Japanese Pharmacopoeia,Sixteenth Edition (1)General Notices (1)General Rules for Crude Drugs (5)General Rules for Preparations (7)General Tests,Processes and Apparatus (25)1.Chemical Methods1.01Alcohol Number Determination (25)1.02Ammonium Limit Test (27)1.03Chloride Limit Test (28)1.04Flame Coloration Test (28)1.05Mineral Oil Test (28)1.06Oxygen Flask Combustion Method (28)1.07Heavy Metals Limit Test (29)1.08Nitrogen Determination(Semimicro-Kjeldahl Method) (30)1.09Qualitative Tests (31)1.10Iron Limit Test (37)1.11Arsenic Limit Test (37)1.12Methanol Test (39)1.13Fats and Fatty Oils Test (39)1.14Sulfate Limit Test (41)1.15Readily Carbonizable Substances Test (41)2.Physical MethodsChromatography2.01Liquid Chromatography (42)2.02Gas Chromatography (45)2.03Thin-layer Chromatography (47)2.04Amino Acid Analysis of Proteins (47)Spectroscopic Methods2.21Nuclear Magnetic ResonanceSpectroscopy (48)2.22Fluorometry (50)2.23Atomic AbsorptionSpectrophotometry (51)2.24Ultraviolet-visible Spectrophotometry (52)2.25Infrared Spectrophotometry (53)Other Physical Methods2.41Loss on Drying Test (55)2.42Congealing Point Determination (55)2.43Loss on Ignition Test (56)2.44Residue on Ignition Test (56)2.45Refractive Index Determination (56)2.46Residual Solvents Test (57)2.47Osmolarity Determination (57)2.48Water Determination(Karl FischerMethod) (58)2.49Optical Rotation Determination (61)2.50Endpoint Detection Methods inTitrimetry (62)2.51Conductivity Measurement (63)2.52Thermal Analysis (65)2.53Viscosity Determination (67)2.54pH Determination (69)2.55Vitamin A Assay (71)2.56Determination of Specific Gravity andDensity (72)2.57Boiling Point and Distilling RangeTest (74)2.58X-Ray Powder Diffraction Method (75)2.59Test for Total Organic Carbon (79)2.60Melting Point Determination (80)3.Powder Property Determinations3.01Determination of Bulk and TappedDensities (82)3.02Specific Surface Area by GasAdsorption (84)3.03Powder Particle DensityDetermination (86)3.04Particle Size Determination (87)4.Biological Tests/Biochemical Tests/Microbial Tests4.01Bacterial Endotoxins Test (92)4.02Microbial Assay for Antibiotics (96)4.03Digestion Test (100)4.04Pyrogen Test (103)4.05Microbial Limit Test (103)4.06Sterility Test (114)5.Tests for Crude Drugs5.01Crude Drugs Test (117)5.02Microbial Limit Test for Crude Drugs (120)6.Tests for Preparations6.01Test for Metal Particles in OphthalmicOintments (126)6.02Uniformity of Dosage Units (127)6.03Particle Size Distribution Test forPreparations (129)6.04Test for Acid-neutralizing Capacity ofGastrointestinal Medicines (129)6.05Test for Extractable Volume ofParenteral Preparations (130)6.06Foreign Insoluble Matter Test forInjections (131)6.07Insoluble Particulate Matter Test forInjections (131)6.08Insoluble Particulate Matter Test forOphthalmic Solutions (134)6.09Disintegration Test (135)6.10Dissolution Test (137)JP XVI Contents6.11Foreign Insoluble Matter Test forOphthalmic Solutions (141)7.Tests for Containers and Packing Materials7.01Test for Glass Containers for Injections..1417.02Test Methods for Plastic Containers (142)7.03Test for Rubber Closure for AqueousInfusions (148)8.Other Methods8.01Sterilization and Aseptic Manipulation (149)9.Reference Standards;Standard Solutions;Reagents,Test Solutions;MeasuringInstruments,Appliances,etc.Reference Standards9.01Reference Standards (150)Standard Solutions9.21Standard Solutions for VolumetricAnalysis (153)9.22Standard Solutions (164)9.23Matching Fluids for Color (166)Reagents,Test Solutions,etc.9.41Reagents,Test Solutions (167)9.42Solid Supports/Column Packings forChromatography (306)9.43Filter Papers,Filters for filtration,Test Papers,Crucibles,etc (308)9.44Standard Particles,etc (308)Measuring Instruments and Appliances,Thermometers,etc.9.61Optical Filters for Wavelength andTransmission Rate Calibration (309)9.62Measuring Instruments,Appliances (309)9.63Thermometers (310)Official Monographs (313)Crude Drugs (1593)Infrared Reference Spectra.....................1775–1961 Ultraviolet-visible Reference Spectra.........1965–2131General InformationG1Physics and ChemistryGuideline for Residual Solvents and Models for the Residual Solvents Test (2135)Inductively Coupled Plasma Atomic Emission Spectrometry (2136)Near Infrared Spectrometry (2141)pH Test for Gastrointestinal Medicine (2144)System Suitability (2145)Test for Trace Amounts of Aluminum inTrans Parenteral Nutrition(TPN)Solutions (2146)Validation of Analytical Procedures (2148)G2Solid-state PropertiesLaser Diffraction Measurement ofParticle Size (2151)Powder Fineness (2154)Powder Flow (2155)Solid and Particle Densities (2158)G3Biotechnological/Biological Products Amino Acid Analysis (2159)Basic Requirements for Viral Safety ofBiotechnological/Biological Productslisted in Japanese Pharmacopoeia (2166)Capillary Electrophoresis (2179)Isoelectric Focusing (2184)Mass Spectrometry of Peptides andProteins (2186)Mycoplasma Testing for Cell Substrates used for the Production of Biotechnological/Biological Products (2188)Peptide Mapping (2191)Qualification of Animals as Origin ofAnimal-derived Medicinal Productsprovided in the General Notices ofJapanese Pharmacopoeia and OtherStandards (2194)SDS-Polyacrylamide Gel Electrophoresis (2196)Total Protein Assay (2201)G4MicroorganismsDecision of Limit for BacterialEndotoxins (2205)Disinfection and Sterilization Methods (2205)Media Fill Test(Process Simulation) (2206)Microbial Attributes of Non-sterilePharmaceutical Products (2209)Microbiological Evaluation of Processing Areas for Sterile PharmaceuticalProducts (2211)Preservatives-Effectiveness Tests (2215)Rapid Counting of Microbes usingFluorescent Staining (2217)Rapid Identification of MicroorganismsBased on Molecular Biological Method (2220)Sterility Assurance for Terminally Sterilized Pharmaceutical Products (2221)Terminal Sterilization and SterilizationIndicators (2225)G5Crude DrugsAristolochic Acid (2227)Purity Tests on Crude Drugs Using Genetic Information (2228)On the Scientific Names of Crude DrugsListed in the JP (2231)G6Drug FormulationTablet Friability Test (2244)G7Containers and PackagePlastic Containers for PharmaceuticalJP XVI ContentsProducts (2244)G8WaterQuality Control of Water for PharmaceuticalUse (2246)Water to be used in the Tests of Drugs (2253)G9OthersInternational Harmonization Implementedin the Japanese Pharmacopoeia SixteenthEdition (2253)AppendixAtomic Weight Table(2010) (2287)Standard Atomic Weights2010 (2288)Index (2291)Index in Latin name (2307)Index in Japanese (2309)PREFACEThe15th Edition of the Japanese Pharmacopoeia (JP)was promulgated by Ministerial Notification No. 285of the Ministry of Health,Labour and Welfare (MHLW)on March31,2006.In July2006,the Committee on JP established the basic principles for the preparation of the JP16th Edi-tion,setting out the roles and characteristics of the JP, the definite measures for the revision,and the date of the revision.At the Committee,the five basic principles of JP, which we refer to as the``five pillars'',were estab-lished as follows:1)Including all drugs which are im-portant from the viewpoint of health care and medical treatment;2)Making qualitative improvement by in-troducing the latest science and technology;3)Pro-moting internationalization;4)Making prompt partial revision as necessary and facilitating smooth adminis-trative operation;and5)Ensuring transparency regarding the revision,and disseminating the JP to the public.It was agreed that the Committee on JP should make efforts,on the basis of these principles,to en-sure that the JP is used more effectively in the fields of health care and medical treatment by taking appropri-ate measurements,including getting the understanding and cooperation of other parties concerned.It was agreed that the JP should provide an official standard,being required to assure the quality of medi-cines in Japan in response to the progress of science and technology and medical demands at the time.It should define the standards for specifications,as well as the methods of testing to assure overall quality of all drugs in principle,and it should have a role in clarifying the criteria for quality assurance of drugs that are recognized to be essential for public health and medical treatment.The JP has been prepared with the aid of the knowledge and experience of many professionals in the pharmaceutical field.Therefore,the JP should have the characteristics of an official standard,which might be widely used by all parties concerned.It should provide information and understanding about the quality of drugs to the public,and it should be conducive to smooth and effective regulatory control of the quality of drugs,as well as promoting and maintaining international consistency and harmoniza-tion of technical requirements.It was also agreed that JP articles should cover drugs,which are important from the viewpoint of health care and medical treatment,clinical results and frequency of use,as soon as possible after they reach the market.The target date for the publication of JP16th Edi-tion(the Japanese edition)was set as April2011.JP Expert Committees are organized with the fol-lowing panels:Panel on the Principles of Revisions; Sub-committee on the Principles of Revisions;Panel on Medicinal Chemicals;Panel on Antibiotics;Panel on Biologicals;Panel on Crude Drugs;Panel on Phar-maceutical Excipients;Panel on Physico-Chemical Methods;Panel on Preparations;Panel on Physical Methods;Panel on Biological Tests;Panel on Nomen-clature;Panel on International Harmonization;Panel on Pharmaceutical Water;and Panel on Reference Standards.Furthermore,working groups are estab-lished under the Panel on Physico-Chemical Methods, Panel on Preparations and Panel on Biological Tests to expedite discussion on revision drafts.In the Committee on JP,Takao Hayakawa took the role of chairman from July2003to December2010, and Mitsuru Hashida from January2011to March 2011.In addition to the regular revision every five years in line with the basic principles for the preparation of the JP it was agreed that partial revision should be done as necessary to take account of recent progress of science and in the interests of international harmonization.In accordance with the above principles,the panels initiated deliberations on selection of articles,and on revisions for General Notices,General Rules for Crude Drugs,General Rules for Preparations,General Tests, Monographs and so on.Draft revisions covering subjects in General Notices, General Rules for Crude Drugs,General Rules for Preparations,General Tests and Monographs,for which discussions were finished between September 2005and March2007,were prepared for a supplement to the JP15.They were examined by the Committee on JP in April2007,followed by the Pharmaceutical Affairs and Food Sanitation Council(PAFSC)in June 2007,and then submitted to the Minister of Health, Labour and Welfare.The supplement was named ``Supplement I to the JP15th Edition'',promulgated on September28,2007by Ministerial Notification No. 316of MHLW,and became effective on October1,i。
国外药典介绍

USP–NF 的不断 修订
修订公告 IRA拟议的修订 说明
勘误表
2019/12/16
美国药典的修订
USP–NF 不断进行修订。 修订包括 USP–NF 年度修订和每年两 次增补,以及 USP 网站上的加速修订。 USP 使用加速修订过程 加快修订美国药典–国家处方集 (USP–NF)。加速修订包括修订公 告、临时修订声明 (IRA) 和勘误表。 是 USP 最快的修订途径,可取代在 USP–NF 及其增补(印刷版 和在线版)中发布的标准。 在 USP 网站上发布的修订公告指示 其正式日期和纳入正式出版物中的日期。 IRA 在 PF 中发布,征求公众意见期为 90 天。 在意见(如果有) 通过审查并且 IRA 得到相关专家委员会的批准后,最终 IRA 将发 布在 USP 网站上。 与修订公告一样,IRA 可取代在印刷版和在线 版的 USP–NF 及其增补中发布的标准。 IRA 被纳入下一个可用的 USP–NF 或增补中。 是指在 USP–NF 或其增补中发布的文字有误,不能准确地反映专 家委员会批准的预期要求。 勘误表发布在网站上,并立即成为正 式版本。 勘误表被纳入下一个可用的正式出版物中。
通则提供各论中所用术语的定义,以及解释各论要求所需 的信息。
2019/12/16
美国药典USP内容介绍
美国药典-国家处方集(USP-NF)是两个法定药品标准 USP 中提供关于原料药和制剂的质量标准。
NF 中提供关于辅料的质量标准。 各论中提到的测试和程序将在 USP-NF 附录中予以详细说明。
2019/12/16
2019/12/16
日本药典简介
简介
《日本药典》(The Jepanese Pharmacopoeia)的名称是《日本药局方 》,英文缩写JP 。由日本药局方编辑委 员会编制,厚生省颁布执行。1886年6月 25号颁布第一版,1887年7月1日开始实
日本药品(PMDA数据库、IF文件、药典(JP))网址信息

日本药品(PMDA数据库、IF文件、药典(JP))网址信息日本的医药学在世界医药学中是重要的组成部分,在某些方面甚至走在了世界的前端,日本医药学和中国的传统医药学有着紧密的联系。
特别是日本的医药化工在全球的地位举足轻重,是传统的制药强国,生产的药品也占有重要地位。
2016年,国家局CFDA发布关于仿制药一致性评价技术指导原则的通告中,将“在欧盟、美国、日本获准上市并获得参比制剂地位的仿制药”定义为国际公认的同种药物,并将与原研药并列作为国内一致性评价的参比制剂,都在说明日本新药具有非常重要的地位。
相较于其它国家不同,日本对上市药品公开信息程度非常高,研究和利用日本医药网站,将有利于了解日本医药领域的研究状况,研发人员引用或借鉴文献资料等。
药融云医药数据库V4.0包含了日本PMDA、日本药典(JP)、日本医药情、原料药、说明书、药品体外溶出试验信息、日本MF注册、日本橙皮书等数据。
一、日本PMDA:PMDA全称为Pharmaceuticals and Medical Devices Agency,其日语名称翻译过来就是独立行政法人医药品医疗器械综合机构,PMDA的一个最重要的职能就是技术审评。
PMDA所行使的职责相当于我国的国家食品药品监督管理局下所属单位国家药典委员会,药品审评中心,审核查验中心里的药品和医疗器械业务,药品评价中心,医疗器械技术审评中心所涵盖的业务内容。
PMDA数据库主要公布了日本上市药品的IF文件、说明书以及相关注册资料。
其中,IF 文件对于仿制药研发者来说,是非常重要的一份资料,其内容主要是公布药品的处方基本信息,药代动力学,临床相关信息等。
IF是Interview form,相当于比较详细的说明书。
日本lF文件查询步骤如下:1.登录PMDA/2.点击“医療用医藻品”(医疗药品)。
3.在“一般名·販壳名(医藻品の名称)”栏输入药品名称(药品名称用日文输入,可先把中文翻译为英文,再英文翻译日文,更容易搜索)。
世界各国药典大汇总

世界各国药典大汇总为了大家方便下载,本帖把中国,美国,英国,日本等主要国家的药典做一个汇总,欢迎大家跟帖补充。
A、中国药典(CHP):不介绍了…… 国家药典委网址/cms/home/B、美国药典/国家处方集(简称USP/NF)U.S. Pharmacopeia / National Formulary:/uspnf/login由美国政府所属的美国药典委员会(The United States Pharmacopeial Convention)编辑出版。
USP于1820年出第一版,1950年以后每5年出一次修订版,到2005年已出至第28版。
NF1883年第一版,1980年15版起并入USP,但仍分两部分,前面为USP,后面为NF。
美国药典是美国政府对药品质量标准和检定方法作出的技术规定,也是药品生产、使用、管理、检验的法律依据。
NF收载了美国药典(USP)尚未收入的新药和新制剂。
美国药典正文药品名录分别按法定药名字母顺序排列,各药品条目大都列有药名、结构式、分子式、CAS登记号、成分和含量说明、包装和贮藏规格、鉴定方法、干燥失重、炽灼残渣、检测方法等常规项目,正文之后还有对各种药品进行测试的方法和要求的通用章节及对各种药物的一般要求的通则。
可根据书后所附的USP 和NF的联合索引查阅本书。
C、英国药典(BP):/《英国药典》是英国药品委员会(British Pharmacopoeia Commission)的正式出版物,是英国制药标准的重要来源。
英国药典不仅为读者提供了药用和成药配方标准以及公式配药标准,而且也向读者展示了许多明确分类并可参照的欧洲药典专著。
英国药典出版周期不定。
BP2004该药典由三卷本组成。
其中两卷为英国药典、一卷为英国兽药典(兽医药品部分)。
各条目均以药品名称字母顺序排列,内容包括药品性质、制法、血产品、免疫产品、电磁药品制法及外科材料等部分。
英国药典书后附有全部内容关键词索引。
Ubenimex-日本药典JP16

1546JP XVIUbenimex /Official Monographs (6)Iron <1.10>—Prepare the test solution with 1.0g of L -Tyrosine according to Method 3,and perform the test ac-cording to Method A.Prepare the control solution with 1.0mL of Standard Iron Solution (not more than 10ppm).(7)Related substances—Dissolve 0.20g of L -Tyrosine in 10mL of diluted ammonia solution (28)(1in 2),add water to make 20mL,and use this solution as the sample solution.Pipet 1mL of the sample solution,add water to make ex-actly 10mL,pipet 1mL of this solution,add water to make exactly 50mL,and use this solution as the standard solution.Perform the test with these solutions as directed under Thin-layer Chromatography <2.03>.Spot 5m L each of the sample solution and standard solution on a plate of silica gel for thin-layer chromatography.Then develop with a mixture of 1-propanol and ammonia solution (28)(67:33)to a distanceof about 10cm,and dry the plate at 809C for 30minutes.Spray evenly a solution of ninhydrin in a mixture of metha-nol and acetic acid (100)(97:3)(1in 100)on the plate,and then heat at 809C for 10minutes:the spot other than the principal spot obtained from the sample solution is not more intense than the spot from the standard solution.Loss on drying <2.41>Not more than 0.3z (1g,1059C,3hours).Residue on ignition <2.44>Not more than 0.1z (1g).Assay Weigh accurately about 0.18g of L -Tyrosine previ-ously dried,dissolve in 6mL of formic acid,add 50mL ofacetic acid (100),and titrate <2.50>with 0.1mol/L perchloric acid VS (potentiometric titration).Perform a blank determi-nation in the same manner,and make any necessary correc-tion.Each mL of 0.1mol/L perchloric acid VS =18.12mg of C 9H 11NO 3Containers and storageContainers—Tight containers.UbenimexウベニメクスC 16H 24N 2O 4:308.37(2S )-2-[(2S ,3R )-3-Amino-2-hydroxy-4-phenylbutanoylamino]-4-methylpentanoic acid [58970-76-6]Ubenimex,when dried,contains not less than 98.5z and not more than 101.0z of C 16H 24N 2O 4.Description Ubenimex occurs as a white crystalline pow-der.It is freely soluble in acetic acid (100),slightly soluble in water,and very slightly soluble in ethanol (99.5).It dissolves in 1mol/L hydrochloric acid TS.Melting point:about 2309C (with decomposition).Identification(1)Determine the absorption spectrum of asolution of Ubenimex (1in 2000)as directed under Ultravio-let-visible Spectrophotometry <2.24>,and compare the spec-trum with the Reference Spectrum:both spectra exhibit simi-lar intensities of absorption at the same wavelengths.(2)Determine the infrared absorption spectrum as di-rected in the potassium bromide disk method under Infrared Spectrophotometry <2.25>,and compare the spectrum with the Reference Spectrum:both spectra exhibit similar inten-sities of absorption at the same wave numbers.Optical rotation <2.49>[a ]20D :-15.5–-17.59(after dry-ing,0.5g,1mol/L hydrochloric acid TS,50mL,100mm).Purity (1)Heavy metals <1.07>—Proceed with 2.0g of Ubenimex according to Method 2,and perform the test.Pre-pare the control solution with 2.0mL of Standard Lead So-lution (not more than 10ppm).(2)Related substances—Dissolve 30mg of Ubenimex in 10mL of the mobile phase A,and use this solution as the sample solution.Pipet 2mL of the sample solution,add the mobile phase A to make exactly 200mL,and use this solu-tion as the standard solution.Perform the test with exactly 20m L each of the sample solution and standard solution as directed under Liquid Chromatography <2.01>according to the following conditions,and determine each peak area of these solutions by the automatic integration method:the area of the peak other than ubenimex obtained from the sample solution is not larger than 1/2times the peak area of ubenimex from the standard solution.Furthermore,the total area of the peaks other than ubenimex is not larger than the peak area of ubenimex from the standard solution.Operating conditions —Detector:An ultraviolet absorption photometer (wave-length:220nm).Column:A stainless steel column 4.6mm in inside diame-ter and 25cm in length,packed with octadecylsilanized silica gel for liquid chromatography (5m m in particle diameter).Column temperature:A constant temperature of about 259C.Mobile phase A:A mixture of diluted 0.1mol/L potas-sium dihydrogen phosphate TS (13in 20)and acetonitrile for liquid chromatography (17:3).Mobile phase B:A mixture of acetonitrile for liquid chro-matography and diluted 0.1mol/L potassium dihydrogen phosphate TS (13in 20)(2:1).Flowing of the mobile phase:Control the gradient by mix-ing the mobile phases A and B as directed in the following table.Time after injection of sample (min)Mobile phase A(vol z )Mobile phase B(vol z )0–20100020–60100→00→10060–70100Flow rate:Adjust the flow rate so that the retention timeof ubenimex is about 14minutes.Time span of measurement:About 5times as long as the retention time of ubenimex,beginning after the solvent peak.System suitability —Test for required detectability:Pipet 1mL of the standard1547 JP XVI Official Monographs/Ubenimex Capsulessolution,and add the mobile phase A to make exactly10 mL.Confirm that the peak area of ubenimex obtained from 20m L of this solution is equivalent to7to13z of that from 20m L of the standard solution.System performance:When the procedure is run with20 m L of the standard solution under the above operating con-ditions,the number of theoretical plates and the symmetry factor of the peak of ubenimex are not less than5000and not more than2.0,respectively.System repeatability:When the test is repeated6times with20m L of the standard solution under the above operat-ing conditions,the relative standard deviation of the peak area of ubenimex is not more than2.0z.Loss on drying<2.41>Not more than0.5z(0.5g,in vacu-um,809C,4hours).Residue on ignition<2.44>Not more than0.1z(1g).Assay Weigh accurately about0.5g of Ubenimex,previ-ously dried,dissolve in60mL of acetic acid(100),and titrate <2.50>with0.1mol/L perchloric acid VS(potentiometric titration).Perform a blank determination in the same man-ner,and make any necessary correction.Each mL of0.1mol/L perchloric acid VS=30.84mg of C16H24N2O4Containers and storage Containers—Tight containers.Ubenimex CapsulesウベニメクスカプセルUbenimex Capsules contain not less than93.0z and not more than107.0z of the labeled amount of ubeni-mex(C16H24N2O4:308.37).Method of preparation Prepare as directed under Cap-sules,with Ubenimex.Identification To a quantity of the contents of Ubenimex Capsules,equivalent to25mg of Ubenimex according to the labeled amount,add water to make50mL,shake well,and filter.Determine the absorption spectrum of the filtrate as directed under Ultraviolet-visible Spectrophotometry<2.24>: it exhibits maxima between250nm and254nm,between255 nm and259nm,and between261nm and265nm. Uniformity of dosage units<6.02>Perform the test accord-ing to the following method:it meets the requirement of the Content uniformity test.To1capsule of Ubenimex Capsules add30mL of a mix-ture of water and acetonitrile(7:3),shake well for30 minutes,and add a mixture of water and acetonitrile(7:3)to make exactly50mL.Centrifuge this solution and filter the supernatant liquid through a membrane filter with a pore size not exceeding0.45m m.Discard the first5mL of the filtrate,pipet V mL of the subsequent filtrate,equivalent to about3mg of ubenimex(C16H24N2O4),add exactly4mL of the internal standard solution,add a mixture of water and acetonitrile(7:3)to make50mL,and use this solution as the sample solution.Separately,weigh accurately about20mg of ubenimex for assay,previously dried at809C for4hours under reduced pressure,and dissolve in a mixture of water and acetonitrile(7:3)to make exactly100mL.Pipet15mL of this solution,add exactly4mL of the internal standard solution,add a mixture of water and acetonitrile(7:3)to make50mL,and use this solution as the standard solution. Perform the test with20m L each of the sample solution and standard solution as directed under Liquid Chromatography <2.01>according to the following conditions,and calculate the ratios,Q T and Q S,of the peak area of ubenimex to that of the internal standard.Amount(mg)of ubenimex(C16H24N2O4)=M S×Q T/Q S×1/V×15/2M S:Amount(mg)of ubenimex for assayInternal standard solution—A solution of ethyl parahy-droxybenzoate in a mixture of water and acetonitrile(7:3) (1in2000).Operating conditions—Proceed as directed in the operating conditions in the Assay.System suitability—System performance:When the procedure is run with20 m L of the standard solution under the above operating con-ditions,ubenimex and the internal standard are eluted in this order with the resolution between these peaks being not less than10.System repeatability:When the test is repeated6times with20m L of the standard solution under the above operat-ing conditions,the relative standard deviation of the ratio of the peak area of ubenimex to that of the internal standard is not more than2.0z.Dissolution<6.10>When the test is performed at50revolu-tions per minute according to the Paddle method using the sinker,using900mL of water as the dissolution medium,the dissolution rate in30minutes of Ubenimex Capsules is not less than70z.Start the test with1capsule of Ubenimex Capsules,with-draw not less than20mL of the medium at the specified minute after starting the test,and filter through a membrane filter with a pore size not exceeding0.45m m.Discard the first10mL of the filtrate,pipet V mL of the subsequent filtrate,add a mixture of water and acetonitrile(7:3)to make exactly V?mL so that each mL contains about11m g of ubenimex(C16H24N2O4)according to labeled amount,and use this solution as the sample solution.Separately,weigh accurately about22mg of ubenimex for assay,previously dried in vacume at809C for4hours,and dissolve in a mix-ture of water and acetonitrile(7:3)to make exactly100mL. Pipet5mL of this solution,add a mixture of water and acetonitrile(7:3)to make exactly100mL,and use this solu-tion as the standard solution.Perform the test with exactly 50m L each of the sample solution and standard solution as directed under Liquid Chromatography<2.01>according to the following conditions,and determine the peak areas,A T and A S,of ubenimex in each solution.Dissolution rate(z)with respect to the labeled amount of ubenimex(C16H24N2O4)=M S×A T/A S×V?/V×1/C×45M S:Amount(mg)of ubenimex for assayC:Labeled amount(mg)of ubenimex(C16H24N2O4)in1 capsule。
Rhubarb-JP16日本药典大黄标准

RhubarbRhei RhizomaダイオウRhubarb is usually the rhizome of Rheum palmatum Linneá , Rheum tanguticum Maximowicz, Rheum officinale Baillon, Rheum coreanum Nakai or their interspecific hybrids (Polygonaceae).It contains not less than 0.25z of sennosides A(C42H38O20: 862.74), calculated on the basis of dried material.Description Ovoid, oblong-ovoid or cylindrical rhizome, often cut crosswise or longitudinally, 4 . 10 cm in diameter, 5 . 15 cm in length. In the case of Rhubarb without most part of cortex, the outer surface is flat and smooth, yellow- brown to light brown in color, and sometimes exhibiting white, fine reticulations; thick and hard in texture. In the case of Rhubarb with cork layer, externally dark brown or reddish black, and with coarse wrinkles; rough and brittle in texture. The fractured surface of Rhubarb is not fibrous; transverse section grayish brown, light grayish brown or brown in color, having patterns of blackish brown tissue complicated with white and light brown tissues; near the cambium, the patterns often radiate, and in pith, consist of whirls of tissues radiated from the center of a small brown circle 1 . 3 mm in diameter and arranged in a ring or scat- tered irregularly.Odor, characteristic; taste, slightly astringent and bitter; when chewed, gritty between the teeth, and coloring the saliva yellow.Under a microscope <5.01>, the transverse section reveals mostly parenchyma cells; small abnormal cambium-rings scattered here and there in the pith; the cambium-rings produce phloem inside and xylem outside, accompanied with 2 to 4 rows of medullary rays containing brown-colored sub- stances, and the rays run radiately from the center of the ring towards the outside forming whirls of tissues; paren- chyma cells contain starch grains, brown-colored substances or crystal druses of calcium oxalate.Identification To 2 g of pulverized Rhubarb add 40 mL ofa mixture of tetrahydrofuran and water (7:3), shake for 30 minutes, and centrifuge. Transfer the supernatant liquid to a separator, add 13 g of sodium chloride, and shake for 30 minutes. Separate the water layer with undissolved sodium chloride, and adjust the pH to 1.5 by adding 1 mol/L hydro- chloric acid TS. Transfer this solution to another separator, add 30 mL of tetrahydrofuran, shake for 10 minutes, sepa- rate the tetrahydrofuran layer, and use this solution as the sample solution. Separately, dissolve 1 mg of Sennoside A RS in 4 mL of a mixture of tetrahydrofuran and water (7:3), and use this solution as the standard solution. Perform the test with these solutions as directed under Thin-layer Chro- matography <2.03>. Spot 40 mL each of the sample solution and standard solution on a plate of silica gel for thin-layer chromatography at 10 mm along the initial line. Develop the plate with a mixture of 1-propanol, ethyl acetate, water and acetic acid (100) (40:40:30:1) to a distance of about 15 cm, and air-dry the plate. Examine under ultraviolet light (main wavelength: 365 nm): one of the spots from the sample solu- tion and a red fluorescent spot from the standard solution show the same color tone and the same Rf value.Purity (1) Heavy metals <1.07>.Proceed with 3.0 g of pulverized Rhubarb according to Method 3, and perform the test. Prepare the control solution with 3.0 mL of Standard Lead Solution (not more than 10 ppm).(2) Arsenic <1.11>.Prepare the test solution with 0.40 gof pulverized Rhubarb according to Method 4, and perform the test (not more than 5 ppm).(3) Raponticin.To 0.5 g of pulverized Rhubarb add ex-actly 10 mL of ethanol (95), heat on a water bath with a reflux condenser for 10 minutes, and filter. Perform the test as directed under Thin-layer Chromatography <2.03>, using the filtrate as the sample solution. Spot 10 mL of the sample solution on a plate of silica gel for thin-layer chromatogra- phy <2.03>. Develop the plate with a mixture of isopropyl ether, 1-butanol and methanol (26:7:7) to a distance of about 10 cm, and air-dry the plate. Examine under ultravio- let light (main wavelength 365 nm): no spot with blue-purple fluorescence is observed at an Rf value between 0.3 and 0.6, though a bluish white fluorescence may appear.Loss on drying <5.01> Not more than 13.0z (6 hours).Total ash <5.01> Not more than 13.0z.Extract content <5.01> Dilute ethanol-soluble extract: not less than 30.0z.Assay Weigh accurately about 0.5 g of pulverized Rhubarb, add exactly 50 mL of a solution of sodium hydro- gen carbonate (1 in 1000), shake for 30 minutes, filter, and use the filtrate as the sample solution. Separately, weigh ac-curately about 10 mg of Sennoside A RS, (separately deter- mine the water) dissolve in a solution of sodium hydrogen carbonate (1 in 1000) to make exactly 50 mL. Pipet 5 mL of this solution, add a solution of sodium hydrogen carbonate (1 in 1000) to make exactly 20 mL and use this solution as the standard solution. Perform the test with exactly 10 mL of the sample solution and standard solution as directed under Liquid Chromatography <2.01> according to the following conditions, and determine the peak areas, A T and A S, of sennoside A.Amount (mg) of sennoside A (C42H38O20)=M S ×A T/A S ×1/4M S: Amount (mg) of Sennoside A RS, calculated on the anhydrous basisOperating conditions.Detector: An ultraviolet absorption photometer (wave- length: 340 nm).Column: A stainless steel column 4 . 6 mm in inside diam- eter and 15 cm in length, packed with octadecylsilanized silica gel for liquid chromatography (5 mm in particle diame- ter).Column temperature: A constant temperature of about409C.Mobile phase: A mixture of diluted acetic acid (100) (1 in 80) and acetonitrile (4:1).Flow rate: Adjust the flow rate so that the retention timeof sennoside A is about 15 minutes.System suitability.System performance: Dissolve 1 mg each of Sennoside A RS and naringin for thin-layer chromatography in a solution of sodium hydrogen carbonate (1 in 1000) to make 10 mL. When the procedure is run with 20 mL of this solution under the above operating conditions, sennoside A and naringin are eluted in this order with the resolution between these peaks being not less than 3.System repeatability: When the test is repeated 6 timeswith 10 mL of the standard solution under the above operat- ing conditions, the relative standard deviation of the peak area of sennoside A is not more than 1.5z.Containers and storage Containers.Well-closed contain- ers.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
日本药典(药局方)标准品汇总信息
序号
名称(中文)
Name(English)
CAS NO.
1
阿奇霉素标准品及杂质对照品
Azithromycin
117772-70-0
2
氨曲南标准品及杂质对照品
Aztreonam
78110-38-0
92636-39-0
61
盐酸头孢甲肟标准品及杂质对照品
CefmenoximeHydrochloride
75738-58-8
62
头孢沙定标准品及杂质对照品
Cefroxadine
51762-05-1
63
头孢呋辛酯标准品及杂质对照品
CefuroximeAxetil
64544-07-6
64
盐酸佐柔比星盐酸柔红霉素
33103-22-9,33137-73-4
14
盐酸土霉素标准品及杂质对照品
OxytetracyclineHydrochloride
2058-46-0
15
硫酸卡那霉素标准品及杂质对照品
KanamycinMonosulfate
25389-94-0
16
卡鲁莫南钠标准品及杂质对照品
CarumonamSodium
10
亚胺培南标准品及杂质对照品
Imipenem
64221-86-9
11
盐酸表柔比星盐酸表阿霉素标准品及杂质对照品
EpirubicinHydrochloride
56390-09-1
12
红霉素标准品及杂质对照品
Erythromycin
114-07-8
13
恩维霉素标准品及杂质对照品
EnviomycinSulfate
83105-70-8
39
磺苄西林钠标准品及杂质对照品
SulbenicillinSodium
28002-18-8
40
头孢克洛头孢克罗标准品及杂质对照品
Cefaclor
53994-73-3
41
头孢曲嗪标准品及杂质对照品
CefatrizinePropyleneGlycolate
51627-14-6
42
头孢羟氨苄标准品及杂质对照品
Siccanin
22733-60-4
32
净司他丁斯酯标准品及杂质对照品
ZinostatinStimalamer
123760-07-6
33
地贝卡星硫酸盐标准品及杂质对照品
DibekacinSulfate
58580-55-5
34
交沙霉素标准品及杂质对照品
Josamycin
16846-24-5
35
交沙霉素丙酸酯标准品及杂质对照品
84
法罗培南钠标准品及杂质对照品
FaropenemSodium
122547-49-3
85
非奈西林钾标准品及杂质对照品
PhenethicillinPotassium
132-93-4
86
硫酸新霉素标准品及杂质对照品
FradiomycinSulfate(混合物)
119-04-0,66-86-4, 1405-10-3
57
头孢曲松钠标准品及杂质对照品
Ceftriaxone sodium
74578-69-1
58
硫酸头孢匹罗标准品及杂质对照品
CefpiromeSulfate
98753-19-6
59
头孢泊肟酯标准品及杂质对照品
CefpodoximeProxetil
87239-81-4
60
头孢米诺钠标准品及杂质对照品
CefminoxSodium
53
头孢妥仑匹酯标准品及杂质对照品
CefditorenPivoxil
117467-28-4
54
头孢地尼标准品及杂质对照品
Cefdinir
91832-40-5
55
头孢磺啶钠标准品及杂质对照品
CefsulodinSodium
52152-93-9
56
头孢他啶标准品及杂质对照品
Ceftazidime
72558-82-8
Hydrochloride
64-73-3
70
盐酸强力霉素标准品及杂质对照品
Doxycycline Hydrochloride
10592-13-9
71
盐酸多柔比星盐酸阿霉素标准品及杂质对照品
Doxorubicin Hydrochloride
25316-40-9
72
妥布霉素标准品及杂质对照品
Tobramycin
123171-59-5
47
头孢地嗪钠标准品及杂质对照品
CefodizimeSodium
86329-79-5
48
头孢唑兰标准品及杂质对照品
CefozopranHydrochloride
113359-04-9
49
头孢替安盐酸盐标准品及杂质对照品
CefotiamHydrochloride
66309-69-1
ChloramphenicolPalmitate
530-43-8
25
硫酸庆大霉素标准品及杂质对照品
Gentamicin Sulfate
1405-41-0
26
粘杆菌素甲基磺酸钠标准品及杂质对照品
ColistinSodiumMethanesulfonate
8068-28-8
27
硫酸粘杆菌素标准品及杂质对照品
MicronomicinSulfate
52093-21-7
91
麦迪霉素标准品及杂质对照品
Midecamycin
35457-80-8
92
醋酸麦迪霉素标准品及杂质对照品
MidecamycinAcetate
55881-07-7
93
盐酸米诺环素标准品及杂质对照品
Minocycline Hydrochloride
50
头孢替安海替酯盐酸盐标准品及杂质对照品
CefotiamHexetilHydrochloride
95789-30-3
51
头孢替坦标准品及杂质对照品Cefotean69712-56-7
52
盐酸头孢卡品酯标准品及杂质对照品
CefcapenePivoxilHydrochloride
147816-24-8
13614-98-7
94
美罗培南标准品及杂质对照品
Meropenem
119478-56-7
95
利福平标准品及杂质对照品
Rifampicin
13292-46-1
96
硫酸核糖霉素标准品及杂质对照品
RibostamycinSulfate
53797-35-6
97
盐酸林可霉素标准品及杂质对照品
LincomycinHydrochloride
7179-49-9
98
仑氨西林盐酸盐标准品及杂质对照品
LenampicillinHydrochloride
80734-02-7
99
罗红霉素标准品及杂质对照品
Roxithromycin
80214-83-1
100
罗他霉素标准品及杂质对照品
Rokitamycin
74014-51-0
37661-08-8
76
杆菌肽标准品及杂质对照品
Bacitracin(混合物)
1405-87-4
77
帕尼培南标准品及杂质对照品
Panipenem
87726-17-8
78
盐酸万古霉素标准品及杂质对照品
VancomycinHydrochloride
1404-93-9
79
盐酸匹美西林标准品及杂质对照品
PivmecillinamHydrochloride
86832-68-0
17
短杆菌肽标准品及杂质对照品
Gramicidin
1405-97-6
18
克拉霉素标准品及杂质对照品
Clarithromycin
81103-11-9
19
灰黄霉素标准品及杂质对照品
Griseofulvin
126-07-8
20
盐酸克林霉素标准品及杂质对照品
Clindamycin Hydrochloride
Cefadroxil
50370-12-2
43
头孢氨苄标准品及杂质对照品
Cefalexin
15686-71-2
44
头孢噻吩钠标准品及杂质对照品
CefalotinSodium
58-71-9
45
Cefixime标准品及杂质对照品
Cefixime
79350-37-1
46
头孢吡肟盐酸盐标准品及杂质对照品
CefepimeDihydrochloride
32887-03-9
80
哌拉西林标准品及杂质对照品
Piperacillin
61477-96-1
81
纳他霉素标准品及杂质对照品
Pimaricin
7681-93-8
82
吡柔比星标准品及杂质对照品
Pirarubicin
72496-41-4
83
吡咯尼林硝吡咯菌素标准品及杂质对照品
Pyrrolnitrin
1018-71-9
3
阿扑西林标准品及杂质对照品
Aspoxicillin
63358-49-6
4
硫酸阿米卡星标准品及杂质对照品
AmikacinSulfate
39831-55-5