GMP计算机系统附录知识培训(CFDAGMP计算机化系统附录、EUGMP附录11、21PART11、GAMP5)2015.12
关于欧盟GMP指南附录11“计算机系统” 的修订 - 概念文件介绍

Revision of the EU GMP Guide Annex 11 "Computerised Systems" -Presentation of Concept Paper关于欧盟GMP指南附录11“计算机系统”的修订- 概念文件介绍The current EU GMP Guidance Annex 11 "Computerised Systems" has been in force since 2011. It has been discussed for a long time to revise this annex in order to meet current technological and regulatory developments. On 16 November 2022, the EMA (European Medicines Agency) published a 5-page "Concept-Paper on the revision of Annex 11 of the guidelines on Good Manufacturing Practice for medicinal products -Computerised Systems". Comments on this concept paper can be submitted until 16 January 2023.当前的欧盟GMP指南附录11“计算机化系统”自2011年起就已经生效。
关于修订该附录以反映最新的技术和法规发展的讨论,已经持续了很长一段时间。
2022年11月16日,EMA(欧洲药品管理局)发表了一份长达5页的“关于修订药品良好生产规范指南附录11-“计算机化系统”的概念文件”。
有关此概念文件的评论可以持续提交,截止到2023年1月16日。
GMP附录-计算机系统

计算机化系统(征求意见稿)第一章 范 围第一条 本附录适用于在药品生产质量管理过程中引入的计算机化系统,它由一系列硬件和软件组成,以满足特定的功能。
第二章 原 则第二条 用计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条 风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证的范围和数据完整性控制的程度。
第四条 企业应当注重计算机化系统供应商的管理,制定相应的操作规程。
供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当能够提供与供应商质量体系和审计信息相关的文件。
第三章 人 员第五条 计算机化系统的“生命周期”中所涉及的各种活动,如验证、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
在职责中涉及使用和管理计算机化系统的人员,应当接受相应的使用和管理培训。
确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等方面进行培训和指导。
第四章 验 证第六条 计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
验证应当贯穿于计算机化系统生命周期的全过程。
第七条 企业应当建立包含所有计算机化系统的清单,标明与药品生产质量管理相关的功能。
清单应当及时更新。
第八条 企业应当指定专人对商业化通用的计算机化系统进行审核,确认其满足用户需求。
在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确保在生命周期内评估系统的质量和性能。
第九条 数据转换格式或迁移时,应当确认数据的数值及含义没有改变。
第五章 系 统第十条 系统应当安装在适当的位置,以防止外来因素干扰。
第十一条 应当有详细阐述系统的文件(必要时,要有图纸),并须及时更新。
CFDA GMP附件 计算机化系统

GMP新附录:计算机化系统(全文)国家食品药品监督管理总局关于发布《药品生产质量管理规范(2010年修订)》计算机化系统和确认与验证两个附录的公告(2015年第54号)根据《药品生产质量管理规范(2010年修订)》第三百一十条规定,现发布《计算机化系统》和《确认与验证》两个附录,作为《药品生产质量管理规范(2010年修订)》配套文件,自2015年12月1日起施行。
特此公告。
附件:1.计算机化系统 2.确认与验证食品药品监管总局2015年5月26日附件1 计算机化系统第一章范围第一条本附录适用于在药品生产质量管理过程中应用的计算机化系统。
计算机化系统由一系列硬件和软件组成,以满足特定的功能。
第二章原则第二条计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证和数据完整性控制的程度。
第四条企业应当针对计算机化系统供应商的管理制定操作规程。
供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当基于风险评估的结果提供与供应商质量体系和审计信息相关的文件。
第三章人员第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
应当明确所有使用和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训。
应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等方面进行培训和指导。
第四章验证第六条计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
应当在计算机化系统生命周期中保持其验证状态。
第七条企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系统清单,标明与药品生产质量管理相关的功能。
欧盟GMP附录11-计算机系统(中英文对照)

EUROPEAN COMMISSION欧盟委员会HEALTH AND CONSUMERS DIRECTORATE-GENERAL卫生与消费者协会Public Health and Risk Assessment公共卫生与风险评估Pharmaceuticals药品Brussels,SANCO/C8/AM/sl/ares(2010)1064599EudraLexThe Rules Governing Medicinal Products in the European Union欧盟药品生产规范Volume 4卷4Good Manufacturing PracticeMedicinal Products for Human and Veterinary Use人用与兽用药品良好生产管理规范Annex 11: Computerised Systems附件11:计算机系统Legal basis for publishing the detailed guidelines:Article 47 of Directive2001/83/EC on the Community code relating to medicinal products for human use and Article 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinal products as laid down in Directive 2003/94/EC for medicinal products for human use and Directive 91/412/EEC for veterinary use.依法发布的具体指导方针:2001/83/EC第47条人用药品规范和2001/82/EC第51条兽用药品规范。
GMP法规附录《计算机化系统》那些事儿-Waters

GMP 法规附录法规附录《《计算机化系统计算机化系统》》那些事儿2015年5月26日,CFDA 正式发布了2010版GMP 法规的新附录之一《计算机化系统》,引起了国内制药行业的广泛讨论和高度关注。
其实许多制药企业对它的内容并不陌生,因为这则法规于2013年作为征求意见稿已经添加到新版GMP 法规附录中。
而现在,它将作为正式的法规于2015年12月1日起执行。
这则法规附录将给国内制药企业带来什么新的挑战?从近两年来CFDA 的一系列举措(频繁的飞行检查,2014年至今已取消近100家药企的GMP 证书)来看,国内GMP 的监管力度是显著增强的。
所以届时如果企业不能满足《计算机化系统》法规的要求,将可能面临十分严重的后果。
CFDA 为何要发布这则法规为何要发布这则法规??国内外GMP 法规有许多差异,而对计算机化系统的要求差异尤为明显。
CFDA 所执行的2010版GMP 法规内容与国际上其他法规机构的cGMP 法规是对等的,如FDA 21 CFR Part 211。
但美国的制药企业除了执行 21 CFR Part 211以外,同时还要遵守21 CFR Part 11法规;欧盟国家的制药企业除了执行欧盟GMP 以外,还要遵循Annex 11法规。
FDA 的21 CFR Part 11与欧盟的Annex 11的内容是类似的,都是针对于制药企业使用计算机化系统的法规要求。
新颁布的《计算机化系统》法规附录是国内法规与国际接轨的重要一步,将填补国内对于计算机化系统要求的法规空白,是实现与国际法规监管机构之间相互认可的前提条件之一。
法规到底讲了些什么法规到底讲了些什么??《计算机化系统》法规附录究竟讲了哪些内容?其实,我们发现内容并不多,全文共24条要求、6页,共计2500字。
我们尝试对这些法规条文作了初步的解读,把所理解的核心内容概括如下:1.CFDA 明确提出进行计算机化系统验证的要求以往,法规对于仪器的确认是一直有要求的,但对计算机软件验证的要求不明确。
GMP附录验证与确认计算机化系统培训试题

GMP附录验证与确认计算机化系统培训试题GMP新增附录培训试题部门: 姓名: 分数一、填空题(每题2分,此题占试卷内容60分):1. 企业应当确定需要进行的确认或验证工作,以证明有关操作的(关键要素)能够得到有效控制。
确认和验证的范围和程度应根据(风险评估)的结果确认。
确认与验证应当贯穿于(产品生命周期)的全过程。
2.所有的确认与验证活动都应当(事先计划)。
确认与验证的关键要素都应在(验证总计划)或同类文件中详细说明。
3.当确认或验证分阶段进行时,只有当(上一阶段)的确认或验证报告得到批准,或者确认或验证活动(符合预定目标)并经批准后,方可进行下一阶段的确认或验证活动。
上一阶段的确认或验证活动中不能满足某项预先设定标准或偏差处理未完成,经评估对下一阶段的确认或验证活动(无重大影响),企业可对上一阶段的确认或验证活动进行(有条件的批准)。
4.企业应当对新的或改造的厂房、设施、设备按照预定用途和本规范及相关法律法规要求制定(用户需求),并经审核、批准。
5. 设计确认应当证明设计符合(用户需求),并有相应的文件。
6.(新的或改造)的厂房、设施、设备需进行安装确认。
7. 安装和运行确认完成并符合要求后,方可进行(性能确认)。
在某些情况下,性能确认可与(运行确认)或(工艺验证)结合进行。
8.工艺验证应当证明一个生产工艺按照规定的工艺参数能够持续生产出符合(预定用途和注册要求)的产品。
工艺验证应当包括(首次验证)、(影响产品质量的重大变更后的验证)、(必要的再验证)以及在产品生命周期中的(持续工艺确认),以确保工艺始终处于验证状态。
9.企业应当有书面文件确定产品的(关键质量属性)、(关键工艺参数)、常规生产和工艺控制中的(关键工艺参数范围),并根据对产品和工艺知识的理解进行更新。
10. 在产品生命周期中,应当进行(持续工艺确认),对商业化生产的产品质量进行监控和趋势分析,以确保(工艺和产品质量)始终处于受控状态。
GMP附录计算机化系统

中洲培训:计算机中化洲系培统训:计算机化系统
• 第四章 验 证 • 第六条 计算机化系统验证包括应用程序的验证和基础架构的确认,其范
围与程度应当基于科学的风险评估.风险评估应当充分考虑计算机化系 统的使用范围和用途. • 应当在计算机化系统生命周期中保持其验证状态. • 第七条 企业应当建立包含药品生产质量管理过程中涉及的所有计算机 化系统清单,标明与药品生产质量管理相关的功能.清单应当及时更新. • 第八条 企业应当指定专人对通用的商业化计算机软件进行审核,确认其 满足用户需求. • 在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确 保在生命周期内评估系统的质量和性能. • 第九条 数据转换格式或迁移时,应当确认数据的数值及含义没有改变.
• 包括系统故障和数据错误在内的所有事故都应当被记录和评估. 重大的事故应当进行彻底调查,识别其根本原因,并采取相应的纠 正措施和预防措施.
• 第二十二条 当采用计算机化系统放行产品时,计算机化系Biblioteka 应当 能明示和记录放行产品人员的身份.
• 第二十三条 电子数据可以采用电子签名的方式,电子签名应当遵 循相应法律法规的要求.
中洲培训:计算中洲机培化训系:统计算机化系统 • 计算机化系统
• 第一章 范 围 • 第一条 本附录适用于在药品生产质量管理过程中
应用的计算机化系统.计算机化系统由一系列硬件 和软件组成,以满足特定的功能.
中洲培训:计算机中化洲系培统训:计算机化系统
• 第二章 原 则 • 第二条 计算机化系统代替人工操作时,应当确保不对产品的质量、
中洲培训:计算机中化洲系培统训:计算机化系统
• 第十九条 以电子数据为主数据时,应当满足以下要求: • (一)为满足质量审计的目的,存储的电子数据应当能够打印
GMP计算机系统附录知识培训(CFDAGMP计算机化系统附录、EUGMP附录11、21PART11、GAMP5)2015.12

1
计算机化系统GMP法规要求
2 3 4
计算机化系统及数据完整性解析
基于风险评估计算机化系统验证
常见问题及讨论
1
计算机化系统GMP法规要求
1.1 计算机化系统相关法规和指南总览 1.2 CFDA《计算机化系统》附录、 EUGMP附 录11、联邦法规21章第11款电子记录;电子 签名框架结构 1.3 《计算机化系统》附录特点介绍 1.4 《计算机化系统》关键词 1.5 MHRA数据完整性定义和指南简介
1
计算机化系统GMP法规要求
(US FDA)工业指南 11部分 电子记录与电子签名‐ 范围和应用 (US FDA) 联邦法规第21篇第210 211部分,成品药 的现行生产质量管理规范 (ISPE)GAMP GPG 良好实践指南,GAMP架构下 的系列良好实践指南 (PIC/S)GMP指南,药用产品良好生产实践指南 (TGA)GMP,药用产品良好生产实践指南 (WHO)GMP 2003, Annex 4 (WHO Technical Repo rt Series, No. 908) (CFDA) GSP附录二附录三《药品经营企业计算机 系统》《温湿度自动监测》 (US FDA) 药品生产中计算机处理系统的验证指南 (February 1983)
本次培训还将结合国内外对计算机化系统
诸如EUGMP附录11,21CFRpart11,GAMP5, MHRA数据完整性指南等国际上有影响力, 有权威性的法规要求、行业标准及参考指 南进行对比介绍 由于能力、经验、水平的欠缺,一些不合 理甚至错误理解和解读的内容希望在座诸 位提出宝贵意见和建议,共同探讨合理的 解决方案,以待提高共同认知水平。
GMP法规基本知识培训——附录9计算机化系统

原文内容: 第二条:计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证
水平造成负面影响,不增加总体风险。
原文解读: 这一条主旨是说计算机化系统可以代替人工操作,提高工作效率,但是需考虑这种替代或变 革随之带来的风险,这种风险可能是来自产品质量的、过程控制的,也可能是整个质量保证 体系的,需慎重、全面考虑这种变革带来的诸多影响,原则上同过去手工操作相比,不增加 总体风险即可。这一条其实给整个附录定了一个基调,就是凡是大到变革、小到变更,只要 有变化都要做好风险识别和风险评估工作,这是一切“变”的前提和刚性要求。
原文内容: 第九条:数据转换格式或迁移时,应当确认数据的数值及含义没有改变。 原文解读: 这一条很好理解,举例来说在做系统升级时,升级后的系统迁移过来的数据的数值和含义必 须是同升级前系统的数据保持一致。这是最基本的要求,否则系统升级就是不成功的。
原文内容: 第十条:系统应当安装在适当的位置,以防止外来因素干扰。 原文解读: 该条款从环境和安全的角度,提出了系统安装的位置,以防止外来因素的干扰,包括室内的 温度、湿度、防尘、防潮、防盗等要求。
原文内容: 第六条:计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于
科学的风险评估。风险评估应当充分考虑计算机化系统的使用范围和用途。应当在计算机化
系统生命周期中保持其验证状态。 原文解读: 验证对象包括应用程序(就是软件)和基础架构(包括硬件和网络)。验证的具体范围和程 度如何确定?答案是基于科学的风险评估,那么风险评估又如何进行呢?答案是要充分考虑 该系统的使用范围和用途。举例来说,某个系统的使用是否同GxP有关或对产品质量有影响, 如果有,就需要进行风险评估,同时还要考虑该系统使用范围大小,范围越大,影响越大, 风险越高。此外,该条款特别强调了系统在生命周期中验证状态的保持,是一项常态工作。
(完整版)新版GMP附录-10-计算机化系统

附录:10计算机化系统第一章范围第一条本附录适用于在药品生产质量管理过程中应用的计算机化系统.计算机化系统由一系列硬件和软件组成,以满足特定的功能。
第二章原则第二条计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证和数据完整性控制的程度.第四条企业应当针对计算机化系统供应商的管理制定操作规程.供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当基于风险评估的结果提供与供应商质量体系和审计信息相关的文件。
第三章人员第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
应当明确所有使用和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训.应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等方面进行培训和指导.第四章验证第六条计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
应当在计算机化系统生命周期中保持其验证状态。
第七条企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系统清单,标明与药品生产质量管理相关的功能。
清单应当及时更新。
第八条企业应当指定专人对通用的商业化计算机软件进行审核,确认其满足用户需求。
在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确保在生命周期内评估系统的质量和性能。
第九条数据转换格式或迁移时,应当确认数据的数值及含义没有改变。
第五章系统第十条系统应当安装在适当的位置,以防止外来因素干扰.第十一条关键系统应当有详细阐述的文件(必要时,要有图纸),并须及时更新。
GMP附录《计算机化系统》那些事儿

GMP附录《计算机化系统》那些事⼉GMP法规附录《计算机化系统》那些事⼉?2015年5⽉26⽇,CFDA正式发布了2010版GMP法规的新附录之⼀《计算机化系统》,引起了国内制药⾏业的⼴泛讨论和⾼度关注。
其实许多制药企业对它的内容并不陌⽣,因为这则法规于2013年作为征求意见稿已经添加到新版GMP法规附录中。
⽽现在,它将作为正式的法规于2015年12⽉1⽇起执⾏。
这则法规附录将给国内制药企业带来什么新的挑战?从近两年来CFDA的⼀系列举措(频繁的飞⾏检查,2014年⾄今已取消近100家药企的GMP证书)来看,国内GMP的监管⼒度是显著增强的。
所以届时如果企业不能满⾜《计算机化系统》法规的要求,将可能⾯临⼗分严重的后果。
CFDA为何要发布这则法规??国内外GMP法规有许多差异,⽽对计算机化系统的要求差异尤为明显。
CFDA所执⾏的2010版GMP法规内容与国际上其他法规机构的cGMP法规是对等的,如FDA?21?CFR?Part? 211。
但美国的制药企业除了执⾏?21?CFR?Part?211以外,同时还要遵守21?CFR?Part?11法规;欧盟国家的制药企业除了执⾏欧盟GMP以外,还要遵循Annex?11法规。
FDA的21?CFR? Part?11与欧盟的Annex?11的内容是类似的,都是针对于制药企业使⽤计算机化系统的法规要求。
新颁布的《计算机化系统》法规附录是国内法规与国际接轨的重要⼀步,将填补国内对于计算机化系统要求的法规空⽩,是实现与国际法规监管机构之间相互认可的前提条件之⼀。
法规到底讲了些什么??《计算机化系统》法规附录究竟讲了哪些内容?其实,我们发现内容并不多,全⽂共24条要求、6页,共计2500字。
我们尝试对这些法规条⽂作了初步的解读,把所理解的核⼼内容概括如下:?1.CFDA明确提出进⾏计算机化系统验证的要求?以往,法规对于仪器的确认是⼀直有要求的,但对计算机软件验证的要求不明确。
中国GMP附录计算机化系统介绍 关于计算机化系统的法规及指南
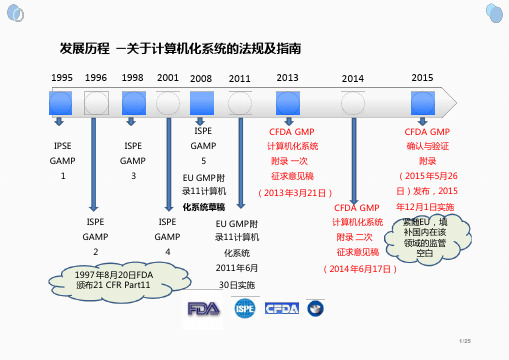
EU GMP Annex 11 是“和”
20/25
系统
第二十条 企业应当建立应急方案,以便系统出现损坏时启用。应急方案启用 的及时性应当与需要使用该方案的紧急程度相关。例如,影响召回产品的相 关信息应当能够及时获得。
7/25
验证
第七条 企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系 统清单,标明与药品生产质量管理相关的功能。清单应当及时更新。 制定计算机化系统的系统清单,内容包括系统名称、系统编号、系统GMP 功能等内容,可以参照或者用系统影响性评估SIA报告的形式,并及时更新。
8/25
验证
第八条 企业应当指定专人对通用的商业化计算机软件进行审核,确认其 满足用户需求。 在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确 保在生命周期内评估系统的质量和性能。
5/25
人员
第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、 管理等,需要各相关的职能部门人员之间的紧密合作。应当明确所有使用 和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训。 应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行 等方面进行培训和指导。
15/25
系统
第十五条 当人工输入关键数据时,应当复核输入记录以确保其准确性。这个 复核可以由另外一个操作人员完成,或采用经验证的电子方式。必要时,系 统应当设置复核功能,确保数据输入的准确性和数据处理过程的正确性。 对人工输入数据的准确性提出复核的要求,复核的方式可以是另外的操作人 员或者是经过验证的电子方式。(比如经过验证的称量配料系统通过报警提 示能够完成“超重”、“欠重”、“批号错误”、“忘记去皮”等关键信息 复 核 , 则 岗 位 无 需 配 备 另 一 名 用 于 复 核 的 操 作 人 员 ) (比如数 据输入的有效性符合:范围和小数点位数,超出指定范围和有效位数的 数据无 法输入)
GMP附录(2015):计算机化系统

3 / 6附件1计算机化系统第一章 范 围第一条 本附录适用于在药品生产质量管理过程中应用的计算机化系统。
计算机化系统由一系列硬件和软件组成,以满足特定的第二章 原 则第二条 计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条 风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证和数据完整性控制的程度。
第四条 企业应当针对计算机化系统供应商的管理制定操作规程。
供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当基于风险评估的结果提供与供应商质量体系和审计信息相关的文件。
第三章人员第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
应当明确所有使用和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训。
应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等第四章验证第六条计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
应当在计算机化系统生命周期中保持其验证状态。
第七条企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系统清单,标明与药品生产质量管理相关的功能。
清单应当及时更新。
第八条企业应当指定专人对通用的商业化计算机软件进行审核,确认其满足用户需求。
在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确保在生命周期内评估系统的质量和性能。
5 / 6第九条数据转换格式或迁移时,应当确认数据的数值及含义第五章 系 统第十条 系统应当安装在适当的位置,以防止外来因素干扰。
第十一条 关键系统应当有详细阐述的文件(必要时,要有图纸),并须及时更新。
GMP附录-计算机化系统(翟铁伟)

本附录共分为七个部分,包括引言、计算机化系统的基本要 求、系统设计和开发、系统实施和运行、系统维护和变更、 系统退役和数据处理以及附则。每个部分都详细阐述了相应 的管理原则和实施要求。
02
计算机化系统概述
定义与特点
定义
计算机化系统是指由硬件、软件、数 据、人员、规程和接口等组成的系统 ,用于指导和控制实验室或生产车间 的活动。
灾难恢复计划
制定完善的灾难恢复计划,确保在意外情况下能 够及时恢复系统运行。
系统监控与报警
建立系统监控机制,实时监测系统运行状态,发 现异常及时报警。
人员培训与素质提升
专业技能培训
针对计算机化系统的特点和要求,开展专业技能培训,提高人员 操作水平。
安全意识教育
加强安全意识教育,提高人员对网络安全、数据安全的重视程度。
建立行业交流平台
鼓励企业之间建立交流平台,分享各自在计算机化系统领 域的实践经验和成功案例。
01
促进产学研合作
推动企业与高校、科研机构之间的合作, 共同研发新技术、新产品,提升行业整 体水平。
02
03
培养专业人才
重视人才培养和引进,建立完善的人 才培养和激励机制,吸引更多优秀人 才投身计算机化系统领域。
明确本附录的编写目的
本附录旨在指导药品生产企业如何建立和实施符合GMP要求的计算机化系统,确保数据的完整性、准确性和可追 溯性,保障药品质量和安全。
汇报范围
计算机化系统的定义和范围
计算机化系统是指用于药品生产、质量控制、物料管理等环 节的所有计算机硬件、软件、网络和数据等组成部分。本附 录适用于药品生产企业中所有与GMP相关的计算机化系统。
06
未来展望与建议
EUGMP附录11:计算机化系统-中文

EUGMP附录11:计算机化系统-中文原则本附录适用于作为《药品生产质量管理规范》活动的一部分所有形式的计算机系统。
计算机化系统是一套软件与硬件组合,以便实现某些特定的功能。
该应用程序应进行验证;信息技术基础设施应该是检定的。
计算机系统代替手工操作,应该降低对产品质量,工艺过程控制和质量保证的影响。
不应该有增加在此过程的整体风险。
总则1、风险管理风险管理应用在计算机系统的整个生命周期内,应该考虑患者安全、数据可靠性和产品质量。
作为一个风险管理系统的一部分,应基于合理的和记录风险评估的计算机系统,决定验证的程度和数据可靠性控制。
2、人员应该有所有相关人员,如流程所有人,系统所有人,质量受权人和IT之间的密切合作。
所有人员应具备相应的资格,访问级别和明确责任,以履行其指定的职责。
3、供应商与服务商3.1 当第三方(如供应商,服务供应商)使用,例如提供,安装,配置,集成,验证,维护(例如,通过远程访问),修改或保留一个计算机系统或相关服务或进行数据处理,必须在制造商和任何第三方之间存在正式的协议,而且这些协议应包括对第三方的责任明确的声明。
信息技术部门应做类似考虑。
3.2 当选择产品或服务提供商的时候,供应商的能力和可靠性是关键因素。
应以风险评估为基础,确定是否需要审计。
3.3 监管的用户应审查所提供现货产品提供的文件,以确认用户的需求得到满足。
3.4 应根据检查员的要求提供质量体系和审核相关的软件和实施系统的供应商或开发人员的相关信息。
项目阶段4、验证4.1 验证文件和报告应涵盖整个生命周期的相关步骤。
生产企业应该能够根据风险评估,以证明他们的标准,协议,可接受标准,程序和记录的适用性。
4.2 验证文件应包括变更控制记录(如有)和验证过程中对任何观察到的偏差报告。
4.3 最新的清单的所有相关系统和GMP功能(目录)应该可用。
对于关键系统的一个最新的系统描述,以详述的物理和逻辑的安排、数据流和接口与其他系统或过程,任何硬件和软件的前提条件,和安全措施。
(完整版)新版GMP附录-10-计算机化系统

附录:10计算机化系统第一章范围第一条本附录适用于在药品生产质量管理过程中应用的计算机化系统。
计算机化系统由一系列硬件和软件组成,以满足特定的功能。
第二章原则第二条计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证和数据完整性控制的程度。
第四条企业应当针对计算机化系统供应商的管理制定操作规程。
供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当基于风险评估的结果提供与供应商质量体系和审计信息相关的文件。
第三章人员第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
应当明确所有使用和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训。
应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等方面进行培训和指导。
第四章验证第六条计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
应当在计算机化系统生命周期中保持其验证状态。
第七条企业应当建立包含药品生产质量管理过程中涉及的所有计算机化系统清单,标明与药品生产质量管理相关的功能。
清单应当及时更新。
第八条企业应当指定专人对通用的商业化计算机软件进行审核,确认其满足用户需求。
在对定制的计算机化系统进行验证时,企业应当建立相应的操作规程,确保在生命周期内评估系统的质量和性能。
第九条数据转换格式或迁移时,应当确认数据的数值及含义没有改变。
—3 —第五章系统第十条系统应当安装在适当的位置,以防止外来因素干扰。
第十一条关键系统应当有详细阐述的文件(必要时,要有图纸),并须及时更新。
GMP新附录:计算机系统

GMP新附录:计算机化系统(全文)2015-06-04来源:蒲公英发布者:admin打印本文国家食品药品监督管理总局关于发布《药品生产质量管理规范(2010年修订)》计算机化系统和确认与验证两个附录的公告(2015年第54号)根据《药品生产质量管理规范(2010年修订)》第三百一十条规定,现发布《计算机化系统》和《确认与验证》两个附录,作为《药品生产质量管理规范(2010年修订)》配套文件,自2015年12月1日起施行。
特此公告。
附件:1.计算机化系统2.确认与验证食品药品监管总局2015年5月26日附件1计算机化系统第一章范围第一条本附录适用于在药品生产质量管理过程中应用的计算机化系统。
计算机化系统由一系列硬件和软件组成,以满足特定的功能。
第二章原则第二条计算机化系统代替人工操作时,应当确保不对产品的质量、过程控制和其质量保证水平造成负面影响,不增加总体风险。
第三条风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患者安全、数据完整性和产品质量。
作为质量风险管理的一部分,应当根据书面的风险评估结果确定验证和数据完整性控制的程度。
第四条企业应当针对计算机化系统供应商的管理制定操作规程。
供应商提供产品或服务时(如安装、配置、集成、验证、维护、数据处理等),企业应当与供应商签订正式协议,明确双方责任。
企业应当基于风险评估的结果提供与供应商质量体系和审计信息相关的文件。
第三章人员第五条计算机化系统生命周期中所涉及的各种活动,如验证、使用、维护、管理等,需要各相关的职能部门人员之间的紧密合作。
应当明确所有使用和管理计算机化系统人员的职责和权限,并接受相应的使用和管理培训。
应当确保有适当的专业人员,对计算机化系统的设计、验证、安装和运行等方面进行培训和指导。
第四章验证第六条计算机化系统验证包括应用程序的验证和基础架构的确认,其范围与程度应当基于科学的风险评估。
风险评估应当充分考虑计算机化系统的使用范围和用途。
中国GMP附录计算机化系统介绍 关于计算机化系统的法规及指南
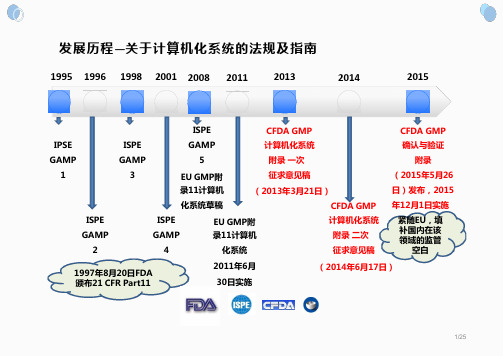
EU GMP Annex 11 是“和”
20/25
系统
第二十条 企业应当建立应急方案,以便系统出现损坏时启用。应急方案启用 的及时性应当与需要使用该方案的紧急程度相关。例如,影响召回产品的相 关信息应当能够及时获得。
3/25
原则
第二条 计算机化系统代替人工操作时,应当确保不对产品的质量、过程 控制和其质量保证水平造成负面影响,不增加总体风险。 第三条 风险管理应当贯穿计算机化系统的生命周期全过程,应当考虑患 者安全、数据完整性和产品质量。作为质量风险管理的一部分,应当根 据书面的风险评估结果确定验证和数据完整性控制的程度。 主要描述了质量风险管理的要求。对于风险管理:一方面是整个生命周 期要实施风险管理,自动代替人工操作不会带来负面影响及总体风险增 加(必要的时候需进行工艺等同性验证来证明);另一方面是验证的范 围和程度要基于风险评估的结果来确定。
12/25
系统
第十二条 软件是计算机化系统的重要组成部分。企业应当根据风险评估 的结果,对所采用软件进行分级管理(如针对软件供应商的审计),评估 供应商质量保证系统,保证软件符合企业需求。 软件分级管理与供应商审计要求(同上所述,参见第四条和第六条解析)。
13/25
系统
第十三条 在计算机化系统使用之前,应当对系统进行全面测试,并确认系统 可以获得预期的结果。当计算机化系统替代某一人工系统时,可采用两个系 统(人工和计算机化)平行运行的方式作为测试和验证内容的一部分。 针对软件的全面测试,根据软件级别的不同其测试的程度也将不同(比如: 针对五类软件系统的源代码审核和模块测试,针对四类软件系统的配置测试, 然后是基于黑盒的功能测试、需求测试,以及包括结合实际工艺或流程的性 能测试)。 EU GMP 附录11 08年版时有“人工和计算机平行运行”要求,11已经正式 去掉。