“Discovery Studio 2.5中基于药效团的药物设计 方法和应用 ”
“Discovery Studio 2.5中基于药效团的药物设计方法和应用 ”

36 Compounds
1150compounds
+ Training set (16) Ligandfit
化合物库筛选
可以构建自己的化合物库
database
安装其他化合物库,CNPD,ACD等 DS中自带的化合物库
构建具有活性预测能力的药效团
• 寻找与化合物活性相关的药效团特性 • 构建的药效团模型具有活性预测功能
实例分析2(先导化合物优化)
Training set:
Test set
Pharmacophore modes
IC50=1.43
IC50=0.004
实例分析3
qualitative
Search 3D database
Dock(Ligandfit)
基于特性结构产生药效团
• 对于训练集要求:
- 输入的分子结构具有多样性
catalyst
Conformer Generation • FAST • BEST
Discovery studio
Conformer Generation • FAST • BEST • CAESAR • Systematic Search • Random Search • Boltzmann Jump
Common Feature Pharmacophore Generation HipHop(quanlitative)
- 化合物数目在2-32个,6个左右比较理想 - 只选用具有活性的分子
- 需要包含Principal和MaxOmitFeat性质
• 方法总结 - 找到一组化合物的公共药效特性
- 不需要SAR的信息
单击此处编辑母版标题样式
DS2.5中基于药效团的药物 单击此处编辑母版文本样式 设计方法和应用 第二级
《药物设计:方法、概念和作用模式》记录

《药物设计:方法、概念和作用模式》读书笔记目录一、药物设计简介 (2)1. 药物设计的定义 (3)2. 药物设计的发展历程 (3)二、药物设计的方法 (5)1. 计算机辅助药物设计 (6)a. 分子对接技术 (7)b. 药效团搜索 (9)c. 药物分子设计 (9)2. 实验室实验 (11)a. 细胞实验 (12)b. 体外实验 (13)c. 体内实验 (14)3. 量子化学计算 (15)a. 量子力学计算 (16)b. 红外光谱学 (17)c. 核磁共振 (19)三、药物设计的概念 (20)1. 药物靶标的发现与验证 (21)2. 药物分子的活性评价 (22)3. 药物的药代动力学和毒性评价 (24)四、药物作用模式 (25)1. 靶向治疗 (26)2. 药物复用 (27)3. 多靶点治疗 (29)五、药物设计的挑战与未来发展趋势 (31)1. 新药研发的挑战 (32)2. 人工智能在药物设计中的应用 (33)3. 疾病机制研究的深入为药物设计提供新的方向 (34)六、结论 (35)1. 药物设计的重要性和意义 (36)2. 药物设计的发展前景 (37)一、药物设计简介药物设计是一门研究药物分子与生物体相互作用机制及如何通过计算机辅助设计药物的科学。
它涵盖了从药物靶标的发现到药物分子的设计、合成和筛选的全过程,旨在为疾病治疗提供有效、安全且具有选择性的药物。
药物设计的核心目标是提高药物的疗效和降低副作用,为了实现这一目标,药物设计师需要充分利用生物学、化学和计算机科学等多学科知识,通过多种手段来优化药物分子的活性、选择性和药代动力学性质。
在药物设计过程中,科学家们通常会运用多种方法和技术,如基于结构的药物设计(SBDD)、计算药剂学(CADD)和组合化学等。
这些方法和技术可以帮助设计师更准确地预测药物分子与靶标的相互作用,从而设计出更为理想的候选药物分子。
药物设计还涉及到药物作用模式的深入研究,药物作用模式是指药物与生物体中特定分子相互作用的方式,它是决定药物疗效和副作用的关键因素之一。
基于药效团的虚拟筛选技术发现全新转化因子β受体1拮抗剂

摘要:作为治疗恶性肿瘤的新药,转化生长因子β受体1(TGFβR1)拮抗剂获得了广泛关注。
作者基于TGFβR1-BMS22的复合物晶体结构和文献报道了抑制剂数据构建了两个药效团模型A02和B10。
应用这两个药效团模型,作者虚拟筛选了DruglikeDiverse、MiniMaybridge和Zinc Drug-like等数据库,发现了两种全新的TGFβR1拮抗剂骨架。
并将筛选出的化合物经过类药五规则和ADMET性质预测。
最终化合物YXY01-03具有新颖的骨架,良好的药物性质,潜在的活性,可能比BMS22具有更高的安全性,这可能对进一步的研究有价值。
MaXFlow生物医药智能创新平台,由创腾科技自主研发,旨为不同领域的一线创新科技工作者提供一个合作共享的B-S架构平台。
以“数据自由,模型自由”为理念,在结构模型与预测模型进行融合的基础上,实现模拟与AI需求的合并,为研发赋能。
药效团模型(Pharmacophore):是指药物活性分子中对活性起着重要作用的“药效特征元素”及其空间排列形式,可以认为是大量活性化合物共同的成药特征。
基于药效团模型,科研人员可以进行快速的药物虚拟筛选工作,寻找结构新颖的活性分子;也可以解释化合物的构效关系,进行化合物的结构优化与改造;此外,还可以进行化合物靶标的识别,实现反向找靶。
Discovery Studio中的Catalyst模块,是经典的药效团模型生成、验证及虚拟筛选的工具,可以基于配体、受体以及复合物结构进行定量、定性的药效团模型研究。
基于药效团的虚拟筛选技术发现全新转化因子β受体1拮抗剂ref:Molecules. 2018 Oct 31;23(11). pii: E2824;IF=3.06链接:doi: 10.3390/molecules23112824作为治疗恶性肿瘤的新药,转化生长因子受体1(TGFβR1)拮抗剂获得了广泛关注。
作者首先对TGFβR1-BMS22复合物结构6B8Y,应用DS_Receptor-Ligand Pharmacophore Genereation模块,生成基于结构的药效团。
基于药效团模型的药物设计与筛选

2020年34期设计创新科技创新与应用Technology Innovation and Application基于药效团模型的药物设计与筛选*刘景陶(河套学院,内蒙古河套015000)1计算机辅助药物设计方法学随着计算机技术与药物设计学科的融合,利用计算机的模拟技术和图形技术进行药物分子筛选成为一门崭新的学科。
计算机辅助药物设计通过对配体和受体间相互作用的原理的研究。
是基于各种分子模拟技术及各种数理统计方法,在缺乏受体结构的情况下,可以进行基于配体小分子的虚拟筛选。
即间接法,从一系列分子中寻找出受体活性位点,再以此设计新的配体。
现代药物研发工作中,基于结构的分子设计已成为临床前药物开发有效策略的标志。
随着对蛋白结构和功能的认识越来越深入,以及基因组学推进了基于结构设计新药的进步,并为该策略应用于新疗法的药物开发提供了巨大的保障。
人类基因组测序结果表明有20000~25000个人类蛋白编码基因,每个基因可以为一种蛋白质编码,这些蛋白负责实现人体内所有的细胞功能。
同时这些蛋白也可以参与病理过程。
为了充分利用新靶点和结构信息,需要深入了解酶的功能、理解分子设计的基本原理、并明确基于结构设计药物活性分子时可能面临的障碍,因而为基于结构的新药设计和新药研发提供独特的机会和挑战。
基于结构设计的方法彻底改变了药物化学实践过程和临床前药物研发过程。
2通过间接法进行药物设计相似的化学结构可能产生相似的生物活性,这就是配体相似性原则,在配体相似性原则指导下,将化学结构相似的一对生物靶标的活性作系统的相关性分析,采用“全局”或“局部”的分子观点,揭示并预测化学结构与药理活性的关系。
化学结构决定着药理作用和成药性,杂泛性关系到药效、药代和安全性等内容,利用靶标的三维结构或根据药物分子的结构的相似性或药效团总结出的规律或模型,可对药物分子的杂泛性进行预测。
基于结构的设计,在许多首创药物发现和开发中的成功应用,产生了药物发现和开发中的新概念和新工具。
Autodock Vina与Discovery Studio在虚拟筛选耐药蛋白抑制剂中的比较

Autodock Vina与Discovery Studio在虚拟筛选耐药蛋白抑制剂中的比较黄勇;陈晨;张志毅;童贻刚;赵勇【期刊名称】《生物信息学》【年(卷),期】2012(10)4【摘要】随着大量与细菌耐药相关的基因的发现和其表达蛋白结构的成功测定,从已有的化合物中通过计算机模拟方法筛选对耐药蛋白靶点有作用的候选化合物,成为了药物发现的一个标准途径.虚拟筛选在耐药基因抑制剂的发现中可以提高效率、降低实验成本.本文介绍了Autodock Vina和Discovery Studio在基于分子对接法的虚拟筛选中的使用,并对比分析其对β-内酰胺酶活性位点的筛选结果.希望通过这种比较促进虚拟筛选在药物设计领域中的应用,提高耐药基因抑制剂的发现速度.%With the increasing discovery of antibiotic resistance gene and determination of the protein structures, it has become a standardized strategy to screen potential protein inhibitors from existing compounds using computer simulation technology. Virtual screening in discoverying antibiotic resistance protein inhibitors heighten the efficiency and reduce experimental cost. In this paper we introduce the using of Autodock Vina and Discovery Stiudio in virtual screening based of molecular docking, and analysis the screening result against β - lactamases active sides. We hope this will help to improve the application of virtual screening in drug design, and to accelarate the finding speed of antibiotic resistance protein inhibitors as well.【总页数】6页(P248-253)【作者】黄勇;陈晨;张志毅;童贻刚;赵勇【作者单位】军事医学科学院微生物流行病研究所,北京100071;军事医学科学院微生物流行病研究所,北京100071;军事医学科学院微生物流行病研究所,北京100071;军事医学科学院微生物流行病研究所,北京100071;国家人类基因组北方研究中心,北京100176【正文语种】中文【中图分类】Q518.2【相关文献】1.基于Discovery Studio软件的本科生创新实验项目设计—TGFβR1抑制剂药效团模型构建 [J], 邓萍; 蒋启华; 孙立力; 蒋君好2.基于生物信息学稻米半胱氨酸蛋白酶抑制剂来源生物活性肽的虚拟筛选及分子对接研究 [J], 石嘉怿;梁富强;张太;张冉3.基于新型冠状病毒SARS-CoV-2主蛋白酶结构的小分子抑制剂虚拟筛选 [J], 徐浩;薛瑞;李娟;李波;陈舟;朴莲花;常珊;孔韧4.基于新型冠状病毒SARS-CoV-2主蛋白酶结构的小分子抑制剂虚拟筛选 [J], 徐浩;薛瑞;李娟;李波;陈舟;朴莲花;常珊;孔韧(指导)5.基于Discovery Studio软件的专业学位CADD研究生课程教学案例设计——抗新冠肺炎中药有效成分山奈酚与靶蛋白的分子对接 [J], 张永红;蒋启华;李艳因版权原因,仅展示原文概要,查看原文内容请购买。
Discovery Studio官方教程--基于MODELER构建抗体模型

抗体3D结构的预测(MODEL ANTIBODIES)教程介绍抗体分子是生物学和医学领域用途最为广泛的蛋白分子。
以肿瘤特异性抗原或肿瘤相关性抗原、抗原独特型决定簇、细胞因子及其受体、激素及一些癌基因产物作为靶分子,利用传统的免疫方法或通过细胞工程、基因工程等技术制备的多克隆抗体、单克隆抗体、基因工程抗体广泛应用于疾病诊断、治疗及科学研究等领域,并以其毒副作用小、天然和高度特异性的疗效,创造出了巨大的社会效益和经济效益。
抗体和抗体-抗原复合物的结构通常被用于了解基于抗体的药物的作用机制,在抗体工程上提供帮助。
X射线晶体学方法有助于抗体结构的解析,但是与计算模拟相比,耗费的经济成本和时间成本太高。
本教程中使用DS基于一个合成人类Fv区域的序列构建3D抗体模型,相对于X射线晶体结构选择性评估模型的质量。
随着大量的抗体Fv区域、Fab区域和高度保守区的结构被解析出来,使用同源模建的方法构建抗体结构成为可能。
构建一个抗体Fv或Fab区域结构模型的一个典型的流程是首先根据已知抗体模板结构构建框架结构,然后必要的时候使用额外的模板优化互补决定区。
在本教程中的任务包括以下几个步骤:♦结构模板的识别♦抗体Framework区模型的构建♦构建抗体Loop区♦模型可靠性的评估抗体序列的分析和识别载入序列。
在本教程中对于序列的分析这一步对于构建抗体结构不是必须的,但是对于序列上述两个序列依次分别为抗体MA5的重链(H)序列和轻链(L)序列。
计算完成之后会自动打开一个序列注释结果窗口(如上图),蓝色的表示轻链可变区,粉红色的表示CDR loop区,同样的结构域在序列注释窗口显示同样的颜色。
如果你在序列注释窗口中选择一段loop区,相应的氨基酸会在下方的序列窗口中以同样的颜色显示出来。
小技巧:鼠标右键点击序列窗口的标尺工具可以选择Residue ID,显示出每个氨基酸的残基号。
同理,可以用同样的方法把重链的序列分析注释显示出来。
计算机辅助药物设计及其在新药研发中的应用

计算机辅助药物设计及其在新药研发中的应用随着科技的不断发展,计算机辅助药物设计已成为新药研发的重要工具之一。
它可以帮助药物研究人员更快地设计出具有高度活性和选择性的化合物,从而加快了新药的研发进程。
本文将从计算机辅助药物设计的概念、方法和应用三个方面进行探讨。
一、计算机辅助药物设计的概念计算机辅助药物设计是利用计算机模拟技术和计算化学方法对分子结构进行模拟和分析,从而快速筛选出具有高选择性、高效率的化合物的过程。
计算机辅助药物设计涉及多个学科领域,其中包括计算机科学、化学、生物学和药学等。
通过计算机辅助药物设计可以高效地预测药物分子的活性和亲和力,加快新药的发现和优化。
二、计算机辅助药物设计的方法计算机辅助药物设计有多种方法,包括分子模拟、药效团分析、构象分析等。
其中,分子模拟是目前最为广泛应用的方法之一。
该方法可以模拟药物分子与目标分子之间的相互作用,从而预测药物的活性。
分子模拟包括分子力场、分子动力学等模拟方法。
药效团分析则是利用化学信息库,从已知的活性化合物中识别出活性团,进而设计新的分子结构。
构象分析则是通过计算化学方法分析分子的结构、构象和物性等特征,为药物设计提供依据。
三、计算机辅助药物设计在新药研发中的应用计算机辅助药物设计已经广泛应用于新药研发的各个环节,从药物分子的筛选、设计、优化到临床试验阶段都能起到重要作用。
计算机辅助药物设计可以大大减少研发周期和成本,提高新药的成功率。
以下是计算机辅助药物设计在新药研发中的具体应用。
1. 药物分子的筛选在众多的候选化合物中,如何快速地筛选出最有前途的活性物质是药物设计中面临的一个重要问题。
计算机辅助药物设计可以通过建立药效团、分子对接和虚拟高通量筛选等方法,加速活性化合物的筛选,从而提高新药研发成功率。
2. 药物设计和优化药物设计和优化是新药研发中至关重要的环节,计算机辅助药物设计可以预测药物的性质和活性,引导化学实验进行进一步的筛选,快速改进药物的质量和效果。
Discovery Studio官方教程--构建基于受体-配体复合物药效团

构建及验证基于受体-配体复合物药效团教程介绍随着X-射线晶体衍射和核磁共振技术的进步,大量的蛋白结构被解析,尤其是如果受体和抑制剂复合物结构已知,则从复合物结构中可以得到抑制剂中对活性贡献较大的基团及其空间分布。
因此,在基于活性配体构建药效团模型,即HipHop和HypoGen方法以及基于受体的药效团模型SBP方法的基础上,从Discovery Studio 3.0版本开始又增加了基于受体-配体晶体复合物构建药效团模型的功能(Receptor-Ligand Pharmacophore Generation)。
研究分子间相互作用对于基于结构的药物设计非常重要。
分子对接是常用的方法之一,在传统的对接方法中,对接的准确性往往要打折扣,因为这些程序可以把化合物放在结合位点的任何位置。
而相应的打分方程往往不能找到最有可能的结合位点。
但是大部分情况下,对一个给定的结合位点来说,哪个相互作用对配体-受体相互作用起关键影响经常是已知的。
对于这种情况,就可以把以经验为主发现的结合位点和已知的结合模式考虑到对接过程中,创建一个用于对接的药效团模型。
这样就可以引导潜在的抑制剂结合到已知的、能量有利的相互作用上。
这个demo主要就是介绍如何根据受体-配体晶体复合物方便的构建药效团模型,并在构建的过程中集成验证功能。
以β-分泌酶以及其抑制剂(Al-Nadaf A , et al. Bioorganic & Medicinal Chemistry, 2010, V olume 18, 3088-115.)作为数据来源进行计算。
本教程包括以下步骤:●药效团模型的构建及验证●药效团结果分析●反向找靶药效团模型的构建及验证本教程采用β-分泌酶与其抑制剂(结构见图1)晶体复合物构建药效团模型。
图1 小分子抑制剂结构1.蛋白的准备在文件浏览器(Files Explorer)中,找到2irz.pdb文件,双击打开在分子窗口中显示(或者点击菜单栏File| Open URL...,输入PDB ID:2irz),分别按下快捷键Ctrl+H和Ctrl+T打开描述此结构的相关树形窗口和表格窗口(图2)图2 蛋白-配体晶体复合物2irz结构在工具浏览器(Tools Explorer)中,展开Macromolecules| Prepare Protein,在工具面板中单击Prepare Protein打开Prepare Protein对话框.设置Input Protein为2irz:2IRZ。
Discovery studio简介

Discovery Studio介绍Discovery Studio™(简称DS), 基于Windows/Linux系统和个人电脑、面向生命科学领域的新一代分子建模和模拟环境。
它服务于生命科学领域的实验生物学家、药物化学家、结构生物学家、计算生物学家和计算化学家,应用于蛋白质结构功能研究,以及药物发现。
为科学家提供易用的蛋白质模拟、优化和药物设计工具。
通过高质量的图形、多年验证的技术以及集成的环境,DS将实验数据的保存、管理与专业水准的建模、模拟工具集成在一起,为研究队伍的合作与信息共享提供平台。
建立在最新的流程管理平台Pipeline Pilot基础上的DS让数据的共享和交流变得更为方便和简洁。
DS中的部分功能流程(protocols)可以在Pipeline Pilot中进行编辑和组合,编辑组合而得的新流程可以导入Discovery Studio中使用,这样使得科研流程的方便共享成为可能。
同时,Pipeline Pilot这个开放平台技术还为使用者整合自己的或第三方的软件工具提供了接口。
科研人员可以在一个统一的平台上完成从基因到先导化合物设计的一系列工作,并且可以通过web形式共享研究成果。
DS的服务器-客户端模式使得科研人员能够最方便且最大限度地实现计算资源共享。
DS目前的主要功能包括:蛋白质的表征(包括蛋白-蛋白相互作用)、同源建模、分子力学计算和分子动力学模拟、基于结构药物设计工具(包括配体-蛋白质相互作用、全新药物设计和分子对接)、基于小分子的药物设计工具(包括定量构效关系、药效团、数据库筛选、ADMET)和组合库的设计与分析等。
DS可以应用于生命科学以下研究领域:新药发现,生物信息学,结构生物学,酶学,免疫学,病毒学,遗传与发育生物学,肿瘤研究。
Discovery Studio功能模块简介- 基本界面和显示模块Discovery Studio StandaloneDiscovery Studio Visualizer Client- 蛋白质模拟模块DS MODELERDS Protein RefineDS Protein HealthDS Protein FamiliesDS Sequence Analysis- 基于结构的药物发现和设计模块DS Flexible DockingDS LigandFitDS LigandScoreDS LibDockDS CDOCKERDS Protein DockingDS LudiDS De Novo EvolutionDS LigandFit CAP/ DS Ludi CAPDS GOLD interface- 基于药效团的药物发现和设计模块DS Catalyst ConformationDS Catalyst HypothesisDS Catalyst SBPDS Catalyst ScoreDS Catalyst ShapeDS Catalyst DB BuildDS Catalyst DB SearchDS De Novo Ligand BuilderHypoDBPCDB (PharmaCoreDB)- 基于小分子的药物发现和设计模块DS QSARGFA ComponentV AMP Descriptors Component/ DMol3 Descriptors ComponentDS Library DesignDS ADMETDS TOPKAT- 分子力学和分子动力学计算模块DS CHARMmDS CHARMm LiteDS CFF (高级II类力场)DS MMFF (Merck Molecular Force Field)- 分析模块DS BiopolymerDS Analysis基本界面和显示模块·Discovery Studio Standalone可视化界面,是利用Discovery Studio软件进行分子设计和模拟的基础,支持服务器-客户端安装在同一台机器上的运行模式。
discoverystudio药物发现与生物大分子计算模拟平台

Discovery Studio 药物发现与生物大分子计算模拟平台个人电脑上的全新分子建模环境,专业的生命科学分子模拟软件Discovery Studio™ (简称DS), 基于Windows/Linux系统和个人电脑、面向生命科学领域的新一代分子建模和模拟环境。
它服务于生命科学领域的实验生物学家、药物化学家、结构生物学家、计算生物学家和计算化学家,应用于蛋白质结构功能研究,以及药物发现。
为科学家提供易用的蛋白质模拟、优化和药物设计工具。
通过高质量的图形、多年验证的技术以及集成的环境,DS将实验数据的保存、管理与专业水准的建模、模拟工具集成在一起,为研究队伍的合作与信息共享提供平台。
建立在最新的流程管理平台Pipeline Pilot基础上的DS让数据的共享和交流变得更为方便和简洁。
DS 中的部分功能流程(protocols)可以在Pipeline Pilot中进行编辑和组合,编辑组合而得的新流程可以导入Discovery Studio中使用,这样使得科研流程的方便共享成为可能。
同时,Pipeline Pilot这个开放平台技术还为使用者整合自己的或第三方的软件工具提供了接口。
科研人员可以在一个统一的平台上完成从基因到先导化合物设计的一系列工作,并且可以通过web形式共享研究成果。
DS的服务器-客户端模式使得科研人员能够最方便且最大限度地实现计算资源共享。
DS目前的主要功能包括:蛋白质的表征(包括蛋白-蛋白相互作用)、同源建模、分子力学计算和分子动力学模拟、基于结构药物设计工具(包括配体-蛋白质相互作用、全新药物设计和分子对接)、基于小分子的药物设计工具(包括定量构效关系、药效团、数据库筛选、ADMET)和组合库的设计与分析等。
DS 可以应用于生命科学以下研究领域:新药发现,生物信息学,结构生物学,酶学,免疫学,病毒学,遗传与发育生物学,肿瘤研究。
一、二、Discovery Studio功能模块简介- 基本界面和显示模块- 蛋白质模拟模块- 基于结构的药物发现和设计模块- 基于药效团的药物发现和设计模块- 基于小分子的药物发现和设计模块- 分子力学和分子动力学计算模块- 分析模块基本界面和显示模块·Discovery Studio Standalone可视化界面,是利用Discovery Studio软件进行分子设计和模拟的基础,支持服务器-客户端安装在同一台机器上的运行模式。
Discovery Studio官方教程--药物设计之片段生长

Discovery Studio Grow Scoffold教程Grow Scoffold –活性位点先导化合物优化介绍先导化合物优化是一个复杂过程,为了得到一个临床前候选药物,通常需要对有前景化合物及骨架不断地进行化学结构优化,以提高化合物的活性、选择性、生物利用度、药效及药代动力学性质,并降低毒性。
该过程通常是药物开发过程中的瓶颈。
通过使用实用且有效的先导化合物优化软件,根据可获取的化合物试剂的快速推荐出容易合成的候选化合物的结构。
基于结构的先导化合物优化主要集中于蛋白靶点的活性位点的化合物结构的设计。
Grow Scaffold工具可以根据蛋白靶点活性位点的特点,通过基于化学反应的原位生长(reaction-based in situ enumeration)方法来找出那些能够产生潜在化合物的试剂,并对它们进行打分排序。
p38α是一个典型的丝氨酸/苏氨酸蛋白激酶,属于丝分裂原活化蛋白激酶(mitogen activated protein kinase)家族。
它在内皮、免疫和炎性细胞中广泛表达,在肿瘤坏死因子和白细胞介素- 1等促炎症细胞因子产生的调控中扮演着重要作用。
实验已经证明,选择性地抑制其中任何一个细胞因子都能够有效治疗各种炎症和免疫疾病,如风湿性关节炎、炎症性肠病,败血性休克,和骨质疏松症。
二芳基脲是p38α的一个先导化合物,BoehringerIngelheim首先对它开展了先导化合物优化,随后Pfizer也开展了相关研究(图1)。
图1 p38α的先导化合物及优化过的先导化合物在本教程中你将使用Grow Scaffold流程来重现BoehringerIngelheim及Pfizer通过结构优化所发现的化合物。
本教程分为两部分:(一)使用基于反应的原位生长方法产生优化过的先导化合物(二)使用自定义反应来产生先导化合物(一)使用基于反应的原位生长方法产生优化过的先导化合物1.执行优化计算图2蛋白质三维结构示意图复合物结构采用PDB号为1KV2的晶体结构,本教程中所采用的该结构文件已经从PDB 库中下载并经过Prepare Protein模块处理。
Discovery Studio3.0中基于药效团的药物设计方法和应用讲义-2011年3月21日zi

+-+ +-+ +-+ +-+ +-+
I I
16
06.05 06.00 05.08
Q(;..25
S701J S300
+ + + +-+
I
+ +
+ +-+
I
t;
IS
oa.6J
03.73 OUili t O.lt 10.34 10.42 I O.m 08.67
0~.00
6000
190 53,500 l:iOU
••
DS3.0保留经典功能
Create
HipHop结果分析
Cl
IFeatures
IRank
ID irect H IPartial Hit 1t [OOlooo 1 000010 looooo1 looooto fiiiil ooo 1 000010 !000010 1 001000 [OOlooo lool()(!l()
HypoGen more than 10 actives and inactives activity values required predictive model wide range of activity maximum of 10 best
Geotrident
ftiJIUHUr Rl ~ Sl
~UUHHi J!IH~ SJ
•••
实例分析一
验证方法三: 随机验证(Fischer)
RP
145
3D QSAR Pharmacophore Generation
Fischer Validation
Discovery Studio官方教程-- 构建基于分子共同特征的药效团模型

构建基于分子共同特征的药效团模型(HipHop)教程介绍Common Feature Pharmacophore Generation protocol (HipHop) 用于发现一系列配体小分子所共有的化学特征,并基于这些共同特性结构的比对叠合自动生成药效团模型,用户可以使用共有的特征药效团去搜索化合物数据库来寻找可能的先导分子。
基于分子共同特征的药效团模型同样也可以用于探索一系列具有相似活性但结构却不同或者结构柔性较大的分子的构效关系。
此教程以6个活性配体小分子所构成的训练集来构建基于分子共同特征的药效团模型,继而用于先导化合物的发现。
本教程包括以下步骤:•基于分子共同特征的药效团模型的构建(训练集)•基于分子共同特征的药效团模型的验证(测试集)•先导化合物的发现(数据库的筛选)Common Feature Pharmacophore的构建1. 训练集分子的准备本教程采用一系列已知的5-HT2c配体(1,2)来构建一个基于分子共同特征的药效团模型用于先导化合物的发现。
5-HT2c受体属于GPCR超家族。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开5HT2c_ligands.sd。
在表格浏览器中可以看到一共有6行,代表了6个分子。
这些分子的Principal和MaxOmitFeat属性都已事先定义。
若无定义,则选择表格浏览器中剩下列的heading,鼠标右键点击选择Add Attributes,打开Add Attributes对话框,添加这两个属性。
Principal属性定义了分子的活性水平:2 有活性参考分子,分子中所有化学特征在构建药效团模型时都要考虑。
1 中等活性定位药效团特征元素时需要考虑该化合物的构象空间。
0 非活性该分子在定位药效团特征元素时不考虑,用于选择性地定义排除体积。
MaxOmitFeat属性定义了每个分子中允许不与药效团模型匹配的特征元素的个数:数值描述内容0 构建的药效团模型中所有特征元素都必需与化合物匹配上。
医药行业智能化药品研发方案

医药行业智能化药品研发方案第1章引言 (3)1.1 背景与意义 (3)1.2 研究目标与内容 (4)第2章医药行业现状分析 (4)2.1 国际医药行业智能化发展概况 (4)2.2 国内医药行业智能化发展现状 (4)2.3 医药行业智能化发展趋势 (5)第3章智能化药品研发技术体系 (5)3.1 人工智能技术 (5)3.1.1 深度学习 (5)3.1.2 机器学习 (5)3.1.3 计算机视觉 (6)3.2 大数据技术 (6)3.2.1 数据挖掘 (6)3.2.2 数据整合与共享 (6)3.2.3 云计算 (6)3.3 生物信息学技术 (6)3.3.1 基因组学分析 (6)3.3.2 蛋白质组学分析 (6)3.3.3 系统生物学 (6)第4章药物靶点发觉与筛选 (7)4.1 药物靶点识别方法 (7)4.1.1 基于生物信息学的方法 (7)4.1.2 基于实验生物学的方法 (7)4.1.3 综合方法 (7)4.2 靶点筛选策略 (8)4.2.1 基于疾病机制的靶点筛选 (8)4.2.2 基于药物作用机制的靶点筛选 (8)4.2.3 基于生物标志物的靶点筛选 (8)4.3 智能化药物靶点筛选平台 (9)4.3.1 数据收集与整合 (9)4.3.2 智能算法与模型 (9)4.3.3 筛选流程优化 (9)4.3.4 靶点验证与评估 (9)4.3.5 知识库与决策支持 (9)第五章分子模拟与药物设计 (9)5.1 分子模拟技术 (9)5.1.1 分子模拟技术原理 (9)5.1.2 分子模拟技术在药物研发中的应用 (10)5.2 药物分子设计方法 (10)5.2.1 基于结构的药物设计(SBDD) (10)5.2.2 基于配体的药物设计(LBDD) (10)5.3 智能化药物设计软件 (10)5.3.1 Schrödinger软件 (10)5.3.2 MOE软件 (11)5.3.3 Discovery Studio软件 (11)5.3.4 AutoDock软件 (11)第6章药物合成与制备 (11)6.1 药物合成方法 (11)6.1.1 有机合成方法 (11)6.1.2 生物合成方法 (11)6.2 制备工艺优化 (12)6.2.1 反应条件优化 (12)6.2.2 催化剂选择 (12)6.2.3 萃取、分离和纯化工艺 (12)6.3 智能化药物合成与制备系统 (12)6.3.1 自动化合成设备 (12)6.3.2 人工智能辅助合成设计 (12)6.3.3 智能化制备工艺优化 (12)6.3.4 智能化质量控制 (12)第7章药物活性评价与筛选 (12)7.1 药物活性评价方法 (12)7.1.1 细胞水平评价 (13)7.1.2 器官水平评价 (13)7.1.3 分子水平评价 (13)7.2 高通量筛选技术 (13)7.2.1 微板技术 (13)7.2.2 自动化设备 (13)7.2.3 数据处理与分析 (13)7.3 智能化药物筛选与评价平台 (13)7.3.1 人工智能在药物筛选中的应用 (13)7.3.2 虚拟筛选技术 (14)7.3.3 个性化药物筛选与评价 (14)第8章药物安全性评价与毒理学研究 (14)8.1 药物安全性评价方法 (14)8.1.1 实验室检测方法 (14)8.1.2 临床试验方法 (14)8.1.3 计算机模拟与预测方法 (14)8.2 毒理学研究内容 (14)8.2.1 急毒性研究 (14)8.2.2 慢毒性研究 (15)8.2.3 遗传毒性、生殖毒性、发育毒性研究 (15)8.2.4 药物代谢与毒物代谢研究 (15)8.3 智能化药物安全性评价系统 (15)8.3.1 智能化实验室检测系统 (15)8.3.2 临床试验数据挖掘与分析系统 (15)8.3.3 计算机辅助药物设计系统 (15)8.3.4 药物安全性监测与预警系统 (15)第9章临床试验与数据分析 (15)9.1 临床试验设计 (15)9.1.1 试验目标与方案 (15)9.1.2 随机对照试验 (16)9.1.3 多中心临床试验 (16)9.2 数据收集与处理 (16)9.2.1 数据收集 (16)9.2.2 数据处理 (16)9.3 智能化临床试验与数据分析 (16)9.3.1 人工智能在临床试验中的应用 (16)9.3.2 数据分析策略 (16)9.3.3 智能化数据解读 (17)第10章医药行业智能化药品研发管理与决策 (17)10.1 研发项目管理 (17)10.1.1 项目规划与组织 (17)10.1.2 项目执行与监控 (17)10.1.3 项目沟通与协调 (17)10.1.4 项目评估与总结 (17)10.2 知识管理与协同创新 (17)10.2.1 知识管理 (17)10.2.2 协同创新 (17)10.3 智能化决策支持系统 (17)10.3.1 数据采集与分析 (18)10.3.2 决策模型构建 (18)10.3.3 决策支持系统应用 (18)第1章引言1.1 背景与意义生物科学与信息技术的飞速发展,医药行业正面临着深刻的变革。
药物设计常用软件DS2.5的下载安装及使用教程
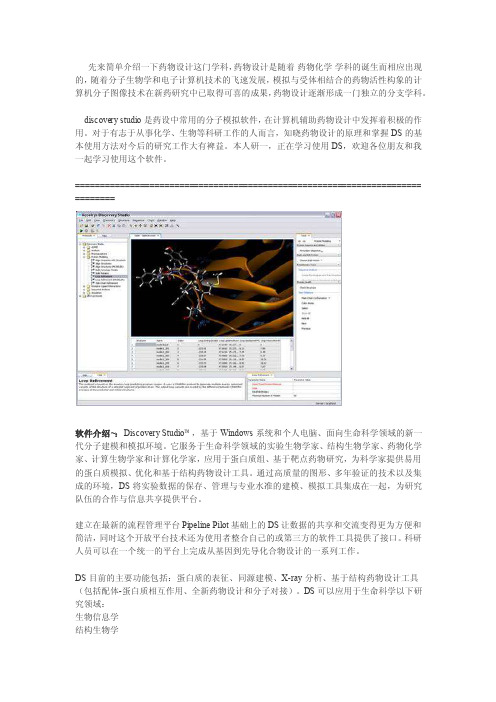
先来简单介绍一下药物设计这门学科,药物设计是随着药物化学学科的诞生而相应出现的,随着分子生物学和电子计算机技术的飞速发展,模拟与受体相结合的药物活性构象的计算机分子图像技术在新药研究中已取得可喜的成果,药物设计逐渐形成一门独立的分支学科。
discovery studio 是药设中常用的分子模拟软件,在计算机辅助药物设计中发挥着积极的作用。
对于有志于从事化学、生物等科研工作的人而言,知晓药物设计的原理和掌握DS的基本使用方法对今后的研究工作大有裨益。
本人研一,正在学习使用DS,欢迎各位朋友和我一起学习使用这个软件。
====================================================================== ========软件介绍~:Discovery Studio™,基于Windows系统和个人电脑、面向生命科学领域的新一代分子建模和模拟环境。
它服务于生命科学领域的实验生物学家、结构生物学家、药物化学家、计算生物学家和计算化学家,应用于蛋白质组、基于靶点药物研究,为科学家提供易用的蛋白质模拟、优化和基于结构药物设计工具。
通过高质量的图形、多年验证的技术以及集成的环境,DS将实验数据的保存、管理与专业水准的建模、模拟工具集成在一起,为研究队伍的合作与信息共享提供平台。
建立在最新的流程管理平台Pipeline Pilot基础上的DS让数据的共享和交流变得更为方便和简洁,同时这个开放平台技术还为使用者整合自己的或第三方的软件工具提供了接口。
科研人员可以在一个统一的平台上完成从基因到先导化合物设计的一系列工作。
DS目前的主要功能包括:蛋白质的表征、同源建模、X-ray分析、基于结构药物设计工具(包括配体-蛋白质相互作用、全新药物设计和分子对接)。
DS可以应用于生命科学以下研究领域:生物信息学结构生物学酶学免疫学病毒学遗传与发育生物学肿瘤研究新药开发下载地址:iso文件,用电驴下载,官网上下的不全: /topics/2819485/ 注意选择系统相应的版本下载,别下错了。
Discovery Studio官方教程(Help-Tutorials) 创建3D QASR模型

采用能量格点作为描述符构建PLS模型(3D-QSAR)教程介绍药物设计即试图发现能够同生物大分子靶标在形状(steric)和电荷(静电势)上互补的小分子。
与2D-QSAR相比,3D-QSAR方法更能间接反映配体小分子和蛋白大分子之间的非键相互作用特征,具有更加丰富的物理化学内涵,因此得到了迅速的发展和广泛的应用。
3D-QSAR模型时基于小分子的立体(steric)和静电(electrostatic)场构建的回归模型,可以用于预测未知配体小分子的活性及观察受体-配体间有利和不利的相互作用。
本教程利用能量格点作为描述符构建了一个偏最小二乘(PLS)模型。
该能量格点是通过两种用于测量静电势和立体效应的探针计算得到的。
本教程包括以下步骤:♦构建3D-QSAR模型♦观察分析结果♦基于3D-QSAR模型预测活性3D QSAR模型的构建在构建3D QSAR模型之前,需要对训练集分子进行叠合,叠合好坏决定了最终模型的可信度。
叠合方式一般有以下几种:♦如果配体分子都来源于晶体结构且都与同一靶标相结合,则可以直接使用晶体结合构象♦如果有配体小分子的药效团模型,则可将配体小分子匹配至药效团模型以实现对配体小分子的叠合♦可直接基于配体小分子的公共骨架进行叠合♦可将配体小分子公共骨架中的药效团特征元素加以提取,用于构建药效团模型,再通过配体药效团的匹配流程(Ligand Pharmacophore Mapping)进行小分子的叠合♦可基于立体场和静电场通过场匹配的方式进行小分子的叠合(Structure | Superimpose | Molecular Overlay…)1. 3D-QSAR的构建在文件浏览器(Files Explorer)中,展开Samples | Tutorials | QSAR,双击trainingset.sd。
在分子窗口中打开一个表格,共14行,即14个训练集分子,该分子事先已进行叠合。
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | QSAR,双击testset.sd。
分子对接 相互作用力 discovery studio

分子对接相互作用力discovery studio分子对接是一种主要用于计算机辅助药物设计的方法,可用于模拟和预测药物与靶标分子之间的相互作用。
在分子对接中,一方是药物分子(ligand),另一方是靶标分子(receptor)。
这两个分子通过非共价相互作用力进行结合,形成稳定复合物。
因此,对于药物发现研究而言,了解药物和靶标分子之间相互作用力的特性至关重要。
Discovery Studio是一种用于计算机辅助药物设计和药物发现的软件平台。
它内置了多种用于分子对接的工具和算法,可以帮助研究人员进行药物分子的筛选和优化,提高研发效率。
本文将以Discovery Studio的分子对接工具为主题,介绍其在药物发现中的应用和相互作用力的计算方法。
一、Discovery Studio分子对接的原理及流程在Discovery Studio中,分子对接的过程可以大致分为以下几个步骤:准备受体结构、准备配体结构、建立格点和计算能量。
1. 准备受体结构受体结构一般是指药物的靶标蛋白,可以从真实蛋白结构数据库中获取。
在进行分子对接之前,需要对受体结构进行优化和准备工作。
这包括去除水分子、修复缺失的原子、填充缺失的氢原子,并对蛋白进行能量最小化的处理。
2. 准备配体结构配体结构是指药物分子或其他小分子化合物。
首先,需要将配体结构进行优化和准备工作。
这包括去除水分子、修复缺失的原子、填充缺失的氢原子,并对配体进行能量最小化的处理。
此外,需要确定配体的荷电状态和形式,如药物分子的离子化状态。
3. 建立格点在分子对接过程中,通常会建立一个三维网格(grid)作为搜索空间,用于计算配体与受体之间的相互作用力。
这个网格的构建需要选择合适的参数和方法。
一般而言,可以使用栅格框(grid box)来定义搜索空间的大小和位置。
4. 计算能量在分子对接过程中,需要计算配体与受体之间的相互作用能量。
这些能量包括范德华力、库仑力、极化等效力等。
基于药效团模型筛选治疗新型冠状病毒肺炎DPP1抑制剂

基于药效团模型筛选治疗新型冠状病毒肺炎DPP1抑制剂钱思彤;杨婷婷;黄健航;梁礼;王长军【期刊名称】《化学研究与应用》【年(卷),期】2023(35)2【摘要】Cathepsin C(CTSC)基因是影响中国人群新型冠状病毒肺炎(Corona Virus Disease 2019,COVID-19)重症化的遗传易感基因之一,其编码的Dipeptidyl peptidase 1(DPP1)蛋白作为潜在的COVID-19治疗靶点,具有重要的临床意义。
本文选取活性好且结构类似的DPP1小分子抑制剂组成训练集,利用Discovery Studio软件构建HipHop药效团。
构建的药效团模型包含6个特征元素,包括3个氢键受体,2个芳香环中心和1个疏水中心。
通过测试集和特征曲线图(ROC)验证,证实药效团模型具有较好的可靠性、稳定性和区分能力。
使用该药效团模型筛选ZINC数据库中306347个小分子化合物,在依次进行类药性规则评判和分子对接研究,分析分子与蛋白的结合模式,选出3个潜在的DPP1抑制剂。
预测上述小分子化合物的药物动力学性质(ADMET)和成药性,发现化合物7(ZINC12503660)最具成药优势,可以开发成治疗COVID-19的候选药物。
同时,本文设计的多层次虚拟筛选方案也为设计和合成新的DPP1小分子抑制剂提供了参考。
【总页数】10页(P379-388)【作者】钱思彤;杨婷婷;黄健航;梁礼;王长军【作者单位】徐州医科大学药学院;中国人民解放军疾病与预防控制中心;中国药科大学药学院【正文语种】中文【中图分类】O641【相关文献】1.基于药效团模型设计合成新型ALS抑制剂2.采用药效团模型和分子对接方法筛选新型的组蛋白甲基转移酶 G9a抑制剂3.基于处方挖掘与药效团模型的新型冠状病毒RdRp抑制成分筛选4.基于药效团模型及虚拟筛选方法发现EphB4全新抑制剂5.基于分子对接及药效团模型的木姜子属木脂素类化合物5-LOX抑制剂筛选研究(Ⅰ)因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
- 化合物数目在 化合物数目在2-32个,6个左右比较理想 个 个左右比较理想 - 只选用具有活性的分子 - 需要包含 需要包含Principal和MaxOmitFeat性质 和 性质 • 方法总结 - 找到一组化合物的公共药效特性 - 不需要 不需要SAR的信息 的信息 - 使用有活性的化合物作为输入结构 - 返回一组公共的药效特性 - 产生的药效团根据打分排序 - 可同时处理大量分子 - 多种不同格式的分子 多种不同格式的分子(*.cpd, *.mol2, *.mmod)
36 Compounds
1150compounds
+ Training set (16) Ligandfit
化合物库筛选
可以构建自己的化合物库
database
安装其他化合物库, 安装其他化合物库,CNPD,ACD等 , 等 DS中自带的化合物库 中自带的化合物库
构建具有活性预测能力的药效团
• • 寻找与化合物活性相关的药效团特性 构建的药效团模型具有活性预测功能 3D-QSAR Pharmacophore Generation HypoGen(quantitative)
2010年其他市场活动 年其他市场活动
– Spotfire Seminar(主题:Spotfire在药物研究中的应用)
• 5月31日上午,北京Seminar • 6月2日上午,上海Seminar • 具体通知请关注我们的网站
筛选 对接 36compounds
1150compounds
模型构建
Training set:
模型构建
如何选择
产生多个药效团模型
产生的模型是否可信
模型验证
Fischer’s randomization method Decoy set
模型验证
Test set
虚拟筛选
MayBridge, Scaffold 160677 compounds
对训练集分子的要求
• • • • • • • 分子结构兼具多样性 活性分子的活性值至少跨越4 活性分子的活性值至少跨越4个数量级 每个活性数量水平的化合物分子数量至 少为3 总数在18 25个 18少为3个,总数在18-25个。 容忍活性水平很小的化合物 结构类似的化合物之间活性相差至少一 个数量级 活性相似的化合物之间结构不同 需要包含Activ Uncert性质 Activ和 需要包含Activ和Uncert性质
单击此处编辑母版标题样式
DS2.5中基于药效团的药物 中基于药效团的药物 单击此处编辑母版文本样式 设计方法和应用 第二级
第三级 第四级 讲座14:00开始 听到声音的老师和同学请举手 开始,听到声音的老师和同学请举手 讲座 开始 第五级
赵冬梅 创腾科技有限公司
2010年网络培训 年网络培训
What’s new in DS2.5.5以及 以及DS2.5Visualizer使用方法 以及 使用方法 DS2.5中基于药效团的药物设计方法和应用 中基于药效团的药物设计方法和应用 生物大分子间相互作用的识别工具及其在生命科学和药物设计中的应用 DS 2.5中的 中的QSAR(2D、3D)方法介绍和应用 中的 ( 、 ) 真正的生物实验数据登录管理系统-BioRegistration 真正的生物实验数据登录管理系统 Spotfire帮您加快药物研发速度 帮您加快药物研发速度 应用于生物信息学中的数据库和分析工具 如何通过Neo-CADD帮助实验人员更快设计和优化药物分子 帮助实验人员更快设计和优化药物分子 如何通过 索取资料途径: 索取资料途径:;market@
排除体积
active inactive
A
B C
A
B C
D
OK
No! D in the Excl Volume!!
• 在D位置同样产生特征元素 • 可通过活性分子与非活性分子之间的结构差别或配体分子和受体 之间的相互作用信息判别
HypoGen and HipHop
HipHop Number of compounds Actives Activity data Type of model Differences in models Number of hypotheses 2-32 all active no data required feature-based structurally diverse user defined HypoGen more than 10 actives and inactives activity values required predictive model wide range of activity maximum of 10 best
HIV-1整合酶抑制剂 5HT再摄取抑制剂 5α-还原酶抑制剂 MC细胞增殖抑制剂 TGFβ受体激酶抑制剂 α4β1拮抗剂 5-HT7受体拮抗剂 CYP17抑制剂 CDK(Pfmrk)抑制剂 鼻病毒coat蛋白抑制剂 FPT抑制剂
界面友好
Catalyst (Linux)
VS
Discovery Studio (Windows & Linux)
pharmacophores ---药效团概述
Pharmacophores(药效团模型) (药效团模型)
• 药物分子与受体靶点发生作用时,分子中的基团对于活性的影响不同。 •1909年,Paul Ehrlich提出,指载有活性必需的特征原子的分子框架。 • 1977年, Peter Gund提出,指分子中的一组能够识别受体,并能形成分子生物 活性的结构特征。 • 泛指药物活性分子中对活性起着重要作用的“药效特征元素”及其空间排列形 式。包括了结合特性、结构和特性约束的信息,以作为数据库检索的提问方式。
- 基于受体-配体结构的药效团:中科院上海药物所 北京大学化学院 中科院上海药物所、北京大学化学院 中科院上海药物所 - 基于配体的药效团:中国医科院药物所 四川大学 华东理工大学 中国医科院药物所、四川大学 中国医科院药物所 四川大学、华东理工大学 - 数据库集成:创腾科技有限公司 创腾科技有限公司
A
B
C
A
B
C
D
药效特征元素
氢键受体:
O O S NH N
氢键给体:
OH
NH2
N H
疏水中心:只要和不带电原子或电负性中心相连的一组连 续的碳原子都可以 形成疏水中心
CH3 C2H5
电荷中心:可能能够与受体形成盐桥或较强的静电相互作用
N
芳环中心:形成π-π相互作用
Catalyst应用实例 应用实例
实例分析2 先导化合物优化) 实例分析 (先导化合物优化)
phore modes
IC50=1.43
IC50=0.004
实例分析3 实例分析
qualitative
Search 3D database
Dock(Ligandfit)
基于特性结构产生药效团
功能添加
catalyst
Conformer Generation • FAST • BEST
Discovery studio
Conformer Generation • FAST • BEST • CAESAR • Systematic Search • Random Search • Boltzmann Jump
结果分析
纵坐标代表5个上市PDE5抑制剂,横坐标代表PCDB中的药效团 模型,包括2个基于PDE的药效团模型(1XOS,2H44),其它 的药效团模型均为INFLAMMATION相关的药效团模型,分值的 大小用不同的颜色表示,红色为高,蓝色为低。
2010年网络培训 年网络培训
What’s new in DS2.5.5以及 以及DS2.5Visualizer使用方法 以及 使用方法 DS2.5中基于药效团的药物设计方法和应用 中基于药效团的药物设计方法和应用 生物大分子间相互作用的识别工具及其在生命科学和药物设计中的应用 蛋白-DNA相互作用 抗原-抗体相互作用;DNA-DNA相互作用 相互作用; 相互作用; (蛋白-DNA相互作用;抗原-抗体相互作用;DNA-DNA相互作用; 多肽药物设计) 多肽药物设计) DS 2.5中的 中的QSAR(2D、3D)方法介绍和应用 中的 ( 、 ) 真正的生物实验数据登录管理系统-BioRegistration 真正的生物实验数据登录管理系统 Spotfire帮您加快药物研发速度 帮您加快药物研发速度 应用于生物信息学中的数据库和分析工具 如何通过Neo-CADD帮助实验人员更快设计和优化药物分子 帮助实验人员更快设计和优化药物分子 如何通过
功能添加
catalyst
HypoGen HipHop HypoRefine&HipHopRefine Shape
Discovery studio
HypoGen HipHop HypoRefine&HipHopRefine Shape Structure-Based Pharmacophore Pharmacophore-Based De Novo Design Ligand Profiler DS2.5
实例分析3 实例分析
qualitative
Search 3D database
Dock(Ligandfit)
排除体积
active inactive
A
B C A
B C
D
OK
OK
• • • •
ABC为活性分子 为活性分子 ABCD 为非活性分子 但ABCD与ABC产生的药效团模型相同 与 产生的药效团模型相同 该药效团模型无法解释ABCD为何无活性 该药效团模型无法解释 为何无活性