线粒体脑肌病2
线粒体遗传病(讲 座)

线粒体肌病的临床分型之二
二、线粒体脑肌病 1、
PEO (进行性眼外肌瘫痪):以慢性进行性眼外肌瘫痪为主 进行性眼外肌瘫痪) KSS (Kearns-Sayre syndrome) (Kearns– 完全型 KSS:眼外肌瘫痪,视网膜色素变性,心脏传导阻滞 KSS:眼外肌瘫痪,视网膜色素变性, 三联征 – 不全型 KSS:眼外肌瘫痪或伴有其他一项。 KSS:眼外肌瘫痪或伴有其他一项。 可伴有身材矮小, 智能低下, 神经性难听, 小脑共济失调, 可伴有身材矮小 , 智能低下 , 神经性难听 , 小脑共济失调 , CSF 蛋白↑,EEG异常,多无家族史 蛋白↑ EEG异常, 发病年龄多在20岁以前 发病年龄多在20岁以前
To be continued...
Mt DNA 的结构特点
人类线粒体的基因组排列非常紧凑, 人类线粒体的基因组排列非常紧凑,除与 mt DNA 复制及 转录有关的一小段区域外,无内含子序列。37个基因间隔 转录有关的一小段区域外,无内含子序列。 37个基因间隔 区总共只有 87 bp,因此,几乎 mt DNA 的任何突变均会 bp,因此, 累及到基因组中一个重要区域。 累及到基因组中一个重要区域 。 线粒体拥有相对独立的 DNA 复制、转录和翻译系统,是半自主性细胞器。重链主 复制、转录和翻译系统,是半自主性细胞器。 要编码2 要编码2个 rRNA,12个多肽及 14个 rRNA;轻链仅编码 rRNA,12个多肽及 14个 rRNA; 一个 NADH 脱氢酶亚单位 4 及8 个 tRNA。 tRNA。
mt DNA 突变
重复 (duplication):指多余的 mt DNA 以数以千计的核苷 (duplication): 酸插入基因组,从而使体积增大。 酸插入基因组,从而使体积增大。 丢失 (depletion):指线粒体内 mt DNA 的拷贝数减少。 (depletion): 的拷贝数减少。
线粒体脑肌病疾病PPT演示课件

家族史
线粒体脑肌病多为母系遗传,因此详细的 家族史询问对于诊断至关重要。
B
诊断标准
结合临床表现、家族史和体征检查,医生可 以初步怀疑线粒体脑肌病。进一步的实验室 检查和辅助检查有助于确诊。
D
鉴别诊断相关疾病
01
多发性硬化症
这是一种中枢神经系统脱髓鞘疾病,与线粒体脑肌病有相似的神经系统
症状,但无肌肉病变和母系遗传特点。
04 患者教育与心理支持
患者教育内容
疾病知识
向患者和家属介绍线粒体脑肌病 的病因、症状、诊断和治疗等方 面的知识,帮助他们更好地理解
和应对疾病。
自我管理
教育患者如何进行自我监测和管理 ,包括观察病情变化、记录症状、 定期随访等,以便及时发现并处理 问题。
生活方式调整
指导患者调整生活方式,如合理饮 食、适当运动、保持充足睡眠等, 以改善身体状况和提高生活质量。
02 03
重症肌无力
这是一种由神经-肌肉接头传递功能障碍引起的自身免疫性疾病,表现 为骨骼肌无力和易疲劳。与线粒体脑肌病不同,重症肌无力无中枢神经 系统受累和母系遗传特点。
其他遗传性肌肉疾病
如肌营养不良症等,这些疾病也可能表现为肌肉无力和萎缩,但通常无 中枢神经系统受累。
实验室检查与辅助检查
血液生化检查
药物选择
根据患者的具体症状,选择合适的药物,如抗癫痫药物、免 疫抑制剂等。
使用注意事项
严格遵守医嘱,注意药物的副作用和禁忌症,及时调整用药 方案。
预后评估及影响因素
预后评估
根据患者的症状、体征、影像学表现 等,综合评估患者的预后情况。
影响因素
患者的年龄、病情严重程度、治疗及 时与否等因素都会影响预后情况。同 时,线粒体脑肌病的预后也与基因突 变类型、线粒体功能障碍程度等因素 有关。
线粒体的功能与疾病

线粒体的功能与疾病人体内有许多微小的细胞器,其中一种重要的细胞器就是线粒体。
线粒体被称为细胞的“能量中心”,是生物合成的关键地点。
线粒体不仅能够供能,而且还参与到细胞的呼吸作用和许多其他生物过程中。
本文将详细阐述线粒体的功能以及与线粒体相关的疾病。
一、线粒体的功能1. ATP合成线粒体最重要的功能之一是合成ATP分子,也就是细胞能量的源泉。
线粒体内的三个关键酶系统通过电子传递链来催化ATP分子的生成,此过程也被称为氧化磷酸化。
在这个过程中,线粒体通过将食物转化为能量而满足细胞对能量的需求。
2. 代谢物的合成除了能量供应外,线粒体还合成一些细胞代谢所必需的催化物质。
例如,线粒体合成尿素的过程是代谢蛋白质和氨基酸时所必需的过程。
3. 脂肪酸氧化线粒体中的另一个关键酶系统是脂肪酸氧化,是体内已知的最重要的能量来源之一。
当身体没有足够的碳水化合物来进行代谢时,线粒体可以利用脂肪酸分子来供能。
4. 钙离子的储存和释放线粒体还可以扮演储存和释放细胞内钙离子的角色。
当细胞内钙离子浓度过高时,线粒体就会吸收和储存这些离子。
一旦细胞需要释放钙离子的时候,线粒体就会释放它们到细胞内。
二、线粒体相关的疾病线粒体与多种疾病的发展过程有着千丝万缕的联系。
大多数的线粒体疾病是由线粒体基因突变引起的,这些基因突变会导致线粒体功能失调。
线粒体疾病的表现形式多种多样,范围广泛,从肌肉无力、癫痫、中风、心肌病到失明、聋等都可能与线粒体有关。
1. 线粒体脑肌病线粒体脑肌病通常是由线粒体DNA的突变引起的,导致线粒体无法正常工作。
这种疾病的症状包括肌无力、表现出代谢性酸中毒、半身不遂等等。
2. 非细胞质性线粒体疾病非细胞质性线粒体疾病是由不遗传线粒体DNA的突变引起的。
这种疾病可以在任何生命阶段诱发,而且在同一家庭中不同的人也可能表现不同的临床症状。
3. 线粒体糖尿病线粒体糖尿病是由线粒体DNA的突变或线粒体功能低下导致的糖尿病类型。
线粒体病(MELAS)

病变区多位于大脑半球后部,即颞、顶、枕叶,亦可同 时见于基底节区。
主要位于皮层,深部白质较少累及, MRI表现为脑回样层状异 常信号区,肿胀较轻,少见出血; 病变形态、大小、范围随病情变化而变化,呈游走性 常多灶性分布,不按血管支配区分布,跨越不同的供血区 脑萎缩和脑室扩大较常见,不具有特异性,但其萎缩程度较明 显,与年龄不相符。 没有大脑前、中、后动脉和基底动脉等较大血管闭塞证据 弥散成像(DWI)的发现病变表观弥散系数(ADC)值升高,提示 存在血管源性水肿,与急性脑梗死因细胞毒性水肿所致ADC值 减低的表现不同。 MRS检查显示病灶内NAA下降,乳酸含量增高。
头颅 CT和 MRI:早期可见与主要脑血管分布不相应的脑软 化灶和多发性类梗塞灶(长T1,长T2信号),后部半球的大 脑皮层(如枕叶)多见,晚期有脑萎缩、脑室扩大和基底 节钙化。
肌活检:光镜下,用改良的 Gomeri三胞色素氧 化酶(COX)染色,肌膜浆下可见大量正常和异形线粒体。
母系遗传,其母多有糖尿病、耳聋、心电图异常和呼吸链复合体Ⅰ、 Ⅲ、Ⅳ活性缺陷。婴儿期正常,3~11岁起病,神经系统受累为主。
突发卒中,偏瘫、偏盲和(或)皮质盲、偏头痛、反复癫痫发 作和呕吐。可有糖尿病、觉感神经性耳聋痴呆和身材矮小。
因异常线粒体不能代谢丙酮酸,导致安静状态下血和脑脊液 中乳酸和丙酮酸浓度升高,最小运动最试验和口服葡萄糖乳 酸刺激试验(+++)。
• 非缺血性神经血管细胞学说
常规检查
血常规:WBC12×109/L,中性比率82%,淋巴 18% 尿便常规正常,肝肾功能正常,电解质检查正常,
血糖7.1mmol/L,心肌酶正常 胸片:双肺纹理增粗,其余未见异常 心电图正常 脑电图: 可见双侧以高幅慢波为主 ,可见棘慢或
线粒体肌病

谢谢
分型
一.伴有破碎红纤维的肌阵挛癫痫常10~20岁发病,主要临床表现为阵发性癫痫.伴 有进行性神经系统障碍(智力倒退、共济失调、意向性震颤),患者肌纤维紊乱、 粗糙,线粒体形态异常并在骨骼肌细胞中积累,用Gomori Trichrome染色显示 为红色,称破碎红纤维。
二.线粒体脑肌病合并乳酸血症及卒中样发作:通常 10〜20岁发病,主要临床表 现为阵发性呕吐、癫痫发作和卒中样发作、血乳酸中毒、近心端四肢乏力等。肌 组织中A3243G突变型mtDNA达40%〜50%时,岀现慢性进行性眼外肌瘫痪、肌病 和耳聋。
治疗
一.药物治疗联合治疗以清除氧自由基、减少毒性产物并补充代 谢辅酶肌酸、肉碱、烟酰胺、硫胺素等。
二.运动疗法通过阻力和耐力训练,提高患儿的肌力。 三.饮食疗法:对不同缺陷的患者应用不同的饮食疗法。 四.基因治疗包括降低突变型mtDNA/野生型mtDNA的比例,使用
错位表达及异质表达,输入其他同源性基因等。 五.细胞移植主要通过定向分化多潜能的干细胞,获得某种特定
线粒体肌病
基本介绍
多在20岁时起病,临床特征是骨骼肌极度不能耐受疲劳,轻度活动即感疲乏,常 伴肌肉酸痛及压痛,肌萎缩少见。易误诊为多发性肌炎、重症肌无力和进行性肌营 养不良等。
该病是一组由于线粒体功能缺陷引起的多系统疾病,以中枢神经和肌肉 系统病变为主,特征是呼吸链酶活性正常的肌纤维与酶活性缺失的肌纤 维混合。患者各种组织内DNA的突变类型、分布各不相同,所以表现出不 同的症状,多表现为肌力低下、易疲劳、小脑失调、耳聋、痴呆、代偿 性高乳酸血症等。
线粒体脑肌病sch

鉴别诊断
需要舆多发性肌炎、重症肌无力、周期性瘫痪和眼咽 型进行性肌营养不良等鉴别
治疗
一般治疗
给予高热量、低脂肪、富含维生素的饮食,特别应限 制长、中链脂肪酸的摄入。少量多餐,或在进行剧烈 运动前后补充高碳水化合物,可预防横纹肌溶解。 丙酮酸羧化酶缺少的患者推荐高蛋白、高碳水化合物 和低脂肪饮食
线粒体脑肌病
孙春华
概述
线粒体肌病和线粒体脑肌病是一组由线粒体DNA或 核DNA缺陷导致线粒体结构和功能障碍、ATP合成 不足所致的多系统疾病,其共同特征为轻度活动后即 感到极度疲乏无力,休息后好转;肌肉活检可见蓬毛 样红纤维。 如病变以侵犯骨骼肌为主,则称为线粒体肌病;如 病变同时累及到中枢神经系统,则称为线粒体脑肌病。 是一种遗传病,遗传方式为母系遗传。
病因及发病机制
线粒体肌病和线粒体脑肌病的病因主要是mtDNA发 生突变、缺失等使编码线粒体在氧化代谢过程中所必 需的酶或载体发生障碍,不能产生足够的ATP。终 因能量不足,不能维持细胞正常的生理功能,产生氧 化应激,诱导细胞凋亡而导致线粒体病。
线粒体脑肌病类型
慢性进行性眼外肌瘫痪(CPEO) 线粒体脑肌病伴乳酸中毒和卒中样发作综合症 (MELAS) 80%的线粒体脑肌病伴高乳酸血症和卒中 样发作是由mtDNA的第3243位点发生A到G的点突 变所致 Kearns-Sayre综合症(KSS) 肌阵挛癫痫伴不整边红纤维综合症(MER-RF)
护理
6、心理护理: 线粒体脑肌病多存在智力下降,反应迟 钝而导致其性格孤僻、社交障碍。该疾病病程长,给 家庭和患者本人带来极大的痛苦。因此心理护理是贯 穿疾病始终的。由于该病作为母系遗传,因此母亲承 受着巨大的家庭压力,同时注重对患者母亲进行心理 护理。
线粒体脑肌病的研究进展2024

线粒体脑肌病的研究进展2024线粒体病是由于线粒体DNA(mitochondrial DNA,mtDNA)或核DNA 缺陷,引起三磷酸腺苷(ATP)合成功能障碍,导致能量来源不足的一组异质性疾病,不包括其他因素导致的继发性线粒体功能障碍性疾病。
其可累及全身各个系统,累及神经系统时称神经系统线粒体病。
成年人mtDNA 突变率为1/5000,核基因突变率为2.9/10万,目前已知的与线粒体基因有关的疾病达270种,且大多有神经系统的表现,国内目前缺乏这方面的详细流行病学统计数据。
线粒体病在神经内科中比较常见,但由于其临床特点比较隐匿且不典型;常常被误诊或延误诊断,因此提高对其临床特征、辅助检查,尤其是核共振成像(MRI)和基因检测结果是十分必要的。
1疾病分型神经系统线粒体病主要分为以下四大类:线粒体脑病、线粒体脑肌病、线粒体神经病、线粒体肌病。
本文主要讨论线粒体脑肌病,其可分为以下四种亚型:①线粒体脑肌病伴高乳酸血症及卒中样发作(Mitochondrial encephalomyopathy with lactate acidosis and stroke-like episodes,MELAS);②肌阵挛性癫痫伴破碎红纤维(Myoclonic epilepsy with ragged redfibers,MERRF);③Kearns-Sayre综合征(Keams-Sayre’s syndrome,KSS);④线粒体神经胃肠脑肌病(Mitochondrial neurogastrointestinal encephalomyopathy,MNGIE)。
2临床表现线粒体脑肌病各个亚型临床表现较为相似,但各亚型又有其特征。
MELAS 发病年龄多在40岁左右,研究表明65%-76%的患者在40岁之前出现症状,大多有母系遗传家族史,其发病机制与一氧化氮(NO)的缺乏有关。
临床表现主要包括发作性头痛、脑卒中样发作(失语、偏瘫、偏盲、偏身感觉障碍等)、癫痫发作、精神行为异常、恶心、呕吐、活动不耐受,患者多伴有身材矮小、智能减退、糖尿病、神经性耳聋,但上述症状缺乏特异性,以上述症状反复发作后可致持续性、进行性听、视、智力低下及运动障碍,最终可导致死亡。
线粒体脑肌病

05
康复支持与心理关怀
康复训练计划制定
01
02
03
个体化评估
针对患者的具体病情和体 能状况,制定个性化的康 复训练计划。
渐进性训练
根据患者的耐受能力,逐 步增加训练强度和时间, 避免过度疲劳。
综合性训练
包括肌力训练、关节活动 度训练、平衡训练等多种 训练方式,全面提高患者 的运动功能。
心理干预策略
心理治疗
线粒体脑肌病患者常伴有心理问题,如焦 虑、抑郁等,心理治疗有助于改善患者心 理状态,提高生活质量。
患者日常管理与教育
定期随访
患者应定期到医院进行随访检查,以 便医生及时了解病情变化,调整治疗 方案。
避免诱发因素
患者应避免感染、饥饿、寒冷等诱发 因素,以免加重病情。
保持良好的生活习惯
患者应保持充足的睡眠时间,避免过 度劳累;保持室内空气流通;注意个 人卫生等。
遗传咨询与宣教不足
线粒体脑肌病为遗传性疾病,但遗传咨询和宣教工作相对 滞后,导致患者及家庭对疾病认知不足,难以有效预防和 控制疾病。
未来发展趋势预测
1 2 3
精准医疗
随着基因测序技术的发展,未来有望实现线粒体 脑肌病的精准诊断和治疗,提高诊疗效果。
新药研发
针对线粒体脑肌病的发病机制,未来有望研发出 更多针对病因的新药,为患者提供更多治疗选择 。
加强自我监测
患者应学会自我监测病情变化,如观 察肌肉力量、感觉异常等,以便及时 发现问题并就医。
04
并发症预防与处理
常见并发症类型及危险因素
常见并发症类型
线粒体脑肌病患者可能出现的并发症包括心脏传导阻滞、心肌病、糖尿病、肾功 能不全、假性肠梗阻等。这些并发症可能单独或同时出现,严重影响患者的生活 质量。
线粒体肌病是什么引起的

线粒体肌病是什么引起的文章目录*一、线粒体肌病是什么引起的*二、线粒体脑肌病的治疗方法*三、线粒体脑肌病的发病机制线粒体肌病是什么引起的1、线粒体肌病是什么引起的线粒体肌病是由线粒体代谢过程中某些酶缺乏所引起的一组遗传性疾病。
肌肉病理的共同特点为破红肌纤维。
2、线粒体肌病的临床表现因酶缺乏种类而异,有下列数种。
进行性眼外肌麻痹,表现为缓慢进行的眼睑下垂和眼外肌麻痹,症状不波动。
Kearns-Sayre 综合征,20岁前起病的进行性眼外肌麻痹、视网膜色素变性和心脏阻滞。
尚可有身材矮小,共济失调,神经性耳聋和甲状腺机能减退等异常。
特点为家族史阴性。
线粒体脑病肌病,病者除有肌无力外,伴发肌阵挛癫痫、共济失调、视神经萎缩、周围神经病以及神经性耳聋和智能低下等症。
部份病者可有卒中样发作,突然出现偏瘫,且有左右交替发作,头颅CT或MRI出现片状低信号。
癫痫发作和发作性呕吐为此型病者最常见症状。
血液乳酸测定可见高乳酸血症。
肌肉活检和乳酸测定常为本病提供诊断依据。
3、线粒体肌病的诊断四肢近端极度不能耐受疲劳、身体矮小和神经性耳聋等,伴各亚型临床特征;血清乳酸、丙酮酸增高和肌肉活组织检查发现RRF,mtDNA缺失或点突变等之结果。
线粒体脑肌病患者CT或MRI 检查可见白质脑病、基底核钙化、脑软化、脑萎缩和脑室扩大等。
但应注意炎症肌病和其他肌病可同时伴存线粒体和糖原堆积之可能。
严防过多过滥诊断线粒体肌病。
线粒体肌病需与多发性肌炎、重症肌无力、周期性瘫痪和眼咽型进行性肌营养不良等鉴别。
线粒体脑肌病的治疗方法1、对于线粒体疾病,目前尚无特别有效的治疗措施。
一般可采用:辅酶Q10,肌内注射或口服。
大剂量B族维生素,如维生素Bl、维生素B2、维生素B6等可改善症状。
能量制剂,如三磷腺苷(ATP)、辅酶A等。
二氯乙酸钠,12.5~100mg/(kg·d),口服。
呼吸链酶复合体Ⅱ+Ⅲ缺陷,可用维生素K3加维生素C治疗。
线粒体脑肌病CT及MRI表现

M M I BIMONTHLY Vol 20 No. 1 Feb 2011—49—线粒体脑肌病的CT 及 MRI 表现常荣1,2 , 2 , 2 ,1马晓文李海燕张明【纲要】目的 : 剖析线粒体脑肌病的脑部 CT 及 MRI 表现,以提升对该病影像特色的认识,为诊疗及鉴识诊疗供给依照。
资料与方法 :回首性剖析8例线粒体脑肌病患者的临床表现、实验室检查及影像检查,总结病变特色。
结果 : 8 例 CT 及 MRI 检查均显示异样,此中 7 例为多发性。
病变在 CT上呈片状略低密度,MR 呈长 T1、长 T2信号,以一侧或双侧颞顶枕叶皮层和皮层下白质最常受累,MRA 血管无狭小。
结论 : MR 对病变的显示优于 CT,可以体现该病的特色,对疾病的诊疗拥有重要的价值。
重点词 :脑疾病线粒体脑肌病磁共振成像线粒体脑肌病 ( Mitochondrial Encephalopathy,ME) 是一组少见的线粒体构造或功能异样所致的以脑和肌肉为主的多系统疾病[1]。
病变以入侵中枢神经系统和骨骼肌为主,临床及影像学表现多样,易与其余病变相混杂,误诊率较高。
本文对 8 例线粒体脑肌病患者的临床、实验室检查及 CT 和 MRI 表现进行回首性剖析,以提升对该病的认识。
1资料与方法1.1 一般资料ME 患者 8 例,此中男 6 例,女 2 例。
年纪 11 至43岁,均匀31 岁。
所有患者均行肌肉活检手术,经病理确诊。
标本取自肱二头肌、三角肌或股四头肌外侧头,冰冻切片做苏木素-伊红染色( HE) 及改进Gomori 三色 ( MGT) 染色,光镜下察看。
1.2 临床表现临床症状主要表现为头痛(7 例)、癫痫 (3 例)、肢体乏力(4 例) 、恶心呕吐 ( 4 例) 、脑卒中样发生(5例)。
查体表现为视力阻碍 (6 例) 肌力降落( 7例) 、中枢性面瘫( 2 例) 、发热 ( 2 例) 、眼球震颤 ( 1例) 。
患者病程 2 月至 1 年。
线粒体脑肌病MELAS综合症 ppt课件

②异质性与阈值效应:线粒体几乎存在于人体的所有细胞中,且每个 细胞中含有成百上千个线粒体,有害的突变基因常常仅影响部分mt DNA,这就使细胞、组织中同时存在野生型DNA和突变型DNA ,这种野生型DNA和突变型DNA同时存在的现象称为异质性。当 mtDNA突变数目达到某一数量时,可引起某组织或器官的功能异 常而出现临床症状,这就是阈值效应。阈值的高低与该组织器官对线 粒体产生的ATP的依赖程度而定。对有氧代谢依赖程度高的组织其 mtDNA突变的病理阈值低于依赖于无氧糖代谢的组织
mtDNA是独立于细胞核染色体之外的又一 个基因组
线粒体脑肌病MELAS综合症
mtDNA与nDNA的主要不同之处包括以下四 个方面
①非孟德尔的母系遗传:mtDNA为母系遗传,发生在生 殖细胞系中的突变能通过女儿传递给子代,引起母系家族性 疾病,而在发育过程中体细胞发生的突变则会引起散发性疾 病和与年龄相关的氧化磷酸化 (OXPHOS)活性降低
线粒体脑肌病MELAS综合症
nDNA改变引起的线粒体功能障碍
呈孟德尔遗传 编码线粒体蛋白的基因缺陷 线粒体蛋白质转运的缺陷 基因组间交流的缺损 大多由nDNA突变造成的线粒体病(包括Leigh综
合征、MNGIE、MDS)都是常染色体隐性的,意 味着这类线粒体病的发生需要父母双方提供的基 因拷贝都含有相同的点突变。
线粒体脑肌病MELAS综合症
线粒体的功能
产生能量 线粒体是细胞的动力工厂,三羧酸循环和氧 化磷酸化均在线粒体内进行
生成自由基 调节细胞凋亡
线粒体脑肌病MELAS综合症
线粒体DNA-mtDNA
线粒体病的临床特征是由mtDNA的遗传特 性决定的
每一个细胞有几百甚至几千个线粒体,而每 一个线粒体平均有5个mtDNA 分子
第十二章 线粒体脑肌病

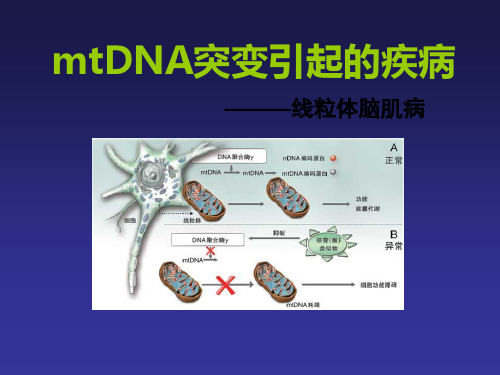
第十二章线粒体脑肌病【概述】线粒体脑肌病(mitochondrial encephalomyopathies)是由于线粒体DNA(mitochondrial DNA,mtDNA)突变,或核基因.(nuclear gene)或核DNA(nuclear DNA,nDNA)改变所致的线粒体呼吸链功能障碍的一组疾病,该组疾病累及身体多种系统。
需高能量供应的器官最易受累,如中枢神经系统和骨骼肌,其次为心、胃肠道、肝、肾等器官。
常见的综合征和名称缩写如下:1.KSS Kearns—Sayre综合征。
2.MELAS 线粒体脑肌病伴乳酸中毒和卒中样发作(mitochon drialencephalomyopathy,lactic acidosis and strokelike episodes)。
3.MERRF肌阵挛癫痫伴破碎红纤维(myoclonic epilepsy with raggedred fibers)。
4.MNGIE或MEPOP线粒体周围神经病、胃肠型脑病(mitOchondrialneuropathy gastrointestinal encephalopathy),或称线粒体脑肌病伴多发周围神经病、眼肌麻痹和假性肠梗阻(mitochondrial neuropathy gastrointestinalencephalopathy with polyneuropathy ophthalrnoplegia and pseudo一0b struction)。
5.NARP 周围神经病、共济失调、色素变性视网膜炎(neurOpathy ataxia and retinitis pigmentosa)。
6.Leber遗传性视神经病(1eber’s hereditary optic neuropathy,LHON)。
7.PEO进行性眼外肌麻痹(progressive external ophthalmoplegia)。
线粒体脑肌病课件

12
MELAS型ME的MRI特点
• 神经影像学是MELAS 诊断的重要手段。
头颅MRI 具有较高的诊断价值.
• 其病变主要集中在左侧颞顶枕叶皮质,病灶单发 ,主要表现为:大脑半球皮质异常信号,病灶常 表现为T1W1低信号,T2W1为高信号,病变不按血 管支配区分布,脑白质受累较轻,脑回肿胀。
• 头颅MR I 的病灶具有反复出现和消退的动态变化
1).清除氧自由基: 辅酶Q10、艾地苯醌、维生素C、维生素E
2).减少毒性产物: 二氯乙酸、二甲辅酶Q10、艾地苯醌、维生素K等
4).补充代谢辅酶:
肌酸、肉碱、核黄线粒素体脑等肌病
19
治疗
• 2. L-精氨酸
L-精氨酸可促进谷氨酰胺的摄取及γ-氨基丁酸的释放, 促进脑代谢,保护大脑。在MELAS急性发作期静脉注射 L-精氨酸有利于降低卒中再发的频率和严重度。
• 脑和骨骼肌等对能量依赖程度较高的器官容易同 时受累。
• 可合并神经肌肉以外的其他系统的症状及体征。
• 进行性的病程,但某些症状可有不同程度的缓解, 甚至完全消失,与此同时又有新的其他症状出现; 同一症状和体征可有多次缓解、复发。
线粒体脑肌病
4
线粒体脑肌病根据临床不同症候群又可分为
• MELAS综合征(为线粒体脑肌病、乳酸血症和卒中样发作) • MERRF综合征(肌阵挛性癫痫发作、小脑共济失调、乳酸血
线粒体脑肌病
线粒体脑肌病
1
定义
• 线粒体脑肌病:是指由线粒体基因或细胞 核基因发生缺失或点突变导致的线粒体结 构和功能异常,引起细胞呼吸链及能量代 谢障碍,主要累及脑及横纹肌的一类疾病 。
• 因其临床表现缺乏特异性,极易误诊为癫痫、脑 梗死、脑炎及脑发育不良等
线粒体脑肌病主要护理问题及措施

线粒体脑肌病主要护理问题及措施
一、概述
线粒体脑肌病病人幼年起就有学习成绩差,运动不耐受等情况,导致患者性格孤僻,社交障碍。
作为他的家人更要从这方面着手帮助他们走出困境。
那么线粒体脑肌病患者日常该怎么护理呢?有什么好的措施帮助其恢复呢?今天,让某同事来跟大家分享关于线粒体脑肌病主要护理问题及措施。
二、步骤/方法:
1、解除患者的思想顾虑,鼓励病人参加集体娱乐活动,鼓励病
人说出自己的心理感受,耐心倾听病人的叙述,,以取得理解并保持
良好的人际关系。
2、保证充足的睡眠,保持精神放松,进行力所能及的活动,必
要时给予氧气吸入,还应观察病人疲劳程度有无缓解并及时给予指导。
3、运动量超负荷,应立即停止活动,卧床休息,根据病情,逐
渐增加活动量,以病人能够耐受为度,病人活动时给予必要的帮助。
4、协助翻身时,避免拖、拉、推等动作;采取保护性措施;保
持皮肤清洁,每晚用温水擦浴,内衣柔软勤更换;加强病人营养,提高肌体抵抗力。
5、给予加床档,防坠床的发生;入厕或外出活动时有人陪护,
防摔倒或其他意外受伤;指导病人进行循序渐进的肢体功能锻炼,运动时机、运动量、运动方法得当,防虚脱、摔伤等意外发生。
三、注意事项:
1、经常巡视病人,提供必要的帮助,避免受伤的可能。
2、避免身体某一部位长期受压,翻身1次/2h。
3、增加营养摄入,给予高蛋白、高糖、高维生素、低脂饮食。
mtDNA引起的脑肌病
病例分析交流
• 患者症状 • 1:步行不稳,易跌跤 • 病例分析 • 属于线粒体脑病 • 原因可能是mtDNA的 突变导致线粒体供能 减少,或者是导致线 粒体数目异常(减 少),抑制了神经冲 动的传导,导致认知 运动障碍。
遗传机制和方式
• 机制:mtDNA遗传,在受精时期,精子上 的线粒体主要集中在尾部,而在进入卵细 胞是,往往是头部进入,所以受精卵中含 来自父方的线粒体很少,可以忽略,绝大 部分都是来自母方。 • 方式:母系遗传。
预防措施
• 预防措施包括避免近亲结婚。
• 推行遗传咨询、携带者基因检测及产前诊 断和选择性人工流产等,防止患儿出生。
小组成员:李程、王效
临床表现
• 以骨骼肌极度不能耐受疲劳为主要特征 • 往往轻微活动后即感疲乏休息后好转常有 肌肉酸痛及压痛 • 但肌萎缩少见易误诊为多发性肌炎重症肌 无力和进行性肌营养不良症等 • 症状可重叠,诊断复杂
病例分析
• 患者,男,7岁。出生时无明显异常。 • 自3岁起开始出现双脚肌无力,走路时呈鸭 形步,步行缓慢,不稳。 • 易跌跤,下蹲后起立困难,症状进行性加 重。
家族史
• 患者的哥哥同患此病,并与15岁时出现下 肢瘫痪,伴有轻度智力低下。 • 先证者的姐姐正常; • 先证者的舅舅、姨表兄弟患此病。
思考
• 1:临床诊断(线粒体脑肌病) • 2:试述该病的一传机理和遗传方式? • 3:如何避免该类患者的出生?
病例分析交流
• 患者症状 • 1:双下肢肌无力,走 路缓慢 • 病例分析 • mtDNA上的3250位点 上的突变。 • 生化缺陷主要为酶复 合体Ⅰ缺乏,也可有 复合体Ⅱ,Ⅲ缺乏。 • 影响了正常的糖类代 谢,导致供能不足。
mtDNA突变引起的疾病 mt的综述
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
参考文献
Walker UA, Collins S, Byrne E. Respiratory chain encephalomyopathies:a diagnostic classification [J]. Eur Neurol, 1996, 36: 260-267 Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol 2001;49:377–83 Chinnery PF, Johnson MA, Wardell TM, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol 2000;48:188–93 Zeviani CT,Antozzi C.Defects of mitochondrial DNA[J].Brain Pathology,1992,(2):121. 马梦华等,MELAS型线粒体脑肌病的MRI诊断价值.中国神经精神疾 病杂志.2007,33(8):484-5
实验室检查
最小运动量实验 应用自行车功量计,于15W功率下令患者运动15min,于运动前,运动后即刻,及运动后 5min分别取静脉血测定乳酸和丙酮酸. 平静时乳酸或丙酮酸增高或运动后大于正常参考值的0.5倍有意义 采用运动前后血乳酸与丙酮酸比值作为判定标准。把运动前后血乳酸/丙酮酸值>17或 <7,或运动后血乳酸/丙酮酸值>22或<7作为阳性标准 电生理 肌电图:约60%表现为肌源性损害,也有少数表现为神经源性损害,一些以脑病为主要表 现的患者,肌电图也有异常表现 脑电图:可见全脑弥漫性异常波,也可见痫性放电(MELAS,MERRF) 心电图检查对KSS具有重要诊断意义,心脏传导阻滞 活检 为了补偿线粒体机能低下会出现线粒体积累,用Gomori Trichrome变色染色法,肌膜下 出现不规则的红色边缘, 这个被染成红色的纤维叫做破碎红纤维(ragged-red fibers,RRF),被用于诊断线粒体脑肌病。 基因检查 Southern 杂交和聚合酶链反应(PCR)
)
2-40岁起病,平均10岁 线粒体tRNA亮氨酸(Leu)基因的3243位点由腺嘌呤A变为鸟嘌呤G,A32 43G点突变出现在80%MELAS患者,称为MELAS点突变 母系遗传,也可以散发 首先表现为运动不耐受,近端肢体无力伴轻度肌萎缩 偏瘫、偏盲、偏身感觉障碍等脑卒中样发作,伴发育异常(身材矮小, 智能低下,神经性耳聋) 发作性头痛、呕吐,癫痫样发作 血乳酸增高 CT或MRI显示分布在顶叶、颞叶及枕叶多发性脑梗塞,不按血管 分布,及基底节钙化、脑萎缩、脑室扩大 骨骼肌活检光镜下发现RRF,电镜下可见异常线粒体和晶格包涵体
多见于儿童 线粒体tRNA赖氨酸(Lys)基因8344位点的腺嘌呤 A被替换 为鸟嘌呤 G(A→G8344)引起 母系遗传 肌阵挛性癫痫 小脑性共济失调 有身材矮小、智能减退、听力丧失等发育异常 血乳酸增高 CT或MRI显示小脑脑干萎缩及白质脑病 脑电图表现为发作性慢波,发作性棘波或棘慢波综合等典 型的癫痫波型 骨骼肌活检光镜下发现RRF,电镜主要表现为异常线粒体数 量明显增多和线粒体内晶格包涵体
T2
T1
FLAIR
FLAIR
FLAIR
T2
影像学上与血管病鉴别:MELAS范围广泛,呈脑回样分布,不按血管支配 区分布,以颞顶枕叶受累为主,MRA大多正常,病情反复发作且病变有进 行性加重和对称性趋势。
肌阵挛性癫痫伴破碎样红纤维(myoclonus epilepsy with ragged-redfiber, MERRF)
主要临床症状
进行性肌病症状,以肢带和面肩肌肉为主,表现为肌无力 和体力活动时耐受力下降 颅神经和其他周围神经损害:慢性进行性眼外肌麻痹,视 网膜变性,感音性耳聋,周围神经病等 血管性头痛,卒中样发作 中枢神经系统病变:发育迟滞,智力低下,癫痫发作,共 济失调 代谢异常:乳酸代谢异常 内脏病变:糖尿病,甲低,心脏病,胃肠病变(假性肠梗 阻)
流行病学
Chinnery 调查发现在线粒体疾病中CPEO and KSS (19%) and MELAS syndrome (15%) 学龄前儿童(<6岁)线粒体脑肌病发病率是1/11000,16岁以 下儿童点患病率是1/21000 小儿发病的中位生存时间是12岁,多在发病后1-2年死亡 近十年国内外报道200例左右
Gomori Trichrome染色下 见RRF
电镜下见线粒体堆积,其 中部分线粒体为空化的线 粒体,部分线粒体出现嗜 锇小体
线粒体脑肌病伴乳酸中毒和卒中样发作(mitochodrial encephalomyopathy with lactic acidosis and stroke-like episodes, MELAS
+
― ― ― ―
+
― ― ― ― ― ―
+ + + + +
―
+ +
―
―
卒中样发作
发作性头痛,癫痫 肌力低下 智能低下 身材矮小 感音性耳聋 高乳酸血症 RRF 基因异常
―
―
+
+ + + + + + + tRNA点突 变
+ + + + + + 缺失
+ + + + + +
主要诊断标准
1符合线粒体脑肌病个综合征的临床表现 2肌活检RRF>2% 3有以下一种或多种呼吸链酶活性受抑制的表现: ①生化或极谱描记的呼吸链复合物活性<20%; ②50 岁以下肌活检细胞色素氧化酶COX(-)肌 纤维>2%;③50 岁以上肌活检C O X (- )肌纤 维>5 % 。 4发现相关的nDNA 或mtDNA 异常。
线粒体脑肌病
概念
线粒体脑肌病(mitochondrial encephalomyopathy)
是指由线粒体基因(mtDNA)或细胞核基因(nDNA)发生缺失或 点突导致的线粒体结构和功能异常,引起机体能量代谢障碍
发病机制
线粒体是提供细胞代 谢所需能量的主要细 胞器,糖、脂、氨基酸 在线粒体被氧化,释 放能量,转变为AT P 受两种基因控制: mtDNA和nDNA 线粒体功能障碍导致 ATP不足,不能满 足器官活动需要,产 生一系列症状
临床分类
骨骼肌和中枢是需要能量最多的组织,最易 受到影响 临床上以骨骼肌受损为主的称为线粒体肌 病 若同时侵犯中枢神经系统称为线粒体脑肌 病
不同部位损害的不同组合构成以下综合征:
1.线粒体脑肌病伴乳酸中毒和卒中样发作(mitochodrial encephalomyopathy with lactic acidosis and stroke-like episodes, MELAS)
CPEO 在临床上与眼肌型重症肌无力非常相似,容易误 诊为重症肌无力,因此,对于症状波动不明显、缓慢进展、 治疗效果欠佳的眼肌型重症肌无力,应考虑到C P E O 的 可能
CPEO(KSS) 家族史 外眼肌麻痹 视网膜色素变性 心传导障碍 脑脊液蛋白高 小脑共济失调 肌阵挛 ±
MERRF
MELAS
诊断标准
明确线粒体脑肌病 2 个主要诊断指标 1 个主要诊断指标+2 个次要诊断指标 可能线粒体脑肌病 1个主要诊断指标+1个次要诊断指标 3个次要诊断指标
治疗
仍无确切的有效治疗 改善能量代谢:大剂量ATP和辅酶A,辅 酶Q10,维生素C,以及B族维生素等 对症支持:肌无力或偏瘫的患者,物理治疗 显得格外重要 ,但是体育锻炼应适度防止 酸中毒;纠正心律失常和心力衰竭 ;抗癫 痫 基因治疗:尚在探索阶段
2.肌阵挛性癫痫伴破碎样红纤维(myoclonus epilepsy with ragged-red-fiber, MERRF) 3.慢性进行性眼外肌瘫痪(progressive external ophthalmoplegia, CPEO)和KSS综合征(Kearn-Sayre syndrome)
次要诊断标准
1完全的线粒体脑肌病的临床表现。 2至少以下一种提示肌肉中线粒体异常的表现:①30~50 岁 肌活检RRF 在 1%~2%;②30 岁以下肌活检出现 R R F; ③电镜下见广泛线粒体异常。 3至少一种呼吸链功能受抑制的表现:①生化或极谱描记的 呼吸链复合物活性在20%~30%;②用免疫方法证实呼吸 链复合物表达减少。 4发现可能相关的mtDNA 异常。 5一种或多种氧化磷酸化受损的表现:①脑脊液或血中乳酸、 丙酮酸和(或)丙氨酸增高;②若疑为KSS,脑脊液蛋白质 增高;③PET 或31P-MRS 证实肌肉或脑代谢降低;④最 大氧分压、平均氧分压或乳酸阈值降低
MERRF患者头MRI轴位相见小脑、脑干萎缩
慢性进行性眼外肌瘫痪(progressive external ophthalmoplegia, CPEO)与 KSS综合征(Kearn-Sayre syndrome)
CPEO: 多在20岁前发病, mtDNA缺失,包括散发、母系、显性、隐性遗传四 种亚型 上睑下垂,眼球活动受限,可伴四肢近端肌无力 (容易与重症肌无力混淆) KSS:一般认为CPEO是一种不完全的KSS 进行性眼外肌瘫痪 视网膜色素变性 心脏传导阻滞 其他神经系统异常,包括智力低下,神经性耳聋,小脑共济失调,周围 神经损害 脑脊液蛋白异常高 CT见基底节钙化,MRI表现为脑萎缩和双侧皮质下损害 骨骼肌活检光镜下发现RRF,电镜下可见异常线粒体和晶格包涵体 一般在30-40岁死亡