分子靶向治疗药物
分子靶向药物概念

分子靶向药物概念近年来,分子靶向药物成为医学领域的热门研究方向,被广泛应用于癌症等疾病的治疗。
分子靶向药物是一类可以直接干预特定分子、基因或信号通路的药物,具有高效、低毒副作用等优势。
本文将介绍分子靶向药物的概念、作用机制以及临床应用情况。
一、分子靶向药物的概念分子靶向药物是指能够靶向特定生物分子(如蛋白质、DNA、RNA 等)进行干预治疗的药物。
与传统化学药物相比,分子靶向药物具有更高的选择性,能够在目标分子上发挥更为显著的作用,减轻对正常细胞的伤害。
它们通常是由小分子化合物、抗体、核酸等构成,通过与特定的分子靶点结合,抑制某种生物过程或促进另一种生物过程的发生。
二、分子靶向药物的作用机制分子靶向药物的作用机制多种多样,根据药物的性质和分子靶点的差异,分子靶向药物可以实现以下几种作用机制:1. 抑制靶点活性:某些分子靶向药物可以与靶点结合,阻断其活性。
例如,基于表皮生长因子受体(EGFR)的靶向药物可以结合EGFR,抑制其信号传导,从而抑制肿瘤细胞的生长和分裂。
2. 诱导细胞凋亡:某些分子靶向药物可以通过与细胞的生存信号通路干扰,诱导肿瘤细胞自身程序性死亡,即细胞凋亡。
这种作用机制能够有效地限制肿瘤细胞的生长和扩散。
3. 阻断血供:一些分子靶向药物可以通过干扰肿瘤的血管生成,阻断其血供,从而导致肿瘤细胞因缺氧而死亡。
这种作用机制被广泛应用于肿瘤治疗中,被称为抗血管生成治疗。
4. 免疫激活:一部分分子靶向药物可以通过激活免疫系统,增强机体对肿瘤的免疫应答。
这些药物可以增加肿瘤细胞被免疫细胞攻击的可能性,提高治疗效果。
三、分子靶向药物的临床应用情况目前,分子靶向药物已经广泛应用于临床治疗中,取得了显著的疗效。
其中,最典型的应用领域就是癌症治疗。
许多分子靶向药物已经成功用于多种癌症的治疗,如乳腺癌、结直肠癌、肺癌等。
这些药物可以针对癌症细胞中的特异性分子靶点,干预其生长和扩散,达到治疗的目的。
此外,分子靶向药物还被应用于其他一些疾病的治疗中,如类风湿性关节炎、糖尿病等。
直肠癌的分子靶向治疗药物(口服) - 易瑞沙 IRESSA (别名 吉非替尼Gefitinib或Geftinat)(主要针对肺癌

Geftinat原产地英文商品名:Geftinat原产地英文药品名:吉非替尼( Gefitinib )中文参考商品译名:易瑞沙( IRESSA ) 艾瑞沙份子结构名:包装规格及销售价:0.25g/片*30片/瓶计价单位:瓶产地国家:印度生产厂家:Natco Pharma Ltd适应症:局部晚期或转移性非小细胞肺癌扩大适应症:其他实体癌易瑞沙处方资料易瑞沙薄膜衣片Iressa成分:吉非替尼Gefitinib包装/剂型:薄膜衣片0.25g x 30 片性状吉非替尼的化学名为:N-(3-氯-4-氟苯基)-7-甲氧基-6-(3-吗啉丙氧基)喹唑啉-4-胺,分子式为:C22H24ClFN4O3,分子量为:446.90。
本药为褐色圆形薄膜衣片;一面印有"IRESSA 250"。
药理作用吉非替尼是一种选择性表皮生长因子受体(EGFR)酪氨酸激酶抑制剂,该酶通常表达于上皮来源的实体瘤。
吉非替尼广泛抑制异种移植于裸鼠的人肿瘤细胞的生长,抑制其血管生成。
在体外,可增加人肿瘤细胞衍生系的凋亡,并抑制血管生成因子的侵入和分泌。
在动物试验或体外研究中已证实,吉非替尼可提高化疗、放疗及激素治疗的抗肿瘤活性。
临床研究两项大型的II期临床研究评估了本品单药治疗局部晚期或转移性非小细胞肺癌(NSCLC)的有效性和安全性。
患者的WHO体力状况评分为0-2,并且必须为既往化疗失败者:IDEAL1(研究0016),既往接受了1或2个化疗方案,并且至少有一个包括铂类治疗(中位年龄为59.6岁[28-85岁] ;n=209)。
IDEAL2(研究0039),既往接受了2个或以上化疗方案,该化疗方案包括同时或先后接受了铂类和多西紫杉醇的治疗(中位年龄为61岁[30-84岁];n=216)。
两个研究设计相似,均为双盲、平行组、多中心,评估了两个吉非替尼口服剂量:250 mg/天和500 mg/天。
患者被随机分配在这两个剂量组。
小分子靶向治疗药物简介
化学抗肿瘤药物经过半个多世纪的发展,已经进入靶向治疗药物时代。
小分子靶向药物在临床上的应用日益增多,在一些肿瘤类别中已经进入一线用药地位,比如肾癌、慢粒白、多发性骨髓瘤等。
本文对小分子靶向治疗药物做一综述。
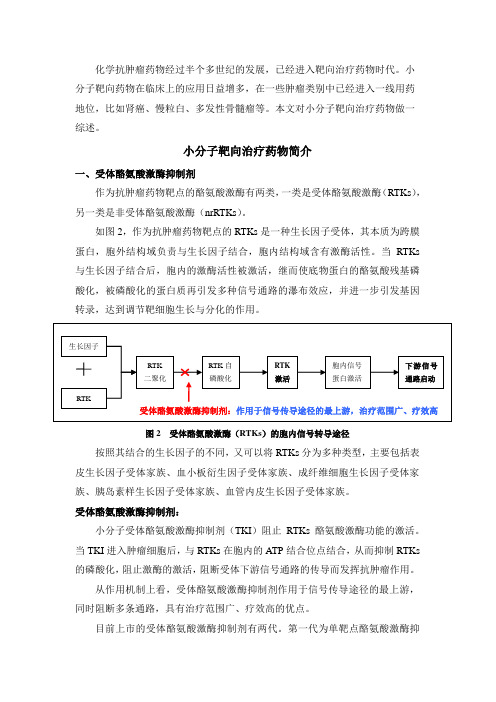
小分子靶向治疗药物简介一、受体酪氨酸激酶抑制剂作为抗肿瘤药物靶点的酪氨酸激酶有两类,一类是受体酪氨酸激酶(RTKs),另一类是非受体酪氨酸激酶(nrRTKs)。
如图2,作为抗肿瘤药物靶点的RTKs是一种生长因子受体,其本质为跨膜蛋白,胞外结构域负责与生长因子结合,胞内结构域含有激酶活性。
当RTKs 与生长因子结合后,胞内的激酶活性被激活,继而使底物蛋白的酪氨酸残基磷酸化,被磷酸化的蛋白质再引发多种信号通路的瀑布效应,并进一步引发基因转录,达到调节靶细胞生长与分化的作用。
图2 受体酪氨酸激酶(RTKs)的胞内信号转导途径按照其结合的生长因子的不同,又可以将RTKs分为多种类型,主要包括表皮生长因子受体家族、血小板衍生因子受体家族、成纤维细胞生长因子受体家族、胰岛素样生长因子受体家族、血管内皮生长因子受体家族。
受体酪氨酸激酶抑制剂:小分子受体酪氨酸激酶抑制剂(TKI)阻止RTKs酪氨酸激酶功能的激活。
当TKI进入肿瘤细胞后,与RTKs在胞内的ATP结合位点结合,从而抑制RTKs 的磷酸化,阻止激酶的激活,阻断受体下游信号通路的传导而发挥抗肿瘤作用。
从作用机制上看,受体酪氨酸激酶抑制剂作用于信号传导途径的最上游,同时阻断多条通路,具有治疗范围广、疗效高的优点。
目前上市的受体酪氨酸激酶抑制剂有两代。
第一代为单靶点酪氨酸激酶抑制剂,如吉非替尼、厄洛替尼。
表已上市的酪氨酸激酶抑制剂注:EGFR:表皮生长因子受体,属HER家族;VEGFR:血管内皮生长因子;PDGFR:血小板衍生因子;HER2:HER家族的一种受体;Abl-Bcr:一种非受体酪氨酸激酶;Raf:酪氨酸激酶的下游信号通路中的一种蛋白;Flt-3:Src:一种非受体酪氨酸激酶;c-kit:Ret:胶质细胞源性神经营养因子的受体吉非替尼为EGFR酪氨酸激酶抑制剂,主要用于非小细胞肺癌,对酪氨酸激酶基因编码区突变型肿瘤的有效率高达80%以上。
常见分子靶向药物治疗幻灯片全文

偶联物的种类
化疗药物 放射性核素 毒素
➢ 单克隆抗体及其偶联技术
以单抗介导的靶向性抗肿瘤药物正成为 肿瘤生物治疗产业化开发的热点。1997 年美国上市的利妥昔单抗(rituximab) 为重组嵌合抗CD20单克隆抗体,用于治 疗淋巴瘤,标志着单抗已进入临床应用 阶段。
单克隆抗体的性质
SGN31 •抗CD4单抗 Zanolimumab
各种靶向治疗药物在中国的上市时间
希罗达
格列卫
Xeloda
Glivec
美罗华
赫赛汀
MabThera
Herceptin
特罗凯
易瑞沙 爱必妥 Tarceva Iressa Erbitux
2000 2001 2002 2003 2004 2005 2006 2007
Avastin 恩度 Endostatin
TARGETS AND INHIBITORS
Targeting Dysregulated Pathways With Novel Agents
Anti-HER1 /2 MAbs
Farnesyl-transferase inhibitors
Tumor-activated chemotherapy
Ras signaling
Vatalanib Sunitinib
Phase III
VEGF TRAP
Cetuximab
HKI-272
Tarceva Gefitinib
Imatinib
Approved Vandetanib
Avastin
Motesanib
Sorafenib
PF-3512676
Talabostat
肿瘤靶向治疗经典药物

肿瘤靶向治疗经典药物
EEGFR(表皮生长因子)抑制剂:
(1)单克隆抗体:Erbitux(爱必妥。
西妥昔单抗,IMC-C225)
(2)作用于TPK系统的小分子靶向药物:
①IreSSa(gefitinib,ZD1839)(易瑞沙,吉非替尼片)可抑制EGFR酪氨酸激酶;是肺癌生物靶点治疗中较为成熟的药物,公认适用于复发、晚期NSCLC二线、三线治疗;IrSSSa治疗临床观察:女性,腺癌,年龄>68岁,不吸烟者疗效较好;
②TarCeVa(erlotinib,0SI-774)(特罗凯,盐酸厄洛替尼片)酪氨酸酶抑制剂。
认为主要对细支气管肺泡癌最有活力,而且在不吸烟中有较好RR;
2、其他酪氨酸激酶抑制剂:
①GIiVeC(格列卫)针对BCR-ABL基因靶目标治疗慢性粒细胞白血病,针对C-Kit基因靶目标从而治疗GlST(胃肠道间质瘤)。
通过与ATP竞争性结合酪氨酸激酶催化部位的核甘酸结合位点,使得激酶不能发挥催化活性,底物的酪氨酸残基不能被磷酸化,使其不能与下游的效应分子进一步作用,导致细胞增殖受抑,诱导细胞凋亡。
②SOrafenib(多激酶抑制剂)索拉非尼
③SUtent(多个受体TK抑制剂)舒尼替尼
3、其他单克隆抗体:
①HerCePtin:赫赛汀,抗HER-2受体单抗;
②Mabthera(美罗华,rituximab利妥昔单抗)抗CD20受体单抗。
4、血管生长抑制剂:AVaStin(贝伐单抗制抗VEGF单抗。
沙利度胺(反应停)。
肿瘤分子靶向药物简介历史及上市药物

肿瘤的靶向药物选择——国内外已经上市的分子靶向(MTT)药物...一、靶向药物(targeted medicine)简介靶向药物是目前最先进的用于医治癌症的药物,是随着今世分子生物学、细胞生物学的发展产生的高科技药物。
靶向药物与常规化疗药物最大的不同在于其作用机理:常规化疗药物通过对细胞的迫害发挥作用,由于不能准确识别肿瘤细胞,因此在杀灭肿瘤细胞的同时也会殃及正常细胞,所以产生了较大的毒副作用。
而靶向药物是针对肿瘤基因开发的,它能够识别肿瘤细胞上由肿瘤细胞特有的基因所决定的特征性位点,通过与之结合(或类似的其他机制),阻断肿瘤细胞内控制细胞生长、增殖的信号传导通路,从而杀灭肿瘤细胞、阻止其增殖。
由于这样的特点,靶向药物不仅效果好,而且副作用要比常规的化疗方式小得多。
靶向药物可以分为以下几类:(一)小分子药物小分子药物一般是信号传导抑制剂,它能够特异性地阻断肿瘤生长、增殖进程中所必需的信号传导通路,从而达到医治的目的。
例如诺华制药生产的格列卫(Gleevec,通用名Imitinib)、阿斯利康生产的易瑞沙(Iressa,通用名Gefitinib)均属此类;(二)细胞凋亡诱导药物通过特异性地诱导肿瘤细胞凋亡,达到医治的目的。
如美国千年制药公司生产的Velcade (通用名bortezomib)、Genta公司生产的Genasense(oblimersen);(三)单克隆抗体例如赫塞汀(Herceptin,通用名Trastuzumab),用于医治HER2基因阳性(过量表达)的乳腺癌。
这种药物是通过抗原抗体的特异性结合来识别肿瘤细胞的。
除上述列举的已经进入临床利用的靶向药物外,另外还有多种靶向药物正在开发中。
二、肿瘤的靶向药物上市历史回顾:■白血病费城染色体开启靶向医治之门早在1960年,美国费城的研究者发现慢性髓性白血病(CML)患者中存在一个染色体异样。
数年后,研究者发现这是9号和22号染色体长臂易位的结果。
分子靶向药物分类

分子靶向药物分类很多人都听说过靶向药物,但是分子靶向药你又知道有哪些吗,分子靶向药物分类是什么?下面是店铺为你整理的分子靶向药物分类的相关内容,希望对你有用!分子靶向药物分类一替伊莫单抗(泽娃灵) Ibritumomab tuixetan(Zevalin)承认时间:2008年1月。
用途:恶性淋巴瘤。
作用:搜索带有CD20标志物的蛋白细胞(B细胞淋巴瘤),阻止其生长,消灭肿瘤细胞。
分子靶向药物分类二伊马替尼(格列卫) Imatinib(Glivec)由日本Novartis Pharma开发的分子靶向药。
用途:以针对治疗慢性髓系白血病和消化道间质瘤被承认使用。
分子靶向药物分类三依维莫司 Everolimus用途:手术无法切除,或转移的肾细胞肿瘤。
作用:抑制mTOR信号通路,防止肿瘤细胞分裂生长。
分子靶向药物分类四厄洛替尼 Erlotinib用途:不能切除的复发/恶化的非小细胞肺癌。
作用:阻碍酪氨酸激酶的活动,抑制肿瘤细胞的增长。
分子靶向药物分类五吉非替尼(艾瑞沙) Gefitinib在世界上,首先被日本承认使用的分子靶向药物。
非常缺乏对此种药物副作用的认识,依然被投入使用,导致患者发生间质性肺炎,相继死亡的问题。
分子靶向药物分类六吉姆单抗奥佐米星 Gemtuzumab ozogamicin利用基因替换产生的单克隆抗体,和抗生物质细胞毒药物刺孢霉素相结合形成的抗癌药物。
用途:复发或较难治疗,CD33抗原阳性的急性髓系白血病。
分子靶向药物分类七索坦(舒尼替尼) Sutent用途:消化道间质瘤(GIST)和肾癌。
作用:以VEGF血管内皮生长因子(VEGF关系着肿瘤血管的生成)和PDGF成长因子受体(PDGF关系着肿瘤细胞的生长)等目标为靶点。
分子靶向药物分类八西妥昔单抗(爱必妥) Cetuximab承认时间2008年7月。
用途:和贝伐单抗(阿瓦斯汀)相同。
作用:针对无法实施根治手术的恶化/复发的结肠癌,所使用的分子靶向药。
常见的靶向治疗药物

⒋ Bcr-Abl酪氨酸激酶抑制剂:
甲磺酸伊马替尼〔Imatinib,mesylate,STI571> : 商品名:格列卫〔美国称Gleevec,欧洲称Glivec 生产商:瑞士诺华〔Novartis
是一种2-苯胺嘧啶的衍生物,是与ATP相关的选择性 Bcr-Abl酪氨酸激酶选择性抑制剂,能够与Abl激酶上的 ATP结合位点相互作用,从而阻止下游蛋白的磷酸化,用于 治疗慢性粒细胞白血病〔CML,单药有效率98%.
主要分子靶向药物的分类
1. 小分子表皮生长因子受体〔EGFR酪氨酸激酶抑制剂:吉非替尼 〔Gefitinib、埃罗替尼〔Erlotinib等.
2. 抗EGFR的单抗:西妥昔单抗〔Cetuximab、帕尼单抗〔Panitumumab、 Matuzumab<EMD 72000>.
3. 抗Her-2的单抗:曲妥珠单抗〔Trastuzumab. 4. Bcr-Abl酪氨酸激酶抑制剂:伊马替尼〔Imatinib、尼洛替尼〔Nilotinib、
⒍ 抗CD20的单抗:
利妥昔单抗Rituximab>商品名:美罗华〔Mabthera 生产商:瑞士豪夫迈·罗氏〔F.Hoffmann-La Roche
1997年11月26日上市,是第1个应用于临床肿瘤的靶向治疗药物.
由小鼠可变区和人恒定区结合的单抗.与CD20抗原特异性结合,诱导 抗体依赖性细胞介导的细胞毒作用〔ADCC和补体介导的溶细胞作用杀伤 靶细胞,从而抑制B细胞增殖,诱导B细胞凋亡,提高肿瘤细胞对化疗的敏 感性.
1、表皮生长因子受体〔EGFR 小分子酪氨酸激酶抑制剂
吉非替尼〔Gefitinib,ZD 1839 商品名:易瑞沙〔Irressa 生产商:英国阿斯利康〔AstraZeneca
肿瘤分子靶向治疗

80
Overall survival (%) R-CHOP 58%
60
40
CHOP 45%
20
0 0
p<0.007
1 2 3 4 5 6 7
Years
Feugier P, et al. J Clin Oncol 2005 23:4117–26
曲妥珠单抗(赫赛汀)
作用机制:抗HER-2受体的单克隆抗体。 适应症:HER-2过表达的乳腺癌
EGF Pathway
EGFR family
EGFR
HER2
HER3
HER4
Adapted from: Ciardiello F, et al. N Engl J Med. 2008;358:1160-1174.
EGF Pathway
Receptor specific ligands
EGF TGFα β-cellulin HB-EGF Epiregulin Amphiregulin NRGs β-cellulin HB-EGF
Slamon D et al. N Engl J Med 2001;344:783-92
曲妥珠单抗联合化疗疗效总结
H+AC AC H+P P H+CT CT (n=143) (n=138) (n=92 ) (n=96) (n=235) (n=234)
mTTP ORR OS
7.8* 56* 26.8
贝伐单抗(癌思停,Avastin)
作用机制:重组的人类单克隆IgG1抗体,通过抑制
人类血管内皮生长因子的生物学活性而起作用。 用法用量:推荐剂量为5mg/kg,每2周静脉输注1次。 在术后28天以后使用,且伤口完全愈合。 临床应用: 转移性结直肠癌(联合5Fu+/-) 转移性乳腺癌(联合紫杉醇) 晚期、非鳞、无脑转移的非小细胞肺癌 最严重的不良反应:胃肠穿孔/伤口并发症,出血, 高血压危象,肾病综合症,充血性心力衰竭。
肺癌分子靶向药物治疗的研究进展

肺癌分子靶向药物治疗的研究进展分子靶向治疗是指针对参与肿瘤发生、发展过程的细胞信号转导和其他生物学途径的治疗手段,具有高效和低不良反应的特点。
随着近年来肿瘤相关研究的不断进步,在恶性肿瘤的个体化治疗和靶向治疗方面取得了令人瞩目的进展。
本文主要针对肺癌的分子靶向治疗研究进展进行概括总结。
标签:肺癌;血管内皮生长因子受体;表皮生长因子受体;肿瘤干细胞;肿瘤抑制基因肺癌是当前发病率和死亡率最高的肿瘤之一,80%以上患者就诊时已处于晚期,失去手术机会。
目前,肿瘤化疗已经处于治疗瓶颈,毒副反应大,有效率低,5年生存率不足15%。
近年来发展起来的靶向治疗,具备高效、低副反应等特点,已成为目前肺癌治疗的研究热点。
其作用靶点包括细胞内信号转导通道中重要的蛋白质、酶、细胞表面的生长因子受体,而广义的分子靶点则包括参与肿瘤细胞分化、凋亡、迁移、浸润、淋巴结转移、全身转移等过程的从DNA到蛋白酶水平的任何亚细胞分子。
1 血管内皮生成因子(VEGF)VEGF是一种细胞因子,它能诱导内皮细胞增生、蛋白酶的表达、抗内皮细胞凋亡和细胞重组,最终形成毛细血管。
在病理血管生成方面,它还能增强血管的通透性,形成不成熟的血管网络。
血管上皮生长因子能够刺激血管内皮细胞的增生,在大多数人体肿瘤组织中,VEGF的表达大大高于其他正常组织[1]。
研究证实贝伐单抗以VEGF作为靶点,具有一定的抗肿瘤作用[2]。
VEGF家族包含6个生长因子(VEGF-A、VEGF-B、VEGF-C、VEGF-D、VEGF-E以及胎盘生长因子)和3个受体(VEGFR-1、VEGFR-2(KDR/FIk.1)和VEGFR-3)。
VEGF 的过度表达与肿瘤进展及不良预后相关。
目前针对VEGF途径的治疗包括抗VEGF单克隆抗体和VEGFR-TKI两大类。
1.1贝伐单抗(Bevacizumab)Bevacizumab即重组人抗VEGF单克隆抗体,可与VEGFR结合,阻断肿瘤血管的细胞信号转导,抑制肿瘤血管生长,抑制肿瘤细胞。
靶向治疗药物在肿瘤治疗中的研究进展

靶向治疗药物在肿瘤治疗中的研究进展摘要:肿瘤的分子靶向治疗药物是指设计出对应靶点的分子治疗药物,在细胞分子水平上,针对已经明确的致癌位点(该位点可以是肿瘤细胞内部的一个蛋白分子,也可以是一个基因片段),在无创或微创条件下以该位点为靶点, 通过精准定、靶向打击,以期能有效控制肿瘤的进展, 同时降低肿瘤周围正常组织细胞损伤为目标的新兴的肿瘤治疗方式。
该治疗方式的发展迅猛,成为近些年肿瘤治疗研究的热点方向,在肿瘤治疗中起到了不可取代的作用,具有很多突出的优势,如:针对性较强、毒副反应小、患者依从性强、便于实施等。
虽然肿瘤的分子靶向治疗带来了之前肿瘤治疗方式所不能比拟的效果,但其也存在自身的局限性,如:高昂的治疗费用、使用对象的局限性、长期用药的耐药性等。
本文就临床上几种常见的恶性肿瘤(肺癌、胃癌、大肠癌)的靶向治疗研究进展进行分析。
关键词:靶向治疗肿瘤治疗研究进展[中图分类号]R735.7 [文献标识码]A [文章编号]1439-3768-(2019)-1-WT 引言:靶向治疗,是目前热门的肿瘤治疗研究方向。
其通过前期的基因检测,筛选出适合使用该方法的患者,将针对目的基因而设计的分子靶向药物送入体内,药物会与致癌位点特异地相结合而对肿瘤进行打击,导致肿瘤细胞特异性死亡,却不会波及肿瘤周围的正常组织细胞,因此分子靶向治疗又被称为“生物导弹”。
靶点定位的准确程度在很大程度上影响着肿瘤靶向治疗的效果,因此前期的基因检测就尤为重要,同时在治疗过程中可靠的制导设备也是靶向治疗不可缺少的重要环节。
在靶向治疗前用计算机确定靶区,制定治疗计划,精确定向引导,实时监测,保证准确地杀死靶区局部的肿瘤细胞,最大限度地减少周围正常组织的损伤,以达到精准杀灭的目的。
1.靶向治疗在肺癌治疗过程中的研究随着其发病率和死亡率也逐年上升,肺癌已跃居我国恶性肿瘤的首位,预计到2025年,我国内肺癌患者将突破100万,成为世界第一肺癌大国。
常见分子靶向药物治疗课件

个体差异可能与患者的基因型、 年龄、性别、体能状态等因素有
关。
针对个体差异,医生需综合考虑 患者的具体情况,制定合适的治 疗方案,并在治疗过程中密切监
测和调整治疗方案。
05 分子靶向治疗的未来展望
新药研发与临床试验
新药研发
随着生物技术的不断发展,新的分子靶向药物不断涌现,为 肿瘤等重大疾病的治疗提供了更多选择。新药研发需要经过 实验室研究、临床前试验、临床试验等阶段,确保药物的安 全性和有效性。
分子靶向治疗的历史与发展
历史回顾
自20世纪90年代起,随着人类基 因组计划的实施,分子靶向治疗
逐渐进入人们的视野。
当前进展
目前已有多种分子靶向药物成功应 用于临床,针对多种癌症、神经性 疾病、心血管疾病等均有显著疗效。
未来展望
随着生物技术的不断发展,分子靶 向治疗将更加精准和个性化,有望 为更多疾病的治疗提供有效手段。
改善了患者的生活质量。
其他癌症
01
其他癌症如肾癌、前列腺癌、卵 巢癌等也开展了分子靶向治疗的 研究和应用。
02
针对不同癌症的特异性靶点,开 发出了多种分子靶向药物,为患 者提供了更多的治疗选择和希望 。
04 分子靶向治疗的疗效与副 作用
疗效评估指标
肿瘤大小变化
通过影像学检查评估肿瘤缩小或消失的情况,是直接反映治疗效果的 指标。
的血栓形成。
蛋白激酶抑制剂(TKI)
蛋白激酶是细胞信号转导过 程中的重要酶类,参与细胞 增殖、分化、凋亡等过程。 蛋白激酶抑制剂(TKI)通过 抑制特定蛋白激酶信号转导, 达到抑制肿瘤生长的目的。
常见TKI包括伊马替尼、舒尼 替尼等,主要用于慢性髓性 白血病、胃肠道间质瘤等的 治疗。
分子靶向药物治疗

分子靶向药物治疗关于《分子靶向药物治疗》,是我们特意为大家整理的,希望对大家有所帮助。
靶向治疗药物是对于于一些癌症末期的大家应用的,它是一种非常好的保守治疗方法,尤其是分子结构靶向治疗药物是如今更为优秀的一种,也是药力最好是的一种,可是这类药品是没有办法除根的,只有是在服食之后具有一个减轻的实际效果,并且一般全是需要长期性应用的药品,否则的话没法在短期内内改进的。
1. 小分子药物小分子药物一般是数据信号传输缓聚剂,它可以特异性地阻隔肿瘤生长发育、繁衍全过程中所必不可少的数据信号传输通道,进而做到医治的目地。
比如诺华制药生产的用以医治漫性粒细胞败血症和胃肠栽培基质瘤的伊马替尼(Gleevec,通用性名Imitinib)、以EGFR为靶标的用以医治非小细胞肺癌的阿斯利康生产的吉非替尼(Iressa,通用性名Gefitinib)和德国默克的厄洛替尼(Tarceva,通用性名Erlotinib)均属该类,并已进到临床医学运用。
英国制药厂生产的Velcade(通用性名bortezomib)是细胞坏死诱导剂,也归属于小分子药物。
2. 单克隆抗体比如用以医治HER2遗传基因呈阳性(过多表述)的乳腺癌的赫塞汀(Herceptin,通用性名Trastuzumab)、以EGFR为靶标的结肠癌和非小细胞肺癌医治药品爱必妥(Erbitux,通用性名Cetuximab)等。
这类药是根据抗原和抗体的特异性融合来鉴别肿瘤体细胞的。
从通用性名的后缀名上看来,单克隆抗体类靶向治疗药物以“-mab”为后缀名,而酪氨酸激酶类靶向治疗药物以“-nib”为后缀名。
除所述例举的早已进到临床医学应用的靶向治疗药物外,此外也有多种多样靶向治疗药物已经开发设计中。
靶向治疗药物与基本化疗药较大的不一样取决于其作用机理:基本化疗药根据对体细胞的危害充分发挥,因为不可以精确鉴别肿瘤体细胞,因而在消灭肿瘤体细胞的另外也会祸及一切正常体细胞,因此造成了很大的毒副作用。
最热门抗肿瘤靶点及小分子靶向药物全景报告

最热门抗肿瘤靶点及小分子靶向药物全景报告抗肿瘤靶点是指对肿瘤生长、转移等过程具有重要调控作用的蛋白分子或通路。
小分子靶向药物是一类能够专一靶向抗肿瘤靶点并抑制其活性的化学物质。
随着抗肿瘤研究的不断深入,越来越多的抗肿瘤靶点及小分子靶向药物被发现并应用于临床。
以下将介绍一些当前最热门的抗肿瘤靶点及小分子靶向药物:1.EGFR(表皮生长因子受体):EGFR是一种跨膜酪氨酸激酶受体,参与肿瘤细胞的生长和分化等过程。
一些小分子靶向药物如吉非替尼和厄洛替尼等通过抑制EGFR的酪氨酸激酶活性,抑制肿瘤细胞生长。
2.HER2(人表皮生长因子受体2):HER2是一种细胞表面受体,参与调节细胞增殖和存活等过程。
一些小分子靶向药物如曲妥珠单抗和拉普替尼等能够靶向结合HER2,抑制其信号传导,减少肿瘤细胞的增殖。
3.ALK(酪氨酸激酶受体):ALK是一种重排基因,其突变被发现与多种肿瘤的发生和发展相关。
小分子靶向药物如克唑替尼和艾尔莎替尼能够抑制ALK的活性,阻断肿瘤细胞的生长和转移。
4.BRAF(B型RAF激酶):BRAF是一种信号转导分子,突变导致了多种恶性黑色素瘤的发生。
例如,维米非尼和达替尼等小分子靶向药物能够抑制BRAF的活性,减少肿瘤细胞的增殖和转移。
5.PD-1(程序性死亡受体1)和PD-L1(程序性死亡配体1):PD-1和PD-L1参与抑制免疫系统对肿瘤的攻击,突变导致肿瘤逃避免疫监视。
一些免疫检查点抑制剂如伊普替尼和纳武利尼等能够靶向PD-1或PD-L1,恢复免疫系统的抗肿瘤活性。
除了上述靶点外,还有许多其他热门的抗肿瘤靶点及小分子靶向药物,如PI3K、FLT3、VEGFR等。
这些靶点及药物的发现和应用为肿瘤治疗提供了新的进展和希望。
需要注意的是,虽然靶向药物在抗肿瘤治疗中具有重要作用,但并非适用于所有患者。
个体化治疗是当前的研究热点,通过检测患者的肿瘤基因和蛋白表达水平来选择最合适的靶向药物,以提高治疗效果和减少不良反应。
肿瘤的分子靶向药物 简介

c-MYC c-JUN
新生血管形成
作用位点
x Cediranib x Bevacizumab x BMS-582664 x Sorafenib x Sunitinib x Thalidomide x TSU-68
Semela D, et al. J Hepatol 2004;41:864–80 Clauss M. Semin Thromb Hemost 2000;26:561–69
• 多靶点抑制剂
– 索拉非尼 (Sorafinib): VEGFR和PDGFR抑制剂,Raf酶抑制剂. – 舒尼替尼(Sunitinib )等。
药物简表
药品名称
商品名 研制公司
药理作用
适应症
备注
拉帕替尼 lapatinib
泰克泊 Tykerb
EGFR/HER2
GSK
表皮生长因子酪氨酸激酶
抑制剂
乳腺癌
曲妥珠单抗
赫赛汀
Transtuzumab Herceptin
Roche
HER2 的人源化单抗
乳腺癌
吉非替尼 gefitinib
埃罗替尼 erlotinib
抗的互补决定区
已在全球 120 多个国 家和地区获批,用于 结直肠癌、非小细胞 肺癌、乳腺癌、恶性 胶质瘤和肾细胞癌等
恶性肿瘤分子靶向治疗

胃肠间质瘤(GIST)
指主要发生于消化管道含有梭形细胞、非普通
型上皮样细胞或含有两种细胞的间叶细胞瘤。
特点:CD117阳性
CD34阳性
2
特异的表达c-kit
3
超过30%是恶性的(即转移性或浸润性)
4
对常规化疗和放疗抗拒
5
能手术切除的病人占很小一部分
STI571 produced response rates of 60% PR and 20% SD > 6 mos in metastatic GIST.
01
靶向治疗:改善症状,提高生活质量,延长寿命。 目标可能是长期带瘤存活。
02
Байду номын сангаас
影像学评价:通过18FDG-PET-CT、CT、MRI等检查方法评价疗效。
近几年微观评价疗效:检测癌变分子异常、细胞生长动力学、血管生长因子、肿瘤细胞标记物、基因的改变。应以延长肿瘤患者生存期和提高生活质量为金标准,不断调整和探索更合理的疗效评价体系。
抗血管内皮生长因子(VEGF) 单抗: 贝伐单抗Bevacizumab(Avastin)
Text
Text
01
靶向治疗药物的作用机制 及常见药物举例
吉非替尼(Iressa,Gefitinib,易瑞沙)
作用机制: 一种口服表皮生长因子受体-酪氨酸激酶(EGFR-TK)拮抗剂,是信号传导干预治疗药物(属小分子化合物)。
我科室曾经现在享受靶向治疗患者
分子靶向治疗药物的分类
分子靶向治疗药物的作用机制
分子靶向诊疗基本步骤
结语
01
单击添加标题
单击此处添加正文
02
单击添加标题
单击此处添加正文
胰腺癌的分子靶向药物治疗

胰腺癌的分子靶向药物治疗胰腺癌是一种病死率极高的恶性肿瘤,早期诊断困难,常常已经转移到体内其他部位才会被发现。
目前,传统的化疗和放疗虽然可以减轻患者的症状,但治疗效果并不理想,而且还带来了很多副作用。
因此,寻找一种高效、低毒副作用的分子靶向药物已成为当今治疗胰腺癌的热门研究领域之一。
一些研究显示,针对胰腺癌干细胞的分子靶向药物能够更好地发挥治疗的作用。
其中,针对肿瘤生长因子受体(EGFR)的分子靶向药物便是其中之一。
由于EGFR在胰岛素样生长因子-1(IGF-1)生长因子通路中起关键作用,因此靶向EGFR也能抑制胰腺癌细胞的生长。
目前,可用于靶向EGFR的药物主要有培美曲塞(Erbitux)、西妥昔单抗(Vectibix)等。
除此之外,还有针对VEGF/VEGFR信号通路和RAS/MAPK信号通路的分子靶向药物。
其中,VEGF通路是参与新生血管的形成,因此对于肿瘤生长起到了非常重要的作用。
目前,可用于靶向VEGF/VEGFR的药物主要有贝伐单抗(Avastin)等。
而RAS/MAPK通路是肿瘤增殖的一个主要信号通路,在常见的肿瘤中都有不同程度的激活。
因此,针对该通路的分子靶向药物也成为了治疗胰腺癌的热门研究领域之一。
可用于靶向RAS/MAPK通路的药物主要有曲妥珠单抗(Erivedge)等。
此外,针对肿瘤免疫调节的分子靶向药物也备受关注。
从前几年的科学研究来看,免疫治疗已经成为了治疗肿瘤的主要方法。
对于胰腺癌而言,因其免疫逃逸的特性,免疫治疗也成为了一种很受关注的治疗手段。
在肿瘤免疫细胞疗法方面,可用于靶向PD-1的药物主要有克唑替尼(Opdivo)等。
总体而言,靶向分子药物是目前治疗胰腺癌最具前景的一种治疗手段,而新的分子靶向药物的不断出现必将会使胰腺癌患者享受更好的治疗效果。
临床应用案例分子靶向药物在肺癌治疗中的疗效观察

临床应用案例分子靶向药物在肺癌治疗中的疗效观察近年来,肺癌作为一种常见的恶性肿瘤疾病,给全球各个国家和地区的卫生保健系统造成了巨大的负担。
在肺癌的治疗过程中,分子靶向药物作为一种新兴的治疗手段,已经取得了一些突破性的进展。
本文将通过几个临床应用案例,观察分子靶向药物在肺癌治疗中的疗效。
案例一:患者为一名52岁的女性,无烟煤矿工工作史。
患者因咳嗽、咳血和胸闷等症状就诊于医院。
肺部CT检查发现左肺下叶大量结节状病灶,胸腔积液明显。
经穿刺活检确认为非小细胞肺癌。
基因检测表明患者KRAS基因突变。
由于患者肺癌处于晚期且存在基因突变,传统的放化疗方法效果较差。
因此,决定在治疗方案中加入分子靶向药物。
经过2个疗程的治疗,患者的症状明显缓解,胸腔积液减少,肺部CT检查显示肿瘤有缩小趋势,治疗疗效良好。
案例二:患者为一名60岁的男性,长期吸烟史。
患者因咳嗽、胸闷和体重下降等症状就诊于医院。
PET-CT检查发现双侧肺部多发结节,肺门淋巴结肿大。
病理活检结果为腺癌。
EGFR基因检测显示EGFR敏感突变。
根据检测结果,患者开始接受分子靶向药物治疗。
经过3个疗程的治疗,患者的症状明显减轻,体重恢复,PET-CT检查显示肿瘤有明显缩小的趋势。
治疗过程中,患者的血液生化指标也逐渐恢复正常水平。
这一例表明分子靶向药物在EGFR敏感突变的肺腺癌治疗中显示出了良好的疗效。
案例三:患者为一名68岁的女性,有吸烟史。
患者因咳嗽、胸痛和呼吸困难等症状就诊于医院。
CT检查发现右上叶肺部肿块,PET-CT显示FDG代谢稍微升高。
病理检查结果为肺鳞癌。
EGFR、ALK和ROS1等常见基因突变检测均为阴性。
鉴于患者基因检测阴性,传统的放化疗被认为是首选治疗方案。
经过放化疗3个疗程后,患者的症状得到了一定程度的缓解,但肿瘤没有显着缩小的趋势。
因此,在治疗方案中引入一种多靶点抗血管生成药物。
治疗过程中,患者的症状持续缓解,CT检查显示肿瘤有轻微缩小的趋势。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子靶向治疗及其药物1971年Suden Follman发现在鼠的角膜上种植1个肿瘤10 d后周围有血管长入,继而肿瘤迅速长大.如将这些血管无论用机械或化学方法阻断,使肿瘤得不到血液供应,肿瘤生长停顿,肿瘤即萎缩。
这一现象给人们所提供了2个信息:一是肿瘤生长发展必须依赖有良好的血液供应;二是肿瘤在原本没有血管的角膜上周围出现大量新生血管,表明肿瘤必然会产生一种促使血管形成的物质,称为血管生成因子(vascular endothelial growth factor,VEGF)。
因而提出了“饥饿疗法”,即让肿瘤得不到血供营养而死亡,并寻找能够抑制对抗阻碍血管生长的物质,至1994年得到第1个具抑制血管生成的物质称为“Angiostatin”。
在2004年美国FDA官员Mc Clellan提出分子靶向治疗(Targeted cancer therapy)已成为结直肠癌除外科手术、放射和化疗的第四种治疗方法。
分子靶向治疗的药物靶点主要包括蛋白酪氨酸激酶、芳香胺酶、拓扑异构酶、细胞周期和凋亡调节因子、法尼基转移酶(FTase)等。
有许多分子靶向药物在研究,其中贝伐单抗、西妥昔单抗和帕尼单抗是得到批准应用于治疗进展性和/或转移性结直肠癌的三个药物。
一、蛋白酪氨酸激酶抑制剂(tyrosine kinase,TKI)(一)蛋白酪氨酸激酶(Protein tyrosinase)蛋白激酶是目前已知的最大的蛋白超家族,主要包括丝氨酸/苏氨酸激酶和酪氨酸激酶,其中的酪氨酸激酶主要与信号通路的转导有关,能催化三磷酸腺苷上的磷酸基转移到许多重要蛋白质的酪氨酸残基上,使其发生磷酸化。
蛋白酪氨酸激酶在细胞内的信号转导通路中占据十分重要的地位,是细胞信号转导机制的中心,调节着细胞体内生长、分化、死亡等一系列生理、生化过程。
蛋白酪氨酸激酶由于突变或重排,可引起信号转导过程障碍或出现异常,导致细胞生长、分化、代谢和生物学行为异常,引发肿瘤。
研究表明,近80%的致癌基因都含有酪氨酸激酶编码。
抑制酪氨酸激酶受体可以有效控制下游信号的磷酸化,从而抑制肿瘤细胞的生长。
酪氨酸激酶受体分为表皮生长因子受体(Epidermal growth factor receptor,EGFR)、血管内皮生长因子受体(Vascular endothelial growth factor receptor,VEGFR)、血小板源生长因子受体(Platelet derived growth factor receptor,PDGFR)等。
TKI的抗肿瘤作用机制可通过以下途径实现:抑制肿瘤细胞的损伤修复、使细胞分裂阻滞在G0期、诱导和维持细胞凋亡、抗新生血管形成等。
此外,TKI还可通过下调肿瘤细胞的血管内皮生长因子,抑制EGFR对肿瘤血管内皮细胞的信号传导以及EGFR和VEGFR两种信号传导通路的“交叉对话”,为临床同时抑制这两种传导通路提供合理的依据。
针对各种受体的酪氨酸激酶抑制剂目前已开发上市的主要为表皮生长因子受体酪氨酸激酶抑制剂( EGFR-TK )、血管内皮细胞生长因子受体酪氨酸激酶抑制剂( VEGFR -TK )和血小板源生长因子受体酪氨酸激酶抑制剂(PDGFR-TK)等。
基于多靶点的酪氨酸激酶抑制剂目前已成为研究重点,包括舒尼替尼和索拉芬尼在内的几个上市新药均获得了良好的临床评价结果。
多数酪氨酸激酶抑制剂通过肝酶CYP3A4代谢,与CYP3A4抑制剂(酮康唑、伊曲康唑)同用,可使伊马替尼、埃罗替尼、吉非替尼的药-时曲线下面积增加,与CYP3A4诱导剂(利福平)同用可使上述药物的药-时曲线下面积降低。
(二)以蛋白酪氨酸激酶为靶点的靶向药物1.表皮生长因子受体酪氨酸激酶抑制剂( EGFR-TK )1.1表皮生长因子受体(EGFR)EGFR是一种跨膜生长因子受体,跨膜糖蛋白,由原癌基因C-erb B1编码。
人EGF受体(human epidermal growth factor receptor,HER)家族由4个结构相近的受体酪氨酸激酶蛋白成员组成,分别称ErbB-1(EGFR或Her-1)、ErbB-2(Her-2)、ErbB-3(Her-3)、ErbB-4(Her-4),是具有配体依赖性的跨膜糖蛋白家族,均属于I型酪氨酸激酶受体基因家族。
除造血干细胞外,存在于大多数细胞中,在多种肿瘤中都有过表达,如CRC(结直肠癌)、乳腺癌、胰腺癌、前列腺癌和非小细胞肺癌等,其中CRC的表达率为25%~77%。
其蛋白质结构可以分为3个区域,即细胞表面的配体结合区、跨膜区和细胞内的酪氨酸激酶区。
EGFR在缺乏特异性配体时以单体形式存在,但与配体结合后发生聚合。
表皮生长因子(epidermel growthfactor,EGF)和转化生长因子α(transforming growth factor α,TGFα)是EGFR 最重要的内源性配体。
当这些配体与EGFR细胞外区结合后,在胞外区域形成受体同源二聚体或异源二聚体,通过跨膜区促进胞内区域二聚化,激活胞内区域酪氨酸激酶活性,通过JAK-STAT和PI3K-Akt等转导途径将信号传递到核内,从而促进细胞增殖,血管生成、转移和抑制细胞凋亡。
在配体诱导下激活后,EGFR能连接很多参与信号转导瀑布反应的细胞内蛋白质,通过不同的受体类型和磷酸化结合的位点,使不同的信号蛋白被激活(图1)。
这个过程使不同的细胞外信号被传导到细胞核中。
EGFR的酪氨酸激酶活性代表了它的有效结构,并且是信号传导的基础,细胞能通过生长因子自体分泌方式发生自身恶变,而为了获得恶性转变,需要活性配体的存在和EGFR的高水平表达。
除了促进增殖外,活化的EGFR 对肿瘤发展起重要作用的生物学反应还包括对细胞运动、细胞黏附、浸润、细胞生存和血管生成等的影响。
EGFR与恶性细胞的生长有关,激活EGFR可以促进肿瘤的生长和进展。
因此,肿瘤患者预后差可能与肿瘤细胞中EGFR表达水平高于正常组织有关。
大量研究报道表明:在很多肿瘤中都存在着EGFR表达或过度表达和戚基因的扩增,这些疾病包括:结直肠癌(CRC)、头颈部鳞癌、乳腺癌、卵巢癌、宫颈癌、食道癌、胰腺癌、膀胱癌、前列腺癌、非小细胞肺癌。
在各种实体肿瘤中EGFR表达率最高的是头颈部肿瘤,达95%~100%。
结直肠癌则为第2位,表达率高达72%~89%。
如果肿瘤表达EGFR,或者合并有EGFR配体表达或其他生长因子受体的表达,则肿瘤恶性度高,侵袭性更强,而且EGFR表达水平的高低与预后相关。
从1984年起,EGFR就作为肿瘤治疗重要的靶分子被重点研究。
目前用于CRC的EGFR 靶向药物主要有两类:一类是单克隆抗体(mortoclonal antibodyies,Mabs),如西妥昔单抗(Cetuximab)、帕尼单抗(Panitumumab)等,主要作用在EGFR的胞外区,通过竞争性抑制配体与EGFR的结合,使受体失去活性;另一类则是小分子的化合物,如吉非替尼(Gefitinib)、埃罗替尼(Erlotinib)等,能进入细胞内,直接作用于EGFR的胞内区,干扰ATP结合,抑制酪氨酸激酶的活性(图2)。
虽然两类药物的作用部位不同,但通过阻止配体介导的受体及下游信号通路的激活,最终产生相似的效果,即阻滞细胞在G0期、促进凋亡、抑制新生血管形成、抑制侵袭和转移,从而起到治疗作用。
小分子抑制剂与单克隆抗体相比,其优势在于分子量小,可口服给药,并且易于化学合成,生产成本比较低廉,缺点在于其半衰期只有几小时,需每天服用:而单抗药物也有其自身优势,例如对肿瘤的靶向性强、半衰期长等,单抗药物的半衰期一般达数天至数周,一般每1~4周给药1次。
1.2 EGFR靶向抑制剂1.2.1 抗EGFR单克隆抗体1.2.1.1西妥昔单抗(Cetuximab,C225,Erbitux,爱比妥)西妥昔单抗是一种经过基因工程修饰过的具有人和鼠双重组件的鼠源抗体,是一种以人EGFR作为靶点的IgG1型单抗,西妥昔单抗与人EGFR胞外区特异性结合,其亲和力高于内源性配体,从而竞争性地与EGFR结合,发挥抑制EGFR的作用,抑制与受体相关的激酶的磷酸化和活化,从而抑制细胞周期进程、诱导凋亡、减少基质金属蛋白酶和血管内皮生长因子的产生,降低浸润和转移扩散。
同时西妥昔单抗可下调凋亡抑制基因Bcl-2水平,上调促凋亡基因Bax水平,引起肿瘤细胞的凋亡。
西妥昔单抗和其他靶向EGFR的药物一般并不认为是抗血管形成的药物,但是,很多研究的结果却认为抗血管形成是其抗肿瘤的机制之一。
因为,体外研究表明,西妥昔单抗能剂量依赖性地抑制VEGF、interleukin-8和bFGF等重要的血管生成因子的表达。
而动物体内试验也发现,西妥昔单抗治疗后这些血管生成因子的下调先于血管退化的出现。
研究表明EGFR表达程度与西妥昔单抗的疗效或总体生存期无必然联系,西妥昔单抗对EGFR表达阴性的转移性结直肠癌(mCRC)患者也同样有效。
对mCRC患者行K-ras基因型检测后发现,西妥昔单抗联合FOLFOX可使K-ras基因野生型患者获益,对突变型患者的益处并不明显。
表明K-ras基因突变与西妥昔单抗的疗效有关。
研究显示对初治晚期结直肠癌,联合西妥昔单抗并未延长中位总生存时间( OS ),中位无病生存期(PFS)甚至更短。
对K-ras野生型患者,联合与不联合西妥昔单抗的中位PFS期相似。
在不同K-ras状态的患者中,联合与不联合西妥昔单抗的中位OS亦无显著性差异,K-ras状态与西妥昔单抗相关的皮肤毒性也无关。
因此目前不推荐在晚期结直肠癌的一线治疗中联合应用靶向药物。
2004年2月获美国FDA批准单药与CPT-11(innotecan,Iri,伊立替康)联合用于进展期/转移性结直肠癌的二线治疗,与CPT-11联合治疗EGFR阳性,含CPT-11方案治疗失败的mCRC的治疗,及单药用于不能耐受的EGFR阳性晚期CRC的治疗。
(FOLFOX4已被批准作为晚期大肠癌一线治疗方案)。
其二线治疗结直肠癌的有效率能达19%;与奥沙利铂+LV5FU2或CPT-11+LV5FU2相比较,西妥昔单抗的加入不但提高了一线治疗的有效率,更为难得的是,西妥昔单抗与CPT-11的联合,能使对CPT-11耐药的肿瘤获得22.9%的有效率。
西妥昔单抗联合FOLFOX4一线治疗EGFR阳性表达的转移性结直肠癌的方案,疗效确切且副作用小。
2008年8月,欧洲药品局(EMEA)已批准爱必妥与化疗联合可作为K-ras野生型的mCRC患者的一线治疗。
西妥昔单抗的主要不良反应为皮疹、虚弱、恶心、呕吐、腹痛、腹泻和发热,其主要毒副作用包括严重输液反应、急性气道阻塞、荨麻疹和低血压。