有机化学人名反应 整理 还原反应
有机人名反应(pdf版)

迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:
反应实例
14
参考文献
[1] [2] [3] [4] E. Beckmann, Ber., 1886, 19,988; 1887, 20, 1507. W. Z. Heldt, Org. Reactions, 1960, 11, 1~156. J. Kenyonn, A. Campbell, J. Chem. Soc., 1946, 25. J. Kenyonn, D. P. Young, J. Chem. Soc., 1941, 263.
Bucherer 反应
反应机理
20
反应实例
参考文献
[1] [2] [3] [4] [5] [6] [7] H. T. Bucherer, J. Prakt. Chem ., 1904, 69(2), 49. H. T. Bucherer, J. Prakt. Chem ., 1904, 70(2), 345. H. T. Bucherer, J. Prakt. Chem ., 1907, 75(2), 249. H. T. Bucherer, J. Prakt. Chem ., 1905, 75(2), 433. N. L. Drake, Org. Reactions, 1942, 1, 105. A. Rieche, H. Seeboth, Ann., 1960, 638, 66, 43, 76. H. Seeboth, Angew. Chem. Int. Ed., Engl., 1967, 6, 307-317.
X
Y Yurev 反应
Z
Zeisel 甲氧基测定法
Arbuzov 反应
亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:
有机化学人名反应

有机化学人名反应
1. Friedel-Crafts反应:由Charles Friedel和James Crafts于1877年首次报道的一种重要的有机化学反应。
2. Grignard反应:法国化学家Victor Grignard于1900年发现的一种有机合成反应。
3. Wolff-Kishner还原反应:德国化学家Kurt Heinrich Wolff和美国化学家Morris Kishner于1913年和1919年分别发现的一种有机还原反应。
4. Birch还原反应:澳大利亚化学家Arthur John Birch于1944年发现的一种有机化学反应。
5. Cannizzaro反应:意大利化学家Stanislao Cannizzaro于1853年发现的一种有机化学反应。
6. Gabriel重氮化反应:德国化学家Siegmund Gabriel于1887年发现的一种有机化学反应。
7. Wurtz反应:法国化学家Charles Adolphe Wurtz于1855年发现的一种有机化学反应。
8. Fries重排反应:德国化学家Karl Fries于1887年发现的一种有机化学反应。
9. Hofmann消去反应:德国化学家August Wilhelm von
Hofmann于1865年发现的一种有机化学反应。
10. Robinson环加成反应:英国化学家Robert Robinson于1925年发现的一种有机化学反应。
有机化学人名反应大全

一、Arbuzov 反应之迟辟智美创作亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次第为:R′I >R′Br >R′Cl.除卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a- 或 b-卤代酸酯、对甲苯磺酸酯等也可以进行反应.当亚酸三烷基酯中三个烷基各不相同时,总是先脱除含碳原子数最少的基团.本反应是由醇制备卤代烷的很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用的卤代烷 R'X 的烷基和亚磷酸三烷基酯 (RO)3P 的烷基相同(即 R' = R),则Arbuzov 反应如下:这是制备烷基膦酸酯的经常使用方法.除亚磷酸三烷基酯外,亚膦酸酯 RP(OR')2和次亚膦酸酯 R2POR' 也能发生该类反应,例如:反应机理一般认为是按 S N2 进行的分子内重排反应:反应实例二、Arndt-Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热获得酸.反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,获得酰基卡宾(2),(2)发生重排得烯酮(3),反应实例三、Baeyer----Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上的一个烃基带着一对电子迁移到-O-O-基团中与羰基碳原子直接相连的氧原子上,同时发生O-O键异裂.因此,这是一个重排反应具有光学活性的3---苯基丁酮和过酸反应,重排产物手性碳原子的枸型坚持不变,说明反应属于分子内重排:分歧毛病称的酮氧化时,在重排步伐中,两个基团均可迁移,可是还是有一定的选择性,按迁移能力其顺序为:醛氧化的机理与此相似,但迁移的是氢负离子,获得羧酸.反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边拔出一个氧原子生成相应的酯,其中三氟过氧乙酸是最好的氧化剂.这类氧化剂的特点是反应速率快,反应温度一般在10~40℃之间,产率高.四、Beckmann重排肟在酸如硫酸、多聚磷酸以及能发生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应获得酰胺.迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:反应实例五、Birch还原芳香化合物用碱金属(钠、钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇)的混合液中还原,苯环可被还原成非共轭的1,4-环己二烯化合物.反应机理首先是钠和液氨作用生成溶剂化点子,然后苯获得一个电子生成自由基负离子(Ⅰ),这是苯环的л电子体系中有7个电子,加到苯环上那个电子处在苯环分子轨道的反键轨道上,自由基负离子仍是个环状共轭体系,(Ⅰ)暗示的是部份共振式.(Ⅰ)不稳定而被质子化,随即从乙醇中篡夺一个质子生成环己二烯自由基(Ⅱ).(Ⅱ)在取得一个溶剂化电子转酿成环己二烯负离子(Ⅲ),(Ⅲ)是一个强碱,迅速再从乙醇中篡夺一个电子生成1,4-环己二烯.环己二烯负离子(Ⅲ)在共轭链的中间碳原子上质子化比末端碳原子上质子快,原因尚不清楚.反应实例取代的苯也能发生还原,而且通过获得单一的还原产物.例如脂肪族羧酸酯可用金属钠和醇还原得一级醇.α,β-不饱和羧酸酯还原得相应的饱和醇.芳香酸酯也可进行本反应,但收率较低.本法在氢化锂铝还原酯的方法发现以前,广泛地被使用,非共轭的双键可不受影响.反应机理首先酯从金属钠获得一个电子还原为自由基负离子,然后从醇中篡夺一个质子转酿成自由基,再从钠得一个电子生成负离子,消除烷氧基成为醛,醛再经过相同的步伐还原成钠,再酸化获得相应的醇.反应实例醛酮也可以用本法还原,获得相应的醇:七、Bucherer反应萘酚及其衍生物在亚硫酸或亚硫酸氢盐存在下和氨进行高温反应,可得萘胺衍生物,反应是可逆的.反应时如用一级胺或二级胺与萘酚反应则制得二级或三级萘胺.如有萘胺制萘酚,可将其加入到热的亚硫酸氢钠中,再加入碱,经煮沸除去氨而得.反应机理本反应的机理为加成消除过程,反应的第一步(无论从哪个方向开始)都是亚硫酸氢钠加成到环的双键上获得烯醇(Ⅱ)或烯胺(Ⅵ),它们再进行下一步互变异构为酮(Ⅲ)或亚胺(Ⅳ):反应实例八、苯基羟胺(N-羟基苯胺)和稀硫酸一起加热发生重排成对-氨基苯酚:在H2SO4-C2H5OH(或CH3OH)中重排生成对-乙氧基(或甲氧基)苯胺:其他芳基羟胺,它的环上的o-p位上未被取代者会起类似的重排.例如,对-氯苯基羟胺重排成2-氨基-5-氯苯酚:反应机理反应实例九、Berthsen,A.Y 吖啶合成法二芳基胺类与羧酸在无水ZnCl2存在下加热起缩合作用,生成吖啶类化合物.反应机理反应机理不详反应实例十、Cannizzaro 反应凡α位碳原子上无活泼氢的醛类和浓NaOH或KOH水或醇溶液作用时,不发生醇醛缩合或树脂化作用而起歧化反应生成与醛相当的酸(成盐)及醇的混合物.此反应的特征是醛自身同时发生氧化及还原作用,一分子被氧化成酸的盐,另一分子被还原成醇:脂肪醛中,只有甲醛和与羰基相连的是一个叔碳原子的醛类,才会发生此反应,其他醛类与强碱液,作用发生醇醛缩合或进一步酿成树脂状物质.具有α-活泼氢原子的醛和甲醛首先发生羟醛缩合反应,获得无α-活泼氢原子的β-羟基醛,然后再与甲醛进行交叉Cannizzaro反应,如乙醛和甲醛反应获得季戊四醇:反应机理醛首先和氢氧根负离子进行亲核加成获得负离子,然后碳上的氢带着一对电子以氢负离子的形式转移到另一分子的羰基不能碳原子上.反应实例十一、Chichibabin 反应杂环碱类,与碱金属的氨基物一起加热时发生胺化反应,获得相应的氨基衍生物,如吡啶与氨基钠反应生成2-氨基啶,如果α位已被占据,则得γ-氨基吡啶,但产率很低.本法是杂环上引入氨基的简便有效的方法,广泛适用于各种氮杂芳环,如苯并咪唑、异喹啉、丫啶和菲啶类化合物均能发生本反应.喹啉、吡嗪、嘧啶、噻唑类化合物较为困难.氨基化试剂除氨基钠、氨基钾外,还可以用取代的碱金属氨化物:反应机理反应机理还不是很清楚,可能是吡啶与氨基首先加成,(Ⅰ),(Ⅰ)转移一个负离子给质子给予体(AH),发生一分子氢气和形成小量的2-氨基吡啶(Ⅱ),此小量的(Ⅱ)又可以作为质子的给予体,最后的产物是2-氨基吡啶的钠盐,用水分解获得2-氨基吡啶:吡啶类化合物不容易进行硝化,用硝基还原法制备氨基吡啶甚为困难.本反应是在杂环上引入氨基的简便有效的方法,广泛适用于各种氮杂芳环,如苯并咪唑、异喹啉、吖啶和菲啶类化合物均能发生本反应.十二、Claisen酯缩合反应含有α-氢的酯在醇钠等碱性缩合剂作用下发生缩合作用,失去一分子醇获得β-酮酸酯.如2分子乙酸乙酯在金属钠和少量乙醇作用下发生缩合获得乙酰乙酸乙酯.二元羧酸酯的分子内酯缩合见Dieckmann缩合反应.反应机理乙酸乙酯的α-氢酸性很弱(pK a-24.5),而乙醇钠又是一个相对较弱的碱(乙醇的pK a~15.9),因此,乙酸乙酯与乙醇钠作用所形成的负离子在平衡体系是很少的.但由于最后产物乙酰乙酸乙酯是一个比力强的酸,能与乙醇钠作用形成稳定的负离子,从而使平衡朝产物方向移动.所以,尽管反应体系中的乙酸乙酯负离子浓度很低,但一形成后,就不竭地反应,结果反应还是可以顺利完成.经常使用的碱性缩合剂除乙醇钠外,还有叔丁醇钾、叔丁醇钠、氢化钾、氢化钠、三苯甲基钠、二异丙氨基锂(LDA)和Grignard试剂等.反应实例如果酯的α-碳上只有一个氢原子,由于酸性太弱,用乙醇钠难于形成负离子,需要用较强的碱才华把酯酿成负离子.如异丁酸乙酯在三苯甲基钠作用下,可以进行缩合,而在乙醇钠作用下则不能发生反应:两种分歧的酯也能发生酯缩合,理论上可获得四种分歧的产物,称为混合酯缩合,在制备上没有太年夜意义.如果其中一个酯分子中既无α-氢原子,而且烷氧羰基又比力活泼时,则仅生成一种缩合产物.如苯甲酸酯、甲酸酯、草酸酯、碳酸酯等.与其它含α-氢原子的酯反应时,都只生成一种缩合产物.实际上这个反应不限于酯类自身的缩合,酯与含活泼亚甲基的化合物都可以发生这样的缩合反应,这个反应可以用下列通式暗示:十三、Claisen—Schmidt反应一个无氢原子的醛与一个带有氢原子的脂肪族醛或酮在稀氢氧化钠水溶液或醇溶液存在下发生缩合反应,并失水获得不饱和醛或酮:反应机理反应实例十四、Claisen 重排烯丙基芳基醚在高温(200°C)下可以重排,生成烯丙基酚.当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要获得邻位产物,两个邻位均被取代基占据时,重排获得对位产物.对位、邻位均被占满时不发生此类重排反应.交叉反应实验证明:Claisen重排是分子内的重排.采纳 g-碳14C 标识表记标帜的烯丙基醚进行重排,重排后 g-碳原子与苯环相连,碳碳双键发生位移.两个邻位都被取代的芳基烯丙基酚,重排后则仍是a-碳原子与苯环相连.反应机理Claisen 重排是个协同反应,中间经过一个环状过渡态,所以芳环上取代基的电子效应对重排无影响.从烯丙基芳基醚重排为邻烯丙基酚经过一次[3,3]s 迁移和一次由酮式到烯醇式的互变异构;两个邻位都被取代基占据的烯丙基芳基酚重排时先经过一次[3,3]s 迁移到邻位(Claisen 重排),由于邻位已被取代基占据,无法发生互变异构,接着又发生一次[3,3]s 迁移()到对位,然后经互变异构获得对位烯丙基酚.取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是Z-构型还是E-构型,重排后的新双键的构型都是E-型,这是因为重排反应所经过的六员环状过渡态具有稳定椅式构象的缘故.反应实例Claisen 重排具有普遍性,在醚类化合物中,如果存在烯丙氧基与碳碳相连的结构,就有可能发生Claisen 重排.十五、Clemmensen 还原醛类或酮类分子中的羰基被锌汞齐和浓盐酸还原为亚甲基:此法只适用于对酸稳定的化合物.对酸不稳定而对碱稳定的化合物可用还原.本反应的反应机理较复杂,目前尚不很清楚.反应实例十六、Combes 喹啉合成法Combes合成法是合成喹啉的另一种方法,是用芳胺与1,3-二羰基化合物反应,首先获得高产率的β-氨基烯酮,然后在浓硫酸作用下,羰基氧质子化后的羰基碳原子向氨基邻位的苯环碳原子进行亲电进攻,关环后,再脱水获得喹啉.反应机理在氨基的间位有强的邻、对位定位基团存在时,关环反应容易发生;但当强邻、对位定位基团存在于氨基的对位时,则不容易发生关环反应.反应实例十七、Cope消除反应叔胺的N-氧化物(氧化叔胺)热解时生成烯烃和N,N-二取代羟胺,产率很高.实际上只需将叔胺与氧化剂放在一起,不需分离出氧化叔胺即可继续进行反应,例如在干燥的二甲亚砜或四氢呋喃中这个反应可在室温进行.此反应条件温和、副反应少,反应过程中不发生重排,可用来制备许多烯烃.当氧化叔胺的一个烃基上二个β位有氢原子存在时,消除获得的烯烃是混合物,可是 Hofmann产物为主;如获得的烯烃有顺反异构时,一般以 E-型为主.例如:这个反应是E2顺式消除反应,反应过程中形成一个平面的五员环过度态,氧化叔胺的氧作为进攻的碱:要发生这样的环状结构,氨基和β-氢原子必需处于同一侧,而且在形成五员环过度态时,α,β-碳原子上的原子基团呈重叠型,这样的过度态需要较高的活化能,形成后也很不稳定,易于进行消除反应.反应实例十八、Cope重排1,5-二烯类化合物受热时发生类似于 O-烯丙基重排为 C-烯丙基的重排反应()反应称为Cope重排.这个反应30多年来引起人们的广泛注意.1,5-二烯在150—200℃独自加热短时间就容易发生重排,而且产率非常好.Cope重排属于周环反应,它和其它周环反应的特点一样,具有高度的立体选择性.例如:内消旋-3,4-二甲基-1,5-己二烯重排后,获得的产物几乎全部是(Z, E)-2,6辛二烯:反应机理Cope重排是[3,3]s-迁移反应,反应过程是经过一个环状过渡态进行的协同反应:在立体化学上,暗示为经过椅式环状过渡态:反应实例十九、Curtius 反应酰基叠氮化物在惰性溶剂中加热分解生成异氰酸酯:异氰酸酯水解则获得胺:反应机理反应实例二十、Crigee,R 反应1,2-二元醇类的氧化产物因所用的氧化剂的种类而分歧.用K2Cr2O7或KMnO4氧化时生成酸类.用特殊氧化剂四乙醋酸铅在CH3COOH或苯等不活泼有机溶剂中缓和氧化,生成二分子羰基化合物(醛或酮).氧化反应也可以在酸催化剂(三氯醋酸)存在下进行.本反应被广泛地应用于研究醇类结构及制备醛、酮类,产率很高.反应机理反应过程中先生成环酯中间产物,进一步C--C键裂开成醛或酮.酸催化的场所,反应历程可以用下式暗示:反应实例二十一、Dakin反应酚醛或酚酮类用H2O2在NaOH存在下氧化时,可将分子中的-CHO基或CH3CO-基被-OH基所置换,生成相对应的酚类.本反应可利用以制备多远酚类.反应机理反应实例二十二、Elbs反应羰基的邻位有甲基或亚甲基的二芳基酮,加热时发生环化脱氢作用,生成蒽的衍生物:由于这个反应通常是在回流温度或高达400-450 °C的温度范围内进行,不用催化剂和溶剂,直到反应物没有水放出为止,在这样的高温条件下,一部份原料和产物发生碳化,部份原料酮被释放出的水所裂解,烃基发生消除或降解以及分子重排等副反应,致使产率不高.反应机理本反应的机理尚不清楚.反应实例二十三、Edvhweiler-Clarke 反应在过量甲酸存在下,一级胺或二级胺与甲醛反应,获得甲基化后的三级胺:甲醛在这里作为一个甲基化试剂.反应机理反应实例二十四、将一元酚类或类似化合物用过硫酸钾在碱性溶液中氧化羟基引入在原有羟基的对位或邻位,生成二元酚类.分子中的醛基或双键等都不影响.产率约20~48%.过硫酸钾的水溶液在加热时放出氧:芳伯胺类如用本试剂氧化时,酿成硝基化合物.反应机理反应实例二十五、Favorskii重排a-卤代酮在氢氧化钠水溶液中加热重排生成含相同碳原子数的羧酸;如为环状a-卤代酮,则招致环缩小.如用醇钠的醇溶液,则得羧酸酯:此法可用于合成张力较年夜的四员环.反应机理反应实例二十六、Friedel-Crafts烷基化反应芳烃与卤代烃、醇类或烯类化合物在Lewis催化剂(如AlCl3,FeCl3,H2SO4, H3PO4, BF3, HF等)存在下,发生芳环的烷基化反应.卤代烃反应的活泼性顺序为:RF > RCl > RBr > RI ; 当烃基超越3个碳原子时,反应过程中易发生重排.反应机理首先是卤代烃、醇或烯烃与催化剂如三氯化铝作用形成碳正离子:所形成的碳正离子可能发生重排,获得较稳定的碳正离子:碳正离子作为亲电试剂进攻芳环形成中间体s-络合物,然后失去一个质子获得发生亲电取代产物:反应实例二十七、Friedel-Crafts酰基化反应芳烃与酰基化试剂如酰卤、酸酐、羧酸、烯酮等在Lewis酸(通经常使用无水三氯化铝)催化下发生酰基化反应,获得芳香酮:这是制备芳香酮类最重要的方法之一,在酰基化中不发生烃基的重排.反应机理反应实例二十八、Fries 重排酚酯在Lewis酸存在下加热,可发生酰基重排反应,生成邻羟基和对羟基芳酮的混合物.重排可以在硝基苯、硝基甲烷等溶剂中进行,也可以不用溶剂直接加热进行.邻、对位产物的比例取决于酚酯的结构、反应条件和催化剂等.例如,用多聚磷酸催化时主要生成对位重排产物,而用四氯化钛催化时则主要生成邻位重排产物.反应温度对邻、对位产物比例的影响比力年夜,一般来讲,较高温度(如室温)下重排有利于形成对位异构产物(动力学控制),较高温度下重排有利于形成邻位异构产物(热力学控制).反应机理反应实例二十九、Fischer,O-Hepp,E重排N-亚硝基芳胺用盐酸或氢溴酸或其乙醇溶液处置时氨基氮上的亚硝基转移到芳核上去形成p-亚硝基芳胺(对位重排):通常发生对位重排,但在奈系化合物中如N-亚硝基-N-加基-2-奈胺则发生邻位重排成1-亚硝基化合物:反应机理在HCl存在下,N-亚硝基化合物首先解离成仲胺及NOCl然后进行亚硝基化:三十、Gabriel合成法邻苯二甲酰亚胺与氢氧化钾的乙醇溶液作用转酿成邻苯二甲酰亚胺盐,此盐和卤代烷反应生成N-烷基邻苯二甲酰亚胺,然后在酸性或碱性条件下水解获得一级胺和邻苯二甲酸,这是制备纯洁的一级胺的一种方法.有些情况下水解很困难,可以用肼解来取代:反应机理邻苯二甲酰亚胺盐和卤代烷的反应是亲核取代反应,取代反应产物的水解过程与酰胺的水解相似.反应实例三十一、Gattermann反应重氮盐用新制的铜粉取代亚铜盐(见)作催化剂,与浓盐酸或氢溴酸发生置换反应获得氯代或溴代芳烃:本法优点是把持比力简单,反应可在较高温度下进行,缺点是其产率一般较低.反应机理见反应实例三十二、Gattermann-Koch 反应芳香烃与等分子的一氧化碳及氯化氢气体在加压和催化剂(三氯化铝及氯化亚铜)存在下反应,生成芳香醛:反应机理反应实例三十三、Gomberg-Bachmann 反应芳香重氮盐在碱性条件下与其它芳香族化合物偶联生成联苯或联苯衍生物:三十四、Hantzsch 合成法两分子b-羰基酸酯和一分子醛及一分子氨发生缩合反应,获得二氢吡啶衍生物,再用氧化剂氧化获得吡啶衍生物.这是一个很普遍的反应,用于合成吡啶同系物.反应机理反应过程可能是一分子b-羰基酸酯和醛反应,另一分子b-羰基酸酯和氨反应生成b-氨基烯酸酯,所生成的这两个化合物再发生Micheal加成反应,然后失水关环生成二氢吡啶衍生物,它很溶液脱氢而芳构化,例如用亚硝酸或铁氰化钾氧化获得吡啶衍生物:反应实例三十五、Haworth 反应萘和丁二酸酐发生然后按标准的方法还原、关环、还原、脱氢获得多环芳香族化合物.反应机理见反应实例三十六、Hell-Volhard-Zelinski 反应羧酸在催化量的三卤化磷或红磷作用下,能与卤素发生a-卤代反应生成a-卤代酸:本反应也可以用酰卤作催化剂.三十七、Hinsberg反应伯胺、仲胺分别与对甲苯磺酰氯作用生成相应的对甲苯磺酰胺沉淀,其中伯胺生成的沉淀能溶于碱(如氢氧化钠)溶液,仲胺生成的沉淀则不溶,叔胺与对甲苯磺酰氯不反应.此反应可用于昆季叔胺的分离与鉴定.三十八、Hofmann烷基化卤代烷与氨或胺发生烷基化反应,生成脂肪族胺类:由于生成的伯胺亲核性通常比氨强,能继续与卤代烃反应,因此本反应不成防止地发生仲胺、叔胺和季铵盐,最后获得的往往是多种产物的混合物.用年夜过量的氨可防止多取代反应的发生,从而可获得良好产率的伯胺.反应机理反应为典范的亲核取代反应(S N1或S N2)反应实例三十九、Hofmann消除反应季铵碱在加热条件下(100--200°C)发生热分解,当季铵碱的四个烃基都是甲基时,热分解获得甲醇和三甲胺:如果季铵碱的四个烃基分歧,则热分解时总是获得含取代基最少的烯烃和叔胺:反应实例四十、Hofmann重排(降解)酰胺用溴(或氯)在碱性条件下处置转酿成少一个碳原子的伯胺:反应机理反应实例四十一、Houben-Hoesch 反应酚或酚醚在氯化氢和氯化锌等Lewis酸的存在下,与腈作用,随后进行水解,获得酰基酚或酰基酚醚:反应机理反应机理较复杂,目前尚未完全说明反应实例四十二、Hunsdieecker 反应干燥的羧酸银盐在四氯化碳中与卤素一起加热放出二氧化碳,生成比原羧酸少一个碳原子的卤代烃:X = Br , Cl , I反应机理反应实例四十三、Kiliani氯化增碳法糖在少量氨的存在下与氢氰酸加成获得a-羟基腈,经水解获得相应的糖酸,此糖酸极易转酿成内酯,将此内酯在含水的乙醚或水溶液中用钠汞齐还原,获得比原来的糖多一个碳原子的醛糖.反应实例四十四、Knoevenagel 反应含活泼亚甲基的化合物与醛或酮在弱碱性催化剂(氨、伯胺、仲胺、吡啶等有机碱)存在下缩合获得a,b-不饱和化合物.反应机理反应实例四十五、Koble 反应脂肪酸钠盐或钾盐的浓溶液电解时发生脱羧,同时两个烃基相互偶联生成烃类:如果使用两种分歧脂肪酸的盐进行电解,则获得混合物:反应机理反应实例四十六、Koble-Schmitt 反应酚钠和二氧化碳在加压下于125-150 ºC反应,生成邻羟基苯甲酸,同时有少量对羟基苯甲酸生成:反应产物与酚盐的种类及反应温度有关,一般来讲,使用钠盐及在较低的温度下反应主要获得邻位产物,而用钾盐及在较高温度下反应则主要得对位产物:邻位异构体在钾盐及较高温度下加热也能转酿成对位异构体:反应机理反应机理目前还不太清楚.反应实例四十七、Kolbe,H.Syntbexis of Nitroparsffini合成将含等摩尔的α-卤代羧酸与亚硝酸钠或钾的水溶液加热时,生成-硝基脂肪酸钠中间体,继续加热起分解作用,失去CO2转酿成硝基烷类及NaHCO3.本方法仅可适用于小量制备碳原子数在以下的硝基烷类(特别适宜于制备硝基甲烷及硝基乙烷).而b-卤代羧酸与亚硝酸钾作用生成产物不能放出CO2,故不能发生此反应.反应机理反应实例四十八、Leuckart 反应醛或酮在高温下与甲酸铵反应得伯胺:除甲酸铵外,反应也可以用取代的甲酸铵或甲酰铵.反应机理反应中甲酸铵一方面提供氨,另一方面又作为还原剂.反应实例四十九、Lossen 反应或其酰基化物在独自加热或在碱、脱水剂(如五氧化二磷、乙酸酐、亚硫酰氯等)存在下加热发生重排生成异氰酸酯,再经水解、脱羧得伯胺:本重排反应后来有过反应机理。
人名反应(类型整理)

人名反应1氧化:1.Baeyer-Villiger 氧化:酮过酸氧化成酯迁移规则:叔>仲>环己基>苄>伯>甲基>氢2.Corey-Kim 氧化: 醇在NCS/DMF 作用后,碱处理氧化成醛酮3.Criegee邻二醇裂解:邻二醇由Pb(OAc)4 氧化成羰基化合物4.Criegee臭氧化: 烯烃臭氧化后水解成醛酮5.Dakin 反应:对羟基苯甲醛由碱性H2O2氧化成对二酚6.Dess-Martin 过碘酸酯氧化: 仲醇由过碘酸酯氧化成酮7. Fleming氧化:硅烷经过酸化,过酸盐氧化,水解以后形成醇8. Hooker氧化:2-羟基-3烷基-1,4-醌被KMnO4 氧化导致侧链烷基失去一个亚甲基,同时羟基和烷基位置互变9.Moffatt 氧化(Pfitzner-Moffatt)氧化:用DCC和DMSO 氧化醇,形成醛酮10.Oppenauer氧化: 烷氧基催化的仲醇氧化成醛酮11. Riley氧化:活泼亚甲基(羰基α位等)被SeO2氧化成酮12. Rubottom氧化:烯醇硅烷经过m-CPBA 和K2CO3处理后α-羟基化13.Sarett 氧化: CrO3·Py络合物氧化醇成醛酮14.Swern氧化:用(COCl)2,DMSO为试剂合Et3N 淬灭的方法将醇氧化成羰基化合物15.Tamao-Kumada 氧化:烷基氟硅烷被KF,H2O2,KHCO3 氧化成醇16.Wacker 氧化: Pd催化剂下,烯烃氧化成酮还原:1..Barton-McCombie 去氧反应:从相关的硫羰基体中间用n-Bu3SnH,AIBN 试剂经过自由基开裂发生醇的去氧作用2.Birch 还原:苯环由Na 单质合液胺条件下形成环内二烯烃(带供电子基团的苯环:双键连接取代基;带吸电子基团的苯环,取代基在烯丙位。
)3.Brown硼氢化:烯烃和硼烷加成产生的有机硼烷经过碱性H2O2氧化得到醇4.Cannizzaro 歧化:碱在芳香醛,甲醛或者其他无α-氢的脂肪氢之间发生氧化还原反应给出醇和酸5.Clemmensen还原:用锌汞齐和氯化氢将醛酮还原为亚甲基化合物6.Corey-Bakshi-Shibata(CBS)还原:酮在手性恶唑硼烷催化下的立体选择性还原7.Gribble 吲哚还原:用NaBH4直接还原会导致N-烷基化,NaBH3CN 在冰醋酸当中还原吲哚双键可以解决8.Gribble 二芳基酮还原:用NaBH4 在三氟乙酸中还原二芳基酮和二芳基甲醇为二芳基甲烷,也可以应用于二杂芳环酮和醇的还原9. Luche还原:烯酮在NaBH4-CeCl3下发生1,2-还原形成烯丙位取代烯醇10.McFadyen-Stevens还原:酰基苯磺酰肼用碱处理成醛11.Meerwein-Ponndorf-Verley 还原:用Al(OPr')3/Pr 'O体H 系将酮还原为醇12.Midland 还原:用B-3-α-蒎烯-9-BBN 对酮进行不对称还原13. Noyori 不对称氢化:羰基在Ru(II)BINAP 络合物催化下发生不对称氢化还原14. Rosenmund还原:用BaSO4/毒化Pd 催化剂将酰氯氢化成醛,如催化剂未被毒化,会氢化为醇15.Wolff-Kishner-黄鸣龙还原:用碱性肼将羰基还原为亚甲基成烯反应:1.Boord反应:β-卤代烷氧基与Zn 作用生成烯烃2.Chugaev消除:黄原酸酯热消除成烯3.Cope消除:胺的氧化物热消除成烯烃4.Corey-Winter olefin 烯烃合成:邻二醇经1,1-硫代羰基二咪唑和三甲氧基膦处理转化为相应的烯5.Doering-LaFlamme 丙二烯合成:烯烃用溴仿以及烷氧化物处理以后生成同碳二溴环丙烷再反应生成丙二烯6.Horner-Wadsworth-Emmons 反应:从醛合磷酸酯生成烯烃.副产物为水溶性磷酸盐,故以后处理较相应的Witting 反应简单的多7.Julia-Lythgoe 成烯反应:从砜合醛生成(E)-烯烃8.Peterson成烯反应:从α - 硅基碳负离子合羰基化合物生成烯烃.也成为含硅的Witting 反应9.Ramberg-Backlund 烯烃合成:Α-卤代砜用碱处理生成烯烃10.Witting 反应:羰基用膦叶立德变成烯烃11.Zaitsev消除:E2消除带来更多取代的烯烃人名反应2偶联反应:Cadiot-Chodkiewicz偶联:从炔基卤和炔基酮合成双炔衍生物Castro-Stephens偶联: 芳基炔合成,同Cadiot-Chodkiewicz 偶联Eglinton 反应:终端炔烃在化学计量 (常常过量) Cu(Oac)2促进下发生的氧化偶联反应Eschenmoser偶联:从硫酰胺和烷基卤生成烯胺Glaser偶联: Cu 催化终端炔烃的氧化自偶联Gomberg-Bachmann偶联:碱促进下芳基重氮盐和一个芳烃之间经自由基偶联生成二芳基化合物Heck 反应: Pb催化的有机卤代物或者三氟磺酸酯和烯烃之间的偶联反应杂芳基Heck 反应:发生在杂芳基受体上的Pd(Ph3P)4,Ph3P,CuI,Cs2CO3催化下的分子内或者分子间Heck 反应Hiyama 交叉偶联反应:Pb 催化有机硅和有机卤代物或者三氟磺酸酯等在诸如F -或者OH-之类的活化剂Pd(Ph3P)4,TBAF 催化剂存在下发生的交叉偶联反应Kumada 交叉偶联( Kharasch交叉偶联) :Ni 和Pd催化下,格氏试剂和一个有机卤代物或者三氟磺酸酯之间的交叉偶联Liebeskind-Srogl偶联:硫酸酯和有机硼酸之间经过Pd 催化发生交叉偶联生成酮McMurry 偶联:羰基用低价Ti,如TiCl3/LiAlH4 产生的Ti(0) 处理得到双键,反应是一个单电子过程Negishi交叉偶联: Pd催化的有机Zn 和有机卤代物,三氟磺酸酯等之间发生的交叉偶联反应Sonogashira反应: Pd/Cu催化的有机卤和端基炔烃之间的交叉偶联反应Stille 偶联: Pd催化的有机Sn 和有机卤,三氟磺酸酯之间的交叉偶联反应Stille-Kelly偶联:双Sn试剂进行Pd催化下二芳基卤代物的分子交叉偶联Suzuki 偶联: Pd催化下的有机硼烷和有机卤,三氟磺酸酯在碱存在下发生的交叉偶联Ullmann 反应:芳基碘代物在Cu 存在下的自偶联反应Wurtz 反应:烷基卤经Na或Mg 金属处理后形成碳碳单键Ymada 偶联试剂: 用二乙基氰基磷酸酯(EtO)2PO-CN 活化羧酸缩合反应:Aldol 缩合: 羰基和一个烯醇负离子或一个烯醇的缩合Blaise反应:腈和α-卤代酯和Zn 反应得到β-酮酯Benzoin 缩合:芳香醛经CN-催化为安息香(二芳基乙醇酮)Buchner-Curtius-Schlotterbeck反应: 羰基化合物和脂肪族重氮化物反应给出同系化的酮Claisen缩合: 酯在碱催化下缩合为β-酮酯Corey-Fuchs反应:醛发生一碳同系化生成二溴烯烃,然后用BuLi 处理生成终端炔烃Darzen 缩水甘油酸酯缩合:碱催化下从α-卤代酯和羰基化合物生成α,β-环氧酯(缩水甘油醛)Dieckmann 缩合:分子内的Claisen缩合Evans aldol反应: 用Evans手性鳌合剂,即酰基恶唑酮进行不对称醇醛缩合Guareschi-Thorpe 缩合(2-吡啶酮合成): 氰基乙酸乙酯和乙酰乙酸在氨存在下生成2-吡啶酮Henry 硝醇反应:醛和有硝基烷烃在碱作用下去质子化产生氮酸酯Kharasch 加成反应:过渡金属催化的CXCl3 对于烯烃的自由基加成Knoevenagel缩合:羰基化合物和活泼亚甲基化合物在胺的催化下缩合Mannnich 缩合(羰基胺甲基化):胺,甲醛,和一个带有酸性亚甲基成分的化合物之间的三组分反应发生胺甲基化Michael 加成:亲核碳原子对α,β-不饱和体系的共扼加成Mukaiyama 醇醛缩合:Lewis 酸催化下的醛和硅基烯醇醚之间的Aldol 缩合Nozaki-Hiyama-KIshi 反应:Cr-Ni 双金属催化下的烯基卤对于醛的氧化还原加成Pechmann 缩合(香豆素合成):Lewis 酸促进的酸和β-酮酯缩合成为香豆素Perkin 反应:芳香醛和乙酐反应合成肉桂酸Prins 反应:烯烃酸性条件下对于甲醛的加成反应Reformatsky反应:有机Zn试剂(从α-卤代酯来)对羰基的亲核加成反应Reimer-Tiemann 反应:从碱性介质当中从酚和氯仿合成邻甲酰基苯酚Schlosser 对Witting 反应的修正:不稳定的叶立德和醛发生的Witting 反应生成Z-烯烃,而改进的Schlosser反应可以得到E-烯烃Stetter 反应(Michael-Stetter 反应):从醛和α ,β-不饱和酮可以得到1,4-二羰基衍生物。
有机合成常用人名反应

有机合成常用人名反应有机合成是化学领域中的一个重要分支,它研究有机化合物的合成方法和反应过程。
在有机合成中,常常会使用一些常用的人名反应,这些反应以人名命名,代表了该反应的发现者或者重要贡献者。
本文将介绍一些常用的人名反应,并对其原理和应用进行阐述。
一、格氏反应(Gattermann Reaction)格氏反应是一种用于合成醛的重要反应。
它是由德国化学家格氏(Gattermann)于1898年发现的。
格氏反应通过在芳香化合物上引入氰基,然后将其加氢还原,得到相应的醛。
格氏反应是一种重要的合成醛的方法,广泛应用于有机合成领域。
二、斯特雷克反应(Strecker Reaction)斯特雷克反应是一种合成α-氨基酸的方法,由德国化学家斯特雷克(Strecker)于1850年发现。
该反应通过使用醛、氰化物和胺,经过缩合和水解反应,合成出具有氨基酸结构的化合物。
斯特雷克反应是合成氨基酸的重要方法之一,广泛应用于生物化学和药物化学领域。
三、沃尔夫-克尼希反应(Wolf-Kishner Reduction)沃尔夫-克尼希反应是一种将醛或酮转化为对应的烷烃的方法。
该反应由德国化学家沃尔夫(Wolf)和克尼希(Kishner)于1912年发现。
沃尔夫-克尼希反应通过使用氨水和氢醇钠,将醛或酮转化为相应的烷烃。
这种还原反应在有机合成中具有重要的应用价值。
四、格里格纳德试剂(Grignard Reagent)格里格纳德试剂是一类由法国化学家格里格纳德(Grignard)于1900年发现的有机金属试剂。
格里格纳德试剂可以与卤代烃反应,生成烷基镁试剂。
这些烷基镁试剂可以与酮、醛、酸等化合物发生加成反应,合成出复杂的有机分子。
格里格纳德试剂是一种重要的有机合成试剂,在有机合成中具有广泛的应用。
五、费舍尔试剂(Fisher Reagent)费舍尔试剂是一种用于合成酮的试剂,由德国化学家费舍尔(Fisher)于1895年发现。
有机化学人名反应总结

有机化学人名反应总结是化学的一个重要分支,研究有机化合物的结构、性质、合成和反应机理。
在的发展过程中,许多学者为该领域做出了巨大的贡献,并被用来命名各种有机反应。
以下是几个人名反应的介绍和总结。
沃尔夫-可克斯反应(Wolf-Kishner reaction)沃尔夫和可克斯是这个反应的共同发现者,这个反应是一种将酮或醛还原为相应的烷烃的方法。
该反应的基本步骤包括酮或醛与叔胺在碱性条件下反应,形成次磺酰胺盐。
随后,在高温下,用氨水还原次磺酰胺盐形成烷烃。
这个反应适用于许多酮和醛的还原,而且产率较高。
亲核取代反应(Nucleophilic substitution reaction)亲核取代反应是一类常见的反应,其中一个亲核试剂通过攻击有机物中的一个电子云丰富的原子,将它替换掉。
亲核试剂可以是氢离子(质子)、一个氢的取代基或一个非常活泼的原子或基团,如氯、溴、碘等。
这个反应在有机合成中广泛应用,可以用来合成醇、酯、酰胺等化合物。
格林纳德试剂反应(Grignard reaction)格林纳德试剂反应是一种重要的有机合成方法,被用来合成新的碳-碳键或存在二氢化碳的间接合成。
它的基本步骤是将卤代烃与镁反应,生成格林纳德试剂。
然后,格林纳德试剂与醛、酮、酸、酯等化合物进行反应,形成相应的醇、醚、羧酸、酮等。
格林纳德试剂反应在有机合成中得到广泛应用,尤其是在构建复杂的有机分子骨架中。
Diels-Alder 反应(Diels-Alder reaction)Diels-Alder 反应是一种重要的环加成反应,其中烯丙烃与二烯或炔烃通过热力学控制和反应物的轨道的对称性控制形成一个六元环。
这个反应在天然产物的全合成、药物合成和高分子材料的合成中得到广泛应用。
Diels-Alder 反应的核心是“四加二”环加成反应,反应条件和底物结构的变化可以使反应具有选择性和灵活性。
柴维克斯基反应(Chichibabin reaction)柴维克斯基反应是一种将氨基化合物转化为吡嗪或嘧啶的方法。
有机化学人名反应大全

一、Arbuzov 反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯与一个新得卤代烷:卤代烷反应时,其活性次序为:R′I >R′Br 〉R′Cl.除了卤代烷外,烯丙型或炔丙型卤化物、a-卤代醚、a—或b-卤代酸酯、对甲苯磺酸酯等也可以进行反应。
当亚酸三烷基酯中三个烷基各不相同时,总就是先脱除含碳原子数最少得基团。
本反应就是由醇制备卤代烷得很好方法,因为亚磷酸三烷基酯可以由醇与三氯化磷反应制得:如果反应所用得卤代烷R’X得烷基与亚磷酸三烷基酯(RO)3P得烷基相同(即R'=R),则Arbuzov反应如下:这就是制备烷基膦酸酯得常用方法。
除了亚磷酸三烷基酯外,亚膦酸酯RP(OR')2与次亚膦酸酯R2POR'也能发生该类反应,例如:反应机理一般认为就是按SN2 进行得分子内重排反应:反应实例二、Arndt—Eister 反应酰氯与重氮甲烷反应,然后在氧化银催化下与水共热得到酸。
反应机理重氮甲烷与酰氯反应首先形成重氮酮(1),(1)在氧化银催化下与水共热,得到酰基卡宾(2),(2)发生重排得烯酮(3),(3)与水反应生成酸,若与醇或氨(胺)反应,则得酯或酰胺。
反应实例三、Baeyer——--Villiger反应反应机理过酸先与羰基进行亲核加成,然后酮羰基上得一个烃基带着一对电子迁移到—O-O-基团中与羰基碳原子直接相连得氧原子上,同时发生O-O键异裂.因此,这就是一个重排反应具有光学活性得3-—-苯基丁酮与过酸反应,重排产物手性碳原子得枸型保持不变,说明反应属于分子内重排:不对称得酮氧化时,在重排步骤中,两个基团均可迁移,但就是还就是有一定得选择性,按迁移能力其顺序为:醛氧化得机理与此相似,但迁移得就是氢负离子,得到羧酸。
反应实例酮类化合物用过酸如过氧乙酸、过氧苯甲酸、间氯过氧苯甲酸或三氟过氧乙酸等氧化,可在羰基旁边插入一个氧原子生成相应得酯,其中三氟过氧乙酸就是最好得氧化剂。
这类氧化剂得特点就是反应速率快,反应温度一般在10~40℃之间,产率高。
有机人名反应大全.

索引:Arbuzov反应Arndt-Eister反应Baeyer-Villiger 氧化Beckmann 重排Birch 还原Bischler-Napieralski 合成法Bouveault-Blanc还原Bucherer 反应Cannizzaro 反应Chichibabin 反应Claisen 酯缩合反应Claisen-Schmidt 反应Clemmensen 还原Combes 合成法Cope 重排Cope 消除反应Curtius 反应Dakin 反应Darzens 反应Demjanov 重排Dieckmann 缩合反应Elbs 反应Eschweiler-Clarke 反应Favorskii 反应Favorskii 重排Friedel-Crafts烷基化反应Friedel-Crafts酰基化反应Fries 重排Gabriel 合成法Gattermann 反应Gattermann-Koch 反应Gomberg-Bachmann 反应Hantzsch 合成法Haworth 反应Hell-V olhard-Zelinski 反应Hinsberg 反应Hofmann 烷基化Hofmann 消除反应Hofmann 重排(降解)Houben-Hoesch 反应Hunsdiecker 反应Kiliani 氰化增碳法Knoevenagel 反应Knorr 反应Koble 反应Koble-Schmitt 反应Leuckart 反应Lossen反应Mannich 反应Meerwein-Ponndorf 反应Meerwein-Ponndorf 反应Michael 加成反应Norrish I和II 型裂解反应Oppenauer 氧化Paal-Knorr 反应Pictet-Spengler 合成法Pschorr 反应Reformatsky 反应Reimer-Tiemann 反应Reppe 合成法Robinson 缩环反应Rosenmund 还原Ruff 递降反应Sandmeyer 反应Schiemann 反应Schmidt反应Skraup 合成法Sommelet-Hauser 反应Stephen 还原Stevens 重排Strecker 氨基酸合成法Tiffeneau-Demjanov 重排Ullmann反应Vilsmeier 反应Wagner-Meerwein 重排Wacker 反应Williamson 合成法Wittig 反应Wittig-Horner 反应Wohl 递降反应Wolff-Kishner-黄鸣龙反应Yurév 反应Zeisel 甲氧基测定法Arbuzov(加成)反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
有机人名反应大全

索引:Arbuzov反应Arndt-Eister反应Baeyer-Villiger 氧化Beckmann 重排Birch 还原Bischler-Napieralski 合成法Bouveault-Blanc还原Bucherer 反应Cannizzaro 反应Chichibabin 反应Claisen 酯缩合反应Claisen-Schmidt 反应Clemmensen 还原Combes 合成法Cope 重排Cope 消除反应Curtius 反应Dakin 反应Darzens 反应Demjanov 重排Dieckmann 缩合反应Elbs 反应Eschweiler-Clarke 反应Favorskii 反应Favorskii 重排Friedel-Crafts烷基化反应Friedel-Crafts酰基化反应Fries 重排Gabriel 合成法Gattermann 反应Gattermann-Koch 反应Gomberg-Bachmann 反应Hantzsch 合成法Haworth 反应Hell-V olhard-Zelinski 反应Hinsberg 反应Hofmann 烷基化Hofmann 消除反应Hofmann 重排(降解)Houben-Hoesch 反应Hunsdiecker 反应Kiliani 氰化增碳法Knoevenagel 反应Knorr 反应Koble 反应Koble-Schmitt 反应Leuckart 反应Lossen反应Mannich 反应Meerwein-Ponndorf 反应Meerwein-Ponndorf 反应Michael 加成反应Norrish I和II 型裂解反应Oppenauer 氧化Paal-Knorr 反应Pictet-Spengler 合成法Pschorr 反应Reformatsky 反应Reimer-Tiemann 反应Reppe 合成法Robinson 缩环反应Rosenmund 还原Ruff 递降反应Sandmeyer 反应Schiemann 反应Schmidt反应Skraup 合成法Sommelet-Hauser 反应Stephen 还原Stevens 重排Strecker 氨基酸合成法Tiffeneau-Demjanov 重排Ullmann反应Vilsmeier 反应Wagner-Meerwein 重排Wacker 反应Williamson 合成法Wittig 反应Wittig-Horner 反应Wohl 递降反应Wolff-Kishner-黄鸣龙反应Yurév 反应Zeisel 甲氧基测定法Arbuzov(加成)反应亚磷酸三烷基酯作为亲核试剂与卤代烷作用,生成烷基膦酸二烷基酯和一个新的卤代烷:卤代烷反应时,其活性次序为:R'I >R'Br >R'Cl。
有机化学人名反应 整理 还原反应

二、还原:
1..Barton-McCombie去氧反应:从相关的硫羰基体中间用n-Bu3SnH,AIBN试剂经过自由基开裂发生醇的去氧作用
2.Birch 还原:苯环由Na单质合液胺条件下形成环内二烯烃(带供电子基团的苯环:双键连接取代基;带吸电子基团的苯环,取代基在烯丙位。
)
3.Brown硼氢化:烯烃和硼烷加成产生的有机硼烷经过碱性H2O2氧化得到醇
4.Cannizzaro歧化:碱在芳香醛,甲醛或者其他无α-氢的脂肪氢之间发生氧化还原反应给出醇和酸
5.Clemmensen还原:用锌汞齐和氯化氢将醛酮还原为亚甲基化合物
6.Corey-Bakshi-Shibata(CBS)还原:酮在手性恶唑硼烷催化下的立体选择性还原
7.Gribble吲哚还原:用NaBH4直接还原会导致N-烷基化,NaBH3CN在冰醋酸当中还原吲哚双键可以解决
8.Gribble二芳基酮还原:用NaBH4在三氟乙酸中还原二芳基酮和二芳基甲醇为二芳基甲烷,也可以应用于二杂芳环酮和醇的还原
9.Luche还原:烯酮在NaBH4-CeCl3下发生1,2-还原形成烯丙位取代烯醇
10.McFadyen-Stevens还原:酰基苯磺酰肼用碱处理成醛
11.Meerwein-Ponndorf-V erley还原:用Al(OPr’)3/Pr’OH体系将酮还原为醇
12.Midland还原:用B-3-α-蒎烯-9-BBN对酮进行不对称还原
13.Noyori不对称氢化:羰基在Ru(II)BINAP络合物催化下发生不对称氢化还原
14.Rosenmund还原:用BaSO4/毒化Pd催化剂将酰氯氢化成醛,如催化剂未被毒化,会氢化为醇
15.Wolff-Kishner-黄鸣龙还原:用碱性肼将羰基还原为亚甲基。
100种有机化学人名反应

120~200℃时热解,顺利产生烯烃,相应醇和氧硫
化碳。黄原酸酯在热解前制备不须离析除黄原酸酯外,其他的酯氨基甲酸酯,碳
酸酯和羧酸酯热解。特别是使用大分子量的羧酸酯(棕榈酸酯)的烯烃提供了有
利条件。因为这些酯本身沸点高,而热解温度较低(约
300℃),在液相中简单加热即可。
250℃
Cl OCH2CH=CH2
;
Cl 醚分子中,
如临位未被取代则不起重排反应,产生复杂的热分解作用。此反应是在苯环引入丙基的
简易方法,因为烯丙基可还原成丙基。
16. Claisen缩合反应(P352~354)
17. Claisen-Schmidt反应(P287)
N+
*
H CH3
CH3 △
H3C
*
CH3
CH2 +(CH3)2NOH
O
H5C6
C6H5
20. Criegee氧化法
乙二醇类在稀醋酸或苯溶液中,室温时用四乙酸铅进行很温和的氧化,两个相连的各带
有游离羟基的碳原子之间的碳链就断裂,得到定量的醛酮类。此法用于研究醇类结构及
此法用来合成呋喃类化合物,在吡啶或氨存在下,α
-氯化羰基化合物或α
,β-氯醚类与
1,3-二羰基化合物发生缩合反应,生成呋喃类化合物。
R C O H2C COOR'
O
COOR'
R
吡啶或氨
-H2O, -HCl
CH2
+
OC
Cl CH3
CH3
解,可得醛类化合物,
R’MgX中
人名反应(类型整理)

人名反应1氧化:1・Baeyer-Villiger氧化:酮过酸氧化成酯迁移规则:叔>仲>环己基>苄>伯〉甲基〉氢2・Corey-Kim氧化:醇在NCS/DMF作用后,碱处理氧化成醛酮3・Criegee邻二醇裂解:邻二醇由Pb(OAc)4氧化成羰基化合物4・Criegee臭氧化:烯烃臭氧化后水解成醛酮5・Dakin反应:对羟基苯甲醛由碱性H2O2氧化成对二酚6・Dess—Martin过碘酸酯氧化:仲醇由过碘酸酯氧化成酮7・Fleming氧化•硅烷经过酸化,过酸盐氧化,水解以后形成醇8・Hooker氧化:2—羟基一3烷基一1,4—醌被KMnO4氧化导致侧链烷基失去一个亚甲基,同时羟基和烷基位置互变9・Moffatt氧化(Pfitzner—Moffatt)氧化:用DCC和DMSO氧化醇,形成醛酮10・Oppenauer氧化:烷氧基催化的仲醇氧化成醛酮11・Riley氧化:活泼亚甲基(羰基a位等)被SeO2氧化成酮12・Rubottom氧化:烯醇硅烷经过m—CPBA和K2CO3处理后a—羟基化KHCO3氧化成醇13・Sarett氧化:CrO3・Py络合物氧化醇成醛酮14・Swern氧化:用(COC1)2,DMSO为试剂合Et3N淬灭的方法将醇氧化成羰基化合物15・Tamao—Kumada氧化:烷基氟硅烷被KF,H2O2,16・Wacker氧化:Pd催化剂下,烯烃氧化成酮还原:1・・Barton—McCombie去氧反应:从相关的硫羰基体中间用n—Bu3SnH,AIBN 试剂经过自由基开裂发生醇的去氧作用2・Birch还原:苯环由Na单质合液胺条件下形成环内二烯烃(带供电子基团的苯环:双键连接取代基:带吸电子基团的苯环,取代基在烯丙位。
)3・Brown硼氢化:烯烃和硼烷加成产生的有机硼烷经过碱性H2O2氧化得到醇4・Cannizzaro歧化:碱在芳香醛,甲醛或者其他无a—氢的脂肪氢之间发生氧化还原反应给出醇和酸5・Clemmensen还原:用锌汞齐和氯化氢将醛酮还原为亚甲基化合物6・Corey—Bakshi—Shibata(CBS)还原:酮在手性恶唑硼烷催化下的立体选择性还原7・Gribble吲哚还原:用NaBH4直接还原会导致N—烷基化,NaBH3CN在冰醋酸当中还原吲哚双键可以解决8・Gribble二芳基酮还原.用NaBH4在三氟乙酸中还原二芳基酮和二芳基甲醇为二芳基甲烷,也可以应用于二杂芳环酮和醇的还原9・Luche还原:烯酮在NaBH4—CeCl3下发生1,2—还原形成烯丙位取代烯醇10・McFadyen—Stevens还原:酰基苯磺酰肼用碱处理成醛11.Meerwein—Ponndorf—Verley还原:用Al(OPr')3/Pr'OH体系将酮还原为醇12・Midland还原:用B—3—a—蒎烯一9—BBN对酮进行不对称还原13・Noyori不对称氢化.羰基在Ru(II)BINAP络合物催化下发生不对称氢化还原14・Rosenmund还原:用BaSO4/毒化Pd催化剂将酰氯氢化成醛,如催化剂未被毒化,会氢化为醇15・Wolff—Kishner—黄鸣龙还原.用碱性肼将羰基还原为亚甲基成烯反应:1・Boord反应:B-卤代烷氧基与Zn作用生成烯烃2・Chugaev消除:黄原酸酯热消除成烯3・Cope消除:胺的氧化物热消除成烯烃4・Corey-Winterolefin烯烃合成:邻二醇经1,1-硫代羰基二咪唑和三甲氧基膦处理转化为相应的烯5・Doering-LaFlamme丙二烯合成:烯烃用溴仿以及烷氧化物处理以后生成同碳二溴环丙烷再反应生成丙二烯6・Horner-Wadsworth-Emmons反应:从醛合磷酸酯生成烯烃.副产物为水溶性磷酸盐,故以后处理较相应的Witting反应简单的多7・Julia-Lythgoe成烯反应:从砜合醛生成(E)-烯烃8・Peterson成烯反应:从a-硅基碳负离子合羰基化合物生成烯烃.也成为含硅的Witting反应9・Ramberg-Backlund烯烃合成:A-卤代砜用碱处理生成烯烃10・Witting反应:羰基用膦叶立德变成烯烃11・Zaitsev消除:E2消除带来更多取代的烯烃人名反应2偶联反应:Cadiot-Chodkiewicz偶联:从炔基卤和炔基酮合成双炔衍生物Castro—Stephens偶联:芳基炔合成,同Cadiot-Chodkiewicz偶联Eglinton反应:终端炔烃在化学计量(常常过量)Cu(Oac)2促进下发生的氧化偶联反应Eschenmoser偶联:从硫酰胺和烷基卤生成烯胺Glaser偶联:Cu催化终端炔烃的氧化自偶联Gomberg—Bachmann偶联:碱促进下芳基重氮盐和一个芳烃之间经自由基偶联生成二芳基化合物Heck反应:Pb催化的有机卤代物或者三氟磺酸酯和烯烃之间的偶联反应杂芳基Heck反应:发生在杂芳基受体上的Pd(Ph3P)4,Ph3P,CuI,Cs2CO3催化下的分子内或者分子间Heck反应Hiyama交叉偶联反应:Pb催化有机硅和有机卤代物或者三氟磺酸酯等在诸如F—或者OH—之类的活化剂Pd(Ph3P)4,TBAF催化剂存在下发生的交叉偶联反应Kumada交叉偶联(Kharasch交叉偶联):Ni和Pd催化下,格氏试剂和一个有机卤代物或者三氟磺酸酯之间的交叉偶联Liebeskind—Srogl偶联:硫酸酯和有机硼酸之间经过Pd催化发生交叉偶联生成酮McMurry偶联•羰基用低价Ti,如TiC13/LiAlH4产生的Ti(0)处理得到双键,反应是一个单电子过程Negishi交叉偶联:Pd催化的有机Zn和有机卤代物,三氟磺酸酯等之间发生的交叉偶联反应Sonogashira反应:Pd/Cu催化的有机卤和端基炔烃之间的交叉偶联反应Stille偶联:Pd催化的有机Sn和有机卤,三氟磺酸酯之间的交叉偶联反应Stille—Kelly偶联:双Sn试剂进行Pd催化下二芳基卤代物的分子交叉偶联Suzuki偶联:Pd催化下的有机硼烷和有机卤,三氟磺酸酯在碱存在下发生的交叉偶联Ullmann反应:芳基碘代物在Cu存在下的自偶联反应Wurtz反应:烷基卤经Na或Mg金属处理后形成碳碳单键Ymada偶联试剂:用二乙基氰基磷酸酯(EtO)2PO-CN活化羧酸缩合反应:Aldol缩合:羰基和一个烯醇负离子或一个烯醇的缩合Blaise反应:腈和a—卤代酯和Zn反应得到B—酮酯Benzoin缩合:芳香醛经CN—催化为安息香(二芳基乙醇酮)Buchner-Curtius-Schlotterbeck反应:羰基化合物和脂肪族重氮化物反应给出同系化的酮Claisen缩合:酯在碱催化下缩合为B—酮酯Corey-Fuchs反应:醛发生一碳同系化生成二溴烯烃》然后用BuLi处理生成终端炔烃Darzen缩水甘油酸酯缩合:碱催化下从a—卤代酯和羰基化合物生成a,B—环氧酯(缩水甘油醛)Dieckmann缩合:分子内的Claisen缩合Evansaldol反应:用Evans手性鳌合剂,即酰基恶唑酮进行不对称醇醛缩合Guareschi—Thorpe缩合(2—吡啶酮合成):氰基乙酸乙酯和乙酰乙酸在氨存在下生成2—吡啶酮Henry硝醇反应:醛和有硝基烷烃在碱作用下去质子化产生氮酸酯Kharasch加成反应:过渡金属催化的CXCl3对于烯烃的自由基加成Knoevenagel缩合:羰基化合物和活泼亚甲基化合物在胺的催化下缩合Mannnich缩合(羰基胺甲基化):胺,甲醛,和一个带有酸性亚甲基成分的化合物之间的三组分反应发生胺甲基化Michael加成:亲核碳原子对a,B-不饱和体系的共扼加成Mukaiyama醇醛缩合:Lewis酸催化下的醛和硅基烯醇醚之间的Aldol缩合Nozaki—Hiyama—KIshi反应:Cr—Ni双金属催化下的烯基卤对于醛的氧化还原加成Pechmann缩合(香豆素合成):Lewis酸促进的酸和B—酮酯缩合成为香豆素Perkin反应:芳香醛和乙酐反应合成肉桂酸Prins反应:烯烃酸性条件下对于甲醛的加成反应Reformatsky反应.有机Zn试剂(从a—卤代酯来)对羰基的亲核加成反应Reimer—Tiemann反应:从碱性介质当中从酚和氯仿合成邻甲酰基苯酚Schlosser对Witting反应的修正不稳定的叶立德和醛发生的Witting反应生成Z—烯烃,而改进的Schlosser反应可以得到E—烯烃Stetter反应(Michael—Stetter反应):从醛和a,B—不饱和酮可以得到1,4—二羰基衍生物。
有机化学人名反应1-50

1. Arndt-Eistert 反应醛、酮与重氮甲烷反应,失去氮并重排成多一个CH2 基的相应羰基化合物,这个反应对于环酮的扩环反应很重要。
2. Baeyer-Villiger 氧化应用过氧酸使酮氧化成酯。
反应中在酮的羰基和相邻的碳原子之间引人一个氧原子。
如由樟脑生成内酯:有时反应能生成二或多过氧化物,但环状酮转变为内酯能得到单一的预期产物。
合适的酸为过硫酸(Caro’s 酸)、过氧苯甲酸、三氟过氧乙酸。
除环酮外,无环的脂肪、芳香酮也可发生此反应。
二酮生成酸酐类、α、β-不饱和酮得到烯醇酯类。
3. Bechamp 还原(可用于工业制备)在铁、亚铁盐和稀酸的作用下,芳香族硝基化合物能还原成相应的芳香胺。
当某些盐(FeCl2、FeCl3、FeSO4、CaCl2 等)存在时,所用酸无论是过量还是少量,甚至在中性溶液中都能够进行这种还原。
此方法适用于绝大部分各种不同结构的芳香族化合物,有时也用来还原脂肪族硝基化合物。
4. Beckmann 重排醛肟、酮肟用酸或路易斯酸处理后,最终产物得酰胺类。
单酮肟重排仅得一种酰胺,混酮肟重排得两种混合酰胺。
但一般质子化羟基的裂解和基团R 的转移是从相反的位置同时进行的。
无论酯酮肟和芳酮肟都会发生此反应。
环酮肟重排得内酰胺,这在工业生产上很重要,利用此反应可帮助决定异构酮肟的结构。
5. Beyer 喹啉类合成法芳香伯胺与一分子醛及一分子甲基酮在浓盐酸或ZnCl2 存在下,反应生成喹啉类化合物。
这是对Doebner-Miller 喹啉合成法的改进。
Doebner-Miller 合成法由芳胺和不饱和醛或酮反应得到喹啉衍生物。
6. Blanc 氯甲基化反应芳香族化合物苯、萘、蒽、菲、联苯及衍生物,在ZnCl2(或NH4Cl、AlCl3、SnCl4、H2SO4、H3PO4 )存在下,用甲醛和极浓盐酸处理,发生芳香化合物的氯甲基化反应。
对于取代烃类,取代基的性质对反应能力影响很亲电取代,烷基,烷氧基一般使反应速度增加,而卤素、羧基特别是硝基用乙醛得到氯乙基化。
有机化学人名反应

有机化学人名反应取代反应:1,加特曼反应:加特曼(GattermannL)发现:用催化量的金属铜代替氯化亚铜或溴化亚铜作催化剂,也可使重氮盐与盐酸或氢溴酸反应制得芳香氯化物或溴化物。
这样进行的反应叫做加特曼反应。
2,加特曼-科赫反应:苯、一氧化碳和氯化氢反应生成苯甲醛,此反应称为加特曼-科赫反应。
3,傅-克反应:芳香化合物芳环上的氢被烷基取代的反应称为傅-克烷基化反应;芳香化合物芳环上的氢被酰基取代的反应称为傅-克酰基化反应;统称傅-克反应。
4,布赫尔反应:萘酚在亚硫酸氢钠存在下与氨作用,转变成相应萘胺的反应称为布赫尔反应。
5,齐齐巴宾反应:吡啶与氨基钠反应,生成-氨基吡啶,如果位已被占据,则得-氨基吡啶,但产率很低。
这个反应称为齐齐巴宾(Chichibabin)反应。
6,刚穆伯—巴赫曼反应:芳香重氮盐中的芳基在碱性条件下与其它芳香族化合物偶联成联苯或联苯衍生物的反应称为刚穆伯(Gomberg)—巴赫曼(Bachmann)反应。
7,柯尔伯—施密特反应:干燥的酚钠或酚钾与二氧化碳在加温加压下生成羟基苯甲酸的反应称为柯尔伯—施密特(Kolbe-Schmitt)反应。
8,威廉森合成法:在无水条件下,醇钠和卤代烷作用生成醚的反应称为威廉森(WilliamonAW)合成法。
9,席曼反应:芳香重氮盐和氟硼酸反应,生成溶解度较小的氟硼酸盐,后者加热分解产生氟苯,这称为席曼(Schiemann)反应。
10,桑德迈耳反应:1884年,桑德迈耳(SandmeyerT)发现:在氯化亚铜或溴化亚铜的催化下,重氮盐在氢卤酸溶液中加热,重氮基可分别被氯或溴原子取代,生成芳香氯化物或溴化物。
这一反应称为桑德迈耳反应。
11,普塑尔反应:一些重氮盐在碱性条件下或稀酸的条件下可以发生分子内的偶联反应。
这个反应是普塑尔(PchorrR)在寻找合成菲环的新方法中首先发现的,故称为普塑尔反应。
12,瑞穆尔—悌曼反应:酚与氯仿在碱性溶液中加热生成邻位及对位羟基醛的反应称为瑞穆尔—悌曼(Reimer—Tiemann)反应。
有机化学人名反应机理

1. Beckma nn 重排* * 肟在酸如硫酸、多聚磷酸以及能产生强酸的五氯化磷、三氯化磷、苯磺酰氯、亚硫酰氯等作用下发生重排,生成相应的取代酰胺,如环己酮肟在硫酸作用下重排生成己内酰胺:反应机理:在酸作用下,肟首先发生质子化,然后脱去一分子水,同时与羟基处于反位的基团迁移到缺电子的氮原子上,所形成的碳正离子与水反应得到酰胺。
迁移基团如果是手性碳原子,则在迁移前后其构型不变,例如:2.Birch 还原芳香化合物用碱金属〔钠"钾或锂)在液氨与醇(乙醇、异丙醇或仲丁醇:)的混合液中旺原,苯坏可被as原成非共純的匕4-坏己二烯化合反应实例取代的苯也能发生还原*并且通常得到单一的还原产物9例如;NaCH3 HaNH3(liq,XEtOH3.Ca nn izzaro 反应4•反应实例4.Chichibab in 反应杂坏碱类,与碱金属的氨基物一起加热时发生胺化反应.得到相应的氨基衍生牺如毗喘与氨基钠反应生成2-氨基毗碇,如果世位已被占据,则得片氨基毗噪,但产率很低*本法是在樂环上引入氨基的简便有敢的方法’广泛适用于各种氮杂芳环,如苯并咪毗异劇t叮喘和菲喘类化合枷均能发生本反应。
哇咻、毗嗪、曙咗、囈醴类化合物反应较为困难。
氨基化试剂除氨基钠、氨基钾外J还可以用取代的碱金属氯优物:f J + Nmc冋--------------- rl反应实例吡啶类化合物不易进行硝化,用硝基还原法制备氨基吡啶甚为困难本反应是在杂环上引入氨基的简便有效的方法,广泛适用于各种氮杂芳环,如苯并咪唑、异喹啉、吖啶和菲啶类化合物均能发生本反应5.Claise n酯缩合反应含有幻氢的酯在醇讷等碱性缩合剂作用下发生縮合作用,失去f子醇得到酸酯。
如2分子乙酸乙酯左金属钠和少量乙醇作用下发生缩含得到乙酰乙酸乙酯。
C a H5ONa2也岁0角论-------- 一GHgCOCH云0心眄75%二元羧酸酯的分子内酯缩合见Dieckma nn 缩合反应。
(完整版)大学有机化学人名反应总结
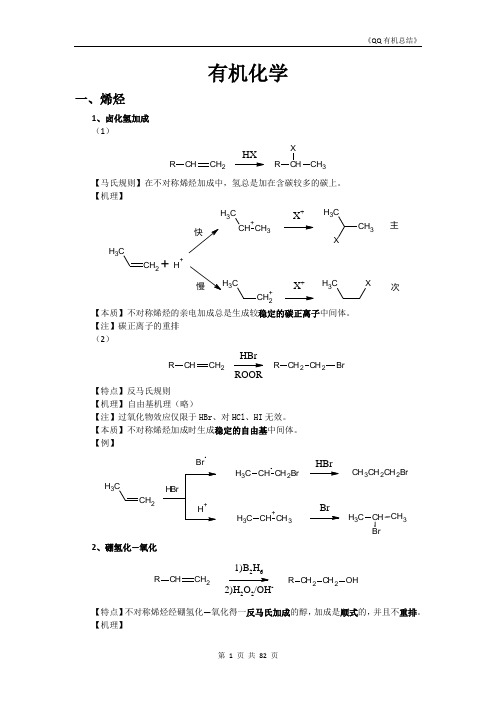
【例】
6、醛(酮)的还原
(1)催化氢化
【注】很多基团都可以催化氢化,如碳碳双键、碳碳三键、硝基、氰基……,所以选择催化氢化还原羰基的时候,要看好化合物是否还有其他可以催化氢化的基团。
(2)用LiAlH4、NaBH4还原
3、醇的氧化
(1)沙瑞特(Sarret)试剂
【注】沙瑞特试剂,是CrO3和吡啶的络合物。它可以把伯醇的氧化控制在生成醛的阶段上,产率比较高,且对分子中的双键无影琼斯试剂是把CrO3溶于稀硫酸中,滴加到醇的丙酮溶液中,在室温下就可以得到很高的产率的酮。同样对分子中的双键无影响。
(格式试剂)
(2)与金属钠反应 武兹(Wurtz)反应
(3)与金属锂反应
【注】二烷基铜锂主要是与卤代烃偶联成烷烃
4、还原反应
5、氯甲基化
五、醇
1、卢卡斯(Lucas)试剂
无水氯化锌与浓盐酸的很合溶液叫卢卡斯试剂,用于鉴别伯、仲、叔醇
2、把羟基变成卤基
(1)、醇与卤化磷(PX5、PX3)
(2)、醇与亚硫酰氯(SOCl2)
4、环氧化合物
(1)开环
①酸性开环
【注】不对称环氧化合物的酸性开环方向是亲核试剂优先与取代较多的碳原子结合。
【例】
②碱性开环
【注】碱性开环,亲核试剂总是先进攻空间位阻较小的,空间效应。
【例】
【注】环氧开环不论酸式还是碱式开环,都属于SN2类型的反应,所以亲核试剂总是从离去基团(氧桥)的反位进攻中心碳原子,得到反式开环产物。这种过程犹如在烯烃加溴时,溴负离子对溴鎓离子的进攻。
【机理】
【注】类似的构型也可发生重排
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
二、还原:
1..Barton-McCombie去氧反应:从相关的硫羰基体中间用n-Bu3SnH,AIBN试剂经过自由基开裂发生醇的去氧作用
2.Birch 还原:苯环由Na单质合液胺条件下形成环内二烯烃(带供电子基团的苯环:双键连接取代基;带吸电子基团的苯环,取代基在烯丙位。
)
3.Brown硼氢化:烯烃和硼烷加成产生的有机硼烷经过碱性H2O2氧化得到醇
4.Cannizzaro歧化:碱在芳香醛,甲醛或者其他无α-氢的脂肪氢之间发生氧化还原反应给出醇和酸
5.Clemmensen还原:用锌汞齐和氯化氢将醛酮还原为亚甲基化合物
6.Corey-Bakshi-Shibata(CBS)还原:酮在手性恶唑硼烷催化下的立体选择性还原
7.Gribble吲哚还原:用NaBH4直接还原会导致N-烷基化,NaBH3CN在冰醋酸当中还原吲哚双键可以解决
8.Gribble二芳基酮还原:用NaBH4在三氟乙酸中还原二芳基酮和二芳基甲醇为二芳基甲烷,也可以应用于二杂芳环酮和醇的还原
9.Luche还原:烯酮在NaBH4-CeCl3下发生1,2-还原形成烯丙位取代烯醇
10.McFadyen-Stevens还原:酰基苯磺酰肼用碱处理成醛
11.Meerwein-Ponndorf-V erley还原:用Al(OPr’)3/Pr’OH体系将酮还原为醇
12.Midland还原:用B-3-α-蒎烯-9-BBN对酮进行不对称还原
13.Noyori不对称氢化:羰基在Ru(II)BINAP络合物催化下发生不对称氢化还原
14.Rosenmund还原:用BaSO4/毒化Pd催化剂将酰氯氢化成醛,如催化剂未被毒化,会氢化为醇
15.Wolff-Kishner-黄鸣龙还原:用碱性肼将羰基还原为亚甲基。