注射剂剂处方工艺研究信息汇总模板
中药注射剂处方点评模板

中药注射剂处方点评模板
一、处方基本信息
1. 处方编号:
2. 处方日期:
3. 处方医生:
4. 处方药名:
5. 处方用量:
6. 处方用法:
7. 患者信息:
二、诊断信息
1. 诊断疾病名称:
2. 诊断依据及病情概述:
3. 治疗方案及目的:
4. 预后评估及注意事项:
三、处方用药合理性评价
1. 药品适应症与诊断是否相符:
2. 药品用法用量是否合理:
3. 药品配伍禁忌及相互作用是否考虑周全:
4. 药品使用是否符合安全性原则:
5. 是否有重复用药情况:
6. 是否考虑患者年龄、性别、病理生理状况等因素:
7. 是否考虑中药注射剂的特殊风险因素(如过敏体质、药物过
敏史等):
8. 是否考虑中药注射剂与其他药物的相互作用:
9. 是否考虑中药注射剂的给药途径、注射速度、滴注时间等参数:
10. 是否考虑中药注射剂的特殊使用方法或注意事项:
四、药学干预建议
1. 是否需要修改药品用量、用法或配伍禁忌:
2. 是否需要提醒医生注意药物不良反应或相互作用:
3. 是否需要建议医生调整治疗方案或更换药品:
4. 其他建议:
五、签名与备注
1. 处方医生签名:
2. 药师签名(如需):
3. 备注(如需):。
奥硝唑注射液的处方及制备工艺研究

半 衰期非 常长 , 具有 良好 的临床 使用 价值 。
5 O 20 5
6
5 O 3o 0
奥硝 唑, g
丙二 醇
限责任公司)WF 5 型微粒分析仪 ( G 一J 天河医疗仪器
有 限公 司 )P A /— 型 安瓿灌 装 封 口机 ( ,G 61 2 上海远 东 机械设备 总厂 ) 。 1 . 2试验 用 药 : 硝 唑原 料 ( 安 博 华 制 药 有 限责 任 奥 西
S 一5 S型药 品稳定 性试 验箱 ( 庆 市永 生实 验仪 HH 10 重 器 厂 )u 10 T 一9 1型紫 外分 光 光 度计 ( 京 普析 通 用有 北
表 1 方 设 计 及 考 查结 果 处
处 方 编 号 1
5 O
2
5 O
3
5 O
4
5 O 2o o
5
本 文对 奥 硝 唑 注射 液 处 方 及 工 艺 进行 初 步研 究 , 以 选 。具 体对 比筛选 过程 见表 1 。 期 得到质 量可 靠 的产 品 , 证 临床 用 药安 全 , 生产 保 为
上处方 改进提供 试验依 据 。 1仪器与试 药 对 上 述 处 方 的样 品暴 露 于 6 ℃环境 中进行 1 d O 0
程 中活性炭在 吸 附杂质 和热源 的 同时 , 主药也 有吸 对
奥硝唑用药安全 、 方便 、 疗效肯定 , 具有 良好 的 附 , 应适 当减少 活性炭 的用 量 。因此在处方 中用冰 故 抗 厌氧 菌和抗 原生 质感 染 作用 , 临床 应用 日趋广 泛 。 醋 酸作 P H调 节剂 , 对 温度 、 并 活性 炭 的用 量 进行筛
注射剂工艺验证模板

注册分类:化学药品4类补充申请(12):新药技术转让注射用XXXXXX(0.1g、0.2g)工艺研究资料——资料项目编号:5-1 (附件2:转让方注射用XXXXXX0.1g工艺验证资料)注册申请机构:XXXXXX注册申请机构地址:XXXXXX2注册申请机构电话:XXXXX注册申请机构负责人:XXXXX原始资料的保存地点:XXXXXX联系人姓名:XXXXX联系电话:XXXXXXX (0)XXXXXXX药品注册申请人名称:XXXXXX验证证书兹有,经按号验证方案实施检查及验证试验,各项结果均合格,通过我公司验证,批准投入使用,特颁此证。
本次验证有效期至:年月日,即在无异常状况下再验证周期为年。
XXXXXX验证委员会主任委员:年月日验证委员会组织机构主任委员:副主任委员:委员:职责:委员会负责本公司GMP验证的日常管理工作及验证方案的起草、实施、审核及批准,委员会实行主任委员会负责制。
验 证 管 理 V ALIDATE ADMINISTER 题目:注射用XXXXXX (0.1g )工艺 颁发部门:质量保证部验证方案制定: 日期: 审核: 日期:批准: 日期:分发:质量保证部、生产组织部、设备动力部1目的为评价注射用XXXXXX (0.1g )冻干粉针剂生产系统要素和生产过程中可能影响产品质量的各种工艺变化因素,特根据GMP 要求制定本验证方案,对其整个生产过程进行验证,以保证在正常的生产条件下,生产出合格、均一、稳定的注射用XXXXXX 冻干粉针剂。
2范围本验证方案使用于在本方案指定的厂房、设施、设备、工艺条件下注射用XXXXXX 冻干粉针剂的生产,当上述条件改变时,应重新验证。
3职责3.1验证委员会3.1.1负责验证方案的审批。
3.1.2负责验证的协调工作,以保证本验证方案规定项目顺利实施。
3.1.3负责验证数据及结果的审核。
3.1.4负责验证报告的审批。
3.1.5负责发放验证报告证书。
3.1.6负责再验证周期的确认。
制剂C T D格式药学研究信息汇总表(4、5.2)
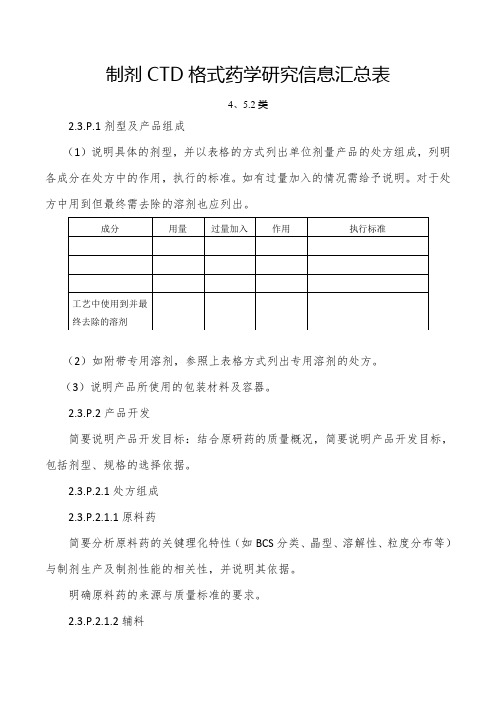
制剂CTD格式药学研究信息汇总表4、5.2类2.3.P.1剂型及产品组成(1)说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
(2)如附带专用溶剂,参照上表格方式列出专用溶剂的处方。
(3)说明产品所使用的包装材料及容器。
2.3.P.2产品开发简要说明产品开发目标:结合原研药的质量概况,简要说明产品开发目标,包括剂型、规格的选择依据。
2.3.P.2.1处方组成2.3.P.2.1.1原料药简要分析原料药的关键理化特性(如BCS分类、晶型、溶解性、粒度分布等)与制剂生产及制剂性能的相关性,并说明其依据。
明确原料药的来源与质量标准的要求。
2.3.P.2.1.2辅料简述辅料及其用量适合所用的给药途径的依据,结合辅料在处方中的作用简述辅料的与制剂性能相关的关键特性。
简述原料药和辅料的相容性试验的情况,包括试验设计、考察指标、试验结果等。
如未进行原料药和辅料的相容性试验,应提供相应的依据。
2.3.P.2.2 制剂研究2.3.P.2.2.1处方开发过程简述处方研究的主要内容。
包括处方开发的基本思路、试验设计、考察指标和方法、试验结果、与原研药的比较研究情况、处方的放大和调整等。
示例如下:某普通片剂的处方研究小结:参考原研药说明书、原辅料相容性试验情况、相关生产经验等,确定了辅料的基本种类;参考原研药的重量和大小、以及各辅料常规用量,确定了辅料的用量范围,以××××为指标,采用××××方法,对××××的种类和用量进行了比较筛选,对××××处方进行了研究,以原研药为对照药,结果显示××××,根据以上研究确定了初步的处方;在以上研究基础上,进行了影响因素稳定性考察,与原研药进行了××××的质量对比;在批量放大过程中,对××××进行了调整,确定了最终处方。
对乙酰氨基酚注射剂的工艺及质量研究

对乙酰氨基酚注射剂的工艺及质量研究对乙酰氨基酚是一种非处方药,常见于家庭药箱中。
对乙酰氨基酚注射剂是一种用于治疗疼痛和发热的处方药物。
本文将从工艺和质量两个方面对对乙酰氨基酚注射剂进行研究。
一、工艺研究1.原料准备对乙酰氨基酚注射剂的主要原料为对乙酰氨基酚和辅料。
对乙酰氨基酚是一种白色结晶性粉末,需要在无菌条件下保存。
辅料包括注射用水、乙醇、羟丙基淀粉、聚山梨酯等,需要按照一定比例进行配比。
2.生产工艺对乙酰氨基酚注射剂的生产工艺主要包括原料准备、混合、灭菌、填充、封口、包装等环节。
其中,混合环节是最为关键的一步,需要精确控制辅料的配比和混合时间;灭菌环节要求对乙酰氨基酚注射剂在防止细菌污染的同时,尽可能减少药物的热力损失。
3.质量评估对乙酰氨基酚注射剂的质量评估主要包括外观、含量、pH值、细菌限度、pyrogen等指标。
其中,含量的检测是最为重要的一项,需要使用特殊的仪器进行检测。
二、质量研究1.含量测定含量测定是评估对乙酰氨基酚注射剂质量的重要指标。
目前常用的含量测定方法有高效液相色谱法、紫外分光光度法等。
本实验采用高效液相色谱法,选用C18色谱柱,以甲醇-水(75:25)为流动相,在波长为254nm处检测峰面积,计算样品中的对乙酰氨基酚含量。
2.pH值测定pH值是衡量对乙酰氨基酚注射剂酸碱性的重要指标。
通常使用数字式pH计在标准条件下进行测定。
对乙酰氨基酚注射剂的pH值通常在4.0-5.5之间。
3.细菌限度测定细菌限度是衡量对乙酰氨基酚注射剂安全性的重要指标。
国家药典规定了不同细菌限度的标准,普通细菌限度不得超过10^2 CFU/mL。
常用的细菌限度检测方法包括真菌细菌总数检测法、限甲氧苄啶嗪菌落计数法等。
4.pyrogen测定pyrogen是一种热性产物,能引起人体炎症反应。
对乙酰氨基酚注射剂中的pyrogen会影响药物的安全性和疗效。
国家药典规定了pyrogen的限度,通常采用大鼠体内试验法或大鼠体外法进行检测。
制剂CTD格式药学研究信息汇总表(1、2、3、5.1类)讲解

制剂CTD格式药学研究信息汇总表1、2、3、5.1类2.3.P.1剂型及产品组成(1)说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成分在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处方中用到但最终需去除的溶剂也应列出。
表1(注:依次编号,下同): 单位剂量产品的处方组成(2)如附带专用溶剂,参照上表格方式列出专用溶剂的处方。
(3)说明产品所使用的包装材料及容器。
2.3.P.2产品开发简要说明产品开发目标,包括剂型、规格的选择依据。
2.3.P.2.1 处方组成2.3.P.2.1.1 原料药简述原料药和辅料的相容性试验结果。
详细信息参见申报资料3.2.P.2.1.1(注明页码)。
简要分析与制剂生产及制剂性能相关的原料药的关键理化特性(如晶型、溶解性、粒度分布等)及其控制。
2.3.P.2.1.2 辅料简述辅料种类和用量选择的试验和/或文献依据。
详细信息参见申报资料3.2.P.2.1.2(注明页码)。
2.3.P.2.2 制剂研究2.3.P.2.2.1 处方开发过程处方的研究开发过程和确定依据参见申报资料3.2.P.2.2.1(注明页码)。
以列表方式说明不同开发阶段(小试、中试、大生产)处方组成的变化、原因以及支持变化的验证研究。
示例如下:表xx:处方组成变化汇总过量投料:过量投料的必要性和合理性依据。
2.3.P.2.2.2 制剂相关特性简要对与制剂性能相关的理化性质,如pH、离子强度、溶出度、再分散性、复溶、粒径分布、聚合、多晶型、流变学等进行分析。
提供自研产品与原研药品在处方开发过程中进行的质量特性对比研究结果,例如:(1)口服固体制剂的溶出度:样品批号、原研药品批号和生产厂;溶出条件,取样点;比较结果。
(2)有关物质:样品批号、原研药品批号和生产厂;测定及计算方法;比较结果。
2.3.P.2.3 生产工艺的开发生产工艺的选择和优化过程参见申报资料3.2.P.2.3(注明页码)。
盐酸艾司洛尔注射液的处方工艺研究

中圉 分 类 号 : Q40. 文献 标 识 码 : 文 章绾 号 :0 636 (o o 一O0 0 .3 T 6 4 A 10 .7 52 l )l 一0 70
Iv sia ino omuaintc n lg f s ll y rc lr e i e t n n e t t nfr lt e h oo yo moo h do ho i jci g o o e d n o
y _ . 82x・ 柚 7 3 RS u r 0 6 : 9 d60 3 qae 8 4: 1 :
节湿度的办法来控制药物的释放度。
参 考 文 献 圉 8 湿 度 .h释放 度的 关 系 2
霉域棱艇 ‘ +b ) 口 t
y= - . 4 g + l 4 4 0 99 掘 0 RS岫r 4 e= . 1 4 0 71 8 6
( ] G. zuk B. P lo , A. hal , n . D es n J 4 A. O tr, 0. a sn T. W et y ad JB. rs s e ma , .
C n r l d Res 1 9 1 2 3. o t ol l. 9 0, 4: 0 e
图 9 湿ቤተ መጻሕፍቲ ባይዱ度 -h释 放 度 的关 系 4
× i u ,I o gs e g C i -hug I tueo e hoo y T i n 1 4 1 C i ) l We- o JA H n-h n ( he s i si t f c n l , ac g2 5 1 , hn g n n n t T g a a
AB TR CT: J CT oN To su yt ep e a aie p o eso s l1h d o ho ieijcin M E S A OB E I td h rp r t r cs fe moo y r c lrd ne t . THODS I v o n
药学研究主要信息汇总表

注:本信息汇总表与国食药监注[2010]387号文中附件中的CTD格式申报主要研究信息汇总表略有不同。
本汇总表适用于已经按照《药品注册管理办法》附件2提交申报资料的品种。
为了提高审评效率,请注册申请人按本汇总表的要求重新整理研究信息并电子提交。
制剂2.3.P.1 剂型及产品组成(1) 说明具体的剂型,并以表格的方式列出单位剂量产品的处方组成,列明各成份在处方中的作用,执行的标准。
如有过量加入的情况需给予说明。
对于处成份用量过量加入作用执行标准工艺中使用到并最终去除的溶剂(2) 如附带专用溶剂,参照上表格方式列出专用溶剂的处方。
(3)说明产品所使用的包装材料及容器。
2.3.P.2 产品开发简要说明产品开发目标,包括剂型、规格的选择依据。
2.3.P.2.1 处方组成2.3.P.2. 原料药简述原料药和辅料的相容性试验结果。
注明信息来源的申报资料编号和页码。
简要分析与制剂生产及制剂性能相关的原料药的关键理化特性(如晶型、溶解性、粒度分布等等)及其控制。
2.3.P.2. 辅料简述辅料种类和用量选择的试验和/或文献依据。
注明信息来源的申报资料编号和页码。
2.3.P.2.2 制剂研究3.2.P.2. 处方开发过程简述处方的研究开发过程和确定依据,包括文献信息(如对照药品的处方信息)、研究信息(包括处方设计,处方筛选和优化、处方确定等研究内容)以及与已上市对照药品的质量特性对比研究结果,并重点说明在药品开发阶段中处方组成的主要变更、原因以及支持变化的验证研究。
注明信息来源的申报资料编号和页码。
以列表方式说明不同开发阶段(小试、中试、大生产)处方组成的变化、原因以及支持变化的验证研究。
示例如下:小试处方中试处方大生产处方主要变化及原因支持依据2.3.P.2. 制剂相关特性简要对与制剂性能相关的理化性质,如pH,离子强度,溶出度,再分散性,复溶、粒径分布、聚合、多晶型、流变学等进行分析。
提供自研产品与已上市对照药品在处方开发过程中进行的质量特性对比研究结果,例如:(1)口服固体制剂的溶出度:样品批号、对照药品批号和生产厂;溶出条件,取样点;说明自研产品与对照药品在不同溶出条件下的溶出曲线比较研究结果,推荐采用f2相似因子的比较方式。
奥硝唑注射液处方工艺研究

・ 6 1・
奥硝 唑注 射液处 方工艺研究
李玉钦 ’ 张 辉 z
( 1 、 哈 尔滨誉衡 药业股份 有限公 司 , 黑龙江 哈 尔滨 1 5 0 0 0 0 2 、 哈 尔滨凯程制 药有 限公 司, 黑龙 江 哈 尔滨 1 5 0 0 0 0 )
摘 要: 目的 : 奥硝 唑 注射 液 样 品 制 备 。方 法: 对 奥 硝 唑 注射 液 处 方 及 工 艺 进行 筛选 。 结 果 : 通过辅料 用量筛选、 活 性 炭 用 量 筛选 、 p H
1 5 ~2 5
表 1灭菌前后变化情况
处方 l
l 5 2
51 2 1
1 5 3 2 4 8 3 毋
4 5 5 5 研
处方 2 2 4 9
处方 3 3 5 5
处方 4 4 5 1 处方 5 5 5 0
与有关物质关系 曲 线
图 1
表 2 活性炭对含量吸漱 明在酸 抬液 中, 温度为 7 0  ̄ C 时 滑l 生炭的吸附效果好 , 本品溶液具有一定黏度, 在温度较高时黏度降低 , 便于脱炭, 奥硝唑对温度 急 定, 因此, 确定吸附、 脱炭时的韫度为 7 0 ℃。 取处方量乙醇 、 聚乙二醇 , 加入注射用水适量 , 搅拌均匀 , 调p H值至 1 . 9 0 , 向其中加奥硝唑 2 5 2 6  ̄调 p H值至 2 . 1 1 , 溶液分成 4份( I、 Ⅱ, Ⅲ, 试样品有关物质略有增加, 含量有所下降 , 但均不高于参 比制剂, 所生产 Ⅳ) , —份 (I ) 未脱炭 , 取三份( Ⅱ, Ⅲ, Ⅳ) , 分别加人 [ 1 1 %、 0 2 %、 O . 5 %( g / m1 ) 的小试样品质量不低于 参 比制 剂; 初步试验结果表明所选用的工艺可行 , 活『 生 炭, 7 0 ℃搅拌 3 0 分钟 , 脱炭, 检查细菌内毒素、 含量, 结果见表 2 。 结果 口 J 以边 步放大生产 。 表明: 用Q 1 %、 0 2 %、 叱 的活 『 生 炭除热原及杂质时, 内毒素检查符合规定 ; 2 . 7中试样品制备及评价 : 按照确定处方及制备工艺制备三批中试样 活 陛炭对奥硝唑均有吸附, 为更好地保证产品质量 , 将活 炭的用量定为 品, 结果表明样品批间—致 激 子 , 产品质量与参E 匕 帝 0 剂等同。 0 L 2 %, 根据活性 断肖 唑吸附量, 确定在生产时按照 1 0 耐 矍 料。 2 8稳定陛砰 劫金 缝 冲 价 :配( . i 黼 I 生砰 罕 . 果表明本品与葡 2 4灭菌条件的筛选。取处方量乙醇、 聚乙二醇, 加入注射用水适量 , 萄糖、 氯化钠配伍良好, 对 中试样品进f 亍 力 口 速6 月试验及长期 1 2 月试验, 搅拌均匀, 调p H值至 1 . 9 8 , 向其中加奥硝唑 5 1 2 舀 调p H值至 2 . 0 4 , 加注射 加速试验及长期试验结果与 0月比铰基本无变化 ,变化趋势与参比制剂 用水 ( 调p H值至 2 3 1 ) 至1 0 0 0 m l , 溶液分成 5 份, —份未灭菌, 其它四份 致, 试殓 目 辕 明本品稳定 好。 分别于 1 0 0  ̄ 2 灭菌 3 0分钟 、 l 1 5 T : 灭菌 3 0分钟 、 1 2 1  ̄ C灭菌 l 5分钟 、 2 9安全 耽 i 胡丝胡 阶: 溶血l 生试验备 探 表 明奥硝唑注射液对 1 2 1 ℃灭菌 3 0 分钟 , 对样品的l 生 状、 无菌 、 有关物质及含量进行考察。与未 家兔红细胞无溶血与凝聚作用 ;血管刺激 f 生 试验结果表明奥硝唑注射液 灭菌相比, 灭菌后无菌检查符合规定 , 有关物质均有所增加 , 含量略有下 对家兔无血管刺激 怍用;过敏l 生 试验结果表明奥硝唑注射液对豚鼠不 降; 1 2 1  ̄ C 灭菌 3 0分钟有关物质增加较大 ,1 1 5 ℃灭菌 3 0 分钟与 1 2 1  ̄ C 产生过敏反应。 灭菌 1 5 分钟所考察指际变化相似,确定本品的灭菌温度为 1 2 1 ℃、 1 5分 质量研究、 稳定陆研究的结果表明三批中试样品的产品质量稳定 , 证
注射剂灭菌工艺研究内容以及资料撰写格式

注射剂灭菌工艺研究内容以及资料撰写格式注射剂按照工艺通常可以分为终端灭菌工艺产品和无菌生产工艺产品,其灭菌/除菌无菌工艺验证的内容也有较大的差别。
下面分别针对终端灭菌工艺产品和无菌生产工艺产品进行阐述。
1、终端灭菌产品的灭菌工艺验证1)热分布试验热原产试验实地考察杀菌过程中杀菌柜各个相同边线的温差状况,为下一步的热反射试验提供数据积极支持。
热原产实地考察通常分成短程热原产和装载热原产实地考察,须要已连续展开三个批次,以保证数据的再现性。
热原产试验主要包含:接收器原产情况;杀菌温度和时间的设置;装载方式和所用样品情况;短程条件下最低温度和最低温度的最小波动数据,冷点的边线和温度;装载条件下最低温度和最低温度的最小波动数据,最冷点和最低温度;试验结论等。
装载热原产试验中应当表明所用样品批号、批量、装载方式等。
例如未使用申报产品展开装载热原产实地考察,应当明晰表明试验所使用的样品的名称、共同组成、规格、外包装形式、装载方式,并评估所使用的样品和装载方式与否能够充分反映样品的实际情况。
2)热反射试验热穿透试验是考察灭菌柜和灭菌程序对待灭菌产品适用性的一项试验,因此热穿透试验一般应采用拟申报的产品进行,并明确产品的批号、批量和装载方式。
在热反射试验中,温度接收器应当填入等待杀菌产品中,挂著温度接收器的产品的放置边线包含热原产试验确认的冷点和高温点。
热反射试验中除了高度关注上述热原产实地考察的内容外,还须要高度关注温度接收器的放置方式,杀菌过程的最小f0值、最轻f0值、平均值f0值等。
3)微生物挑战试验应明确微生物挑战试验所采用的生物指示剂的来源、种类、规格(d值与菌数量)、试验结果,应说明试验中所采用的生物指示剂的耐热性及数量对灭菌工艺是否构成必要的挑战。
生物指示剂的耐热性应大于生产环境和产品中常见污染菌的耐热性。
4)灭菌前微生物负荷的控制除了对杀菌过程的监控外,还必须表明杀菌步骤之前实行了哪些措施监控药液的微生物负荷,并提供更多有关的检验数据;应当提供更多杀菌前产品中的污染菌及其耐热性的监控方法与测量结果;药物溶液在杀菌前的最久置放时间等等。
注射剂的制备研究报告

注射剂的制备研究报告一、引言注射剂是一种常用的药物剂型,用于将药物直接注射到人体血液或组织中。
与其他剂型相比,注射剂具有快速、准确和高生物利用度的特点,适用于需要迅速发挥药效或无法通过口服途径给药的情况。
因此,注射剂的制备研究对于药物研发和临床应用具有重要意义。
二、注射剂的制备技术1. 药物选择和配伍注射剂的制备首先需要选择适用的药物。
药物的选择要考虑其药物相容性、稳定性和生理活性等因素。
此外,当需要同时给予多种药物时,还需要考虑药物的相互作用和配伍稳定性,以避免发生不良反应或降低疗效。
因此,药物选择和配伍是注射剂制备的关键步骤。
2. 剂量确定和溶解制备注射剂时,需要确定合适的药物剂量。
这需要综合考虑患者的年龄、性别、病情和体重等因素,以达到最佳疗效。
同时,某些药物可能需要进行溶解,以便更好地通过注射途径给药。
对于难以溶解的药物,可以考虑使用辅助溶剂或改良剂型,以提高药物的溶解度和稳定性。
3. 质量控制和灭菌在注射剂的制备过程中,质量控制是至关重要的。
制备过程中需要进行严格的质量检测,包括药物纯度、配方准确性、质量稳定性等。
此外,注射剂还需要进行灭菌处理,以消除可能存在的微生物污染。
灭菌方法可以选择热灭菌、辐射灭菌或滤过灭菌等方法。
4. 包装和储存完成制备的注射剂需要进行适当的包装和储存,以确保其质量和稳定性。
包装材料应具有良好的密封性和防漏性,以防止药物的污染和失效。
同时,注射剂的储存条件也需要控制在一定的温度和湿度范围内,以延长其有效期。
三、注射剂制备研究的挑战和发展方向1. 制备工艺的优化注射剂的制备过程中存在许多工艺参数,如溶解度、溶解温度、溶解时间、配方比例等。
优化这些制备工艺参数可以提高药物的溶解度和稳定性,从而提高注射剂的质量和疗效。
2. 新型载体和输送系统的研发随着药物的多样化和个体化需求的增加,需要开发新型的载体和输送系统以满足不同的药物给药要求。
这包括纳米颗粒、脂质体、聚合物等,可以提高药物的生物利用度和目标输送效果。
中药注射剂类药品生产工艺和处方核查工作汇总表

中药注射剂类药品生产工艺和处方核查工作汇总表
1. “中药注射剂”是指批准文号为“国药准字Z”格式的所有注射剂。
2.表内各项仅针对中药注射剂,包括静脉给药者。
每个药品批准文号作为一个品种计数。
3.“未曾生产品种数”是指获得国家药品注册或地标升国标后未曾批量生产的品种数。
4.“停止生产品种数”是指获得国家药品注册或地标升国标后曾经批量生产、现因停止生产未予核查的品种数。
5.“存在来源或评估问题的品种”是指专题列明有关遗留问题、需审核确认的品种。
6. 本表由省(区、市)局汇总填写并加盖公章后报送,原已报送总结的亦须填报。
可将本表传真至:************,并发电子邮件至:************.cn。
甘草酸二铵注射液工艺处方的研究

甘草酸二铵注射液工艺处方的研究目的探讨甘草酸二铵注射液的处方筛选、制备工艺、影响因素试验,确定产品的最佳处方和工艺。
方法通过对不同pH值、药用炭用量的筛选确定最佳处方,并对所生产的产品进行影响因素试验。
结果确定甘草酸二铵注射液在pH为6.5~7.5,活性炭用量为0.1%,按此工艺生产的产品对光照和高温均比较稳定。
结论本研究研制的甘草酸二铵注射液的工艺处方合理,质量稳定。
[Abstract] Objective To explore the optimum formulation and preparation process of diammonium glycyrrhizinate injection by studying prescription screening,preparation technology and impact factors. Methods To determine the best prescription according to different pH and carbo activatus dosage screening,and investigate the stability of the product through influence factor experiments. Results When the pH was adjusted between 6.5-7.5,and 0.1% activated carbon was used,the products prepared using confirmed formulation and preparation process proved good stability under light and heat. Conclusion This study developed two ammonium glycyrrhetate injection process prescription is reasonable,the quality is stable.[Key words] Diammonium glycyrrhizinate injection;Prescription screening;Preparation process;Influence factor甘草酸二铵是从中药甘草中高效分离,经差向异构筛选出的α体的甘草酸二铵盐,具有较强的抗炎作用。
注射剂处方工艺变更主要研究报告信息汇总模板

注射剂处方工艺变更主要研究信息汇总模板一、品种概述1.1同品种上市背景信息:包括品种国内外上市情况、国家标准和国内外药典收载情况,已上市产品的剂型、规格和适应症。
1.2申报品种获准上市的信息,包括规格、批准文号、批准时间、执行标准、有效期、包材,以及最近一次再注册的情况等内容。
1.3简述变更事项简述变更事项。
若非首次申报且未被批准,应简述未获批准的原因。
二、立题合理性根据同品种上市背景信息及本品种最新的研究进展,对该化合物、剂型、规格的立题合理性进行自我评价。
三、变更内容及变更理由3.1处方变更以文字或列表方式说明变更前处方组成、变更后处方组成,主要变化及原因。
范例:3.2工艺变更以文字或列表方式说明变更前生产工艺、变更后生产工艺,主要变化(包括批量、设备、工艺参数等的变化)及原因范例:对于工艺变更的补充申请,应同时提供处方,明确说明处方是否发生变更。
3.3关联变更说明关联变更的具体事项和理由。
四、变更研究4.1 变更的合理性评价和风险分析根据《已上市化学药品变更研究的技术指导原则》,基于具体问题具体分析的原则对变更内容进行风险分析。
分析变更对药品质量的影响,确定变更的合理性和变更风险。
4.2 处方变更研究4.2.1 原料药提供原料药的供应商、批准文号、质量标准,说明原料药的来源和质量控制是否变更。
简述与注射剂处方工艺变更相关的原料药的理化性质,如溶解性、稳定性(光照、温度、不同pH 值等)等,分析对注射剂生产过程控制和质量控制的影响。
4.2.2 辅料提供辅料来源、级别、批准文号、质量标准,重点列出变更后新增辅料信息。
对于特殊辅料,应简述安全应用限度和相关依据。
4.2.3 处方筛选研究结合变更情况和原研产品处方,简述处方的研究开发过程和确定依据,包括辅料种类和用量的筛选工作等。
4.3 工艺变更研究4.3.1 生产工艺完整描述变更后的生产工艺。
4.3.2 工艺筛选研究结合变更情况,简述生产工艺的选择和优化过程,列出相应的关键工艺步骤及工艺参数控制范围、中间体质量控制标准。
复方甘草酸苷注射液处方及工艺研究

加强该注射液的性能, 治疗更多患有慢性肝 炎的患者 。
l仪 器 与试 药
1 . 1仪 器 L a b A l l i a n c e高效液相色谱仪 , Y B一 2澄 明度检测仪 , B C 一 5 0 1 低温光照仪、 F Z H一 1 0小型定量灌装机 、 R F F 一 0安瓿熔封机。 1 . 2试药 草酸单铵盐 , 甘氨 酸, D I . 一 蛋氧酸 , 无 水亚硫酸钠 , 氯化钠 , 活性炭。
2方 法 与结 果
的检测方法 是对药物 的酸碱度进行检测 , 将其 中药物 的含量一一的测定 出 来, 看其是否满足 国家 的相应规定。 在完成上述工序后, 如果测得 的结果符 合标准, 就可以进行灌装 了, 分别将试剂装入相应 的经过灭菌的安瓿瓶 中, 通常安瓿瓶的容积为 2 0 ml ,最后将装有试剂 的安瓿瓶 再次放 入相关的设 备中进行杀菌 的处理 , 在检验完毕后 , 装入库房等待装配 。 这就是该试剂的 具体工艺, 每一环 节对于试剂 的效 果都具有 明显 的影响 , 要想 有效的保证 该试剂的性 能, 就要严格对试 剂的加工工艺进行监督与处理 。
响, 下面针对该注射 液进 行详细的介绍 , 从相 关的处方到具体 的工艺过 程 开展分析 , 希望 能够 为相关的工作人员提 供帮助, 在此基础上进 一步
一 一
意对试剂的剂量进行严格 的测量 , 如果剂量 的检测不准确 , 将会对药物造
成直接的影响, 不仅无法达到治疗的 目的, 反而极有可能起 到副作用。 按照 定 的顺序将各种试剂配入 到器皿中后 , 进行均匀 的搅拌 , 直至溶液得到 完全的溶解 , 并加入适量的水进行稀释 , 随即使用药物炭进行过滤的处理 。 在这一过程 中, 要 时常对药物的性能进行检测 , 防止 出现严重的情况 , 具体
注射剂的制造工艺与配方研究

注射剂的制造工艺与配方研究注射剂是一种常用的药物制剂形式,广泛应用于临床治疗和药物研发领域。
随着科技进步和制造工艺的不断改进,注射剂的制造工艺与配方研究也在不断地进行着。
一、制造工艺注射剂制造工艺是注射剂研发和生产的重要环节,其过程包括原料准备、制剂配方、加工工艺、灭菌、灌装和包装等多个环节。
1.原料准备原料准备是注射剂生产的第一步,要求原料的质量稳定、纯度高、无毒性、易于加工等特点。
2.制剂配方制剂配方是注射剂生产的关键环节,其样品应经过多次反复试验才能成功确定最终配方。
3.加工工艺加工工艺是注射剂生产中的一个重要环节。
该过程主要包括混合、调整pH值、浓缩、干燥等步骤。
4.灭菌灭菌是制造注射剂过程中不可或缺的环节。
该过程旨在杀死注射剂中可能存在的细菌和微生物。
5.灌装和包装灌装和包装是注射剂生产的最后一步。
灌装后的注射剂应进行检测,以确保其满足相关质量标准和要求。
最后再进行包装,以防止注射剂的变质和污染。
二、配方研究注射剂的配方研究也是注射剂研发和生产中的重要环节。
注射剂的配方研究需要全面考虑药剂的物理化学特性和药代动力学等因素,制定合理的药品配方,同时注重药品与人体的相容性和安全性。
在注射剂的配方研究中,应根据药物的性质和机理,选择不同的添加剂。
例如:某些药物对渗透压敏感,因此需要添加渗透压调整剂;某些药物易发生光降解,因此需要添加光稳定剂来稳定药物结构。
同时,注射剂配方研究也需要考虑药品的制剂类型和应用方式。
比如:根据药物的溶解度和吸收机制,选择适当的配方以保证药物在体内的吸收和分布。
在配方研究中,药品的稳定性也是一个重要的问题。
注射剂中往往含有具有化学反应活性的成分,因此要考虑药品与容器、溶剂和其他成分的相互作用,以确保药品的稳定性和卫生安全性。
总之,注射剂的制造工艺和配方研究是注射剂生产过程中的重要环节,需要综合考虑药剂的物理化学特性和药代动力学等因素,制定合理的药品配方,并确保制剂质量和安全性。
氨溴索注射乳剂的处方及工艺研究

氨溴索注射乳剂的处方及工艺研究张会丽【摘要】Objective To determine the formulation and preparation technology of Ambroxol Injectable Emulsion. Methods Ambroxol In-jectable Emulsion was prepared by using soybean oil as the oil phase, soybean phospholipid and poloxamer as the complex emulsifying agent. The isoosmotic adjusting agent was also added into the formulation, the formulation and preparation technique were selected by changing the oil phase ratio, emulsifying agent, preparation temperature, dispersing time, preparation pressure and sterilization meth-ods. Results The optimal formulation was 0. 25% of ambroxol, 10% of oil phase, 1. 2% of emulsifying agent, 1. 0% of stabilizing agent and 2. 25% of glycerol, adding water to the total amount. The optimal technology was the mixture and emulsification of the water phase and the oil phase at 70℃, which was homogenized for 5 min at 19 000 r/min, the final emulsion was obtainedby microfluidizer for 6 times under 60 psi pressure, sterilized with nitrogen for 30 min. Conclusion The physical stability of the prepared Ambroxol Injectable Emulsion is good, which provides a basis for the development of the product.%目的:确定氨溴索注射乳剂的处方及制备工艺。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
注射剂处方工艺研究信息汇总模板
一、品种概述
1.1同品种上市背景信息:包括品种国内外上市情况、国家标准和国内外药典收载情况,已上市产品的剂型规格和适用症。
1.2 申报品种获批上市的信息,包括规格、批准文号、批准时间、执行标准、有效期,以及最近一次再注册的情况等内容。
1.3简述变更事项
简述变更事项。
若非首次申报且未被批准,应简述未批准原因。
二、立题合理性
根据同品种上市背景信息及本品种最新的研究结果,对该化合物,剂型变更处方工艺的立题合理性进行自我评价。
三、变更内容及理由
3.1 处方变更
以文字或列表方式说明变更前后处方组成,主要变化及原因。
列表方式如下:范例:
以文字或列表方式说明变更前后生产工艺,主要变化(包括批量,设备,工艺参数等的变化)及原因。
列表方式如下:
范例:
3.3 关联变更
说明关联变更的具体事项和理由。
四、变更研究
4.1 变更的合理性评价和风险分析
根据《已上市化学药品变更研究的技术指导原则》,基于具体问题具体分析的原则对变更内容进行风险分析,分析变更对药品质量的影响,确定变更的合理性。
4.2 处方变更研究
4.2.1 原料药的主要理化性质
提供原料的供应商、批准文号、质量标准,说明原料的来源和质量控制是否变更。
简述处方工艺变更相关的原料药的主要理化性质,如溶解性、稳定性(光照、温度、不同pH值等),分析对制剂生产过程和质量控制的影响。
4.2.2 辅料的来源、级别、质量标准
提供辅料的来源、级别、批准文号、质量标准,重点列出变更后新增辅料的详细信息,对于特殊的辅料应说明安全应用限度及其依据。
4.2.3 处方筛选研究
结合变更情况和原研产品处方,简述处方的研究开发过程和确定依据,
包括辅料种类和用量的筛选工作。
4.3 工艺变更研究
4.3.1 生产工艺
完整描述变更后的生产工艺。
4.3.2 工艺筛选研究
结合变更情况,简述生产工艺的选择和优化过程,列出关键的工艺步骤及工艺参数控制范围、中间体质量控制标准。
4.3.3 生产工艺验证和灭菌工艺验证
结合生产工艺变更情况,简述生产工艺验证情况小结。
如果涉及到生产工艺的局部变更,可重点对变更部分进行研究验证,如果涉及的生产工艺的整体变更,应对完整的生产工艺进行研究和验证。
结合灭菌工艺变更情况,简述灭菌工艺验证情况小结。
范例:
本品采用XX灭菌柜,灭菌工艺为XXX,灭菌工艺验证包括空载热分布、满载热分布、热穿透、生物指示剂试验,验证采用样品为XXX,规格XXX,采用XX包装,装载方式为XXX,共设置XX个温度探头,分布位置为XXX,空载热分布结果显示冷点为XX,热点为XX,各点与平均温度的差最高为XX,符合XXX的要求。
满载热分布结果显示冷点为XX,热点为XX,各点与平均温度的差最高为XX,符合XXX的要求。
热穿透试验结果显示F0值为XX,各点与平均F0值的差最高为XX,符合XXX的要求。
生物指示剂试验采用XXX菌,规格为XX,来源为XX,试验结果为XX。
4.4 包装材料/容器
说明包装材料/容器是否变更,如变更,以文字或列表方式说明变更前后说明包装材料/容器的变化情况、原因以及支持变化的研究验证情况。
具此部分可参照《包装材料/容器变更研究信息汇总模板》的相关内容进行。
4.5 质量研究
4.5.1 质量标准
说明企业原执行标准和本次拟定的新标准,标准中有无因处方工艺变更所导致的关联变更项目。
说明该品种国内外药典收载情况,以文字或列表方式对原执行标准、现行版中国药典标准以及现行版国外主流药典标准进行比较。
对原执行标准/拟定标准是否符合现行技术要求进行自我评价。
如不符合现行技术要求,应进行系统的方法学研究和修订。
范例:
4.5.2 方法学研究与验证
根据处方工艺变更及拟定质量标准的变更情况,对变更的项目进行相应的方法学研究和验证,对有关物质、溶出度、含量等项目的检验方法的适用性进行分析,决定是否需要进行方法学验证。
应对进行的验证工作能否支持检测方法的可行性进行自我评价。
应在杂质谱分析的基础上评价有关物质方法的合理性,以列表的方式列明可能存在的杂质,如涉及到方法学的变更,应提供方法学筛选和验证的总结信息。
范例:
本品的杂质谱分析如下:
筛选,结果显示BP2014版XXX标准的有关物质方法对已知杂质的分离度良好,杂质检出量和检测个数较多。
因此采用该方法作为本品有关物质的检测方法,并进一步进行验证,如下表:
4.5.3.1 质量对比样品
说明质量对比研究中所采用的参比制剂(应为原研),以及参比样品的生产商、批号、规格等信息,如参比样品非原研品,应说明参比样品的选择依据。
说明资质样品的批次、批量、批号、生产地点等相关信息。
4.4.3.2 质量对比研究
根据变更的具体情况,剂型特性和药物性质,参考质量标准,选择适当的项目与参比样品进行质量比较研究。
杂质对比研究应列出杂质谱比较研究结果。
范例:
4.5 批检验报告
提供三个连续批次(批号:)的检验报告,应提供样品的生产时间、地点、批量等信息。
4.6 稳定性研究
以文字或列表方式与变更前样品或变更前样品的稳定性数据(如方法学适用)进行比较研究情况,应说明变更前后样品批号和批量,对关键项目如有关物质、含量等,列出具体检测数据,比较变化趋势。
根据已上市样品的内包装材料、贮藏条件、有效期等,结合变更后样品的稳定性研究情况、变更前后样品稳定性的比较研究情况、已上市产品的贮藏条件和有效期等,说明变更后样品的内包装材料、贮藏条件和有效期的确定依据。