实验3 两条序列比对与多序列比对
实验3 两条序列比对与多序列比对
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
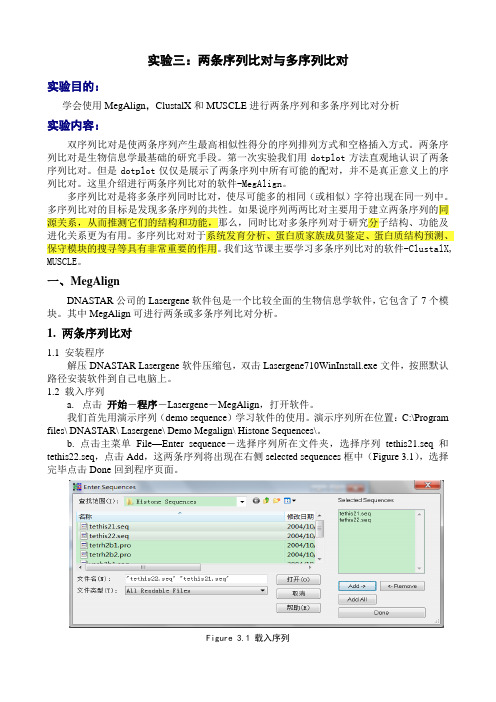
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
多序列比对方法

[编辑] 全局比对 全局比对是指将参与比对的两条序列里面的所有字符进行比对。全局比对主要被用来寻找关系密切 的序列。由于这些序列也都很易通过本地比对方法找到,现在全局比对也有些被认为只是一种技 巧。另外,全局比对在应用于分子进化时也有些问题(比如domain shuffling -见下),这也限制 了这种方法的可用性。
发博文
博文 搜×索
人-机-地的时空交互
/gistime [订阅] [手机订阅]ຫໍສະໝຸດ 首页 博文目录 图片 关于我
个人资料
Geoinformatics
Qing
微博
正文
字体大小:大 中 小
序列分析(序列比对) (2012-02-15 18:32:40)
标签: 校园 分类: 工作篇
序列分析是指通过一定的方法确定DNA上核苷酸排列的顺序,包括序列比对。序列分析是分子生物学 的重要技术之一。
参考条目 l 序列比对
外部链接 l Sequence analysis - 123 Genomics l Nucleic sequence analysis - 巴斯德研究院
加好友 写留言
发纸条 加关注
艺术类期刊《金田》编辑部征稿 正规期刊论文
材料作文“树根的命运”写作指 柳栖士
更多>>
推荐博文
美女大学生激励球队的背后(图) 昕薇
全美国实习薪水最高的10家技术 北外网院
那年,我的饭香四溢的高三 春黛同安
盘点2013加拿大最受学子青睐 启德教育集团
【留澳需知】承认高考成绩的12 启德教育集团
【原创】拿下美国大学offer 启德北京
实习四:多序列比对(Multiple alignment)

实习四:多序列比对(Multiple alignment)学号姓名专业年级实验时间提交报告时间实验目的:1. 学会利用MegAlign进行多条序列比对2. 学会使用ClustalX、MUSCLE 和T-COFFEE进行多条序列比对分析3. 学会使用HMMER进行HMM模型构建,数据库搜索和序列比对实验内容:多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似区域只能通过将多个序列同时比对才能识别。
只有在多序列比之后,才能发现与结构域或功能相关的保守序列片段,而两两序列比对是无法满足这样的要求的。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻以及PCR引物设计等具有非常重要的作用。
作业:1.Align the orthologous nucleotide and protein sequences from 5 organisms you found from first practice with MegAlign. Describe the sequences you used (the title of each sequence), explain whether the phylogenetic tree is consistent with the species tree from NCBI taxonomy database. Set the alignment report to show consensus strength and decorate the residues different from consensus with green shade.(Hint: use the taxonomy common tree from NCBI to get the evolutionary relationship among the organisms. Save your organism name in a text file with each organism name in a line, and upload the file, choose Add from file, and you will see the relationship among the specified organisms) /Taxonomy/CommonTree/wwwcmt.cgiHint 2:Change the accession number in your fasta or genPept format sequence file to organism name, so that the phylogenetic tree can be easily understood.方法与结果:打开Megalign,选择FILE下的Enter sequence ,打开之前保存的来自于五个物种的蛋白(或核酸)序列;首先选择打分矩阵,点击“Align”,选择Set residue Weight Table 选择矩阵:PAM100(核酸则设为weighted),通过“method parameters”查看参数,使用Clustal V的默认值;其次进行序列的比对,选择Align下的“by Clustal V Method”开始比对,再次待其结束后,进行比对结果的显示,选择view下的“Phylogenetic Tree”,显示出树形图;(图)与NCBI上找到的树形图进行对比(图);接下来点击View 下的“Alignment reports ”,选择OPTIONS下的“Alignment report contents”勾中“show consensus strength”,即在序列中显示出相似性条块;在OPTIONS下选择“New decorations”对decoration parameters 下选“shade—residues differing from—the consensus”把字符选择现实的颜色为绿色,结果显示如下:(图)同法可以得到核酸的树形图:(图)分析:系统发育树与NCBI上的物种树有很大的差异,因为可能这些物种间含有很多同源序列,我们不能单凭几条相似序列的同源关系来判断物种的亲缘关系,而应该考虑到物种更多相似序列的同源关系。
第三章 序列两两比对

序
言
序列相似(similarity)与序列一致(identity)
序列比对中用到的另一对相关术语是序列相似与序列一致。这两个 概念对于核苷酸序列是同义的。而对于蛋白质序列,这两个概念是非常 不同的。在蛋白质序列比对中,序列一致是指待比对的两条序列中相同 残基匹配的比例;序列相似是指待比对的两条序列中很容易彼此替换具 有相似理化性质残基匹配的比例。有两种方法计算序列相似/一致度。 一种方法是用两条序列的全部长度,而另一种是利用较短的序列进 行标准化。第一种方法用如下公式计算序列相似度: S=[(Ls*2)/(La+Lb)]*100 其中S是序列相似的百分比,Ls是相似的残基数目,La和Lb分别是两条 序列的长度。
3
序
进化基础
言
DNA和蛋白质是进化的产物。它们可以被认为是编码数百万年 进化史的分子化石。在进化史上,这些分子经历了随机变化过程,期 中一些被进化所选择而保留了下来。这些被选择的序列逐渐积累突变 和交叉,进化的痕迹在序列的某些部分被保留下来从而可以识别它们 共同的祖先。进化痕迹的存在是由于一些对序列结构和功能起关键作 用的残基倾向于被自然选择所保留;而另一些不起关键作用的残基倾 向于频繁的改变。例如,一个酵母家族的活性位点残基倾向于被保存 下来是由于它们对催化功能起作用。所以,通过序列比对,保守的和 改变了的序列模式就能被识别出来。在比对中序列的保守度体现了不 同序列之间的进化关系。反之,序列之间的差别反映了在进化的过程 中序列以替换、插入和删除残基的形式发生了变化。
15
序列比对的方法
16
序列比对的方法
17
序列比对的方法
点阵方法
点阵法有许多变形。例如,一条序列可以和它自身比对以识别内部 重复元素。在自比对当中会存在一条主对角线以表示其完美匹配。如果 内部重复元素存在,会观察到在主对角线的上方或下方有短的对角线。 DNA序列的自补(也叫反向重复),例如那些存在发夹结构的家族,也 能用点距阵法识别。在这种情况下,一条DNA序列与它的反向补序列进 行比较。平行的对角线代表反向重复。为了比较蛋白质序列,必须使用 一个权重系统来描述氨基酸残基的相似度。
多序列比对

的序列,结果不理想。
• 目前应用最广的多序列比对算法。
34 /93
ClustalW2
• ( /Tools/msa/clustalw2 )目前应用 最广的多序列比对工具。 • 3个步骤:
1.) Construct pairwise alignments(构建双序列比对)
0 1 A 0 1 A 1 -2 A 2 T 3 T 3 G 3 -4 C 4 C
x coordinate y coordinate
--
A
T
G
C
•
13 /93
Alignment Paths
• Align 3 sequences: ATGC, AATC,ATGC
0 1 A 0 1 A 0 0 1 -2 A 1 2 T 3 T 2 3 G 3 -3 4 C 4 C 4
之前我们进行的是序列与序列的比对。 Can we align a sequence against a profile?
Can we align a profile against a profile?
Profile和pattern都可以表示多序列比对,哪个更好?
29 /93
Multiple Alignment: 贪婪算法
v1 v2 v3 v4
v1,3 v1,3,4 v1,2,3,4
Calculate: = alignment (v1, v3) = alignment((v1,3),v4) = alignment((v1,3,4),v2)
• 之前进行的是双序列比对
• 如果多个序列进行比对呢? 如何进行?
4 /93
Multiple Alignment versus Pairwise Alignment
Clustalx多序列比对-生物信息学

Clustalx多序列比对-生物信息学实验三:多条序列比对——Clustalx实习目的:了解掌握Clustalx软件的应用,学会做多条序列比对并分析。
实习内容:一、ClustalX的使用Clustal是一种利用渐近法(progressive alignment)进行多条序列比对的软件。
即从多条序列中最相似(距离最近)的两条序列开始比对,按照各个序列在进化树上的位置,由近及远的将其它序列依次加入到最终的比对结果。
1. 准备要比对的序列请查找至少存在于5个物种中的同源序列(核酸或蛋白质皆可),并保存为fasta格式,存为文本文件(所有的序列请粘贴到同一个文本文件中)。
选择NM、XM或NP打头的序列,不要选择NC或NW打头的序列,那是全基因组序列。
建议关键词:hemoglobin,trypsin, peroxidase, p53, Superoxide Dismutase, h5n1, etc.2. 打开clustalX程序开始菜单,程序,clustalX2- clustalX23. 载入序列点最上方的File菜单,选择Load Sequence-选择你刚保存的序列文件,点打开。
”后的字符。
注意:ClustalX程序无法识别汉字,无法识别在左侧窗口里是fasta格式序列的标识号,取自序列第一行“>带空位的文件夹名,如 my document。
各位同学的序列文件不要保存在桌面上或带汉字的文件夹中,推荐保存在D盘根目录下。
4. 比对参数的选择可以对两条序列比对的参数和多条序列比对的参数进行设置。
a. 两条序列比对的参数设置点击Alilgnment菜单,选择Alignment Parameters,再选择Pairwise Alignment Parameters。
首先可以选择比对的效果,是slow/accurate 还是fast/approximate。
第一种模式采用的是动态规划算法进行比对的,第二种模式采用的是启发式的算法。
第四章:双序列比对

Finding k-tups
position 1 2 3 4 5 6 7 8 9 10 11 protein 1 n c s p t a . . . . . protein 2 . . . . . a c s p r k position in offset amino acid protein A protein B pos A - posB ----------------------------------------------------a 6 6 0 c 2 7 -5 k 11 n 1 p 4 9 -5 r 10 s 3 8 -5 t 5 ----------------------------------------------------Note the common offset for the 3 amino acids c,s and p A possible alignment is thus quickly found protein 1 n c s p t a | | | protein 2 a c s p r k
比对的算法
Needleman-Wunsch Smith-Waterman算
算法适用于整体水平 上相似性程度较高的 2个序列。是整体比 对算法,其结果反映 了两个序列中所有残 基地整体相似性。
法在识别局部相似性 时,具有很高的灵敏 度,但只是寻找序列 中一些小的、具有局 部相似性的片断。
Basic Pairwise Alignment
列片断,称为k-tuple. 用于蛋白质序列比对时,k- tuple长度为1~2个 残基,用于DNA序列比对时, k- tuple长度最多 为6个碱基。 通过比较2个序列中断片断及其相对位置可以构 成一个动态规划矩阵地对角线方向上的一些匹 配片断 期望值E:E值越接近0,表明2序列第匹配不大 可能是由随机因素造成的,即E值越低,置信 度越高。
实验3两条序列比对与多序列比对

实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
第3章序列比对[1]
![第3章序列比对[1]](https://img.taocdn.com/s3/m/82e92b74a417866fb84a8ec0.png)
contents
3.1概述 3.2两条序列比对方法 3.3多条序列比对方法
3.1概述
3.1.1序列比对的概念 3.1.2生物序列之间的关系
3.1.1序列比对的概念
⑴序列比对(Sequence
alignment)
序列比对是序列相似性分析的常用方法,又称序 列联配。 通过将两个或多个核酸序列或蛋白序列进行比 对,显示其中相似的结构域,这是进一步相似性 分析的基础。通过比较未知序列与已知序列的一 致性或相似性,可以预测未知序列功能。
Query: 181 catcaactacaactccaaagacacccttacacccactaggatatcaacaaacctacccac 240 |||||||| |||| |||||| ||||| | ||||||||||||||||||||||||||||||| Sbjct: 189 catcaactgcaaccccaaagccacccct-cacccactaggatatcaacaaacctacccac 247
一致性(identity)
Identity: The extent to which two (nucleotide or amino acid) sequences are invariant. 当两条序列同源时,它们的氨基酸序列或核苷酸序列通常 有显著的一致性(identity)。 一致性反映的是两个氨基酸序列(或核苷酸序列)之间相 同的程度。 因此,同源性是序列同源或不同源的一种论断,而一致性 和相似性是一种描述序列相关性的量。
⑵同源性、相似性、一致性
同源性(homology)
Homology: Similarity attributed to descent from a common ancestor.
多序列比对

对于数目较少且较短的序列来说都不 切实际
多维的动态规划算法
Sequence 1
Sequence 2
分而治之方法
分而治之 (Divide and Conquer, DCA)方法 将MSA的空间复 杂度减小 DCA在线MSA
http://bioweb.pasteur.fr/seqanal/int erfaces/dca-simple.html
So in effect …
Sequence 1
Sequence 2
SP(Sum of Pairs)方法
为了找到最佳比对,并解决动态 规则算法的计算复杂问题, Carrillo & Lipman (1988)发明了 SP(Sum of Pairs)方法
进行所有序列间的双序列比对
基于双序列比对分数产生一个相邻连 接进化树(neighbor-join tree) 根据进化树提供的序列间关系按顺序 对序列进行比对 比对可以用以下两种方法: - slow/accurate - fast/approximate
CLUSTALW
******** CLUSTAL W (1.8) Multiple Sequence Alignments ******** 1.Sequence Input From Disc 2. Multiple Alignments 3. Profile / Structure Alignments 4. Phylogenetic trees S. Execute a system command H. HELP X. EXIT (leave program)
多序列比对

实验六:多序列比对- Clustal、MUSCLE西北农林科技大学生物信息学中心实验目的:学会使用Clustal 和MUSCLE 进行多序列比对分析。
实验内容:多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似区域只能通过将多个序列同时比对才能识别。
只有在多序列比对之后,才能发现与结构域或功能相关的保守序列片段,而两两序列比对是无法满足这样的要求的。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守motif 的搜寻等具有非常重要的作用。
我们这节课主要学习两个广泛使用的多序列比对软件-Clustal、MUSCLE。
一、Clustal/Clustal 是一种利用渐近法(progressive alignment)进行多条序列比对的软件。
即先将多个序列两两比较构建距离矩阵,反应序列之间的两两关系;随后根据距离矩阵利用邻接法构建引导树(guide tree);然后从多条序列中最相似(距离最近)的两条序列开始比对,按照各个序列在引导树上的位置,由近及远的逐步引入其它序列重新构建比对,直到所有序列都被加入形成最终的比对结果为止(Figure 6.1)。
Clustal 软件有多个版本。
其中Clustalw 采用命令行的形式在DOS 下运行;Clustalx 是可视化界面的程序,方便在windows 环境下运行;Clustal omega 是最新的版本,优点是比对速度很快,可以在短短数小时内比对成百上千的序列,同时由于采用了新的HMM 比对引擎,它的比对准确性也有了极大的提高,有DOS 命令行和网页服务器版。
我们今天主要学习clustalx 的使用。
范例1. 采用clustalx 进行多序列比对。
多序列比对的实验报告

一、实验目的1. 掌握多序列比对的基本原理和方法。
2. 熟悉使用BLAST、CLUSTAL W等工具进行多序列比对。
3. 分析比对结果,了解序列间的进化关系。
二、实验原理多序列比对是指将两个或多个生物序列进行排列,以揭示序列间的相似性和进化关系。
通过比对,可以识别保守区域、功能域和结构域,为生物信息学研究和进化生物学研究提供重要依据。
多序列比对的方法主要包括以下几种:1. 动态规划法:通过构建一个动态规划表,计算最优比对路径,实现序列的比对。
2. 人工比对法:通过分析序列结构、功能域等信息,人工进行比对。
3. 基于启发式算法的比对:通过寻找序列间的相似性,快速进行比对。
三、实验材料1. 仿刺参EGFR基因氨基酸序列(Fasta格式)。
2. 同源序列数据库(如NCBI)。
3. 多序列比对软件(如BLAST、CLUSTAL W)。
四、实验步骤1. 使用BLAST工具进行同源序列搜索。
(1)在NCBI网站上,选择“BLAST”功能。
(2)将仿刺参EGFR基因氨基酸序列粘贴到“Query Sequence”框中。
(3)选择合适的比对参数,如“MegaBLAST”。
(4)点击“BLAST”按钮,等待结果。
(5)在结果页面,找到相似度最高的几个序列,下载下来。
2. 使用CLUSTAL W进行多序列比对。
(1)将下载的同源序列整合到一个Fasta格式的文本文件中。
(2)在CLUSTAL W软件中,选择“Multiple Sequence Alignment”功能。
(3)上传Fasta格式的文本文件。
(4)选择合适的比对参数,如“Gap Penalty”和“Gap Reward”。
(5)点击“Align”按钮,等待结果。
3. 分析比对结果。
(1)观察比对结果,分析序列间的相似性和进化关系。
(2)绘制系统进化树,展示序列的进化历程。
五、实验结果与分析1. 使用BLAST工具,找到与仿刺参EGFR基因氨基酸序列相似度最高的几个序列,如Anopheles gambiae、Nasonia vitripennis等。
双序列比对的方法

Match = 1 Mismatch = 0
Window size = 5
Stringency = 3
G C G A T G C A T
T G A G T A T C A T A
21
A T A C T A C A A G A C A C G T A C C G
Match = 1 Mismatch = 0
i -x
Si –1, j- 1 + s(ai , bj)
Si - x,j - wx
Si,j这个位置的
i -1 i
分数为图中箭 头所示三个方 向值中最大的 一个
Si, j - y - wy
i -y j -1 j
Si, j
32
动态规划算法的数学形式
Sij=max{Si-1,j-1,+s(aibj), max x≥1 (Si-x,j-wx), max y ≥ 1 (Si,j-y-wy) }
5
序列比对两种类型
6
空位罚分(Gap Penalties)
空位为了获得两个序列最佳比对,必须使用空 位和空位罚分 空位罚分分类:
空位开放罚分(Gap opening penalty) 空位扩展罚分(Gap extension penalty)
最优的序列比对通常具有以下两下特征: 尽可能多的匹配 尽可能少的空位 插入任意多的空位会产生较高的分数,但找到 的并不一定是真正相似序列
T
-8
-3 2 7 8 6
C
-10
-5 0 5 6 7
G
-12
-7 -2 3 4 9
A C
-2 -4 -6 -8 -10
T
A G
回溯
多序列比对-生物信息学

>SIV MSMSSSNITSGFIDIATFDEIEKYMYGGPTATAYFVREIRKSTWFTQVPVPLSRNTGNAAFGQEWSVSIS RAGDYLLQTWLRVNIPPVTLSGLLGNTYSLRWTKNLMHNLIREATITFNDLVAARFDNYHLDFWSAFTVP ASKRNGYDNMIGNVSSLINPVAPGGTLGSVGGINLNLPLPFFFSRDTGVALPTAALPYNEMQINFNFRDW HELLILTNSALVPPASSYVSIVVGTHISAAPVLGPVQVWANYAIVSNEERRRMGCAIRDILIEQVQTAPR QNYVPLTNASPTFDIRFSHAIKALFFAVRNKTSAAEWSNYATSSPVVTGATVNYEPTGSFDPIANTTLIY ENTNRLGAMGSDYFSLINPFYHAPTIPSFIGYHLYSYSLHFYDLDPMGSTNYGKLTNVFVVPAASSAAIS AAGGTGGQAGSDYAQSYEFVIVAVNNNIVRIENSLVRNRRRWSREGPMVMVC >TIV MSMSSSNITSGFIDIATFDEIEKYMYGGPTATAYFVREIRKSTWFTQVPVPLSRNTGNAAFGQEWSVSIS RAGDYLLQTWLRVNIPPVTLSGLLGNTYSLRWTKNLMHNLIREATITFNDLVAARFDNYHLDFWSAFTVP ASKRNGYDNMIGNVSSLINPVAPGGTLGSVGGINLNLPLPFFFSRDTGVALPTAALPYNEMQINFNFRDW HELLILTNSALVPPASPYVPIVVGTHISAAPVLGPVQVWANYAIVSNEERRRMGCAIRDILIEQVQTAPR QNYVPLTNASPTFDIRFSHAIKALFFAVRNKTSAAEWSNYATSSPVVTGATVNYEPTGSFDPIANTTLIY ENTNRLGAMGSDYFSLINPFYHAPTIPSFIGYHLYSYSLHFYDLDPMGSTNYGKLTNVSVVPQASPAAIA AAGGTGGQAGSDYPQNYEFVILAVNNNIVRISGGETPQNYIAVC >WIV MSMSSSNITSGFIDIATFDEIEKYMYGGPTATAYFVREIRKSTWFTQVPVPLSRNTGNAAFGQEWSVSIS RAGDYLLQTWLRVNIPQVTLNPLLAATFSLRWTRNLMHNLIREATITFNDLVAARFDNYHLDFWSAFTVP ASKRTGYDNMIGNVSSLINPVAPGGNLGSTGGTNLNLPLPFFFSRDTGVALPTAALPYNEMQINFNFRDW TELLVLQNSALVAPASPYVPIVVPTHLTVAPVLGPVQVWANYAIVSNEERRRMGCAIRDILIEQVQTAPR QNYTPLTNASPTFDIRFSHAIKALFFSVRNKTSASEWSNYATSSPVVTGATVNFEPTGSFDPIANTTLIY ENTNRLGAMGSDYFSLINPFYHAPTIPSFIGYHLYSYSLHFYDLDPMGSTNYGKLTNVSVVPQASPAAVN AASGAGGFPGSDYPQSYEFVIVAVNNNIVRISGGETPQNYLSGSFVTLLNRRKWSREGPMIMVQ >CzIV MSMSSSNITSGFIDIATFDEIEKYMYGGPTATAYFVREIRKSTWFTQVPVPLSRNTGNAAFGQEWSVSIS RAGDYLLQTWLRVNIPQVTLNAQLGPTFGLRWTRNFMHNLIREATITFNDLVAARFDNYHLDFWSAFTVP ASKKIGYDNMIGNISALTNPVAPGGSLGSVGGINLNLPLPFFFSRDTGVALPTAALPYNEMQINFNFRDW PELLILTNTALVPPASPYVPIVVGTHLSAAPVLGAVQVWANYAIVSNEERRRMGCAIRDILIEQVQTAPR QNYTPLTNAMPTFDIRFSHAIKALFFSVRNKTSSAEWSNYATSSPVVTGQLVNYEPPGAFDPISNTTLIY ENTNRLGAMGSDYFSLINPFYHAPTIPSSIGYHLYSYSLHFFDLDPMGSTNYGKLTNVSVVPQASPAAVT AAGGSGAAGSGADYAQSYEFVIIGVNNNIIRISGGALGFPVL >CIV MSISSSNVTSGFIDIATKDEIEKYMYGGKTSTAYFVRETRKATWFTQVPVSLTRANGSANFGSEWSASIS RAGDYLLYTWLRVRIPSVTLLSTNQFGANGRIRWCRNFMHNLIRECSITFNDLVAARFDHYHLDFWAAFT TPASKAVGYDNMIGNVSALIQPQPVPVAPATVSLPEADLNLPLPFFFSRDSGVALPTAALPYNEMRINFQ FHDWQRLLILDNIAAVASQTVVPVVGATSDIATAPVLHHGTVWGNYAIVSNEERRRMGCSVRDILVEQVQ TAPRHVWNPTTNDAPNYDIRFSHAIKALFFAVRNTTFSNQPSNYTTASPVITSTTVILEPSTGAFDPIHH TTLIYENTNRLNHMGSDYFSLVNPWYHAPTIPGLTGFHEYSYSLAFNEIDPMGSTNYGKLTNISIVPTAS PAAKVGAAGTGPAGSGQNFPQTFEFIVTALNNNIIRISGGALGFPVL
序列比对多序列试验的流程和方法

序列比对多序列试验的流程和方法下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor.I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!序列比对:多序列试验的流程与方法在生物信息学领域,序列比对是一种关键的技术,用于比较和分析不同生物序列之间的相似性和差异性。
3多重序列比对

Sreal − Srand Snorm = Sident − Srand
据此计算距离相似性得分DistanceAB为 据此计算距离相似性得分 DistanceAB=-log(Snorm) 完全不相似: Snorm=0 导出 DistanceAB=∞ 完全不相似: ∞ 完全相似: 完全相似: Snorm=1 导出 DistanceAB=0
3.生物信息学可以分成三个层次: 3.生物信息学可以分成三个层次: 生物信息学可以分成三个层次 第一个层次是实验者的应用, 第一个层次是实验者的应用,主要是根据实验工作者具体的需要 应用相应的网站来分析其实验中获取的数据, 应用相应的网站来分析其实验中获取的数据,如找新的蛋白质的功能 位点等。 位点等。 其次是一些从事生物信息学方法研究的需要某个生物信息学方法 的结果,在些基础上建立新的方法,如应用PSIBLAST方法构建相应 的结果,在些基础上建立新的方法,如应用 方法构建相应 的位置专一化得分矩阵即PSSM,这就需要将相应的软件下载并组合 的位置专一化得分矩阵即 , 到某个程序中; 到某个程序中; 其三是重建该方法,找出其中的不足, 其三是重建该方法,找出其中的不足,然后对它作出相应的改进
构建导向树方法: 构建导向树方法:
有邻位加入法( 有邻位加入法(Neighbour-Joining Method) ) 算术平均非加权配对组法( 算术平均非加权配对组法(Unweighted Pari Group Method of Arithmetic Averages UPGMA) )
W1=1.0+(3.25-1.0)/2=2.125 ( ) W2=1.0+(3.25-1.0)/2=2.125 W3=2.25+(3.25-2.25)/3=2.583 W4=1.5+(2.25-1.5)/2+(3.25-2.25)/3=2.208 W5=1.5+(2.25-1.5)/2+(3.25-2.25)/3=2.208
多序列比对

2.同步法
同步法实质是把给定的所有序列同时进行比对, 而不是两两比对或分组进行比对。 其基本思想是将一个二维的动态规划矩阵扩展 到三维或多维。矩阵的维数反映了参与比对的序 列数。这类方法对于计算机的系统资源要求较高, 通常是进行少量的较短的序列的比对
3.步进法 这类方法中最常用的就是Clustal,它是由Feng和 Doolittle于1987年提出的(Feng和Doolittle,1987)。由 于对于实际的数据利用多维的动态规划矩阵来进行序列的 比对不太现实,因此大多数实用的多序列比对程序采用启 发式算法,以降低运算复杂度。 Clustal的基本思想是基于相似序列通常具有进化相关性 这一假设。比对过程中,先对所有的序列进行两两比对并 计算它们的相似性分数值,然后根据相似性分数值将它们 分成若干组,并在每组之间进行比对,计算相似性分数值。 根据相似性分数值继续分组比对,直到得到最终比对结果。 比对过程中,相似性程度较高的序列先进行比对,而距离 较远的序列添加在后面。作为程序的一部分,Clusal可以 输出用于构建进化树的数据。
一般来说,对于具有较高相似性的一组序列之间的比 对,自动比对方法是很有效的。一旦序列的亲缘关系变 得较远,所得结果就不那么可信。若要得到比较可靠而 又具有明确生物学意义的比对结果,比较有效的方法是 对比对结果进行手工编辑和调整。这对于构建二次数据 库是非常重要的信息。在选择现有的序列模式或序列模 体公开数据库构建自己的数据库系统时,对这些现有数 据库的可靠性必须采取谨慎的态度
比对方法
1.手工比对方法 手工比对方法在文献中经常看到。因为难免加入 一些主观因素,手工比对通常被认为有很大的随意 性。其实,即使用计算机程序进行自动比对,所得 结果中的片面性也不能予以忽视。在运行经过测试 并具有比较高的可信度的计算机程序基础上,结合 实验结果或文献资料,对多序列比对结果进行手工 修饰,应该说是非常必要的
实习四:多序列比对(Multiple alignment)
实习四:多序列比对(Multiple alignment)学号姓名专业年级实验时间提交报告时间实验目的:1. 学会利用MegAlign进行多条序列比对2. 学会使用ClustalX、MUSCLE 和T-COFFEE进行多条序列比对分析3. 学会使用HMMER进行HMM模型构建,数据库搜索和序列比对实验内容:多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似区域只能通过将多个序列同时比对才能识别。
只有在多序列比之后,才能发现与结构域或功能相关的保守序列片段,而两两序列比对是无法满足这样的要求的。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻以及PCR引物设计等具有非常重要的作用。
作业:1.Align the orthologous nucleotide and protein sequences from 5 organisms you found from first practice with MegAlign. Describe the sequences you used (the title of each sequence), explain whether the phylogenetic tree is consistent with the species tree from NCBI taxonomy database. Set the alignment report to show consensus strength and decorate the residues different from consensus with green shade.(Hint: use the taxonomy common tree from NCBI to get the evolutionary relationship among the organisms. Save your organism name in a text file with each organism name in a line, and upload the file, choose Add from file, and you will see the relationship among the specified organisms) /Taxonomy/CommonTree/wwwcmt.cgiHint 2:Change the accession number in your fasta or genPept format sequence file to organism name, so that the phylogenetic tree can be easily understood.方法与结果:打开Megalign,选择FILE下的Enter sequence ,打开之前保存的来自于五个物种的蛋白(或核酸)序列;首先选择打分矩阵,点击“Align”,选择Set residue Weight Table 选择矩阵:PAM100(核酸则设为weighted),通过“method parameters”查看参数,使用Clustal V的默认值;其次进行序列的比对,选择Align下的“by Clustal V Method”开始比对,再次待其结束后,进行比对结果的显示,选择view下的“Phylogenetic Tree”,显示出树形图;(图)与NCBI上找到的树形图进行对比(图);接下来点击View 下的“Alignment reports ”,选择OPTIONS下的“Alignment report contents”勾中“show consensus strength”,即在序列中显示出相似性条块;在OPTIONS下选择“New decorations”对decoration parameters 下选“shade—residues differing from—the consensus”把字符选择现实的颜色为绿色,结果显示如下:(图)同法可以得到核酸的树形图:(图)分析:系统发育树与NCBI上的物种树有很大的差异,因为可能这些物种间含有很多同源序列,我们不能单凭几条相似序列的同源关系来判断物种的亲缘关系,而应该考虑到物种更多相似序列的同源关系。
序列比对

最常见的比对是蛋白质序列之间或核酸序列之间的两两比对,通过比较两个序列之间的相似区域和保守性位点,寻找二者可能的分子进化关系。
进一步的比对是将多个蛋白质或核酸同时进行比较,寻找这些有进化关系的序列之间共同的保守区域、位点和profile,从而探索导致它们产生共同功能的序列模式。
此外,还可以把蛋白质序列与核酸序列相比来探索核酸序列可能的表达框架;把蛋白质序列与具有三维结构信息的蛋白质相比,从而获得蛋白质折叠类型的信息。
序列比对的理论基础是进化学说,如果两个序列之间具有足够的相似性,就推测二者可能有共同的进化祖先,经过序列内残基的替换、残基或序列片段的缺失、以及序列重组等遗传变异过程分别演化而来。
序列相似和序列同源是不同的概念,序列之间的相似程度是可以量化的参数,而序列是否同源需要有进化事实的验证。
在残基-残基比对中,可以明显看到序列中某些氨基酸残基比其它位置上的残基更保守,这些信息揭示了这些保守位点上的残基对蛋白质的结构和功能是至关重要的,例如它们可能是酶的活性位点残基,形成二硫键的半胱氨酸残基,与配体结合部位的残基,与金属离子结合的残基,形成特定结构motif的残基等等。
但并不是所有保守的残基都一定是结构功能重要的,可能它们只是由于历史的原因被保留下来,而不是由于进化压力而保留下来。
因此,如果两个序列有显著的保守性,要确定二者具有共同的进化历史,进而认为二者有近似的结构和功能还需要更多实验和信息的支持。
通过大量实验和序列比对的分析,一般认为蛋白质的结构和功能比序列具有更大的保守性,因此粗略的说,如果序列之间的相似性超过30%,它们就很可能是同源的。
早期的序列比对是全局的序列比较,但由于蛋白质具有的模块性质,可能由于外显子的交换而产生新蛋白质,因此局部比对会更加合理。
通常用打分矩阵描述序列两两比对,两条序列分别作为矩阵的两维,矩阵点是两维上对应两个残基的相似性分数,分数越高则说明两个残基越相似。
因此,序列比对问题变成在矩阵里寻找最佳比对路径,目前最有效的方法是Needleman-Wunsch动态规划算法,在此基础上又改良产生了Smith-Waterman算法和SIM算法。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。
Figure 3.1 载入序列此时程序窗口分为三部分,最左侧较窄的是sequence name,中间显示的是序列起始位置,最右侧显示序列末尾部分,可以通过拖动窗口底部滚动条,查看序列其它部分(Figure 3.2)。
若想改变字体显示方式,点击主菜单OPTIONS,选择Font改变字体,选择Size改变字号大小。
若要移除序列,选中sequence name的序列名,右击,选clear。
Figure 3.2 载入序列后(注意标注的绿色箭头,即为坐标位置)1.3 设定序列比对位置MegAlign允许使用者选择序列的一部分进行比对分析,例如,可以根据GenBank格式的序列中Features部分关于编码区(CDS)位置的描述,设定只对此编码区进行分析。
a. 点击最左侧Sequence Name框中的第一条序列tethis,然后选择主菜单OPTIONS-Set sequence limits-from feature table。
(Figure 3.3)此时根据feature内容,出现四个可以选择的片段,第一个为全长,从序列起始到末尾(1-906),其它三个则只包括序列的一部分,选择最后一个Histone H2B-1—CDS,点击Change the Reset,点击OK,同样对第二条序列进行上述操作,回到主界面工作区,此时窗口中的序列起始和终止位置已经发生了变化。
(Figure 3.4)Figure 3.3 利用Feature Table选择序列特定部分Figure 3.4 选择序列特定部分b. 我们还可以通过设定序列坐标进行部分序列比对,首先选定序列,选择主菜单OPTIONS-Set sequence limits-by coordinates,输入起始和终止位置坐标来选择部分序列进行分析。
注意:只有genbank格式的序列才可以Set sequence limits from feature table,fasta格式的序列因为没有feature那一项内容,只可以Set sequence limits by coordinates。
1.4 进行两条序列比对如果输入两条序列后不设置序列起始和终止位置,默认是全长序列进行比对。
按住Shift选择序列tethis21和tethis22,然后点击主菜单Align-One pair,由于目前输入的是核酸序列,此时有两个选项,Wilbur-Lipman Method和Martiner NW Method。
如果输入的是蛋白质序列,这两个选项将是灰色,只能用Lipman-Pearson Method进行比对。
Wilbur-Lipman Method是一种以word为单位的(word-based)启发式局部比对方法;Martiner NW Method是一种改进了的全局动态规划算法。
Lipman-Pearson Method是序列相似度搜索软件Fasta的比对算法,也是一种以word为单位的快速启发式算法。
选择其中一个,出现比对参数设定窗口(Figure 3.5),选择默认参数不做更改,直接点击OK即可。
Figure 3.5 Wilbur-Lipman比对方法参数设定这时出现一个新窗口,即为比对结果。
可以选择OPTION-size,放大字号观察比对结果。
可以看到在窗口上部显示的是比对方法名称,所用参数,两条序列各自的起止位置,相似度值,比对结果中空位数目,长度和一致序列的长度。
随后就是比对结果部分,其中第一行是第一条序列,它上面的v70是标尺,其中的“V”的位置对应的是第一条序列的第70个核苷酸所在位置;第三行是第二条序列,它下方的数字同样对应该序列位置坐标;中间那行是根据两条序列比对结果中匹配部分推断出来的一致序列(consensus sequence),错配或空位显示为空白(Figure 3.6)。
Figure 3.6 Wilbur-Lipman方法比对结果设置比对结果显示方式:点击比对结果窗口最左侧的按钮,出现Alignment View Options窗口,可以选择匹配,错配和一致序列的字符颜色和其它显示选项。
推荐使用设置:选择match为红色,mismatch为绿色,consensus为蓝色,并选择show identities as vertical bars (一致序列显示为竖线),则得到Figure 3.7。
还可以尝试选中或不选show header, show ruler,show names,show contest四个选项,看看显示结果有何变化。
Figure 3.7 Alignment View OptionsTIP:MegAlign分析自己下载的序列时要注意序列扩展名如果是从NCBI直接下载的fasta格式文件,可以象上面一样,用enter sequence直接将序列读入程序。
但是如果序列文件是复制粘贴到txt文档中的,MegAlign程序是无法识别扩展名为txt的文件。
此时可将每条序列文件(fasta或genbank格式皆可)扩展名改为MegAlign可以识别的类型(核酸序列为seq,蛋白质序列为pro),即可从File-Enter sequence 载入。
更改文件扩展名的方法:找到你要更改扩展名的文件,将.txt改为.seq或 .pro,此时会弹窗口,提示“如果改变文件扩展名,可能会导致文件不可用。
确实要更改吗?”选择“是”,文件图标会变成MegAlign特定图标,说明修改成功。
若扩展名自动隐藏,打开文件夹,点击窗口上的主菜单工具-文件夹选项,在打开的页面选择选项卡查看,去掉“隐藏已知文件类型的扩展名”前面的对勾,确定退出。
然后再用上述方法更改扩展名。
2. 多序列比对2.1 载入序列进行多条序列比对的演示序列(demo sequence)在c:\program files\ dnastar\ lasergene\ demo megalign\ Calmodulin Sequences\ 文件夹里。
点击主菜单File-Enter Sequence-根据路径到达Calmodulin Sequences文件夹,点击Add All,此时14条序列全都出现在右侧的selected sequences框中,点击Done,回到主程序工作区。
(Figure 3.8)这是来自14个物种的钙调蛋白。
Figure 3.8载入14条序列2.2 序列比对第一步,选择比对所用的打分矩阵。
点击主菜单Align-Set residue Weight Table,由于钙调蛋白比较保守,我们选择PAM100作为打分矩阵,点击OK结束设定(Figure 3.9)。
Figure 3.9 选择打分矩阵此时还可以通过点击Align-Method Parameters设定比对所用的其它参数。
打开的新窗口中包含三个选项卡,Jotun Hein、Clustal V和Clustal W,对应程序中多条序列比对可用的三种算法。
推荐大家不做修改,使用默认参数即可。
第二步,比对。
点击Align-by Clustal V Method,此时出现窗口显示比对进度,比对结束后,回到原来工作窗口,显示比对结果。
注意序列上方彩色条块,颜色代表对应列中相似程度,相似度由低到高,依次以深蓝、浅蓝、绿、黄、桔、红几种颜色代表。
(Figure 3.10)Figure 3.10 比对后结果2.3 查看比对结果此时可以通过几种方式观察比对结果。
a.点击View-Sequence Distances出现新窗口,显示两两序列percent identity(上半部分)和divergence(下半部分)。
Figure 3.11 比对结果-一致度(identity)b.点击View-Residue Substitutions出现新窗口,显示比对中所有替换的类型和数目。
Figure 3.12 比对结果-替换情况c.点击View-Phylogenetic Tree出现新窗口,显示根据14条序列比对结果构建出的进化树。
Figure 3.13 比对结果-进化树d.点击View-Alignment Reports出现新窗口,显示比对结果报告。
点击OPTIONS-Alignment report contents,选中show consensus strength,其它不变,点击OK。
在序列上方出现条块,显示每一列序列的相似程度。
Figure 3.14选择show consensus strength显示结果设置比对结果显示方式:突出显示匹配或错配的氨基酸。