表达序列标签的应用现状及分析方法研究
表达序列标签(est)在基因组学研究中的应用

表达序列标签(est)在基因组学研究中的应用序列标签(Sequence Tag)是指由DNA或RNA片段构成的一系列序列标记,它可以在遗传疾病、基因表达、信号转导等生命科学研究中发挥重要作用。
其中,表达序列标签(EST)是一种简单而有效的标记技术,它的应用在基因组学领域得到了广泛关注。
一、EST技术简介EST技术是一种由测序技术支持的高通量筛选技术,其主要原理是通过随机挑选某些不同的cDNA克隆来获得所需的不同的EST序列。
它的应用可以大大降低生物学研究的难度,也可以在较短的时间内获得大量的基因序列。
二、EST在基因组学研究中的应用(一)基因组注释及功能预测基因组注释是指对基因组序列进行生物信息学分析,以确定其中的基因区域和基因的结构。
EST技术可以通过对基因组序列的全长编码区域进行建库、测序和组装,从而确定基因的结构和位置,从而实现基因组注释。
(二)基因家族的发现和分类EST技术可以应用于表达的基因家族的发现和分类,例如受体基因、酶基因和转录因子基因家族等。
EST序列可以用作启动点,通过比对模式,可将同源序列聚类形成基因家族,并进一步研究其与环境、生长过程或其它生物学过程的关系。
(三)隐形基因的寻找传统的基因克隆方法主要寻找已知的基因进行克隆,而隐形基因(也称未知基因)的寻找则需要更为深入的研究。
EST技术可以通过测序与注释,从全基因组的角度分析,实现隐形基因的发现。
这可以为了解未知疾病的发病机制、发病率等方面提供重要支持和信息。
(四)基因调控机理的研究EST技术不仅可以应用于基因组学研究,还可以应用于表观遗传学——研究基因调控机理。
EST序列常常用于测定细胞特异性基因表达、基因表达的时间和空间分布等方面。
它对于异等基因的表达差异、组织特异性基因表达模式、静态和动态转录调控等具有重要的科研价值。
同时,它还可以用于筛选差异表达基因,并进一步研究其相关的信号传导机制、生长发育机制等。
三、结论在基因组学研究中,EST技术可以为高通量测序提供支持,并更好地揭示基因组结构和基因调控机制。
基因差异表达分析方法及其在作物遗传育种中的应用

基因差异表达分析方法及其在作物遗传育种中的应用苏在兴高闰飞李强【摘要】植物基因的差异表达是细胞形态和功能多样性的根本原因,也是各种生理及病变过程的物质基础.分析基因差异表达是近30年来分子生物学研究的重点,研究方法也从最早的差减杂交、差异显示PCR和cDNA代表性差异分析等,不断地发展到基于测序的表达系列标签和转录组测序技术,其中高通量测序技术的应用,使得分子生物学进入后基因组时代,特别是转录组测序可高效率、大批量地获取差异表达基因.通过基因差异表达分析,可挖掘农作物的优异农艺性状、高品质、抗性以及杂种优势等相关基因,辅助常规育种,提高农作物的品质、产量、抗性等综合性状,并为探究其机理、机制奠定基础.【期刊名称】《江苏师范大学学报:自然科学版》【年(卷),期】2017(035)001【总页数】8页(P38-45)【关键词】基因差异表达;转录组测序;农艺性状;品质性状;抗性;杂种优势【作者】苏在兴高闰飞李强【作者单位】[1]江苏徐淮地区徐州农业科学研究所/农业部甘薯生物学与遗传育种重点实验室,江苏徐州221131;[2]中国农业科学院甘薯研究所,江苏徐州221131;[3]江苏师范大学生命科学学院,江苏徐州221116【正文语种】中文【中图分类】Q786植物基因差异表达是在转录水平上对基因的表达情况进行研究,包括2个及2个以上材料之间存在差异基因或者差异基因在相同环境条件下具有不同的表达模式,以及同一材料在不同处理下,同一基因呈现不同的表达模式2种情况.在真核生物基因组中,仅约10%~15%的基因在细胞中表达,而且在不同发育阶段、不同生理状态和不同类型的细胞中基因表达也不同[1].基因的差异性表达是细胞形态及功能多样性的根本原因,也是植物生长发育和各种生理及病变的物质基础[2].通过基因差异表达,分离新的功能基因、挖掘和鉴定差异表达基因的新功能等,对作物遗传改良具有十分重要的意义.目前,分子生物学技术逐步应用到作物遗传育种中,分子标记辅助育种、转基因育种以及分子设计育种正在成为作物遗传改良的重要手段[3].1990年代开始,基因差异表达分析方法逐渐得到发展[4-12],并在挖掘新的功能基因以及揭示基因的新功能方面表现出优势.随着研究的深入,对差异性表达基因的富集程度要求更高,从而促使基因差异表达的筛选方法不断得以丰富和改进,尤其是测序技术的发展,使得差异表达基因的获得更加便捷,数量更多,效率更高[13].本实验室也采用基因差异表达分析技术,解析徐薯18和徐781 2个甘薯品种在新陈代谢、抗逆性和碳水化合物积累等方面的机理机制,已获得一批与新陈代谢、抗逆性、物质积累等相关的功能基因.本文简要综述不同基因差异表达分析方法的特点、原理及优缺点,进一步阐述基因差异表达分析技术在作物农艺性状分析、品质性状分析、抗性分析以及杂种优势分析等方面的应用,以期对后续的研究工作有所裨益.1.1 基因差异表达分析方法1.1.1 差减杂交(subtractive hybridization,SH) 最初由Lamar等[4]于1984年报道,用于分离老鼠Y染色体的特异性探针.该方法也叫扣除杂交或减法杂交.差减杂交是对2种遗传背景大致相同而性状有差异的材料进行研究,基因组DNA或者mRNA(反转录成cDNA)经特定的核酸限制性内切酶消化后,在一定的条件下进行分子杂交,选择性地去除2部分共有基因杂交后形成的复合物,将含有目的基因的未杂交部分收集后装入载体,从而构建差减文库.佘卫炜等[14]用该方法成功地分离到6条与藏红花苷合成相关的特异性表达cDNA片段.该方法克服了示差筛选技术的局限性,灵敏度较高,也能有效检测转录丰度低的基因[15],但操作难度大,费时费力,重复性较差,并且在酶切不彻底等情况下很难得到满意的结果[16].1.1.2 mRNA差异显示逆转录PCR(differential display of reverse transcriptional PCR,DDRT-PCR) 1992年,Liang等[5]根据高等生物成熟的mRNA具有poly(A)尾巴的特性,建立了mRNA差异显示逆转录PCR.该方法利用含Oligo(dT)n的寡聚核苷酸作为锚定引物,通过逆转录酶的催化,将真核生物细胞中全部表达的mRNA逆转录为cDNA,通过PCR扩增,利用变性聚丙烯酰胺凝胶电泳将有差异的片段分开,从而筛选出差异表达基因.张弛等[17]利用该方法研究水稻77-170(Oryza Sativa var. Japoinca)及其耐盐突变体M-20在盐胁迫下基因表达的差异,克隆到13个与盐诱导相关的cDNA片段,其长度范围在200~600 bp 之间.该方法具有技术应用成熟、效率高、灵敏度高的优点,实验每一步均可检测,无需实验结束,但假阳性率高,最高达70%,所得的cDNA片段较短,很难扩增到ORF(open reading frame)内部[18-19].1.1.3 cDNA代表性差异分析(cDNA-RDA) 在Lisitsyn等[20]建立的DNA代表性差异分析(representational difference analysis,RDA)方法的基础上,1994年,Hubank等[6]建立了cDNA代表性差异分析技术.该技术对2组材料的cDNA 进行酶切消化,并为酶切片段连接特异寡聚核苷酸接头,进行PCR扩增,分别获得实验组(T)和对照组(D)的扩增子.再次酶切2组扩增子并对T组扩增子添加新接头,然后将T组扩增子与富余的D组扩增子混合,形成杂交体,用与新接头互补的特异引物对杂交体进行PCR扩增,其中T/T杂交体进行指数扩增,T/D杂交体进行线性扩增,D/D杂交体不扩增.对差异产物进行多轮PCR后,可用普通琼脂糖凝胶检测差异表达条带[21-22].Ling等[23]将该技术运用于分离大豆不同萌发期子叶中的差异表达基因,并成功克隆到CysP1和CysP2 2个编码半胱氨酸蛋白酶的新基因.1.1.4 表达系列标签(serial analysis of gene expression,SAGE) 1995年,Velculescua等[7]首先提出基因表达系列分析技术,该方法通过限制性酶切含有生物素标记的cDNA,产生能够代表其相应转录物的cDNA短标签(9~14 bp),然后随机连接并进行测序分析.单一转录体由其特异性的短标签所代替,用SAGE软件定量分析标签的丰度,代表转录体的表达水平.Song等[24]采用SAGE法分析超级杂交稻LYP9及其亲本93-11、PA64s在不同时期、不同组织部位的差异表达基因,获得12种主要的基因表达模式,其中406个基因上调表达,469个基因下调表达,这些基因可能与水稻的杂种优势有关.该方法可以将多个短标签串联测序,能够寻找低丰度的转录物,但其依赖已测序的基因序列,过短的序列标签所涵盖的信息无法被准确注释到基因组上[25-26].1.1.5 抑制差减杂交(suppression subtractive hybridization,SSH) 1996年,Diatchenko等[8]提出抑制差减杂交,也叫抑制性消减杂交,结合了抑制PCR和差减杂交技术,利用抑制性PCR,选择性地扩增目的cDNA片段,显著增加了低丰度差异表达cDNA获得的概率.Tirumalaraju等[27]应用SSH技术从抗花生根结线虫和感花生根结线虫2份材料中获得70个差异表达ESTs,并证实各种非生物、生物(含根结线虫)胁迫和植物应答此类胁迫时与水杨酸(SA)、茉莉酸(JA)及乙烯信号传导之间的关系.这些差异表达候选基因为获得抗根结线虫种质资源并培育优良抗性花生新品种提供可能.该方法简单、成熟、易操作,且效率高,筛选周期短,通常3~4 d可获得基因差异表达片段.但是SSH技术得到的cDNA是限制酶消化的cDNA,不是全长cDNA;材料之间最好是存在细微差异,小片段缺失时也不能有效检测;实验中酶切后的cDNA与接头连接的效率是该方法的关键,若连接效率低,有些差异表达的基因就会漏检[18].1.1.6 cDNA限制性片段长度多态性分析(cDNA-AFLP) 在Botstein等[28]建立的限制性片段长度多态性(restriction fragment length polymorphism,RFLP)方法的基础上,1995年,Vos等[29]结合PCR扩增提出一种新的DNA指纹技术,即扩增片段长度多态性(amplification fragment length polymorphism,AFLP).1996年,Bachem等[9]结合RT-PCR和AFLP提出cDNA-AFLP技术,用于对转录组表达情况进行分析.该技术采用2种不同的内切酶切割cDNA片段,并添加含有与引物序列互补的人工接头,进行PCR预扩增后用聚丙烯酰胺凝胶区分差异条带.Nie等[30]运用cDNA-AFLP技术从玉米亲本和杂交种的叶、根和成熟胚中分别分离到180、170和108个差异表达基因,为揭示玉米杂种优势提供了线索.cDNA-AFLP 技术具有很好的重复性,假阳性比较低,不需要预先知道基因的序列信息,能够通过扩增条带显色强度判断基因表达量的差异[31].1.1.7 基因芯片(DNA Chips)技术是指把大量核酸片段固定在载体上,组成密集的按序排列的探针群,通过与标记样品的核酸杂交,判断靶核苷酸的有无或数量多少的一项技术,主要包括芯片的制备、杂交与检测等3个步骤.常见的芯片可分为2大类:一种是原位合成,适用于寡核苷酸;另一种是直接点样,多用于大片段DNA.姜兆远等[32]将Affymetrix表达谱芯片运用于水稻与稻瘟病不同小种的互作研究,水稻与稻瘟病菌非亲和互作的基因表达谱及其亲和互作的基因表达谱之间存在较大差异,将基因芯片筛选到的差异表达基因通过GO注释,明确了差异基因的分子功能及信号通路,有利于进一步了解植物抗病机制,并可能为稻瘟病防治提供新的途径.该方法同时将大量的探针固定于支持物上,可以同时对大量序列进行检测,克服了传统的核酸印迹杂交操作复杂、自动化程度低,且检测序列数量少等缺点.但该方法所用仪器及软件价格较昂贵,探针的合成和固定比较复杂,难以检测低丰度表达的基因[33].1.1.8 半定量RT-PCR和实时荧光定量PCR 半定量逆转录多聚合酶链式反应(reverse transcription polymerase chain reaction,RT-PCR)是探究基因差异表达的有效手段之一[10].采用PCR技术同时对2组或多组材料的目的基因和内参基因(internal reference genes)进行扩增,运用琼脂糖凝胶电泳PCR扩增产物,并调节内参基因条带强度一致,便可直观地呈现出目的基因在不同组织或者不同材料中是否表达,且能对比其表达丰度[11,34].1993年,Higuchi等[35]根据PCR延伸阶段随着DNA双链的生成,含有荧光的EB(ethidium bromide)染料能嵌入DNA链内部而激发荧光,提出实时荧光定量PCR(real time quantitative RT-PCR,qRT-PCR)的概念.荧光定量PCR具有很好的特异性,重复性好,操作简单快捷,全反应过程在一个封闭的PCR管中进行,可以实时地进行监测,而且扩增结束后不需要进一步处理.Applied Biosystems、Bio-RAd等公司推出实时荧光定量PCR配套的仪器和试剂,使得该技术在研究基因表达方面逐渐成为主流手段[36]. Fu等[37]采用SSH法从3份水稻材料中获得一批抗旱相关的基因,并用半定量RT-PCR和实时荧光定量PCR对300多条特异条带进行确证,为完善水稻抗旱相关QTLs及获得候选功能基因奠定基础.1.1.9 转录组测序(RNA-Seq)技术转录组(transcriptome),广义上指特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的总和,主要包括信使RNA(message RNA,mRNA)、核糖体RNA(ribosome RNA,rRNA)、转运RNA(transport RNA,tRNA)和非编码RNA(non-coding RNA,ncRNA).狭义上,一般指特定组织或细胞中转录的全部mRNA[25].转录组测序就是利用高通量技术对转录组进行测序分析,并对获得的读段进行过滤、组装以及生物信息学分析.RNA-Seq需要将mRNA反转录成cDNA,并对合成的cDNA作末端修复、加poly(A)尾巴及连接测序接头,片段化为测序平台所需的长度,PCR扩增,构建测序文库,利用相应的测序平台进行序列测定.对于有参考基因组序列的物种,可根据其参考序列(reference assembly)组装,没有参考基因组序列的物种,则进行从头组装(denovo assembly)[12,38].根据组装情况,以单位长度的转录物上覆盖的读段数来衡量基因的表达水平(reads per kilo bases per million reads,RPKM).RNA-Seq 主要用于研究2个及以上样本中基因的差异表达情况,如正常条件下的棉花幼苗和盐胁迫下的棉花幼苗等[39].转录组测序技术具有较高的灵敏度,可以同时获得组织内的全部转录本;能检测出SNP等单个核苷酸的差异,具有很高的精确度;通过组装分析能得出基因家族中的不同拷贝或可变剪接.随着测序仪器的升级,RNA-Seq 费用逐渐下降,除了从测序数据中挖掘差异表达基因外,还可以挖掘SSR、SNP信息以及组装出尽可能完整的Unigenes序列,为后续的基因克隆和功能验证奠定坚实基础[40-45].1.1.10 基因编辑技术近年来,锌指核酸酶(zinc-finger nucleases,ZFNs)、类转录激活样效应核酸酶(artificial transcription activator-like effector nucleases,TALENs)和CRISPR-Cas9等[46]基因编辑技术(gene editing)逐步发展并得到广泛应用.基因编辑技术能在基因组水平上对DNA序列进行剪辑或插入,从而导致目的基因的表达受到抑制或表达产物失去相应的功能.Piffanelli等[47]发现,在与小麦亲缘关系较近的大麦中,MLO基因功能的缺失突变使其对白粉病产生广谱和持久的抗性.Wang等[48]采用TALEN和CRISPR-Cas9技术对小麦MLO基因进行编辑,已经获得具有广谱抗白粉病的小麦材料.Qi等[49]结合qRT-PCR检测NgAgo酶与不同引导序列组合作用下目标基因fabp11a的差异表达情况,表明NgAgo技术在降低基因表达水平方面表现出优异的特性.1.2 基因差异表达方法的特点比较SH、DDRT-PCR、cDNA-RDA等方法都是研究基因差异表达的有效工具(表1),其中SAGE、cDNA-AFLP等5种技术能检测出差异表达基因的表达丰度,而其他4种方法则不能;除SH外,其他基因差异表达分析方法均基于PCR技术.应用DDRT-PCR时,结合PCR扩增,可检测出低丰度的mRNA样品,而cDNA-RDA、SSH和cDNA-AFLP等需要经过2~3次PCR扩增,高度富集差异表达基因,保证有较高的特异性,减少假阳性率;SH、cDNA-RDA和SSH技术需要在2个材料之间进行杂交,故仅能检测2组mRNA的差异表达,其他方法可以同时比较多组材料;SAGE 和RNA-seq需要结合测序以及相应软件分析,才能获取差异表达片段以及各自的表达量,其他技术则通过扩增或杂交即可;DNA Chips和RT-PCR/qRT-PCR分别在设计杂交探针和扩增引物时需要预先知道基因序列信息,其他方法均不需要[8,11,28].2.1 挖掘重要农艺性状相关基因农艺性状是指农作物的株高、生育期、育性及产量等可以代表作物特点的重要因子,是作物育种重要考察指标.Firon等[41]通过分析甘薯起始膨大根(initiating storage roots,ISRs)和纤维根(fibrous roots,FRs)的转录组信息,发现至少2.5倍的表达差异短片段8 353个,采用qRT-PCR法对其中Sporamin、AGPase和GBSS1等9个基因进行检测,表明这些差异表达基因参与碳水化合物的代谢和淀粉合成,促使储藏根的形成.Tao等[43]利用Illumina paired-end(PE)转录组测序技术,结合重头组装策略对甘薯7个不同组织的转录组进行分析,为甘薯组织特异表达基因和非生物逆境基因的研究奠定基础.程立宝等[50]对莲藕进行转录组测序分析,发现86个可能与莲藕根茎膨大相关基因,得到10 个贮藏蛋白合成和5 个淀粉合成相关基因,其中Lrplp8和Lrgbss对莲藕根状茎的膨大起到重要作用.育性是有性繁殖作物重要的农艺性状.雄性不育性的发现及三系配套育种、光温不育等概念的提出及成功运用,为新品种的培育和推广带来了极大的方便[51].黄鹂等[52]利用拟南芥ATH1基因芯片与3种不同类型的白菜不育系及其共同保持系的花蕾的mRNA进行杂交,发现各不育系与保持系的花蕾中基因表达存在巨大差异,不同类型不育系之间花蕾转录组的组成特征也有差异.由于3种不育系与保持系花蕾的差异仅表现在花粉的形成和绒毡层的发育上,而其他花器官均无差别,从而推断这些差异表达的基因可能与花粉花药的发育有关.刘冬梅等[53]用陆地棉洞A 的不育株和可育株小孢子单核早期花药进行转录组测序,获得51个激素相关差异表达基因,首次分析小孢子时期激素相关基因在转录组水平上的差异,并对其中2个关键基因进行验证,为深入研究陆地棉洞A的不育机理和挖掘关键基因奠定了基础.2.2 挖掘重要品质性状相关基因随着农作物新品种的更迭以及栽培技术的革新,我国的粮食产量已达到比较理想的水平,人均收入逐步提高的同时,人们的食物消费开始转向有营养、益健康且口感佳的方向,所以对农产品的外观品质和营养品质等要求更高.外观品质是农产品商品价值的重要指标,如水稻种子灌浆不充分、胚乳中的淀粉粒等营养物质排列疏松导致垩白,影响稻米的外观品质[54].Chen等[55]采用RNA-Seq法,在垩白率及胚乳垩白度均低的籼稻品种PYZX和垩白率及胚乳垩白度均高的粳稻品种P02428中发现5 552个差异表达基因,与PYZX相比,P02428中表达量较高的基因有3 603个,较低的基因1 949个;而与2亲本的高垩白重组自交系(recombinant inbred lines,RIL)混样相比,低垩白RIL混样中有88个基因表达量较高,623个基因表达量较低,从中分析确定33个可能与垩白相关的候选差异表达基因,为后续的基因功能验证和育种应用奠定了基础.营养品质包括淀粉及可溶糖等碳水化合物、蛋白质、脂肪酸等,不同加工用途对营养成分的要求不尽相同[56].小麦、甘薯等是重要的淀粉类作物,利用基因差异表达技术分析淀粉合成相关的基因,对育种研究至关重要.小麦材料CB037A具有A 型(直径>10 μm)、B型(直径5~10 μm)和C型(直径<5 μm)3种淀粉粒,而PI330483仅有A型淀粉粒,Cao等[57]采用qRT-PCR法对这2份小麦材料的淀粉粒大小与AGPase大亚基、AGPase小亚基、SSⅠ、SSⅡa和SBEⅠ等淀粉合成相关基因的表达模式进行研究,发现SBEⅡa、SBEⅡb、WaxyD1和AGPase大亚基基因在2份材料中呈截然不同的表达模式.2.3 挖掘耐逆相关基因全球气候逐渐恶化,极端天气逐渐增多,其中干旱是非常普遍的现象,正考验着农业生产.Li等[58]利用基因芯片对玉米抗旱相关小RNA的基因差异表达进行分析,得到miR156、miR159、miR319等3个与抗旱相关的家族基因.Deng等[59]用差异表达的方法从耐旱玉米品系中分离到4个差异表达cDNA片段,并用实时荧光定量PCR分析这4个基因在干旱胁迫下的6个玉米近交系中的表达模式,证实候选基因在耐旱品系中呈上调表达,而在干旱敏感品系则相反.现代农业的投入逐渐加大,而农药、除草剂、化肥以及工业废弃物等各种形式的土地污染严重影响我国的粮食和其他经济作物的产出,植物功能基因的差异表达使其能最大限度地耐受逆境胁迫.Gao等[60]通过转录组测序技术获得紫花地丁镉处理与非镉处理条件下892个差异表达基因,且随机选取15个DEGs进行qRT-PCR 结果验证,为进一步研究其耐镉胁迫机制提供遗传学基础.印莉萍等[61]比较正常供铁和缺铁胁迫下铁高效型小麦(京-411)和铁低效型小麦(三属麦-3)的基因表达差异模式,获得ATP结合转运体(ATP binding cassette,ABC)的cDNA片段并进行Northern杂交,证明它的基因表达受缺铁胁迫的抑制.Kato等[62]利用基因芯片分析硝酸铵诱导下拟南芥和水稻中eIF6(eukaryotic translation initiation factor 6)基因的差异表达,发现该基因在这2种植物中呈现出不同的表达模式,表明eIF6基因在不同的物种中具有表达特异性.除了非生物胁迫外,病虫害等生物胁迫也给农业生产造成巨大的损失,所以挖掘生物胁迫应答基因,辅助选育抗病虫新品种,能有效地缓减农药的使用,增加农民收入和提高生产效率.Evers等[63]以抗马铃薯晚疫病品系Solanum phureja和感晚疫病双单倍体S. tuberosum subsp. tuberosum为材料,用差异显示mRNA法,获得与抗病性、胁迫应答、初级新陈代谢和次级新陈代谢相关的基因.2.4 挖掘与作物杂种优势相关的基因作物杂种优势是杂种后代在表型上优于亲本的现象,涉及作物病虫抗性、高产、高油以及高蛋白等多个方面.杂种优势在自然界比较普遍,但对其具体机理却知之甚少.近年来研究者试图运用基因差异表达技术揭示杂种优势的成因,并取得一定的进展.Zhao等[64]用棉花杂交种及其亲本进行杂种优势研究,发现其中差异表达基因有定量和定性的区别,定性差异是在亲本中高表达或低表达的基因在杂交种中显著高表达;而定量差异有4种基本模式,即在双亲中表达,但后代不表达(BPnF1);其中一个亲本表达,后代不表达(UPnF1);亲本均不表达,后代表达(UF1nP);双亲之一有表达,同时后代也表达(UPF1).在亲本及其后代整个生长期叶片中观察到的基因差异表达可能是杂种优势现象的成因.Wang等[65]通过分析12个玉米近交系及其配组的33个杂交系的基因差异表达情况,发现基因在双亲及其杂种后代中均表达的模式占大多数,故杂种优势不仅与基因表达与否有关,还与基因的表达丰度有关;在玉米雌幼穗发育初期,杂交种的基因表达与双亲的基因表达差异最大;另外,某些基因在杂种中不表达,可以促进籽粒的发育并抑制幼穗中小花发育.利用基因差异表达分析技术,能挖掘新的功能基因,揭示基因的新功能等,为探究农作物的农艺性状、品质性状以及抗逆性等方面的机理机制奠定基础.随着生命科学进入后基因组时代,通过测序及功能注释将对DNA序列、基因表达通路、蛋白质结构及其互作关系等进行初步的鉴定.高通量测序技术和生物信息学的运用,结合qRT-PCR验证提高研究的准确性,也加快了该领域的研究进程.本课题组采用转录组测序技术,对比分析甘薯徐薯18和徐781的转录组信息,在一定程度上解释2种材料的淀粉含量差异和抗性差异(数据未发表),但其具体的调控机制有待进一步研究.未来,从基因差异表达分析入手获得相关功能的候选基因,采用基因编辑技术对目标基因进行敲除或降低其表达量,可逐步实现分子设计育种的目标[66-67].*通讯作者:李强,男,研究员,博士,主要从事甘薯遗传与分子育种研究,E-mail:****************.【相关文献】[1] 吴乃虎.基因工程原理[M].2版.北京:科学出版社,1998.[2] 刘凯,曾继吾,夏瑞,等.mRNA差异显示技术及其在园艺植物上的应用(综述)[J].亚热带植物科学,2009,38(1):78.[3] 黎裕,王建康,邱丽娟,等.中国作物分子育种现状与发展前景[J].作物学报,2010,36(9):1425.[4] Lamar E E,Palmer E.Y-encoded,species-specific DNA in mice:evidence that the Y chromosome exists in two polymorphic forms in inbred strains[J].Cell,1984,37(1):171. [5] Liang P,Pardee A B.Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction[J].Sci,1992,257(5072):967.[6] Hubank M,Schatz D G.Identifying differences in mRNA expression by representational difference analysis of cDNA[J].Nucl Acids Res,1994,22(25):5640.[7] Velculescu V E,Zhang L,Vogelstein B,et al.Serial analysis of geneexpression[J].Sci,1995,270(5235):484.[8] Diatchenko L,Lau Y F,Campbell A P,et al.Suppression subtractive hybridization:a method for generating differentially regulated or tissue-specific cDNA probes and libraries[J].Proc Natl Acad Sci USA,1996,93(12):6025.[9] Bachem C W,van der Hoeven R S,de Bruijn S M,et al.Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP:analysis of gene expression during potato tuber development[J].Plant J,1996,9(5):745.[10] Cottrez F,Auriault C,Capron A,et al.Quantitative PCR:validation of the use of a multispecific internal control[J].Nucl Acids Res,1994,22(13):2712.[11] 金凤媚,薛俊,郏艳红,等.半定量RT-PCR技术的研究及应用[J].天津农业科学,2008,14(1):10.[12] 张春兰,秦孜娟,王桂芝,等.转录组与RNA-Seq技术[J].生物技术通报,2012,28(12):51.[13] 白根本,沈昕,王沙生.差减杂交方法的原理和应用[J].生物工程进展,1998,18(6):54.[14] 佘卫炜,郭志刚,刘瑞芝.用扣除杂交法分离藏红花苷合成相关基因的克隆[J].清华大学学报(自然科学版),2004,44(12):1592.[15] 李捷,印莉萍,刘维仲.示差扣除杂交法及其在分子生物学中的应用[J].生物技术通报,1999,15(3):9.[16] 白根本,沈昕,王沙生.胡杨盐诱导基因与盐抑制基因的差减杂交显示研究[J].林业科学,2003,39(2):168.[17] 张弛,陈受宜.利用DDRT-PCR技术分析在盐胁迫下水稻耐盐突变体中特异表达的基因[J].中国科学(B辑),1995,25(8):840.。
EST (Expressed Sequence Tag)表达序列标签

EST (Expressed Sequence Tag)表达序列标签EST (Expressed Sequence Tag)表达序列标签—是从一个随机选择的cDNA 克隆,进行5’端和3’端单一次测序挑选出来获得的短的cDNA 部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20 到7000bp 不等,平均长度为360 ±120bp。
由于cDNA文库的复杂性和测序的随机性,有时多个EST代表同一基因或基因组,将其归类形成EST 簇(EST cluster)原理:EST是从一个随机选择的cDNA 克隆进行5’端和3’端单一次测序获得的短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20 到7000bp 不等,平均长度为360 ±120bp。
EST 来源于一定环境下一个组织总mRNA 所构建的cDNA 文库,因此EST也能说明该组织中各基因的表达水平。
技术路线:首先从样品组织中提取mRNA,在逆转录酶的作用下用oligo (dT) 作为引物进行RT-PCR合成cDNA,再选择合适的载体构建cDNA 文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST 序列的产生过程。
应用:EST作为表达基因所在区域的分子标签因编码DNA 序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比更可能穿越家系与种的限制,因此EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有用。
同样,对于一个DNA 序列缺乏的目标物种,来源于其他物种的EST也能用于该物种有益基因的遗传作图,加速物种间相关信息的迅速转化。
具体说,EST的作用表现在:⑴用于构建基因组的遗传图谱与物理图谱;⑵作为探针用于放射性杂交; ⑶用于定位克隆;⑷借以寻找新的基因; ⑸作为分子标记;⑹用于研究生物群体多态性;⑺用于研究基因的功能;⑻有助于药物的开发、品种的改良;⑼促进基因芯片的发展等方面。
利用表达序列标签电子克隆cDNA全序列的策略

利用表达序列标签电子克隆cDNA全序列的策略孙淼;赵茂林【摘要】基因组计划的进展及表达序列标签数据的迅速扩增使得电子克隆方法孕育而生,为进行基因克隆开辟了一条新的路径.介绍了表达序列标签和电子克隆的原理及过程,重点分析电子克隆过程中遇到的问题及解决方法,展望其在新基因功能研究中的作用.【期刊名称】《生物技术通报》【年(卷),期】2010(000)001【总页数】4页(P49-52)【关键词】表达序列标签;电子克隆;聚类;叠连群【作者】孙淼;赵茂林【作者单位】首都师范大学生命科学学院,北京,100048;北京市农林科学院农业生物技术研究中心,北京,100097【正文语种】中文Abstract: The progress of genome project and the rapid expansion of expressed sequence tags(ESTs),make in silico cloning brought on,which for us has opened a new path to gene cloning.In this article,an overview of EST,the principle andmethod of in silico cloningwere discussed,focusing on analysis of problems and solutions during in silico cloning process,also,it prospected the roles in the study of the new gene function.Key words: Expressed sequence tags In silico cloning Clustering Contig随着基因组计划的深入进行,很多实验室采用cDNA文库大规模测序、差异显示PCR(different display PCR,DDRT-PCR)、代表性差异分析 (representation difference analysis,RDA)及抑制性消减杂交(suppression subtractive hybridization,SSH)等技术发现了大量具有潜在应用价值的新基因片段。
表达序列标签技术及其应用

( n r l o t ie st fF r sr & Te h oo y,Ch n s a 4 0 0 Ce ta u h Unv ri o o e ty S y c n lg a g h 1 0 4,Hu a n n,Chn ) i a
r i w ev e ห้องสมุดไป่ตู้
表 达序 列 标 签 ( x rs e u n etg ,以下 简 称 为 E T) c E p essq e c a s S 是 DNA 的 部分 序 列 , 是将 mRNA 反 转 录成 c DNA 并克 隆到 载体 构建 成 c DNA 文 库 后 , 规模 随机 挑 选 c NA 克 隆 , 其 5 或 3 端进 行 一步 法 测序 , 大 D 对 ’ ’ 并
它 序列特 征 等研 究领 域 , 且取 得 了显 著 成效 。 并
型 和不 同发 育 阶段 的表达 基 因序 列 的数 目急剧 上 升 。E T 技 术被 广泛 应用 于分 子标 记 、 S 分离 鉴定 新 基 因、 因 基
表 达谱 分 析 、 因组 功 能 注释 、 因电 子 克隆 、 备 D 基 基 制 NA 芯 片 、 NAiR nefrn e 技 术 的研 究 、 找其 R ( NA itree c ) 寻
将 所获序 列 与基 因数 据库 中已知 序列进 行 比较 , 而 获得 对生 物体 生长 发 育 、 殖 分化 、 从 繁 遗传 变异 、 老死 亡等 衰
一
系列 生命 过程认 识 的技 术 。 近 年来 , 随着 E T 计 划在 不 同物种 问 的展 开 和研 究 内容 的深 入 , 源于 不 同物种 、 同组织 、 同细胞 类 S 来 不 不
表达序列标签(EST)及其在抗孢囊线虫大豆研究中的应用

收稿 日 : 0 5 2 3 期 2 0 —1 - 1 基金项 目:{ 龙江省教育厅项 目(0 4 0 8 I l I 1 5 18 ) 第一作着筒 介:毕髟东( 9 4 , , 1 7 一)男 黑龙江省 望誊县人 。 士, 事分 子遗传学研究 .T l0 5 —5 7 8 1 ・ 硕 从 el4 1 9 8 4 0 E—me Id i0  ̄1 3 i y b3 8 6 . l
维普资讯
黑龙 汪农业科 学 20 t3 =0 3 0 6 () 9  ̄9
He o gi gAg i l r l c n e i n j n r ut a S i c s l a e u e
表达序列标签 ( S 及其在抗孢囊线虫 E T) 大豆研究 中的应用
b a t e i a c h o b a y t e td S N)wees mmai di hsp p r e nwi rss net tesy e nc s n mao e( C h t O r u r e t i a e. z n
Ke od : x rse eunetg E T) sy en o ba yt e td S N) yw rs epesdsq e c as( S ; ob a ;ຫໍສະໝຸດ y encs nmao e( C
L ii I - n .WA G X a -ig . AI o g u : Jl N iopn ¨ B n - n Y j
(. ilg e at n f Habn Noma Unv ri .Habn 1 0 8 ; . e in A r u ca 1B oo y D p rme to r i r l ies y t r i 5 0 0 2 B ia g i h rl l c S ri e trKeh n Ke h n 1 1 3 evc C ne . s a . s a 6 6 3 e l
基因表达系列分析(Serial Analysis of Gene Expression,SAGE)技术
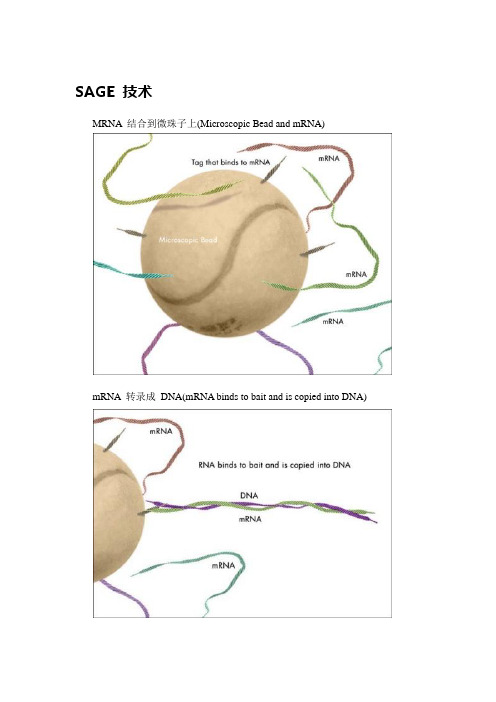
SAGE 技术MRNA 结合到微珠子上(Microscopic Bead and mRNA)mRNA 转录成DNA(mRNA binds to bait and is copied into DNA)用酶切开DNA的一小段(An enzyme cuts the DNA)另一个酶定在DNA末端以便切下一小段(An enzyme locks onto the DNA and cuts off a short tag),这一小段就被视为这个基因的标签两个标签连在一起(Two tags are linked together)在末端的定位分子被切掉(Enzymes cut off the "Docking Molecules")都连成一条线(Di-Tags are combined into large concatemers)DNA上所携带的遗传信息,需要通过RNA为中介体,合成出组织和正常生理功能所需要的蛋白质,这个过程被称为基因的表达。
在生物体中不同的组织和器官所表达的基因群是不一样的,我们把基因群的表达状况称为基因表达谱。
目前,高通量地研究基因表达谱的方法主要有两种,即生物芯片和基因表达串联分析(serial analysis of gene expression, SAGE)。
基因芯片所能检测的基因必须是已知的基因,放在芯片上几种基因的探针就只能检测这几种基因的表达谱;相比之下,SAGE能以远高于DNA芯片的精确度和重复性来检测在病理条件下基因表达谱的改变,而不必考虑所检测的基因是已知的还是未知的。
因此在检测疾病相关的新基因,特别是无法用基因芯片进行检测的低表达量致病基因时,SAGE是目前的最佳手段,无可取代。
SAGE技术为Genzyme公司所拥有的专利技术。
其技术简介如下:SAGE技术得以建立的理论基础首先,一段来自于任一转录本特定区域的"标签"(Tag),即长度仅9-14bp的短核苷酸序列,就已包含足够的信息以特异性地确定该转录本。
基因表达数据分析方法及其应用研究共3篇

基因表达数据分析方法及其应用研究共3篇基因表达数据分析方法及其应用研究1随着技术的不断发展,基因表达数据分析在生命科学研究中扮演着越来越重要的角色。
基因表达数据分析是研究基因功能的关键一步,它使得科学家可以了解基因在特定情况下的表达水平。
在本文中,我们将讨论基因表达数据分析的方法及其应用。
1.基因表达数据的来源和类型基因表达数据是通过分析转录组和基因芯片等数据获得的。
转录组技术通过测量RNA浓度,包括RNA-seq和microarray。
而基因芯片就是一种将成千上万的基因测量并呈现的芯片。
基因表达数据存在多种类型,包括原始数据、表达矩阵、差异表达矩阵、注释文件和元数据等等。
2. 基因表达数据分析的方法(1)数据清理数据清理是数据分析过程中的第一步。
它包括数据预处理、去除冗余数据、去除噪声和填补数据空缺等操作。
(2)正则化正则化的目的是调整不同基因表达数据之间的差异,消除数据中的计量误差和探测效率的误差。
几种正则化方法包括平滑、归一化和标准化。
(3)差异分析差异分析是研究基因表达数据中各基因在不同样品之间差异的方法。
常用的差异分析方法包括t-test、ANOVA、FDR和q值等。
(4)聚类分析聚类分析是将数据根据观察指标相似度进行分类的方法。
在基因表达数据上,它通常用于发现不同条件下的基因表达模式。
(5)变异分析变异分析是一种寻找表达值变异的基因的方法。
通常,基因的变异程度与其在癌症和其他疾病中的作用有关。
(6)功能注释功能注释是将基因表达数据与已知基因功能相结合的方法,从而获得数据更深层次的信息。
它通常用于解释基因表达数据的生物学意义,如基因表达数据和肿瘤发展的相关性等。
3.应用研究基因表达数据分析可应用于许多研究领域,包括基因表达和调控、单细胞分析和肿瘤生物学等。
(1)基因表达和调控基因表达数据分析可用于挖掘基因之间的相互关系以及调控通路。
这些信息可以在理解细胞生物学、发育及疾病发生机制的过程中发挥重要作用。
鹅卵泡差异显示表达序列标签(ESTs)的筛选及分析的开题报告

鹅卵泡差异显示表达序列标签(ESTs)的筛选及分析的开题报告一、研究背景与意义:鹅卵泡是女性生殖系统中负责生成卵子的基本单位,其发育过程受到多种内外因素的影响。
为了更好地理解鹅卵泡发育的分子机制,近年来研究重点逐渐转向基因表达调控层面。
ESTs技术可以对特定组织或细胞中的部分基因进行快速分析,寻找分子调控网络中的关键因子和信号通路,从而探究鹅卵泡发育的分子调控机制。
因此,建立鹅卵泡ESTs数据库和研究表达序列标签的筛选及分析方法具有重要的理论和实践意义。
二、研究内容和方法:1. 构建鹅卵泡ESTs数据库。
该数据库包括单个鹅卵泡组织中的全部ESTs 序列,通过高通量测序技术获取 ESTs 数据库,并进行基本注释和序列比对,建立 ESTs 序列库。
2. ESTs 序列筛选。
通过 ISAAC3.0 软件对 ESTs 序列进行自动序列清洗,去掉低质量序列,同时选择出序列长度大于200 bp 的清洗后的ESTs 序列,作为后续分析的数据源。
3. ESTs 序列比对和聚类分析。
采用 BLASTX 程序将 ESTs 序列比对至 NCBI NR 数据库,获得ESTs序列的功能注释和分类,然后根据序列比对结果进行序列聚类分析,以获得一组 ESTs标签集合。
4. ESTs 序列表达量分析。
采用 RSEM 工具进行 ESTs 序列的表达量估算,包括 Reads 数等指标,以简化 ESTs 序列表达量研究的数据分析流程。
同时,采用基因差异表达分析,筛选保守区和差异表达区域。
5. 功能注释和途径富集分析。
通过 GO 和 KEGG 数据库对 ESTs 序列进行功能注释和途径富集分析,识别出差异表达的基因和分子途径,并对 ESTs 序列的生物学意义进行进一步解释和说明。
三、预期研究结果:通过以上研究内容和方法,我们期望获得鹅卵泡 ESTs 序列的有关信息和特征,得到一组鹅卵泡 ESTs 标签,包括表达量、功能注释和途径富集等。
表达序列标签研究进展及其在甲壳动物中的应用概况

表达序列标签研究进展及其在甲壳动物中的应用概况摘要:随着生物信息学的发展,表达序列标签(EST)在分子标记开发、新基因分离鉴定、基因表达谱分析、基因组功能注释、基因电子克隆等方面具有重要作用。
简要介绍了EST分析的原理及其在基因识别、基因预测、物理图谱的构建、DNA芯片制备等方面的应用概况。
综述了甲壳动物EST的研究现状,并对EST的应用前景进行了展望。
关键词:表达序列标签(EST);甲壳动物;生物信息学Abstract:Withthedevelopmentofbioinformatics,expressedsequencetag(EST)played animportantroleinmolecularmarkersdevelopment,newgenesisolationandidentification,geneexpressionprofileanalysis,genomefunctionalannotationandsilicogenecloning.TheprincipleofESTanalysisanditsapplicationsingeneidentification,geneprediction,physicalmapconstructionandDNAchippreparationwas briefly introduced.Inaddition,theresearchstatusofcrustaceanESTandthe prospectofESTapplicationwerealsosummarized.Keywords:expressedsequencetag(EST);crustacean;bioinformatics表达序列标签(Expressedsequencetag,EST)是从一个随机选择的cDNA克隆进行5’端和3’端单一次测序获得的短的cDNA部分序列。
基于表达序列标签(EST)的基因克隆和基因表达分析研究进展

腹 果蝇 ( Dr o s p h i l a me l a n n o g a s t e r ) L 1 、 拟南 芥 ( Ar a — b i d o p t h d l i d d ) [ i s 等 模 式 生 物 基 因 组 测 序 的 完 成, 这 些 生 物 的 基 因得 以初 步 确 定 。据 估 计 , 人 类 约 有 3万 余 个 基 因 , 线虫有 1 . 9万 个 基 因 , 果蝇有 1 . 3 6 万个基 因, 拟南芥有 2 . 5万 多 个 基 因 。 这 些 基 因 都 已 被 收 录 到公 共 数 据 库 中 。 由于 E S T 代 表 着 一 段 表 达
[ 中图 分类 号] Q7 8
[ 文 献标识 码 ] A
[ 文 章编 号] 1 0 0 0 2 7 8 2 ( 2 0 0 2 ) 0 4 — 0 1 4 1 — 0 5
在 人 类基 因组 计划 ( Hu ma n Ge n o me P r o j e c t ,
育、 繁殖分化 、 遗 传变异 、 疾 病发生 、 衰 老 死 亡 等 生 命
Ge n B a n k /i md e x . h t m1 ) 、 欧 洲 分 子 生 物 学 室 验 室
维普资讯
第 3 O卷
第 4期
西北农 林 科技大 学 学报 ( 自然 科 学 版 )
V O1 . 3O N O.4 A ug. 2 002
EST文库构建原理和应用

参考资料:
• • • • 生物技术通报 2004年第一期 作物杂志 2007年 中国知网 EST技术流程应用
• EST作为表达基因所在区域的分子标签因 编码DNA 序列高度保守而具有自身的特殊 性质,与来自非表达序列的标记(如AFLP、 RAPD、SSR 等) 相比更可能穿越家系与 种的限制,因此EST标记在亲缘关系较远 的物种间比较基因组连锁图和比较质量性 状信息是特别有用。同样,对于一个DNA 序列缺乏的目标物种,来源于其他物种的 EST也能用于该物种有益基因的遗传作图, 加速
1. 2. 3. 4. 5. EST概述 EST序列原理 EST产生原因 EST的应用 展望
EST概述:
• 表达序列标签(expressed sequence tags,ESTs)是指从不同组织来源的cDNA 序列,指的是一组cDNA的部分序列,一般 长度为150~500bp,是由大规模随机挑取的 cDNA克隆测序得到的组织或细胞基因组的 表达序列标签。一个EST代表生物某一时 期的某种组织或细胞的一个表达基因。
表达序列标签 (expressed sequence tags,ESTs)
EST的制备程序
• 某特异组织→总RNA的提取 →Poly(A)+mRNA→cDNA→两 端加接头→与λzap载体连接、包 装、裂解→转染E.coli的XLI— Blue细胞,进行in vivo excision 用含有x-gal和IPTG的氨苄平板筛 选→随机挑取白色菌落→提取质粒 →M13活T7通用引物测序。
的约150500bp的一段cDNA序列。
EST产生的原因:
• 1990年人类基因组计划 (HGP)及其它模式生物的 基因组计划实施以来 ,人们对生物体生命现象的 研究已不仅仅局限于局部某个或几个基 因,而是 把目光投向整个基因组 ,从整体水平去考虑基因 的存在 ,结构与功能,以及基因之 间的相互关系 等.而如何快速、高效地从基因组中获取生物学信 息,已成为一个急迫而富有挑战性的课题摆在我 们面前 。 EST技术就是基于这种认识而发展起来 的,并成为基因组学研究的一个强有力的工具。
《2024年数据标注研究综述》范文

《数据标注研究综述》篇一一、引言随着人工智能技术的不断发展,数据标注作为其重要一环,在机器学习、深度学习等领域得到了广泛应用。
数据标注是指对原始数据进行加工、处理、标记等操作,以便于机器学习算法进行训练和模型优化。
本文旨在综述数据标注的研究现状、方法、应用及未来发展趋势,为相关领域的研究者提供参考。
二、数据标注的研究现状近年来,数据标注技术在学术界和工业界均得到了广泛关注。
学者们针对数据标注的方法、工具和流程进行了深入研究,为相关领域的实践提供了重要支撑。
当前,数据标注的研究已经涉及到了图像、文本、语音等多个领域,并且在不同的应用场景中发挥着重要作用。
三、数据标注的方法数据标注的方法主要包括人工标注和自动标注两种方式。
其中,人工标注是通过专业人员对数据进行标记、分类和注释等操作,具有准确度高、可靠性强的优点。
然而,人工标注成本高、效率低,难以满足大规模数据处理的需求。
因此,自动标注技术逐渐得到了广泛关注。
自动标注是通过机器学习算法对数据进行自动标记和分类,具有处理速度快、成本低的优点。
但是,由于机器学习算法的局限性,自动标注的准确性和可靠性还有待进一步提高。
四、数据标注的应用数据标注在多个领域中得到了广泛应用。
在计算机视觉领域,数据标注被广泛应用于图像分类、目标检测、语义分割等任务中。
在自然语言处理领域,数据标注被用于文本分类、情感分析、机器翻译等任务中。
此外,在智能驾驶、医疗影像分析等领域中,数据标注也发挥着重要作用。
通过数据标注,可以有效地提高机器学习算法的准确性和性能,为相关领域的实际应用提供了有力支持。
五、数据标注的流程及工具数据标注的流程通常包括数据预处理、标记和验证等步骤。
首先需要对原始数据进行清洗、去噪和标准化等预处理操作,以便于后续的标记工作。
然后,根据具体任务的需求,对数据进行分类、注释和标签等操作。
最后,需要对标记后的数据进行验证和评估,以确保其准确性和可靠性。
目前,已经出现了多种数据标注工具和平台,如LabelImg、VOTAPlay等图像标注工具,以及TextRazor等文本标注工具。
植物功能基因组的主要研究方法及其应用

植物功能基因组的主要研究方法及其应用摘要概述了植物基因功能的主要研究方法,并论述了主要技术如cDNA微阵列与基因芯片技术、反向遗传学技术、表达序列标签(EST)、蛋白质组学、生物信息学等及其应用。
关键词植物功能基因组;方法;应用基因组学(genomics)指对所有基因进行基因组作图、核苷酸序列分析、基因定位和基因功能分析的一门科学[1,2]。
许多生物全基因组的破译,使基因组学的研究有了一次质的突破:从结构基因组学开始过渡到功能基因组学。
结构基因组学(structural genomics)是通过基因作图、核苷酸序列分析以确定基因组成、基因定位的一门科学。
功能基因组学(functional genomics)代表基因组分析的新阶段,被称为后基因组学(post genomics),旨在利用结构基因组学丰富的信息资源,应用高通量、大规模的实验分析方法,结合统计和计算机分析来研究基因的表达、调控与功能,基因间、基因与蛋白质、蛋白质与底物、蛋白质与蛋白质之间的相互作用以及生物的生长、发育等规律[3]。
传统的遗传学的方法已不能适应现在基因组学的发展,cDNA微阵列(cDNA micro-array)和基因芯片(gene chip)法、反向遗传学、表达序列标签(expressed sequence Tag,EST)、蛋白质组学、生物信息学等方法相继诞生,为基因组学的研究奠定了坚实的基础。
1cDNA微阵列与基因芯片法cDNA微阵列和基因芯片都是基于Reverse Northern杂交以检测基因表达差异的技术。
二者的基本原理是利用光导化学合成、照相平板印刷以及固相表面化学合成等技术,在固相支持物上固定成千上万个cDNA、EST或基因特异的寡核苷酸探针,并与放射性同位素或荧光标记的靶DNA进行杂交,然后用相应的检测系统进行检测,根据杂交信号强弱及探针的位置和序列,即可确定靶DNA的表达情况以及突变和多态性的存在。
该技术优点在于可以同时对大量基因,甚至整个基因组基因的表达差异进行对比分析。
生物信息学 第6章 表达序列标签

➢使用合适的比对参数,大于90%的已经注释的基因都能
在EST库中检测到。
精品课件
(二) ESTs与基因表达谱的构建
➢表达量比较分析:不同组织或发育阶段基因表达量比较 ➢EST来源于不同的组织,那么就可以对不同来源的基因 表达进行比较
精品课件
(三) ESTs与新基因预测
➢由于EST的一个基因的 部分序列。
从已建好的cDNA库中随机取出一个克隆,从 5′末端或3′末端对插入的cDNA片段进行一轮单 向自动测序,所获得的约60-500bp的一段cDNA序 列。
精品课件
二、EST数据分析方法
随机挑取克隆进行5′或3′端测序 序列前处理 聚类和拼接
基因注释及功能分类 后续分析
精品课件
(一)序列前处理
➢ 去除低质量的序列(如使用Phred)
精品课件
五、常用的EST数据库
数据库名称
网址
说明
dbEST
/dbEST/ 综合
UniGene /unigene 综合
Gene Indices /tgi/ 综合
电子PCR克隆,指利用已经有的片段进行 全长基因序列的分析。
5
3
5
3
精品课件
四、EST数据的不足
➢ESTs很短,没有给出完整的表达序列; ➢低丰度表达基因不易获得; ➢由于只是一轮测序结果,出错率达2%~5%; ➢有时有载体序列和核外mRNA来源的cDNA污染或是基 因组DNA的污染; ➢有时出现镶嵌克隆; ➢序列的冗余,导致所需要处理的数据量很大。
精品课件
(三)序列注释和分析
➢ 序列注释 ➢ 后续分析
精品课件
三、EST的用途
基因识别 基因表达谱的构建 发现新基因 SNP(single nucleotide polymorphism)发 现
基因表达标签测序技术
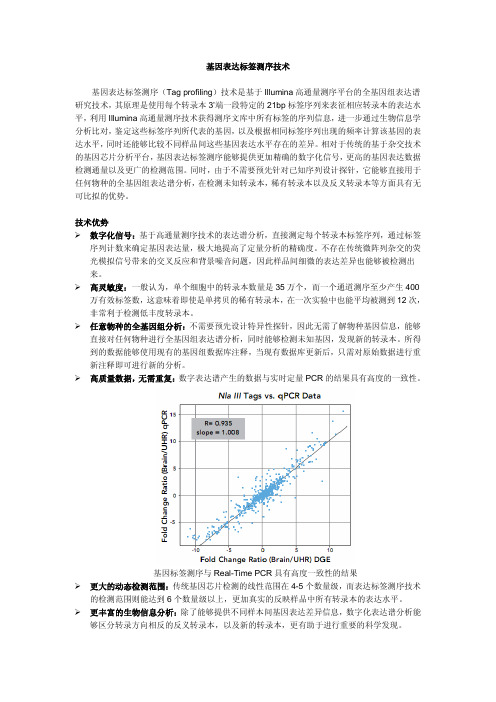
基因表达标签测序技术基因表达标签测序(Tag profiling)技术是基于Illumina高通量测序平台的全基因组表达谱研究技术,其原理是使用每个转录本3’端一段特定的21bp标签序列来表征相应转录本的表达水平,利用Illumina高通量测序技术获得测序文库中所有标签的序列信息,进一步通过生物信息学分析比对,鉴定这些标签序列所代表的基因,以及根据相同标签序列出现的频率计算该基因的表达水平,同时还能够比较不同样品间这些基因表达水平存在的差异。
相对于传统的基于杂交技术的基因芯片分析平台,基因表达标签测序能够提供更加精确的数字化信号,更高的基因表达数据检测通量以及更广的检测范围。
同时,由于不需要预先针对已知序列设计探针,它能够直接用于任何物种的全基因组表达谱分析,在检测未知转录本,稀有转录本以及反义转录本等方面具有无可比拟的优势。
技术优势¾数字化信号:基于高通量测序技术的表达谱分析,直接测定每个转录本标签序列,通过标签序列计数来确定基因表达量,极大地提高了定量分析的精确度。
不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题,因此样品间细微的表达差异也能够被检测出来。
¾高灵敏度:一般认为,单个细胞中的转录本数量是35万个,而一个通道测序至少产生400万有效标签数,这意味着即使是单拷贝的稀有转录本,在一次实验中也能平均被测到12次,非常利于检测低丰度转录本。
¾任意物种的全基因组分析:不需要预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行全基因组表达谱分析,同时能够检测未知基因,发现新的转录本。
所得到的数据能够使用现有的基因组数据库注释,当现有数据库更新后,只需对原始数据进行重新注释即可进行新的分析。
¾高质量数据,无需重复:数字表达谱产生的数据与实时定量PCR的结果具有高度的一致性。
基因标签测序与Real-Time PCR具有高度一致性的结果¾更大的动态检测范围:传统基因芯片检测的线性范围在4-5个数量级,而表达标签测序技术的检测范围则能达到6个数量级以上,更加真实的反映样品中所有转录本的表达水平。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
表达序列标签的应用现状及分析方法研究王晓娜,卢欣石(北京林业大学草地资源与生态实验室,北京100083)摘要:表达序列标签是由大规模随机挑取的cDN A克隆测序得到的组织或细胞基因组的表达序列标签。
1个表达序列标签(EST)代表生物某一时期的某种组织或细胞的1个表达基因。
数量迅速增加的表达序列标签已经成为开发分子标记的重要资源。
介绍了EST原理、基因表达分析的方法比较、基因测序聚类分析的3个数据库比较及详细方法,表明EST在发现新基因及基因组研究中的应用具有良好的前景。
关键词:表达序列标签;聚类;分析方法中图分类号:Q78 文献标识码:A 文章编号:1001 0629(2010)05 0076 09表达序列标签EST(Ex pressed Sequence Tag)是从一个随机选择的cDNA克隆进行5 端和3 端单一次测序获得的短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20~7000bp不等,平均长度为360 120bp。
EST作为表达基因所在区域的分子标签因编码DNA序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比更可能穿越家系与种的限制,因此EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息上是特别有用的。
另外,由于EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此EST也能说明该组织中各基因的表达水平。
EST s已经被广泛地应用于基因识别,研究发现EST s的数目比GenBank中其他的核苷酸序列多,研究人员更容易在EST库中搜寻到新的基因[1]。
由于EST 测序只是测定部分序列,也不需要对克隆进行排序,因而完成EST测定所需要的人力、物力消耗与基因组测序和全长cDNA测序相比要少的多,具有经济和高效的特点。
由于DNA测序技术的不断更新和大规模测序技术的出现,在DNA测序中逐步实现了工厂化和流水作业,因此测序费用大幅度降低[2]。
近年来,表达序列标签数据增长迅速。
在GenBank102版本数据中,EST序列已经占用了2/3的记录[3]。
美国国立生物技术信息中心(Na tional Center for Biotechno logy Information,NC BI)对EST进行了聚类分析,按基因划分EST,组成UniGene数据库。
还有一些网站开发了基于Internet的EST延伸服务,如Labonw eb网站的IRACE(http://ww bonw ),Bio sino 网站的BioEclone(: 9090/bio eclone.htm l)等[4 5]。
因此对EST的技术要求及应用进行归纳分析,有利于对研究对象分析不同基因的表达水平,为挖掘和克隆基因提供理论支撑。
1 EST特点及其应用EST计划作为植物基因组计划的一个重要组成部分,已经在多种植物物种中开展起来。
相关标记包括EST SSR、EST PCR、EST SNP、EST AFLP、EST RFLP等[6]。
近年来,EST的应用已经深入到生物学的领域,其中表达序列标签微卫星(EST SSR)技术的发展和应用较为普遍,根据SSR的来源可将其分为基因组SSR和76-84 05/2010草 业 科 学PRA T A CU L T U RA L SCI EN CE27卷05期V ol.27.N o.05收稿日期:2009 10 15基金项目: 863 高产、多抗、优质苜蓿新品种分子聚合育种项目(2008AA10Z149)作者简介:王晓娜(1986 ),女,河北衡水人,在读硕士生,主要从事牧草分子标记辅助育种研究。
E_mail:xiaotaoyan5070@通信作者:卢欣石 E_m ail:luxins hi304@EST SSR[7]。
EST SSR标记狭义上是指位于EST序列上的或者基于EST序列开发的SSR标记,也被称为eSSR标记。
目前较为常用的核酸序列数据库有:美国国家信息中心的GenBank,欧洲分子生物学实验室的EMBL,日本国家数据库DDBJ,这3个数据库是收录范围最广并完全向公众开放的数据库,在它们中均含有EST子数据库dbEST。
在核酸序列数据库中,EST的量要占65%以上[8]。
由于EST是功能基因的一部分,不同基因组间,基因编码区序列的保守性远远高于非编码区,与基因组SSR相比EST SSR表现出较好的物种之间的可转移性[9 10]。
作为一种新型分子标记,EST SSR来自表达基因,因而除具备传统基因组来源的SSR标记的所有优势外,可能与基因功能表达具有直接或间接关系,从而强化了SSR 标记在遗传研究中的应用[11]。
在种质资源遗传多样性方面,张鹏等[12]利用SRAP和EST SSR分子标记对192份国内外芝麻S esamum ind icum进行分析。
发现我国南部地区芝麻品种遗传多样性较中部和北部地区丰富。
Eujayl等[13]利用EST SSR等3种不同类型的微卫星标记对64个硬粒小麦T riticum aesti vum品种的遗传多样性进行评价,表明EST SSR 可在硬粒小麦中揭示较高的多态性。
在基因连锁方面,利用分群分析法对多花黑麦草L olium p erenne抗叶斑病进行EST CA PS 标记,得到位点p56位于第5遗传连锁群,所处的基因为编码多花黑麦草天冬酰胺合成酶基因[14]。
在基因功能方面,郭久峰等建立沙冬青A m mop ip tan thus mongolicus的cDNA文库并通过EST分析技术研究其抗逆机理,得到的313个已知功能的基因标签中抗逆相关的有48条[15]。
杨成君等[16]建立了药用植物人参Panax qinseng的EST SSR标记。
陈士林等[17]构建了西洋参P.quinquef olius的cDNA文库,经EST 分析获得与水分胁迫相关的基因7个,与受伤诱导相关的基因2个,编码抗氧化酶相关的基因6个。
并在根系的EST文库中发现抗病基因12个,62个EST是其他物种尚未报道的新基因。
佘玮等[18]以生长中期的苎麻Boehmer ia nivea茎皮为材料构建cDNA文库,并进行EST分析,随即测序得到275个有效序列,约53.5%的EST 序列可能是未报道的新基因序列。
综上所述,EST为种质资源的保护利用和遗传育种工作提供科学依据,同时作为功能基因组研究的重要手段,在功能基因的开发与研究中也发挥重要作用。
2 EST在苜蓿中的研究近几年,苜蓿M edicago sativa分子水平的研究有所深入,利用RAPD分子标记研究苜蓿种质资源遗传多样性[19]及其他相关基因的克隆序列分析等研究相对较多,如蒺蒺藜状苜蓿中MtERF 6基因的克隆及序列分析[20]。
但EST的研究相对较少,闫娟等[21]利用EST SSR标记分析了我国北部和中部地区天蓝苜蓿M.lup ulina 的遗传多样性和遗传结构,推测中等水平的遗传多样性和高度的居群间遗传分化主要受它的自交特性和分布方式影响。
在Genbank数据库中进行搜索,得出测序最多的20个物种中,除经济类作物玉米Zea may s、水稻Ory z a sativa、小麦等序列较多外,大部分物种为动物,蒺藜状苜蓿排在最后,序列条数为409757。
在表达序列标签数据库(dbEST)中进行搜索,测序最多的前20个物种中,没有和苜蓿相关的物种序列(总序列45660524条)。
由以上数据可以看出,苜蓿基因的测序分析研究相对较少,只有蒺藜状苜蓿得到的EST 较多,而紫花苜蓿和黄花苜蓿M.f alcata有待深入的研究。
3 EST获取及分析过程3.1EST的获取过程 构建生物某一发育阶段的cDNA文库,然后大规模、随机地挑选cDNA 文库中的克隆或通过某种方法筛选cDNA中的某些克隆,最后对cDNA克隆的5 及3 进行测序,进而得到一个EST[22]。
7705/2010草 业 科 学(第27卷05期)3.2EST分析过程3.2.1利用ESTs大规模分析基因表达水平 一般认为,组织和细胞分化依赖于基因特异性的时空表达,而生物体在某一时期的基因表达数量通常只占全部基因的15%[23]。
因为EST序列是从某种特定组织的cDNA 文库中随机测序而得到的,所以可以利用未经标准化和差减杂交的cDNA文库EST分析特定组织的基因表达谱。
标准化的cDNA文库和经过差减杂交的cDNA文库则不能反应基因表达的水平。
为研究癌症的分子机理,美国国家癌症研究所NCI的癌症基因组解析计划(Cancer Genome Anatomy Pr oject,CGAP)构建了很多正常的或是癌症前期的和癌症后期的组织的cDNA文库,并进行了大规模的EST测序,其中大部分的文库未经标准化或差减杂交处理。
CGAP网站提供了多种工具用以分析不同文库间基因表达的差异,如:Digital Gene Expressio n Displayer(DGED)和cDNA x Pro filer。
3.2.2基因表达系列分析(Serial Analy sis of Gene Expressio n,SAGE) 随着公用数据库中EST数据的急剧增加,基因表达研究可以利用数字化分析方法来实现[24 25],即从能够代表相应组织或器官基因表达情况的cDNA文库中获得大量EST,经过软件聚类拼接后依据代表基因的EST及其出现频率的信息进行基因表达分析。
同样原理,也可以利用代表基因3 端表达信息的SA GE标签或近来出现的代表基因5 端信息的CA GE标签来进行。
有学者把这种基于表达标签的基因表达水平定量分析方法称为数字化方法(digital metho d)或者数字化No rthern(digital Northern),而将传统的与cDN A克隆阵列和Oli g o芯片杂交分析称为模拟方法(analo g meth o d)[26]。
Velculescu等[27]1995年提出基因表达系列分析是一种用于定量、高通量基因表达分析的实验方法。
SAGE的原理就是分离每个转录本的特定位置的较短的单一的序列标签(约9~14个碱基对),这些短的序列被连接、克隆和测序,特定的序列标签的出现次数就反映了对应的基因的表达丰度。
技术流程如图1。
3.2.3DNA微阵列或基因芯片的研究 随着EST s数据的扩大,用EST s文库制备的DNA芯片将使测序过程简化并有力促进功能基因组学研究[28]。
高密度寡核苷酸cDNA芯片或cDNA微阵列是一种新的大规模检测基因表达的技术,具有高通量分析的优点。
在许多情况下,cDNA芯片的探针来源于3 EST[29],所以EST序列的分析有助于芯片探针的设计。