EST (Expressed Sequence Tag)表达序列标签
基于est-ssr标记鉴定枇杷品种的方法

一、引言枇杷是一种常见的中药材和水果,其品种繁多,形态特征变异较大。
鉴别枇杷品种对种植、销售及科研具有重要意义。
传统的鉴定方法主要依靠形态学特征和生物学特性,但存在主管专家有限、耗时、费力、受环境和经验等因素影响的缺点。
近年来,基于现代分子生物学方法的est-ssr标记技术已经被广泛应用于枇杷品种的鉴定,具有高效、快速、准确的优势。
二、est-ssr标记技术的原理est-ssr(expressed sequence tag-derived simple sequence repeat)标记是利用EST数据(表达序列标签)进行简单重复序列的分析,并进行标记。
est-ssr标记是一种分子标记技术,利用基因组中的微卫星序列进行检测,并通过PCR扩增技术进行鉴定。
由于EST数据是从不同组织、不同生长阶段的cDNA样本中获得的,因此est-ssr 标记具有较强的系统进化、物种特异性和表达特异性。
三、est-ssr标记技术在枇杷品种鉴定中的应用1. 枇杷品种鉴定样本的准备在进行枇杷品种鉴定之前,首先需要收集不同品种的叶片或幼芽作为样本。
为了保证est-ssr标记技术的高效性和准确性,样本的选择和处理非常重要。
样本应选择来自不同地区或不同时间的品种,避免同一地区、同一种植基地或同一批次的样本。
样本的保存和处理过程中需要注意避免DNA的污染和降解。
2. est-ssr标记技术的实验步骤est-ssr标记技术主要包括DNA提取、PCR扩增、电泳分析等步骤。
首先需要从样本中提取DNA,可以采用CTAB法或商用DNA提取试剂盒进行提取。
提取的DNA需要经过质量检测,确保其完整性和纯度。
接下来是PCR扩增反应,选择合适的est-ssr引物进行扩增,PCR扩增条件需要进行优化,以获得清晰、特异的条带。
最后进行聚丙烯酰胺凝胶电泳分析,根据PCR扩增产物大小和样品条带图谱的差异,进行品种鉴定和分析。
3. est-ssr标记技术的数据分析和结果解读通过PCR扩增和电泳分析得到样品的est-ssr标记图谱,根据条带长度和数量对不同品种进行鉴别。
EST之详细介绍

Expressed Sequence Tags(ESTs)何谓Expressed Sequence Tags(ESTs)?从一特定细胞族群之mRNA转录而成的一群cDNA,经过single-pass的定序过程,而得到的一组序列。
此一细胞族群可以是特定的组织、器官,或是处于某特定发育状态或环境的细胞。
--- A set of single-pass sequenced cDNAs from an mRNA population derived from a specified cell population (e.g. a specific tissue, organ, developmental state or environmental condition).〈2〉ESTs之发展演进快速产生大量低质量的cDNA之概念在1980年代晚期被提出,此方法所能带来的利益,在当时并未能被普遍地认同。
提倡者认为这些cDNA序列能让很多新的protein-coding gene很快地被发现。
批评者则提出反驳,认为这些cDNA 序列将会遗漏掉许多原本能在genimic DNA中被找到的重要调控要素(regulatory elements)。
最后,还是由提倡cDNA定序的人赢得了胜利。
1991年时,有609个Expressed Sequence Tags(ESTs)首度被描述,而公用数据库(public databases)中ESTs的数量,更是呈现戏剧性的成长,到了1995年中,GenBank里ESTs records 的数量已超过非ESTs records的数量;2000年六月,四百六十万的ESTs records 已占了GenBank里所有序列的百分之六十二。
一开始,ESTs的来源只有人类;现在NCBI的EST database(dbEST)已包含了超过250种生物来源的ESTs,包括小鼠(mouse)、大鼠(rat)、Caenorhabditis elegans和黄果蝇(Drosophila melanogaster)等。
EST介绍

表达序列标签(expressed sequence tags,ESTs)是指从不同组织来源的cDNA序列。
这一概念首次由Adams等于1991年提出。
近年来由此形成的技术路线被广泛应用于基因识别、绘制基因表达图谱、寻找新基因等研究领域,并且取得了显著成效。
在通过mRNA差异显示、代表性差异分析等方法获得未知基因的cDNA部分序列后,研究者都迫切希望克隆到其全长cDNA序列,以便对该基因的功能进行研究。
克隆全长cDNA序列的传统途径是采用噬斑原位杂交的方法筛选cDNA文库,或采用PCR的方法,这些方法由于工作量大、耗时、耗材等缺点已满足不了人类基因组时代迅猛发展的要求。
而随着人类基因组计划的开展,在基因结构、定位、表达和功能研究等方面都积累了大量的数据,如何充分利用这些已有的数据资源,加速人类基因克隆研究,同时避免重复工作,节省开支,已成为一个急迫而富有挑战性的课题摆在我们面前,采用生物信息学方法延伸表达序列标签(ESTs)序列,获得基因部分乃至全长cDNAycg,将为基因克隆和表达分析提供空前的动力,并为生物信息学功能的充分发挥提供广阔的空间。
文本将就EST技术的应用并就其在基因全长cDNA克隆上的应用作一较为详细的介绍。
1、ESTs与基因识别EST技术最常见的用途是基因识别,传统的全基因组测序并不是发现基因最有效率的方法,这一方法显得即昂贵又费时。
因为基因组中只有2%的序列编码蛋白质,因此一部分科学家支持首先对基因的转录产物进行大规模测序,即从真正编码蛋白质的mRNA出发,构建各种cDNA文库,并对库中的克隆进行大规模测序。
Adams等提出的表达序列标签的概念标志着大规模cDNA测序时代的到来。
虽然ESTs序列数据对不精确,精确度最高为97%,但实践证明EST技术可大大加速新基因的发现与研究。
Medzhitov等通过果蝇黑胃TOLL蛋白进行dbEST数据库检索,该蛋白已证实在成熟果蝇抗真菌反应中发挥重要作用,通过同源分析的方法,找到相应的人类同源EST(登录号为H48602),这为接下来研究人类TOLL同源蛋白的功能提供了很好的条件。
生物信息分析经常使用名词说明

生物信息分析经常使用名词说明生物信息学(bioinformatics):综合运算机科学、信息技术和数学的理论和方式来研究生物信息的交叉学科。
包括生物学数据的研究、存档、显示、处置和模拟,基因遗传和物理图谱的处置,核苷酸和氨基酸序列分析,新基因的发觉和蛋白质结构的预测等。
基因组(genome):是指一个物种的单倍体的染色体数量,又称染色体组。
它包括了该物种自身的所有基因。
基因(gene):是遗传信息的物理和功能单位,包括产生一条多肽链或功能RNA所必需的全数核苷酸序列。
基因组学:(genomics)是指对所有基因进行基因组作图(包括遗传图谱、物理图谱、转录图谱)、核酸序列测定、基因定位和基因功能分析的科学。
基因组学包括结构基因组学(structural genomics)、功能基因组学(functional genomics)、比较基因组学(Comparative genomics)宏基因组学:宏基因组是基因组学一个新兴的科学研究方向。
宏基因组学(又称元基因组学,环境基因组学,生态基因组学等),是研究直接从环境样本中提取的基因组遗传物质的学科。
传统的微生物研究依托于实验室培育,元基因组的兴起填补了无法在传统实验室中培育的微生物研究的空白。
蛋白质组学(proteomics):说明生物体各类生物基因组在细胞中表达的全数蛋白质的表达模式及功能模式的学科。
包括鉴定蛋白质的表达、存在方式(修饰形式)、结构、功能和彼此作用等。
遗传图谱:指通过遗传重组所取得的基因线性排列图。
物理图谱:是利用限制性内切酶将染色体切成片段,再依照重叠序列把片段连接称染色体,确信遗传标记之间的物理距离的图谱。
转录图谱:是利用EST作为标记所构建的分子遗传图谱。
基因文库:用重组DNA技术将某种生物细胞的总DNA 或染色体DNA的所有片断随机地连接到基因载体上,然后转移到适当的宿主细胞中,通过细胞增殖而组成各个片段的无性繁衍系(克隆),在制备的克隆数量多到能够把某种生物的全数基因都包括在内的情形下,这一组克隆的整体就被称为某种生物的基因文库。
重大生物学考研题库-分子名词解释(3)

1、前导链(leading strand):在DNA复制过程中,与复制叉运动方向相同,以5’→3’方向连续合成的链被称为前导链。
2、基因组(genome):一个细胞或病毒所携带的全部遗传信息或整套基因,包括每一条染色体和所有亚细胞器的DNA序列信息。
3、分子杂交(molecular hybridization):在退火条件下,不同来源的DNA互补区形成双链或DNA单链与RNA单链的互补区形成DNA-RNA杂合双链的过程。
4、锚定PCR(anchored PCR):用于在体外扩增未知序列的DNA片段的方法,一般的PCR 必须预先知道欲扩增DNA片段两侧的序列,但人们经常需要分析一端序列未知的基因片段,此时就可利用锚定PCR。
该法的基本原理是在基因未知序列端添加同聚物尾,人为赋予未知基因末端序列信息,再用人工合成的与多聚尾互补的引物作为锚定引物,在与基因另一侧配对的特异引物参与下,扩增带有同聚物尾的序列。
锚定引物PCR对分析未知序列基因有特殊用途。
5、基因工程(genetic engineering):是对携带遗传信息的分子进行设计和施工的分子工程,包括基因重组、克隆和表达,其核心技术是基因重组。
6、反式作用因子(transacting factor):是指能够结合在顺式作用元件上调控基因表达的蛋白质或者RNAs。
7、核内不均一RNA(hnRNA,heterogeneous nuclear RNA):即mRNA的前体,经过5’加“帽”和3’酶切加多聚腺苷酸,再经过RNA的剪接,编码蛋白质的外显子部分就连接成为一个连续的可译框,作为蛋白质合成的模板。
8、移码突变(frameshift mutation):指一种突变,其结果可导致核苷酸序列与相对应蛋白质的氨基酸序列之间的正常关系发生改变。
移码突变是由删去或插入一个核苷酸的“点突变”构成的,突变位点之前的密码子不发生改变,但突变位点之后的所有密码子都发生变化,编码的氨基酸出现错误。
生物信息学主要英文术语及释义

生物信息学主要英文术语及释义Coding region of DNA. See CDS.Expressed Sequence Tag (EST) (表达序列标签)Randomly selected, partial cDNA sequence; represents it's corresponding mRNA. dbEST is a large database of ESTs at GenBank, NCBI.FASTA (一种主要数据库搜索程序)The first widely used algorithm for database similarity searching. The program looks for optimal local alignments by scanning the sequence for small matches called "words". Initially, the scores of segments in which there are multiple word hits are calculated ("init1"). Later the scores of several segments may be summed to generate an "initn" score. An optimized alignment that includes gaps is shown in the output as "opt". The sensitivity and speed of the search are inversely related and controlled by the "k-tup" variable which specifies the size of a "word". (Pearson and Lipman)Extreme value distribution(极值分布)Some measurements are found to follow a distribution that has a long tail which decays at high value s much more slowly than that found in a normal distribution. This slow-falling type is called the extreme value distribution. The alignment scores between unrelated or random sequences are an example. These scores can reach very high value s, particularly when a large number of comparisons are made, as in a database similarity search. The probability of a particular score may be accurately predicted by the extreme value distribution, which follows a double negative exponential function after Gumbel.False negative(假阴性)A negative data point collected in a data set that was incorrectly reported due to a failure of the test in avoiding negative results.False positive (假阳性)A positive data point collected in a data set that was incorrectly reported due to a failure of the test. If the test had correctly measured the data point, the data would have been recorded as negative.Feed-forward neural network (反向传输神经网络)Organizes nodes into sequence layers in which the nodes in each layer are fully connected with the nodes in the next layer, except for the final output layer. Input is fed from the input layer through the layers in sequence in a “feed-forward” direction, resulting in output at the final layer. See also Neural network.Filtering (window size)During pair-wise sequence alignment using the dot matrix method, random matches can be filtered out by using a sliding window to compare the two sequences. Rather than comparing a single sequence position at a time, a window of adjacent positions in the two sequences is compared and a dot, indicating a match, is generated only if a certain minimal number of matches occur.Filtering (过滤)Also known as Masking. The process of hiding regions of (nucleic acid or amino acid) sequence having characteristics that frequently lead to spurious high scores. See SEG and DUST.Finished sequence(完成序列)Complete sequence of a clone or genome, with an accuracy of at least 99.99% and no gaps.Fourier analysisStudies the approximations and decomposition of functions using trigonometric polynomials.Format (file)(格式)Different programs require that information be specified to them in a formal manner, using particular keywords and ordering. This specification is a file format.Forward-backward algorithmUsed to train a hidden Markov model by aligning the model with training sequences. The algorithm then refines the model to reduce the error when fitted to the given data using a gradient descent approach.FTP (File Transfer Protocol)(文件传输协议)Allows a person to transfer files from one computer to another across a network using an FTP-capable client program. The FTP client program can only communicate with machines that run an FTP server. The server, in turn, will make a specific portion of its tile system available for FTP access, providing that the client is able to supply a recognized user name and password to the server.Full shotgun clone (鸟枪法克隆)A large-insert clone for which full shotgun sequence has been produced.Functional genomics(功能基因组学)Assessment of the function of genes identified by between-genome comparisons. The function of a newly identified gene is tested by introducing mutations into the gene and then examining the resultant mutant organism for an altered phenotype.gap (空位/间隙/缺口)A space introduced into an alignment to compensate for insertions and deletions in one sequence relative to another. To prevent the accumulation of too many gaps in an alignment, introduction of a gap causes the deduction of a fixed amount (the gap score) from the alignment score. Extension of the gap to encompass additional nucleotides or amino acid is also penalized in the scoring of an alignment. Gap penalty(空位罚分)A numeric score used in sequence alignment programs to penalize the presence of gaps within an alignment. The value of a gap penalty affects how often gaps appear in alignments produced by the algorithm. Most alignment programs suggest gap penalties that are appropriate for particular scoring matrices.Genetic algorithm(遗传算法)A kind of search algorithm that was inspired by the principles of evolution. A population of initial solutions is encoded and the algorithm searches through these by applying a pre-defined fitness measurement to each solution, selecting those with the highest fitness for reproduction. New solutions can be generated during this phase by crossover and mutation operations, defined in the encoded solutions.Genetic map (遗传图谱)A genome map in which polymorphic loci are positioned relative to one another on the basis of the frequency with which they recombine during meiosis. The unit of distance is centimorgans (cM), denoting a 1% chance of recombination.Genome(基因组)The genetic material of an organism, contained in one haploid set of chromosomes.Gibbs sampling methodAn algorithm for finding conserved patterns within a set of related sequences. A guessed alignment of all but one sequence is made and used to generate a scoring matrix that represents the alignment. The matrix is then matched to the left-out sequence, and a probable location of the corresponding pattern is found. This prediction is then input into a new alignment and another scoring matrix is produced and tested on a new left-out sequence. The process is repeated until there is no further improvement in the matrix.Global alignment(整体联配)Attempts to match as many characters as possible, from end to end, in a set of twomore sequences. Gopher (一个文档发布系统,允许检索和显示文本文件)Graph theory(图论)A branch of mathematics which deals with problems that involve a graph or network structure. A graph is defined by a set of nodes (or points) and a set of arcs (lines or edges) joining the nodes. In sequence and genome analysis, graph theory is used for sequence alignments and clustering alike genes.GSS(基因综述序列)Genome survey sequence.GUI(图形用户界面)Graphical user interface.H (相对熵值)H is the relative entropy of the target and background residue frequencies. (Karlin and Altschul, 1990).H can be thought of as a measure of the average information (in bits) available per position that distinguishes an alignment from chance. At high value s of H, short alignments can be distinguished by chance, whereas at lower H value s, a longer alignment may be necessary. (Altschul, 1991)Half-bitsSome scoring matrices are in half-bit units. These units are logarithms to the base 2 of odds scores times 2.Heuristic(启发式方法)A procedure that progresses along empirical lines by using rules of thumb to reach a solution. The solution is not guaranteed to be optimal.Hexadecimal system(16制系统)The base 16 counting system that uses the digits O-9 followed by the letters A-F.HGMP (人类基因组图谱计划)Human Genome Mapping Project.Hidden Markov Model (HMM)(隐马尔可夫模型)In sequence analysis, a HMM is usually a probabilistic model of a multiple sequence alignment, but can also be a model of periodic patterns in a single sequence, representing, for example, patterns found in the exons of a gene. In a model of multiple sequence alignments, each column of symbols in the alignment is represented by a frequency distribution of the symbols called a state, and insertions and deletions by other states. One then moves through the model along a particular path from state to state trying to match a given sequence. The next matching symbol is chosen from each state, recording its probability (frequency) and also the probability of going to that particular state from a previous one (the transition probability). State and transition probabilities are then multiplied to obtain a probability of the given sequence. Generally speaking, a HMM is a statistical model for an ordered sequence of symbols, acting as a stochastic state machine that generates a symbol each time a transition is made from one state to the next. Transitions betweenstates are specified by transition probabilities.Hidden layer(隐藏层)An inner layer within a neural network that receives its input and sends its output to other layers within the network. One function of the hidden layer is to detect covariation within the input data, such as patterns of amino acid covariation that are associated with a particular type of secondary structure in proteins.Hierarchical clustering(分级聚类)The clustering or grouping of objects based on some single criterion of similarity or difference.Anexample is the clustering of genes in a microarray experiment based on the correlation between their expression patterns. The distance method used in phylogenetic analysis is another example.Hill climbingA nonoptimal search algorithm that selects the singular best possible solution at a given state or step. The solution may result in a locally best solution that is not a globally best solution.Homology(同源性)A similar component in two organisms (e.g., genes with strongly similar sequences) that can be attributed to a common ancestor of the two organisms during evolution.Horizontal transfer(水平转移)The transfer of genetic material between two distinct species that do not ordinarily exchange genetic material. The transferred DNA becomes established in the recipient genome and can be detected by a novel phylogenetic history and codon content com-pared to the rest of the genome.HSP (高比值片段对)High-scoring segment pair. Local alignments with no gaps that achieve one of the top alignment scores in a given search.HTGS/HGT(高通量基因组序列)High-throughout genome sequencesHTML(超文本标识语言)The Hyper-Text Markup Language (HTML) provides a structural description of a document using a specified tag set. HTML currently serves as the Internet lingua franca for describing hypertext Web page documents.HyperplaneA generalization of the two-dimensional plane to N dimensions.HypercubeA generalization of the three-dimensional cube to N dimensions.Identity (相同性/相同率)The extent to which two (nucleotide or amino acid) sequences are invariant.Indel(插入或删除的缩略语)An insertion or deletion in a sequence alignment.Information content (of a scoring matrix)A representation of the degree of sequence conservation in a column of ascoring matrix representing an alignment of related sequences. It is also the number of questions that must be asked to match the column to a position in a test sequence. For bases, the max-imum possible number is 2, and for proteins, 4.32 (logarithm to the base 2 of the number of possible sequence characters).Information theory(信息理论)A branch of mathematics that measures information in terms of bits, the minimal amount of structural complexity needed to encode a given piece of information.Input layer(输入层)The initial layer in a feed-forward neural net. This layer encodes input information that will be fed through the network model.Interface definition languageUsed to define an interface to an object model in a programming language neutral form, where an interface is an abstraction of a service defined only by the operations that can be performed on it. Internet(因特网)The network infrastructure, consisting of cables interconnected by routers, that pro-vides global connectivity for individual computers and private networks of computers. A second sense of the word internet is the collective computer resources available over this global network.Interpolated Markov modelA type of Markov model of sequences that examines sequences for patterns of variable length in order to discriminate best between genes and non-gene sequences.Intranet(内部网)Intron (内含子)Non-coding region of DNA.Iterative(反复的/迭代的)A sequence of operations in a procedure that is performed repeatedly.Java(一种由SUN Microsystem开发的编程语言)K (BLAST程序的一个统计参数)A statistical parameter used in calculating BLAST scores that can be thought of as a natural scale for search space size. The value K is used in converting a raw score (S) to a bit score (S').K-tuple(字/字长)Identical short stretches of sequences, also called words.lambda (λ,BLAST程序的一个统计参数)A statistical parameter used in calculating BLAST scores that can be thought of as a natural scale for scoring system. The value lambda is used in converting a raw score (S) to a bit score (S').LAN(局域网)Local area network.Likelihood(似然性)The hypothetical probability that an event which has already occurred would yield a specific outcome. Unlike probability, which refers to future events, likelihood refers to past events. Linear discriminant analysisAn analysis in which a straight line is located on a graph between two sets of data pointsin a location that best separates the data points into two groups.Local alignment(局部联配)Attempts to align regions of sequences with the highest density of matches. In doing so, one or more islands of subalignments are created in the aligned sequences.Log odds score(概率对数值)The logarithm of an odds score. See also Odds score.Low Complexity Region (LCR) (低复杂性区段)Regions of biased composition including homopolymeric runs, short-period repeats, and more subtle overrepresentation of one or a few residues. The SEG program is used to mask or filter LCRs in amino acid queries. The DUST program is used to mask or filter LCRs in nucleic acid queries.Machine learning(机器学习)The training of a computational model of a process or classification scheme to distinguish between alternative possibilities.Markov chain(马尔可夫链)Describes a process that can be in one of a number of states at any given time. The Markov chain is defined by probabilities for each transition occurring; that is, probabilities of the occurrence of state sj given that the current state is sp Substitutions in nucleic acid and protein sequences are generally assumed to follow a Markov chain in that each site changes independently of the previous history ofthe site. With this model, the number and types of substitutions observed over a relatively short period of evolutionary time can be extrapolated to longer periods of time. In performing sequence alignments and calculating the statistical significance of alignment scores, sequences are assumed to be Markov chains in which the choice of one sequence position is not influenced by another.Masking (过滤)Also known as Filtering. The removal of repeated or low complexity regions from a sequence in order to improve the sensitivity of sequence similarity searches performed with that sequence.Maximum likelihood (phylogeny, alignment)(最大似然法)The most likely outcome (tree or alignment), given a probabilistic model of evolutionary change in DNA sequences.Maximum parsimony(最大简约法)The minimum number of evolutionary steps required to generate the observed variation in a set of sequences, as found by comparison of the number of steps in all possible phylogenetic trees.Method of momentsThe mean or expected value of a variable is the first moment of the value s of the variable around the mean, defined as that number from which the sum of deviations to all value s is zero. The standard deviation is the second moment of the value s about the mean, and so on.Minimum spanning treeGiven a set of related objects classified by some similarity or difference score, the mini-mum spanning tree joins the most-alike objects on adjacent outer branches of a tree and then sequentially joins less-alike objects by more inward branches. The tree branch lengths are calculated by the same neighbor-joining algorithm that is used to build phylogenetic trees of sequences from a distance matrix. The sum of the resulting branch lengths between each pair of objects will be approximately that found by the classification scheme.MMDB (分子建模数据库)Molecular Modelling Database. A taxonomy assigned database of PDB (see PDB) files, and related information.Molecular clock hypothesis(分子钟假设)The hypothesis that sequences change at the same rate in the branches of an evolutionarytree.Monte Carlo(蒙特卡罗法)A method that samples possible solutions to a complex problem as a way to estimate a more general solution.Motif (模序)A short conserved region in a protein sequence. Motifs are frequently highly conserved parts of domains.Multiple Sequence Alignment (多序列联配)An alignment of three or more sequences with gaps inserted in the sequences such that residues with common structural positions and/or ancestral residues are aligned in the same column. Clustal W is one of the most widely used multiple sequence alignment programsMutation data matrix(突变数据矩阵,即PAM矩阵)A scoring matrix compiled from the observation of point mutations between aligned sequences. Also refers to a Dayhoff PAM matrix in which the scores are given as log odds scores.N50 length (N50长度,即覆盖50%所有核苷酸的最大序列重叠群长度)A measure of the contig length (or scaffold length) containing a 'typical' nucleotide. Specifically, it isthe maximum length L such that 50% of all nucleotides lie in contigs (or scaffolds) of size at least L. Nats (natural logarithm)A number expressed in units of the natural logarithm.NCBI (美国国家生物技术信息中心)National Center for Biotechnology Information (USA). Created by the United States Congress in 1988, to develop information systems to support thebiological research community.Needleman-Wunsch algorithm(Needleman-Wunsch算法)Uses dynamic programming to find global alignments between sequences.Neighbor-joining method(邻接法)Clusters together alike pairs within a group of related objects (e.g., genes with similar sequences) to create a tree whose branches reflect the degrees of difference among the objects.Neural network(神经网络)From artificial intelligence algorithms, techniques that involve a set of many simple units that hold symbolic data, which are interconnected by a network of links associated with numeric weights. Units operate only on their symbolic data and on the inputs that they receive through their connections. Most neural networks use a training algorithm (see Back-propagation) to adjust connection weights, allowing the network to learn associations between various input and output patterns. See also Feed-forward neural network.NIH (美国国家卫生研究院)National Institutes of Health (USA).Noise(噪音)In sequence analysis, a small amount of randomly generated variation in sequences that is added to a model of the sequences; e.g., a hidden Markov model or scoring matrix, in order to avoid the model overfitting the sequences. See also Overfitting.Normal distribution(正态分布)The distribution found for many types of data such as body weight, size, and exam scores. The distribution is a bell-shaped curve that is described by a mean and standard deviation of the mean. Local sequence alignment scores between unrelated or random sequences do not follow this distribution but instead the extreme value distribution which has a much extended tail for higher scores. See also Extreme value distribution.Object Management Group (OMG)(国际对象管理协作组)A not-for-profit corporation that was formed to promote component-based software by introducing standardized object software. The OMG establishes industry guidelines and detailed object management specifications in order to provide a common framework for application development. Within OMG is a Life Sciences Research group, a consortium representing pharmaceutical companies, academic institutions, software vendors, and hardware vendors who are working together to improve communication and inter-operability among computational resources in life sciences research. See CORBA.Object-oriented database(面向对象数据库)Unlike relational databases (see entry), which use a tabular structure, object-oriented databases attempt to model the structure of a given data set as closely as possible. In doing so, object-oriented databases tend to reduce the appearance of duplicated data and the complexity of query structure often found in relational databases.Odds score(概率/几率值)The ratio of the likelihoods of two events or outcomes. In sequence alignments and scoring matrices,the odds score for matching two sequence characters is the ratio of the frequency with which the characters are aligned in related sequences divided by the frequency with which those same two characters align by chance alone, given the frequency of occurrence of each in the sequences. Odds scores for a set of individually aligned positions are obtained by multiplying the odds scores for each position. Odds scores are often converted to logarithms to create log odds scores that can be added to obtain the log odds score of a sequence alignment.OMIM (一种人类遗传疾病数据库)Online Mendelian Inheritance in Man. Database of genetic diseases with references to molecular medicine, cell biology, biochemistry and clinical details of the diseases.Optimal alignment(最佳联配)The highest-scoring alignment found by an algorithm capable of producing multiple solutions. This is the best possible alignment that can be found, given any parameters supplied by the user to the sequence alignment program.ORF (开放阅读框)Open Reading Frame. A series of codons (base triplets) which can be translated into a protein. There are six potential reading frames of an unidentifed sequence; TBLASTN (see BLAST) transalates a nucleotide sequence in all six reading frames, into a protein, then attempts to align the results to sequeneces in a protein database, returning the results as a nucleotide sequence. The most likely reading frame can be identified using on-line software (e.g. ORF Finder).Orthologous(直系同源)Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function. A pair of genes found in two species are orthologous when the encoded proteins are 60-80% identical in an alignment. The proteins almost certainly have the same three-dimensional structure, domain structure, and biological function, and the encoding genes have originated from a common ancestor gene at an earlier evolutionary time. Two orthologs 1 and II in genomes A and B, respectively, may be identified when the complete genomes of two species are available: (1) in a database similarity search of all of the proteome of B using I as a query, II is the best hit found, and (2) I is the best hit when 11 is used as a query of the proteome of B. The best hit is the database sequence with the highest expect value (E). Orthology is also predicted by a very close phylogenetic relationship between sequences or by a cluster analysis. Compare to Paralogs. See also Cluster analysis.Output layer(输出层)The final layer of a neural network in which signals from lower levels in the network are input into output states where they are weighted and summed togive an outpu t signal. For example, the output signal might be the prediction of one type of protein secondary structure for the central amino acid in a sequence window.OverfittingCan occur when using a learning algorithm to train a model such as a neural net or hid-den Markov model. Overfitting refers to the model becoming too highly representative of the training data and thus no longer representative of the overall range of data that is supposed to be modeled.。
表达序列标签EST概要

表达序列标签EST概要摘要:随着EST研究的开展、深入,以及相关研究技术和分析手段的不断改进并走向成熟,EST 数据资源不断丰富,而其本身又具备独特的优势和多方面的利用价值。
本文介绍了EST序列的获取、加工、储存、分配、分析和释读的相关研究。
关键词:EST 表达序列标签聚类cDNA文库生物信息学从事对生物信息的获取、加工、储存、分配、分析和释读,并综合运用数学、计算机科学和生物学工具,以达到理解数据中的生物学含义的目的。
随着人类基因组计划在世界范围内的开展,生物信息学作为一门热门交叉学科,不断地完善和发展起来作为一种强有力的工具,它在帮助我们对巨量的生物信息进行归纳和理解,从而揭示生命的奥妙的过程中发挥了重要的作用。
然而信息的爆炸增长,面对复杂和庞大的数据库,如何有效地地获取我们所需要的信息,充分利用这些已有的数据资源,加速基因克隆研究已成为一个富有挑战性的课题。
表达序列标签的广泛应用,为大规模进行基因克隆和表达分析提供了强大的动力,也为生物信息学功能的充分发挥提供了广阔的空问表达序列标签(EST,Expressed Sequence Tag)是指从一个随机选择的cDNA 克隆进行5’端和3’端单一次测序获得的短的cDNA 部分序列,代表了一个完整基因的一小部分。
Adams等人在1991年提出了EST技术,宣布了cDNA大规模测序时代的开始。
随着大规模的测序,EST数据呈指数级增长。
到了1995年中,GenBank里ESTs的数量已超过非ESTs的数量;2000年6月,将近460万的ESTs 已占了GenBank里所有序列的62%。
ESTs序列不止来源于人类,NCBI的dbEST (EST database)中已包含了超过250种生物来源的ESTs,包括小鼠、大鼠、秀丽线虫和黄果蝇等。
除此之外,也有许多商业性的机构保存了一些属于机构内部不公开的ESTs 序列。
EST序列的制备EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此EST也能说明该组织中各基因的表达水平。
基因表达分析

基因表达分析1、EST(Expressed Sequence Tag)表达序列标签(EST)分析1、EST基本介绍1、定义:EST是从已建好的cDNA库中随机取出一个克隆,进行5’端或3’端进行一轮单向自动测序,获得短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20到7000bp不等,平均长度为400bp。
EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此,EST也能说明该组织中各基因的表达水平。
2、技术路线:首先从样品组织中提取mRNA,在逆转录酶的作用下用oligo(dT)作为引物进行RT-PCR 合成cDNA,再选择合适的载体构建cDNA文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST序列的产生过程。
3、EST数据的优点和缺点:(1)相对于大规模基因组测序而言,EST测序更加快速和廉价。
(2)EST数据单向测序,质量比较低,经常出现相位的偏差。
(3)EST只是基因的一部分,而且序列里有载体序列。
(4)EST数据具有冗余性。
(5)EST数据具有组织和不同时期特异性。
4、EST数据的应用EST作为表达基因所在区域的分子标签因编码DNA序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比,更可能穿越家系与种的限制。
因此,EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有用的。
同样,对于一个DNA序列缺乏的目标物种,来源于其他物种的EST也能用于该物种有益基因的遗传作图,加速物种间相关信息的迅速转化。
具体说,EST的作用表现在:(1)用于构建基因组的遗传图谱与物理图谱;(2)作为探针用于放射性杂交;(3)用于定位克隆;(4)借以寻找新的基因;(5)作为分子标记;(6)用于研究生物群体多态性;(7)用于研究基因的功能;(8)有助于药物的开发、品种的改良;(9)促进基因芯片的发展等方面。
分子生物学简答题

试述乳糖操纵子的阻遏作用、诱导作用及正调控。
阻遏作用:阻遏基因lacl转录产生阻遏物单体,结合形成同源四体,即阻遏物。
它是一个抗解链蛋白,当阻遏物与操纵基因O结合时,阻止DNA形成开放结构,从而抑制RNA聚合酶的功能。
lacmRNA的转录起始受到抑制。
诱导作用:按照lac操纵子本底水平的表达,每个细胞内有几个分子的β-半乳糖苷酶和β-半乳糖苷透过酶。
当加入乳糖,在单个透过酶分子的作用下,少量乳糖分子进入细胞,又在单个β-半乳糖苷酶的作用下转变为诱导物异构乳糖,诱导物通过与阻遏物结合,改变它的三维构象,使之因不能与操纵基因结合而失活,O区没有被阻遏物占据从而激发lacmRNA的合成。
调控作用:葡糖糖对lac操纵子的表达的抑制是间接的,不是葡萄糖本身而是其降解产物抑制cAMP的合成。
cAMP-CAP复合物与启动子区的结合是lacmRNA转录起始所必须的,因为该复合物结合于启动子上游,能使DNA双螺旋发生弯曲。
有利于形成稳定开放型启动子-RNA 聚合酶结构。
如果将葡萄糖和乳糖同时加入培养基中,lac操纵子处于阻遏状态,不能被诱导试述 E.coli的RNA聚合酶的结构和功能。
2个α亚基、一个β亚基、一个β’亚基和一个亚基组成的核心酶,加上一个亚基后则成为聚合酶全酶α亚基:核心酶组装、启动子识别β和β’亚基:β和β’共同形成RNA合成的催化中心因子:存在多种因子,用于识别不同的启动子试述原核生物DNA复制的特点。
1.原核只有一个起始位点。
2.原核复制起始位点可以连续开始新的复制,特别是快速繁殖的细胞。
3.原核的DNA聚合酶III复制时形成二聚体复合物。
4.原核的DNA聚合酶I具有5'-3'外切酶活性DNA解旋酶通过水解ATP 产生能量来解开双链DNA单链结合蛋白保证被解链酶解开的单链在复制完成前保持单链结构DNA拓扑异构酶消除解链造成的正超螺旋的堆积,消除阻碍解链继续进行的这种压力,使复制得以延伸真核生物hnRNA必须经过哪些加工才能成为成熟的mRNA,以用作蛋白质合成的模板?(1)、在5’端加帽,5’端的一个核苷酸总是7-甲基鸟核苷三磷酸(m7Gppp)。
第6章 分子生物学研究方法(下)

④具有良好的机械性能
非特异吸附少
4.2 常用的固相支持物 ①硝酸纤维素膜:优点是吸附能力强,杂交信号本 底低。缺点是DNA分子结合不牢固 ②尼龙膜:优点是结合单链,双链DNA的能力比硝酸 纤维素膜强;缺点:杂交信号本底高 ③化学活化膜:优点:DNA与膜共价结合;对不同 大小的DNA片段有同等结合能力;缺点:结合能 力较上述两种膜低
(2)电转法 利用电场的电泳作用将凝胶中的DNA转移到 固相支持物上。
(3)真空转移法
此法原理与毛细管虹吸法相同,只是以滤膜在下,凝
胶在上的方式将其放臵在一个真空室上,利用真空作用
将转膜缓冲液从上层容器中通过凝胶和滤膜抽到下层真 空室中,同时带动核酸片段转移到凝胶下面的尼龙膜或
硝酸纤维素膜上。
各种转移方法的比较:
6.1.2 RNA选择性剪接技术
RNA的选择性剪接是指用不同的剪接方式从 一个mRNA前 体产生不同的mRNA剪接异构 体的过程。可分为:平衡剪 切、 5’ 选择性剪切、 3’ 选择性剪切、外显子遗漏型剪切及 相互排斥性剪切。常用RT-PCR法研究某个基 因是否存在 选择性剪切。 果蝇 Dscam 基 因可 以通过可变 剪接产生38000多 种 可 能 的 mRNA 异构体
Illumina Solexa 测序 Workflow
Illumina Sequencing Technology
有参考基因组序列生物信息分析
• 基因结构优化
• 鉴定基因可变剪接 • 预测新转录本 • SNP 分析 • 基因融合鉴定
5.4.1 RACE 法克隆基因全长 (Rapid Amplification of cDNA Ends)
4.3 Southern印迹的常用方法 (1)毛细管虹吸印迹法
EST大规模表达序列标签
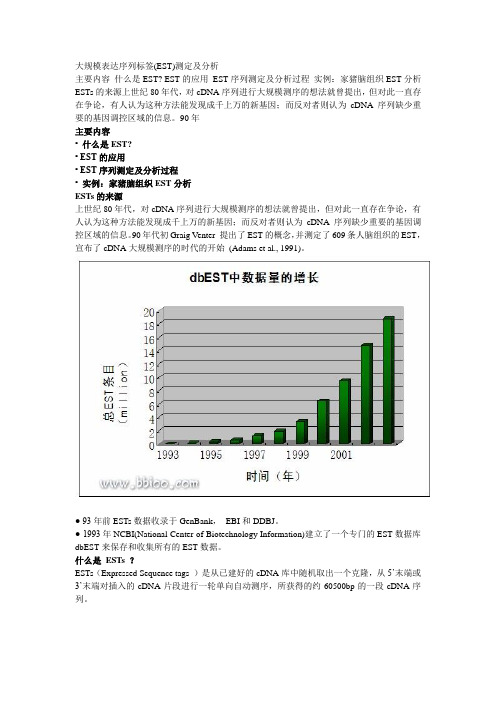
大规模表达序列标签(EST)测定及分析主要内容什么是EST? EST的应用EST序列测定及分析过程实例:家猪脑组织EST分析ESTs的来源上世纪80年代,对cDNA序列进行大规模测序的想法就曾提出,但对此一直存在争论,有人认为这种方法能发现成千上万的新基因;而反对者则认为cDNA序列缺少重要的基因调控区域的信息。
90年主要内容• 什么是EST?• EST的应用• EST序列测定及分析过程• 实例:家猪脑组织EST分析ESTs的来源上世纪80年代,对cDNA序列进行大规模测序的想法就曾提出,但对此一直存在争论,有人认为这种方法能发现成千上万的新基因;而反对者则认为cDNA序列缺少重要的基因调控区域的信息。
90年代初Graig Venter 提出了EST的概念,并测定了609条人脑组织的EST,宣布了cDNA大规模测序的时代的开始(Adams et al., 1991)。
● 93年前ESTs数据收录于GenBank,EBI和DDBJ。
● 1993年NCBI(National Center of Biotechnology Information)建立了一个专门的EST数据库dbEST来保存和收集所有的EST数据。
什么是ESTs ?ESTs(Expressed Sequence tags )是从已建好的cDNA库中随机取出一个克隆,从5‘末端或3‘末端对插入的cDNA片段进行一轮单向自动测序,所获得的约60500bp的一段cDNA序列。
ESTs与基因识别ESTs已经被广泛的应用于基因识别,因为ESTs的数目比GenBank中其它的核苷酸序列多,研究人员更容易在EST库中搜寻到新的基因(Boguski et al., 1994).● 在同一物种中搜寻基因家族的新成员(paralogs)。
● 在不同物种间搜寻功能相同的基因(orthologs)。
● 已知基因的不同剪切模式的搜寻。
【注:不过很难确定一个新的序列是由于交替剪切产生的或是由于cDNA文库中污染了基因组DNA序列(Wolfsberg et al., 1997)】ESTs与基因图谱的绘制EST可以借助于序列标签位点(sequence-tagged sites)用于基因图谱的构建. STS本身是从人类基因组中随机选择出来的长度在200-300bp左右的经PCR检测的基因组中唯一的一段序列。
EST数据分析

http://pbil.univ-lyon1.fr/cap3.php p p y p p p
电子克隆
对contig再次比对后未发 现新的高相似度EST序列
电子克隆
为获取较多的EST序列,除blastn外,还 可以选择tblastx及tblastn程序。 EST序列质量对拼接结果有影响。EST序 序列质量对拼接结果有影响。 序 列的E值要尽量小,并且数量要适度。 利用 Univec数据库进行 U i 数据库进行 blast比对来发现 bl t比对来发现 并去除载体序列。 电子克隆结果尚需要采用RT-PCR并配合 Northern Blot或RACE等方法加以验证。 Blot或RACE等方法加以验证
基因序列分析技术
伦永志
大连大学医学院
目录
第一讲 生物信息学导论 第二讲 序列的获取 第三讲 序列的相似性查询 第四讲 多序列比对 第五讲 分子进化分析 第六讲 EST数据分析 第七讲 基因结构分析 第八讲 基因功能注释 第九讲 蛋白质结构分析
电子克隆 基因定位与表达谱分析
电子克隆
电子克隆
表达序列标签 (Expressed Sequen段,长 度一般约为60-500bp。 EST数据具有简便易得的优势,通常作为 EST数据具有简便易得的优势 通常作为 基因标签应用于绘制物理图谱和转录图 谱、识别基因、电子克隆、预测基因和分 析基因表达水平等。 析基因表达水平等
电子克隆
电子克隆是利用生物信息学技术组装延伸 EST序列,获得目的基因的部分乃至全长 cDNA序列。 序列。 首先以一条EST数据作为查询探针,在数 据库中进行同源性查询 获取高度同源的 据库中进行同源性查询,获取高度同源的 EST数据进行序列拼装以构建重叠群,然 后以此重叠群(contig)为种子序列重复 搜索 拼接使序列获得延伸 搜索、拼接使序列获得延伸。
表达序列标签研究进展及其在甲壳动物中的应用概况

表达序列标签研究进展及其在甲壳动物中的应用概况摘要:随着生物信息学的发展,表达序列标签(EST)在分子标记开发、新基因分离鉴定、基因表达谱分析、基因组功能注释、基因电子克隆等方面具有重要作用。
简要介绍了EST分析的原理及其在基因识别、基因预测、物理图谱的构建、DNA芯片制备等方面的应用概况。
综述了甲壳动物EST的研究现状,并对EST的应用前景进行了展望。
关键词:表达序列标签(EST);甲壳动物;生物信息学Abstract:Withthedevelopmentofbioinformatics,expressedsequencetag(EST)played animportantroleinmolecularmarkersdevelopment,newgenesisolationandidentification,geneexpressionprofileanalysis,genomefunctionalannotationandsilicogenecloning.TheprincipleofESTanalysisanditsapplicationsingeneidentification,geneprediction,physicalmapconstructionandDNAchippreparationwas briefly introduced.Inaddition,theresearchstatusofcrustaceanESTandthe prospectofESTapplicationwerealsosummarized.Keywords:expressedsequencetag(EST);crustacean;bioinformatics表达序列标签(Expressedsequencetag,EST)是从一个随机选择的cDNA克隆进行5’端和3’端单一次测序获得的短的cDNA部分序列。
EST—SSR分子标记在茶树品种鉴别中的应用

EST—SSR分子标记在茶树品种鉴别中的应用摘要:从GenBank数据库中龙井43的EST序列中筛查EST-SSR位点,设计并获得了65对茶树(Camellia sinensis)EST-SSR引物。
以鄂茶1号、龙井43、龙井群体、云抗10号、雪芽100、矮丰的基因组DNA为模板,对所设计的65对EST-SSR引物进行筛选。
结果表明,65对引物均能扩增出清晰条带,有效扩增率为100%;其中有4对引物的扩增产物在6个样本间具有多态性,能用于6个茶树品种的品种鉴别。
关键词:茶树(Camellia sinensis);表达序列标签(EST);简单序列重复(SSR);品种鉴别Abstract:EST-SSR loci were screened from EST sequences of Longjing 43 in the NCBI public database,65 pairs of EST-SSR primers were designed using Primer 5.0 software. The genomic DNA of tea varieties Hubei No.1 tea,Longjing 43,Longjing groups,Yunkang No.10,Xueya 100 and Aifeng were extracted and used as a template to select EST-SSR primers. The results showed that targeted bands of all the 65 pairs of primers were amplified,the amplification rate was 100%. 4 pairs of EST-SSR primers showed polymorphism in the 6 samples,could be used for identification of the 6 tea varieties.Key words:tea(Camellia sinensis);expressed sequence tags(EST);simple sequence repeats(SSR);variety identification茶(Camellia sinensis)是深受广大消费者喜爱的传统农产品,是中国重要的经济作物之一,在国际贸易中也占有较大份额。
18-硕士研究课-现代分子生物学-电子克隆-胡忠

基于染色体DNA的电子克隆优缺点
缺点和局限:
1. 必须经实验验证 2. 不适用以下种类的基因预测 A. 种间保守性差的基因 B. 外显子数目多而且每个外显子短的基因
3)电子克隆获得的基的完整cDNA序列和该序列所编码的氨基酸顺序。根 据待测生物的偏爱密码子反推合适的碱基顺序,设计引物
Rice root Oryza sativa cDNA clone R2345, mRNA sequence
AU184451
RICR2584A
99AS825
C25822
D004D07
AAAAAA
RICS1291A
ATG
C25822
AU184451
D004D07 99AS825
TAA AAAAAA
RICR2584A RICS1291A
est walking 的优点和局限
优 点:
快速,无须实验操作
局 限:
1. est库不均一;
2. est库测序精度不高(最高为97% ) 3. est库中有不完全剪切产物
CLONE INFO Clone Id: S20127_2Z DNA type: cDNA RIMERS PolyA Tail: Unknown SEQUENCE GATATGCGGCTANTATAGCCAGCATGCCATATGAGGGGCTTTTAGCATTAGAAGAGCAGA TTGGNGATGTAAATACTGGTCTGGCAAAAAGCTACATTGTAGAGAAATTGAAGACTAGCT TATTTGTNCCAGGATCATCCTGCATGTCTAATAAGTCTTCTGAATCTTCCATGGAGAATG ATGCTTGCATAATATGCCAGGAAGAGTATCAGGTTAAAGAATGCATTGGAACCCTTGACT GTGGCCACAGGTACCACGAAGATTGCATAAAACAATGGTTGATGGTAAAGAATTTATGCC CCATCTGCAAGACGACAGCTTTGTCAACCGGAAGAAGAAGTGGATAACGAACAGGAATAA TCTTATTAGTTATTTACTTCCGACAAATATTCAGCTCAATTTTGTATATAAGAAACGGTA GACCATTCTGCTACCTGTATTTGTTGCTCACTTTGTTGTGATCCGGGAGTAACTCAGCTT CCTAAACTGTACAGCCATAACATTGATCATTTTCTTCGGTGTAGAATATTTTAAATTACT CAGTTCGCCCCCATCTGTATCATAAGGCGGACCGACAAAAAAACTCACAATGTCATTTCT AGGCAAACATTGTATCTACCATCAGATTAAAAATCAGAACAGAACATGTGCTCTTCTGTN CAAAAAAAAAAAAAAAAAAAAAAAAAAAA
基因组学名解问答题重点

基因组作图 genomicmapping:确定界标或基因在构成基因组的各条染色体上的位置,以及染色体上各个界标或基因之间的相对距离,绘制遗传连锁图或物理图。
1)同源基因(homologous gene) 系指起源于同一祖先但顺序已经发生变异的基因成员, 分布在不同物种间的同源基因又称直系(orthologous)基因. 同一物种的同源基因则称水平(paralogous)基因, 水平基因由重复后趋异产生.基因同源性(homology)只有“是”和“非”的区别, 无所谓百分比.
管家基因 house-keepinggene:所有细胞中均要稳定表达的一类基因,其产物是对维持细胞基本生命活动所必需的,始终保持着低水平的甲基化并且一直处于活性转录状态的基因。
宏基因组/宏基因组学metagenome/metagenomics:研究一类在特殊或极端的环境下共栖生长微生物的混合基因组。
15.基因的元件、结构、特征以及功能
①在5′端转录起始点上游约20~30个核苷酸的地方,有TATA框(TATA box)。 TATA框是一个短的核苷酸序列,其碱基顺序为TATAATAAT。TATA框是启动子中的一个顺序,它是RNA聚合酶的重要的接触点,它能够使酶准确地识别转录的起始点并开始转录。当TATA框中的碱基顺序有所改变时,mRNA的转录就会从不正常的位置开始。
2) 因克隆载体自身的限制或DNA序列特殊的组成等原因造成某些序列丢失或未能克隆, 这类间隙称为物理间隙.
引物两两配组扩增基因的DNA.
11.为何着丝粒区和近端粒区DNA测序非常困难?
est序列名词解释

est序列名词解释
EST(Expressed Sequence Tag)是指表达序列标签,是一段短的DNA 序列,通常是从 cDNA 文库中通过随机测序得到的。
EST 序列通常包含一个基因的部分编码区域,可以用来鉴定和筛选新的基因。
EST 序列的获得通常是通过对 cDNA 文库进行随机测序得到的。
在测序过程中,会得到大量的短序列,这些序列可能来自于不同的基因。
通过对这些序列进行分析和比较,可以鉴定出一些新的基因,并了解它们的表达模式和功能。
EST 序列的应用非常广泛,包括基因鉴定、基因表达谱分析、基因组注释、药物研发等。
EST 序列也可以用于构建基因芯片,用于高通量的基因表达分析。
生物信息学 第6章 表达序列标签

➢使用合适的比对参数,大于90%的已经注释的基因都能
在EST库中检测到。
精品课件
(二) ESTs与基因表达谱的构建
➢表达量比较分析:不同组织或发育阶段基因表达量比较 ➢EST来源于不同的组织,那么就可以对不同来源的基因 表达进行比较
精品课件
(三) ESTs与新基因预测
➢由于EST的一个基因的 部分序列。
从已建好的cDNA库中随机取出一个克隆,从 5′末端或3′末端对插入的cDNA片段进行一轮单 向自动测序,所获得的约60-500bp的一段cDNA序 列。
精品课件
二、EST数据分析方法
随机挑取克隆进行5′或3′端测序 序列前处理 聚类和拼接
基因注释及功能分类 后续分析
精品课件
(一)序列前处理
➢ 去除低质量的序列(如使用Phred)
精品课件
五、常用的EST数据库
数据库名称
网址
说明
dbEST
/dbEST/ 综合
UniGene /unigene 综合
Gene Indices /tgi/ 综合
电子PCR克隆,指利用已经有的片段进行 全长基因序列的分析。
5
3
5
3
精品课件
四、EST数据的不足
➢ESTs很短,没有给出完整的表达序列; ➢低丰度表达基因不易获得; ➢由于只是一轮测序结果,出错率达2%~5%; ➢有时有载体序列和核外mRNA来源的cDNA污染或是基 因组DNA的污染; ➢有时出现镶嵌克隆; ➢序列的冗余,导致所需要处理的数据量很大。
精品课件
(三)序列注释和分析
➢ 序列注释 ➢ 后续分析
精品课件
三、EST的用途
基因识别 基因表达谱的构建 发现新基因 SNP(single nucleotide polymorphism)发 现
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
EST (Expressed Sequence Tag)表达序列标签
EST (Expressed Sequence Tag)表达序列标签—是从一个随机选择的cDNA 克隆,进行5’端和3’端单一次测序挑选出来获得的短的cDNA 部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20 到7000bp 不等,平均长度为360 ±120bp。
由于cDNA
文库的复杂性和测序的随机性,有时多个EST代表同一基因或基因组,将其归类形成EST 簇(EST cluster)
原理:
EST是从一个随机选择的cDNA 克隆进行5’端和3’端单一次测序获得的短的cDNA
部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20 到7000bp 不等,平均长度为360 ±120bp。
EST 来源于一定环境下一个组织总mRNA 所构建的cDNA 文库,因此EST也能说明该组织中各基因的表达水平。
技术路线:
首先从样品组织中提取mRNA,在逆转录酶的作用下用oligo (dT) 作为引物进行RT
-PCR合成cDNA,再选择合适的载体构建cDNA 文库,对各菌株加以整理,将每一个菌株的插入片段根据载体多克隆位点设计引物进行两端一次性自动化测序,这就是EST 序列的产生过程。
应用:
EST作为表达基因所在区域的分子标签因编码DNA 序列高度保守而具有自身的特殊
性质,与来自非表达序列的标记(如AFLP、RAPD、SSR等)相比更可能穿越家系与种
的限制,因此EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息是特别有用。
同样,对于一个DNA 序列缺乏的目标物种,来源于其他物种的EST也能用于
该物种有益基因的遗传作图,加速物种间相关信息的迅速转化。
具体说,EST的作用表现在:⑴用于构建基因组的遗传图谱与物理图谱;⑵作为探针用于放射性杂交; ⑶用于
定位克隆;⑷借以寻找新的基因; ⑸作为分子标记;⑹用于研究生物群体多态性;⑺用于研究基因的功能;⑻有助于药物的开发、品种的改良;⑼促进基因芯片的发展等方面。
正是因为EST 表现出了这些巨大潜能,使其得到了充分的利用与发展。
在人类基因组研究中,有一个区别于“全基因组战略”的“cDNA战略”,既只测定转录的DNA序列,也就是测定基因转录产物mRNA反转录产生的互补DNA---cDNA.cDNA代表了基因中编码蛋白质的序列。
EST则是cDNA的一个片段,一般长200-400个核苷酸对。
一个全长的cDNA分子可以有许多个EST,但特定的EST有时可以代表某个特定的cDNA分子。
两端有重叠的共有序列的EST可以组装成一个叠连群(contig),直到装配成全长的cDNA 序列,这样就等于是克隆了一个基因的编码序列。
将EST定位在基因组,也可作为基因组作图时的一种标记序列。